

Abstract
Human GLTP on chromosome 12 (locus 12q24.11) encodes a 24 kD amphitropic lipid transfer protein (GLTP) that mediates glycosphingolipid (GSL) intermembrane trafficking and regulates GSL homeostatic levels within cells. Herein, we provide evidence that GLTP overexpression inhibits the growth of human colon carcinoma cells (HT-29; HCT-116), but spares normal colonic cells (CCD-18Co). Mechanistic studies reveal that GLTP overexpression arrested the cell cycle at the G1/S checkpoint via upregulation of cyclin-dependent kinase inhibitor-1B (Kip1/p27) and cyclin-dependent kinase inhibitor 1A (Cip1/p21) at the protein and mRNA levels, and downregulation of cyclin-dependent kinase-2 (CDK2), cyclin-dependent kinase-4 (CDK4), cyclin E and cyclin D1 protein levels. Assessment of the biological fate of HCT-116 cells overexpressing GLTP indicated no increase in cell death suggesting induction of quiescence. However, HT-29 cells overexpressing GLTP underwent cell death by necroptosis as revealed by phosphorylation of human mixed lineage kinase domain-like protein (pMLKL) via receptor-interacting protein kinase-3 (RIPK-3), elevated cytosolic calcium, and plasma membrane permeabilization by pMLKL oligomerization. Overexpression of W96A-GLTP, an ablated GSL binding site mutant, failed to arrest the cell cycle or induce necroptosis. Sphingolipid assessment (ceramide, monohexosylceramide, sphingomyelin, ceramide-1-phosphate, sphingosine, and sphingosine-1-phosphate) of HT-29 cells overexpressing GLTP revealed large decreases (>5-fold) in sphingosine-1 -phosphate with minimal change in 16:0-ceramide, tipping the ‘sphingolipid rheostat’ (S1P/16:0-Cer ratio) towards cell death. Depletion of RIPK-3 or MLKL abrogated necroptosis induced by GLTP overexpression. Our findings establish GLTP upregulation as a previously unknown suppressor of human colon carcinoma HT-29 cells via interference with cell cycle progression and induction of necroptosis.
Keywords: programmed cell death, necroptosis, sphingolipidomics, sphingolipid rheostat, glycolipid transfer protein, colon cancer
1. Introduction
Colorectal cancer (CRC) is the third most common cancer in United States. In spite of substantial improvement in early diagnosis and relatively successful prevention strategies, annual deaths from CRC continue to exceed 50,000 people [1,2]. Sphingolipids (SLs) play pivotal roles in cell growth and metabolism and their emerging importance in colon cancer is becoming more and more evident [3–7]. SL metabolite upregulation that stimulate apoptosis (e.g. ceramide) has been investigated as an avenue to supplement and enhance chemotherapeutic-induced apoptosis. Yet, it has become clear that CRC along with many cancers develop resistance to apoptosis [8]. An emerging strategy for circumventing apoptosis resistance is to identify and explore ways to induce alternate pathways of programmed cell death to combat cancer progression [9–12].
Human glycolipid transfer protein (GLTP) is a small (24kD) amphitropic protein encoded at chromosome 12 (locus 12q24.11) that mediates the non-vesicular trafficking of various glycosphingolipids (GSLs) [13–15], some which have established roles in surface adhesion, neurodegeneration, differentiation, drug resistance, and apoptosis [16]. GLTP possesses a unique all α-helical conformation, arranged in a two layer ‘sandwich motif’ that forms a single ‘pocket-like’ glycolipid binding site, i.e. GLTP-fold, and defines the GLTP superfamily [15,17,18]. In vivo changes in GLTP expression alter not only the homeostatic levels of certain GSLs involved in cell-cell contact and surface adhesion, but also modulate cell shape changes in certain human cancer cell lines [19]. Cell shape changes often serve as phenotypic indicators of programmed cell death processes (e.g. apoptosis, autophagy, pyroptosis, necroptosis).
Necroptosis, a regulated form of necrosis, has generated much recent interest in cancer therapeutics primarily as a potential means for induction of the caspase-independent cell death pathway when apoptosis induction is compromised [20,21]. Necroptosis triggers tumor necrosis factor-alpha (TNFα), a multifunctional cytokine with a long history of involvement in cell death and survival, immunity, inflammation, and cancer treatment [22]. Necroptosis is regulated by the kinase activities of receptor-interacting proteins 1 (RIPK-1) and 3 (RIPK-3), the latter being a major component of the necrosome complex [23]. Necrosome assembly results in RIPK-3 activation via phosphorylation at Ser227. Activated RIPK-3 phosphorylates Thr357 and Ser358 of human mixed lineage kinase domain-like (MLKL) protein, stimulating MLKL oligomerization and translocation to the plasma membrane, resulting in pore formation and vesicle ‘bubbling’ that compromises membrane barrier integrity to enable osmotically-driven cell death, i.e. necroptosis [24–27]. MLKL phosphorylation and translocation to the plasma membrane appear to depend upon an early rise in cytosolic Ca2+ via activated ion channels that may include Transient Receptor Potential Melastatin 7 (TRPM7) [26]. The early uptick in Ca2+ levels also triggers Ca2+-calmodulin kinase (CaMK) II-mediated phosphorylation of RIPK-1 [28].
SLs as such glucosylceramide, certain gangliosides, sphingosine-1-phosphate (S1P), ceramide-1-phosphate (C1P), and ceramide have long been implicated in Ca2+ regulation within cells [29,30]. Robust cytosolic and mitochondrial Ca2+ elevations reportedly are elicited by elevated lyso-galactosylceramide (pyschosine) and accompanied by mitochondrial reactive oxygen species (ROS) production in oligodendrocytes. Extracellular Ca2+ chelation or antioxidant treatment decreased intra-mitochondrial ROS production and increased cell viability [31]. Given the important role of Ca2+ in necroptosis, emerging links between sphingolipid intracellular levels and necroptosis are not surprising [16,32–34]. FTY720, a sphingosine analogue, induces necroptotic cell death and suppresses lung tumor growth [33]. TNFα-induced ceramide upregulation associated with necroptosis, and not apoptosis, is mitigated by the antioxidant, butylated hydroxyanisole [35]. TNFα-induced necroptosis is also antagonized by docosahexaenoic acid in part by attenuation of cellular ceramide levels [34]. In such ways, SLs help regulate cellular longevity and modulate cell death, i.e. apoptosis and necroptosis [36,37].
In the present study, we provide evidence that overexpression of human GLTP inhibits colon cancer cell proliferation by interfering with cell cycle progression. Unlike HCT-116 cells which became quiescent, HT-29 cells overexpressing GLTP exhibited RIPK-3-mediated phosphorylation of MLKL, increased intracellular Ca2+ levels and induction of cell death via necroptosis. The discovery of necroptosis induction by changes in GLTP expression could potentially provide a new avenue for combating cancer progression.
2. Materials and Methods
2.1. Cell lines and treatments
Human colon cancer cell lines [HT-29 (ATTC HTB-38) and HCT-116 (ATTC CCL-247)] as well as normal colon cell line CCD-18Co (ATCC, CRL-1459) were obtained from the American Type Culture Collection (ATCC), Manassas, VA. Cells were grown in DMEM medium (HyClone GE Healthcare, SH30022.01) supplemented with 10% fetal bovine serum (FBS; Thermo Scientific, 10082147) and penicillin/streptomycin (Thermo Scientific, 15140122) at 5% CO2 and 37°C. GLTP overexpression and depletion were confirmed by western blot analyses (Supplemental Fig. S1). TurboGFP-GIPZ lentiviral shRNAmir constructs for GLTP, RIPK-3, MLKL and non-targeting (scrambled) control shRNAs were supplied by the University of Minnesota Genomics Center (http://genomics.umn.edu/rna-interference.php).
2.2. Cell growth and viability assays
Cells were plated at 5000 cells/cm2 and transfected with different vectors using Lipofectamine 2000 (Invitrogen, 11668-019). At 24, 48 and 72 h post transfection, total cells were collected by brief trypsinization and washed with PBS. Total cell number was determined by counting each sample in duplicate using a hemocytometer under an inverted microscope. Cell viability was measured using trypan blue exclusion assay. Briefly, cells were plated in a six well plate and were subjected to treatment/transfection following day. At 24, 48 and 72 h post transfection, cells were harvested by trypsinization and were resuspended in phosphate-buffered saline (PBS). The cell suspension was then mixed with an equal volume of 0.4% trypan blue solution prepared in PBS. The number of live cells (unstained) over the total number of cells was calculated as the percentage of viability. Each treatment and time point involved three independent plates. The data shown are the mean of three independent experiments.
2.3. FACS analysis for cell cycle distribution
Seeded cells were harvested, fixed with ice-cold 70% ethanol, and kept overnight at −20°C. Cells were then washed twice with PBS followed by incubation with 200 μg/ml RNase A and 20 μg/ml propidium iodide in PBS at room temperature for 30 min in the dark. The samples were subjected to flow cytometry using the FACS Calibur flow cytometer and data were analyzed using ModFit LT (Verity Software House, Inc., Topsham, ME).
2.4. Quantitative real-time PCR analysis
To evaluate mRNA transcript levels, qPCR was performed as previously described [38]. Briefly, total RNA was isolated using Trizol kits (Invitrogen, 15596026). RNA (1 μg) was reverse transcribed using Superscript III reverse transcriptase (Invitrogen, 18080093) and forward and reverse primers (Table S1). Transcript levels were quantified using qPCR SYBR® green assay (Applied Biosystems, 4367659) and primer sequences specific to genes.
2.5. Western blot analysis and reagents
Semiconfluent cells were collected by manual scraping, lysed in RIPA buffer (Thermo Scientific, 89900), pelleted at 4°C by centrifugation at 16,000 × g, for 15 min, and boiled in Laemmli sample buffer (Bio-Rad, 1610737). Proteins were separated on 12% discontinuous SDS-PAGE gels (Bio-Rad, 4561043EDU), transferred to PVDF membranes (Bio-Rad, 1620177), and immunolabeled using commercial antibodies. Antibodies used were: CDK2 (Cell Signaling Technology, 2546), CDK4 (Cell Signaling Technology, 12790), Kip1/p27 (Rockland Immunochemicals Inc.,600-401-K08S), Cip1/p21 (Cell Signaling Tech., 2946), GLTP (Proteintech Inc., 10850-I-AP), Cyclin D1 (Cell Signaling Tech., 2978), Cyclin E (Santa Cruz Biotech., sc-247), Actin (BD Biosciences, 612656) Caspase3 (Cell Signaling Tech., 9662), cleaved CASP3-Asp175 (Cell Signaling Tech., 9661), PARP (Cell Signaling Tech., 9542), cleaved PARP-Asp214 (Cell Signaling Tech., 9541), p-MLKL (ab187091; Abcam), MLKL (GeneTex Inc. GTX107538), RIPK-3 (Cell Signaling Tech., 95702), Tubulin (Cell Signaling Tech., 86298). Immunoreactive bands were detected by chemiluminescence (ImageQuant LAS4000 system, GE Healthcare, MA, USA).
2.6. Cell death analyses by flow cytometry
Cells were evaluated at 24 and 48 h post-transfection using a FACSCalibur flow cytometer (BD Biosciences, CA, USA) with the ANXA5/Annexin V-allophycocyanin/APC detection kit (BD Biosciences, 561012) according to the manufacturer’s recommendations. Briefly, cells were trypsinized, washed with 2.5% FBS in PBS, and incubated with ANXA5-allophycocyanin plus 7-AAD/7-amino actinomycin D PE/phycoerythrin-Cy5/cyanine 5 in binding buffer at room temperature for 5 min in the dark. Stained cells were analyzed for PS exposure via ANXA5-APC signal in the FL1 detector (660/20 bandpass filter) and for dead cell DNA binding via 7-AAD PE-Cy5 signal in the FL2 detector (670/14 bandpass filter). Early apoptotic cells stained positive for ANXA5 and negative for 7AAD (lower right quadrant) whereas late apoptosis or necroptosis stained positive for both ANAXA5 and 7AAD (upper right quadrant). Distinction of necroptosis versus apoptosis was confirmed by treatment of cells with either with pan caspase inhibitor z-VAD-FMK (Sigma Aldrich, 627610) or necroptosis inhibitor necrosulfonamide (Tocris, 5025).
2.7. Immunofluorescence staining and analysis
Cells were grown on polylysine (Sigma-Aldrich, P4707)-coated glass coverslips as described earlier [74]. Briefly, after transfection with plasmids encoding FLAG-GLTP, mock transfected Control vector or TSZ treatment, cells were labelled with p-MLKL antibody (ab187091; Abcam) followed by secondary antibodies coupled to Alexa Fluor 594 (Thermo Scientific, A-11037) as described in the figure legends, washed with PBS and fixed for 20 min at room temperature with 4%paraformaldehyde (Sigma-Aldrich, P6148). Cells were then counter-stained with DAPI (Sigma-Aldrich, 10236276001), washed and mounted in 10% PBS, 90% glycerol (Sigma-Aldrich, G5516) on glass slides. The fixed cells were imaged with a 63× /1.4 NA apochromatic CS oil-immersion objective on a Apotome.2 imaging system (Carl Zeiss Microscopy, NY) and analyzed using ZEN 2.3 lite software from Zeiss. Necroptosis was assessed in a minimum of 20 cells per preparation in 3 independent experiments.
2.8. Calcium measurement by FIuo-3/AM ester
For Ca2+ measurement, cells were incubated post transfection with 5 μM Fluo-3/AM ester (Sigma-Aldrich, 39294) for 30 min at 37 °C. To analyze for Ca2+ origin (intra- or extracellular), cells were pretreated either with BAPTA-AM or EDTA (Sigma-Aldrich, 196419 or E6511).
2.9. Sphingolipidomics analyses
Sphingolipids were prepared and analyzed as previously described [39–41]. Briefly, a 200 μl aliquot of PBS was added to the cell pellet and transferred to glass screwcap vials. After cell pellet transfer, 10 μl of 250 pmol/μl sphingolipid internal standard (Avanti Polar Lipids) was added to each sample (d18:1/12:0 species to sphingomyelin, ceramide, and monohexosylceramide), (d17:1 sphingosine to sphingosine and sphingosine-1-phosphate, and d17:0 sphinganine to sphinganine and sphinganine-1-phosphate), prior to extraction using a modified Bligh-Dyer protocol (1 ml CH3OH) + 0.5 mol CHCl3). The resulting mixtures were sonicated to disperse aggregates followed by incubation for 6 h at 48°C. After incubation, the extracts were transferred to a new glass tube, dried down, resuspended in CH3OH (500 μl) by sonication, centrifuged, and transferred to 1.5 ml LC injection vials.
Sphingolipids were separated using a Shimadzu Nexera X2 LC-30AD ultra-performance LC (UPLC) column coupled to a SIL-30AC auto injector, coupled to a DGU-20A5R degassing unit in the following way. An 8 min. reversed phase LC method utilizing an Acentis Express C18 column (5 cm × 2.1 mm, 2.7 μm) was used to separate the sphingolipids at a 0.5 ml/min flow rate at 60°C. The column was equilibrated with 100% Solvent A [methanol:water:formic acid (58:44:1, v/v/v) with 5mM ammonium formate] for 5 min and then 10 μl of sample was injected. 100% Solvent A was used for the first 0.5min of elution. Solvent B [methanol:formic acid (99:1, v/v) with 5mM ammonium formate] was increased in a linear gradient to 100% Solvent B from 0.5min to 3.5min. Solvent B was held constant at 100% from 3.5 min to 6 min. From 6 min to 6.1 min min solvent B was reduced to 0%, and solvent A returned to 100%. Solvent A was held constant at 100% from 6.1 min to 8 min.
Sphingolipids were analyzed via mass spec using an AB Sciex Triple Quad 5500 Mass Spectrometer. Q1 and Q3 were set to detect distinctive precursor and product ion pairs. Ions were fragmented in Q2 using N2 gas for collisionally induced dissociation. Analysis used multiple-reaction monitoring in positive-ion mode. Sphingolipids were monitored using precursor → product MRM pairs [41]. The mass spectrometer parameters used were: Curtain Gas: 30 psi; CAD: Medium; Ion Spray Voltage: 5500 V; Temperature: 500 °C; Gas 1: 60 psi; Gas 2: 40 psi; Declustering Potential, Collision Energy, and Cell Exit Potential vary per transition.
2.9. Statistical analyses
Data are expressed as the mean ± SE of at least 3 separate experiments performed in triplicate. The Student t test and one-way ANOVA (analysis of variance) were performed using GraphPad Prism 5 (GraphPad Software, Inc. La Jolla, CA) to compare differences between groups. P values < 0.05 were considered statistically significant. Significant differences are indicated by asterisks (*P < 0.05, **P < 0.01, ***P < 0.001) obtained using the Student t-test.
3. Results
3.1. GLTP overexpression inhibits proliferation of human colon carcinoma cells, but spares normal colonic cells
Changes in GLTP expression are known to influence cell morphology and intracellular sphingolipids levels in human cells [19,42]. Because previous studies showed a strong correlation between altered sphingolipid levels and the initiation and progression of colon cancer [5,9], we tested the effect of altered GLTP expression in HT-29 and HCT-116 colon cancer cell (Supplemental Fig. S1). wtGLTP overexpression was found to significantly reduce the proliferation of HT-29 (Fig. 1A) and of HCT-116 (Supplemental Fig. S2) cells, but did not affect the growth rate of normal human colon cells, CCD-18Co (Fig. 1B). No significant change in HT-29 cell proliferation was produced by overexpression of W96A-GLTP mutant with ablated glycolipid binding and transfer ability [18] or wtGLTP depletion by siGLTP.
Fig. 1. GLTP overexpression inhibits growth in human colon carcinoma cell lines but spares normal colon cells.
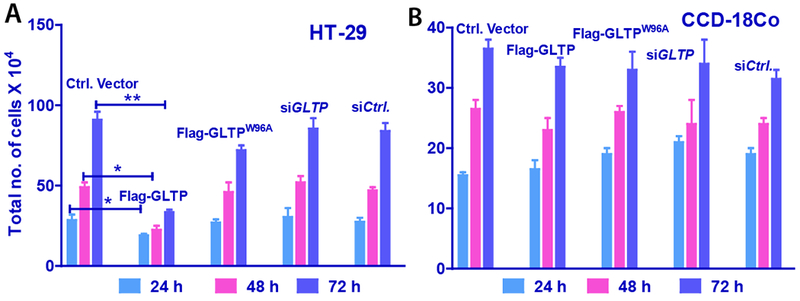
(A) HT-29 and (B) CCD-18Co cells analyzed for cell growth following 24, 48 and 72 h post transfection with FLAG-tagged empty vector (Control Vector), GLTP overexpression vector (FLAG-GLTP), W96A-GLTP mutant with an ablated glycolipid binding site vector (FLAG-W96AGLTP), GLTP depletion vector (siGLTP) and non-targeting siRNA control vector (siCtrl). Total cells were collected and counted by hemocytometry. Experiments were done in triplicate and values represent means ± sem. *P<0.05, **P<0.01, **P<0.001 Student t-test. (see Supplemental Fig. S2 for HCT-116 cell data)
3.2. GLTP overexpression inhibits cell cycle progression of human colon carcinoma cell lines
We next examined the effect of GLTP overexpression on cell cycle progression. HT-29 and HCT-116 cells overexpressing FLAG-GLTP were stained with saponin/propidium iodide (PI) and subjected to FACS analysis. Figs. 2A and 2B show that G0/G1 cell populations increased at the expense of the S phase cell populations after 48 h of GLTP overexpression. Similar findings also were observed at 24 h post transfection (Supplemental Fig. S3).
Fig. 2. Inhibition of cell cycle progression in human colon carcinoma cells by GLTP overexpression.
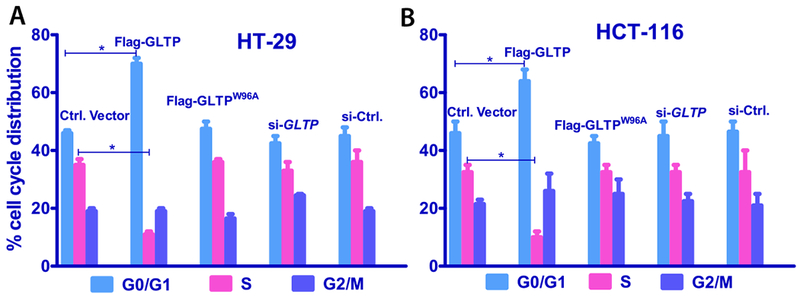
(A) HT-29 and (B) HCT-116 cells were analyzed for percent cell cycle distribution following 48 h post transfection with FLAG-tagged empty vector (Ctrl Vector), GLTP overexpressing vector (FLAG-GLTP), GLTP mutant (W96A, with an ablated glycolipid binding site) overexpressing Vector (FLAG-W96AGLTP), reduced GLTP expressing vector (siGLTP) and non-targeting siRNA control vector (siCtrl). Overexpression of GLTP (FLAG-GLTP) slowed cell growth in HT-29 and HCT-116 cells by increasing G0/G1 populations at the expense of S phase populations. Experiments were done in triplicate and values were means ± sem. *P<0.05, **P<0.01, **P<0.001 Student t-test. (see Suppl. Fig. S3 for 24 h data).
3.2. GLTP overexpression upregulates Kip1/p27 and Cip1/p21 levels and modulates CDKs
In most cancer cells, high expression of cyclins accompanies the increased CDK activity that is a major contributor to the deregulated and unchecked cell proliferation [43,44]. Accordingly, we analyzed whether GLTP overexpression modulates the expression of G0/G1 CDKs and cyclins in HT-29 cells. Fig. 3A shows western blot analyses that illustrate the moderate-to-strong decreases in the expression of cyclin D1 and cyclin E found in HT-29 cells overexpressing GLTP. In contrast, the changes were minimal when mutant GLTP (W96A) was overexpressed and nonexistent with RNAi-induced GLTP depletion compared to control cells.
Fig. 3. Upregulation of Kip1/p27 and Cip1/p21 levels and modulation of CDKs by GLTP overexpression.
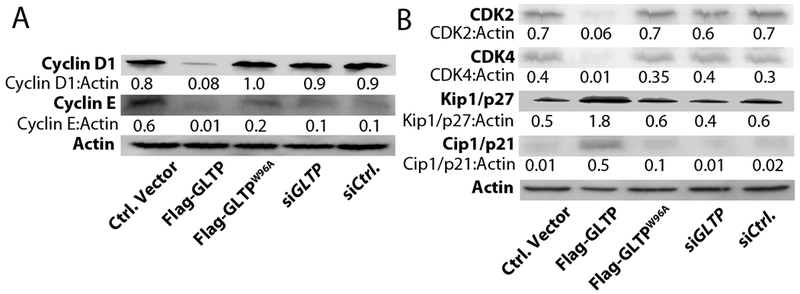
Western blot images of HT-29 cells at 48 h post transfection with indicated vectors. (A) Decreased levels of Cyclin D1 and Cyclin E induced by wtGLTP overexpression are shown. By contrast, alterations are minimal with mutant GLTP (W96A) overexpression and nonexistent with GLTP depletion (siGLTP) compared to vector-control cells. (B) Increased levels of Kip1/p27 and Cip1/p21 and decreased levels of cyclin-dependent kinases (CDK), CDK2 and CDK4 induced by wtGLTP overexpression. Quantitative insights are provided by ratiometric comparisons of band intensities to Actin (loading ctrl.)
To gain additional insights into the molecular mechanism of the observed cell cycle arrest, we examined Kip/Cip protein family involvement by evaluating the expression levels of cyclin-dependent kinase (CDK) inhibitor 1B (Kip1/p27) and 1A (Cip1/p21) [43]. Kip1/p27 binds to and prevents activation of the cyclin E-CDK2 or cyclin D-CDK4 complexes, thus controlling cell cycle progression at G1 phase. As shown by western blot analyses (Fig. 3B), overexpressed GLTP significantly decreased CDK2 and CDK4 levels while strongly upregulating Kip1/p27 protein expression, which is in accord with the observed cell cycle arrest by GLTP (Fig. 2). The effect of GLTP overexpression on cyclin-dependent kinase inhibitor 1A (Cip1/p21), a universal inhibitor of cell cycle progression [43] that is upregulated in cells arrested at G0/G1, was also analyzed and found to be strongly increased (Fig. 3B).
3.4. GLTP induces necroptosis in HT-29 cells
Cell cycle arrest at G0/G1 need not reflect only diminished DNA replication (S-phase), but can also be indicative of programmed cell death onset [45–47]. To assess whether programmed cell death was induced in colon carcinoma cells following GLTP overexpression, HT-29 cells were transfected with GFP-GLTP overexpression vector and analyzed by flow cytometry using 7-amino-actinomycin D (7AAD) and Annexin-V staining at 24 h post transfection. 7AAD discriminates live and dead cells by being efficiently excluded from intact viable cells, while labeling DNA in permeabilized dead cells. Annexin-V efficiently labels phosphatidylserine (PS) that has become exposed in the external face of the plasma membrane during onset of programmed cell death. Notably, PS exposure is not restricted to apoptosis but also occurs during shedding of damaged plasma membrane in necroptosis [48–50]. Figures 4A and 4B show that GLTP overexpression, but not GLTP depletion, led to increased Annexin V+/7AAD− and Annexin V+/7AAD+ labeling in HT-29 cells consistent with onset of cell death. However, similar changes were not observed for HCT-116 cells (Supplemental Figs. S4A and S4B). To delineate whether GLTP-induced cell death in HT-29 cells reflected apoptosis or necroptosis, cells were treated with either pan caspase inhibitor zVAD-FMK (Z) or necroptosis inhibitor necrosulfonamide (NSA) prior to GLTP overexpression and analyzed for Annexin V/7AAD staining (Figs. 4A & 4B). Treatment with NSA, but not zVAD-FMK, reduced AnnexinV+/7AAD+ and Annexin V+/7AAD− staining in HT-29 cells overexpressing GLTP suggesting cell death via necroptosis (Figs. 4A & 4B). TSZ treatment (TNFα, SMAC mimetic, and zVAD-FMK combination) was used as a positive control for necroptosis induction [21,51]. Immunoblotting (Fig. 4C) confirmed the necroptotic nature of GLTP mediated cell death by showing that HT-29 cells overexpressing GLTP fail to produce caspase 3 and PARP cleavage fragments indicative of apoptosis (e.g. caspase3 p17 and PARP p89). Yet, phosphorylation of MLKL occurs in HT-29 cells overexpressing GLTP (Fig. 4D) but not in cells pretreated with NSA (Fig. 4E). In contrast, in HCT-116 cells overexpressing GLTP, neither induction of cell death nor MLKL phosphorylation is observed (Supplemental Fig. S4).
Fig. 4. GLTP induces necroptosis in HT-29 cells.
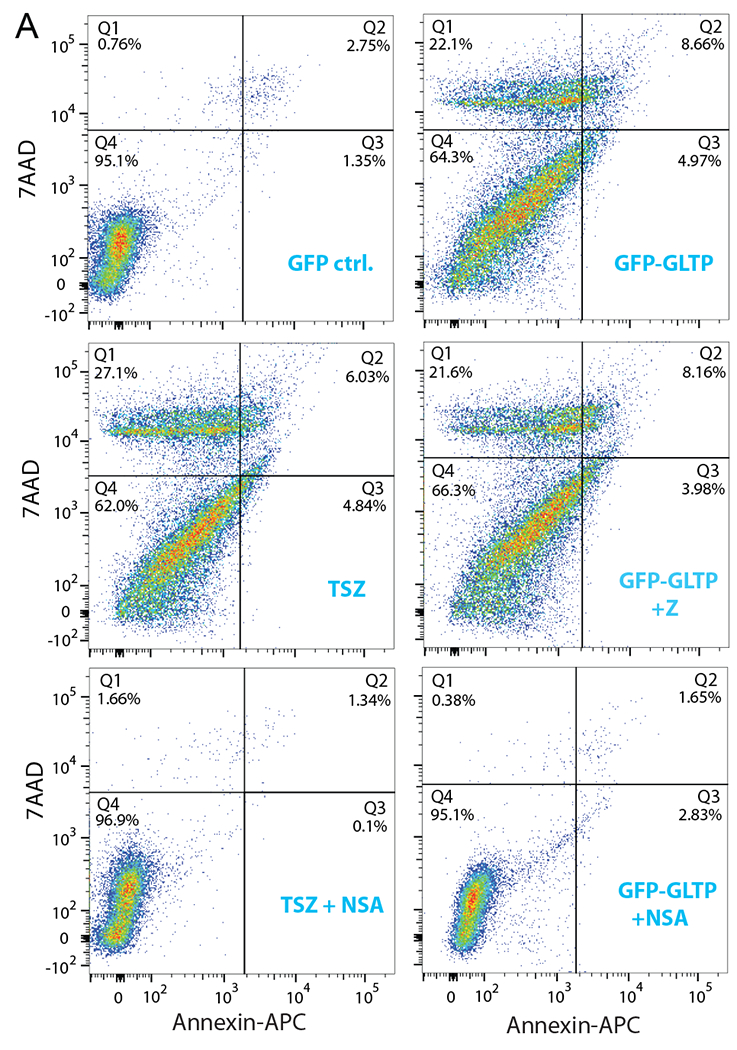
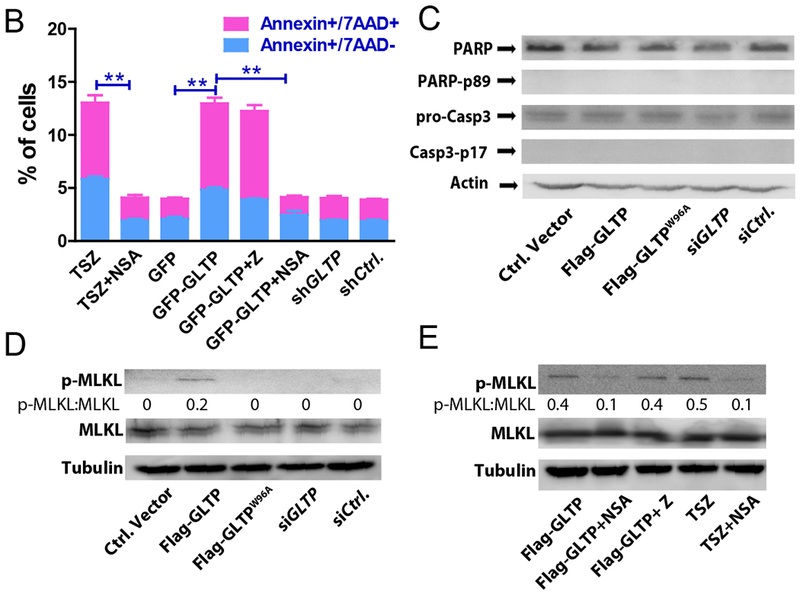
(A) Cell viability was measured by flow cytometry using AnnexinA5/7AAD staining. GLTP overexpression (GFP-GLTP) leads to increased levels of Annexin A5+/7AAD− and AnnexinA5+/7AAD+ cell populations compared with mock transfected controls. Necroptosis inhibitor necrosulfonamide (NSA) but not pan caspase apoptotic inhibitor z-VAD-FMK (Z) mitigated the GLTP induced cell death. TSZ (combination of TNFα, SMAC mimetic, and zVAD-FMK) treatments served as positive control to induce necroptotic cell death. (B) Quantification of scatter plot data presented in panel (A). (C) Western blot images of HT-29 cells transfected with indicated vectors showing levels of cleaved PARP and caspase 3. No indication of apoptosis was observed based on cleavage of PARP and caspase 3. (D) and (E) show WB images of p-MLKL and MLKL proteins in HT-29 cells transfected with indicated vectors and/or treated with positive control TSZ . Some cells were pretreated with either pan caspase inhibitor (Z) or necroptosis inhibitor (NSA) to ascertain the cell death pathway induced by GLTP overexpression. Quantitative insights are provided by ratiometric comparisons of band intensities. Actin and tubulin served as loading controls.(see Suppl. Fig. S4 for HCT-116 cell data)
In necroptosis, RIPK3 is required for phosphorylation of MLKL [23,27]. pMLKL oligomerization and translocation to the plasma membrane depend upon an early rise in cytosolic Ca2+ via activated ion channels such as Transient Receptor Potential Melastatin 7 (TRPM7) [26]. The ensuing loss of membrane barrier integrity induced by pMLKL leads to cell swelling and death [26,28]. We analyzed whether GLTP overexpression induces intracellular Ca2+ level changes within HT-29 cells using Fluo-3/AM fluorescent probe in conjunction with flow cytometry and detected intracellular accumulation of Ca2+ (Fig. 5A). Pretreatment with the intracellular Ca2+ chelator, BAPTA-AM, blocked the Fluo-3/AM response. By contrast, the intracellular Ca2+ levels of HT-29 cells were not affected by GLTP depletion induced via siGLTP or by expression of GLTPW96A mutant with an ablated GSL binding site (Fig. 5A). Considering that extracellular Ca2+ influx is often a major mechanism for increasing cytosolic Ca+2 levels [52], we assessed the effect of EDTA pretreatment on the GLTP-triggered Ca2+ accumulation. Pretreatment with EDTA did not significantly reduce the cellular Ca+2 levels detected in HT-29 cells overexpressing GLTP (Fig. 5A) consistent with the cytosolic Ca2+ accumulation originating from intracellular Ca2+ stores rather than from extracellular Ca2+ influx. Notably, BAPTA-AM chelation of intracellular Ca2+, but not EDTA chelation of extracellular Ca2+, abrogated the loss of HT-29 cell viability induced by GLTP overexpression (Fig. 5B). In contrast, HCT-116 cell viability was affected neither by GLTP overexpression nor by Ca2+ availability from internal or external stores (Supplemental Fig. S5). Also, BAPTA-AM pretreatment blocked MLKL phosphorylation in HT-29 cells overexpressing GLTP; whereas EDTA pretreatment exhibited no blocking of pMLKL formation (Fig. 5C). These findings emphasize the key role played by elevated cytosolic Ca2+ levels from internal Ca2+ stores for induction of pMLKL oligmomerization after upregulation by RIPK3 and permeabilization of the plasma membrane that kills HT-29 cells.
Fig. 5. GLTP overexpression releases Ca2+ from internal stores to drive necroptosis.
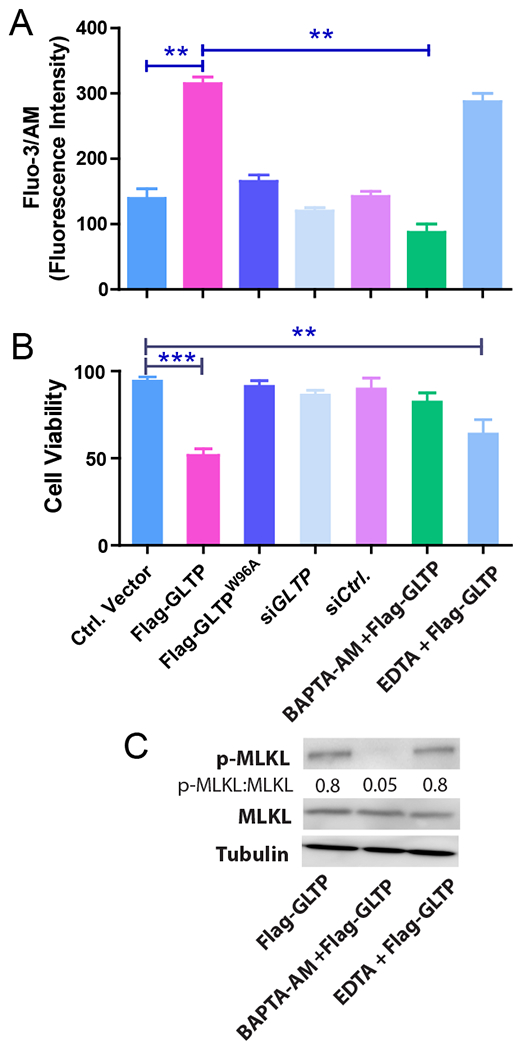
(A) Intracellular Ca2+ levels were measured using cell permeable Fluo-3/AM fluorescent probe in conjunction with flow cytometry of HT-29 cells. Following GLTP overexpression, intracellular Ca2+ accumulation was observed which was significantly reduced by pretreatment with BAPTA-AM (1μM), an intracellular Ca2+ chelator but not with EDTA (25 μM). siGLTP treatment or overexpression of GLTPW96A mutant (Flag-GLTPW96A) did not change Ca2+ levels compared with mock transfected empty vector ctrl or non-targeting siRNA ctrl. (B) Chelation of intracellular Ca2+ by BAPTA-AM, but not extracellular Ca2+ by EDTA, significantly abrogated HT-29 cell viability loss induced by GLTP overexpression as determined using trypan blue analyses. Experiments were done in triplicates and values reported are means ± sem. *P<0.05, **P<0.01, **P<0.001 using Student t-test. (C) WB analysis of p-MLKL and MLKL levels in HT-29 cells transfected with GLTP overexpressing vectors either alone or pretreated with BAPTA-AM or EDTA. Quantification is provided by ratiometric comparisons of band intensities. Tubulin = loading ctrl. (see Supplemental Fig. S5 for HCT-116 cell data)
To further ascertain RIPK-3 dependence of GLTP-induced cell death in HT-29 cells, we depleted RIPK-3 using shRNA and analyzed p-MLKL status (Fig. 6A). As a control, we also depleted MLKL in cells overexpressing GLTP. The preceding conditions abrogated the expression of pMLKL despite GLTP overexpression. Moreover, dramatically increased levels of p-MLKL puncta in HT-29 cells overexpressing GLTP compared to control cells were observed by immunocytochemical microscopy (Figs. 6B and 6C).
Fig. 6. Necroptosis induced by GLTP overexpression is RIPK3 dependent and leads to p-MLKL punctae in HT-29 cells.
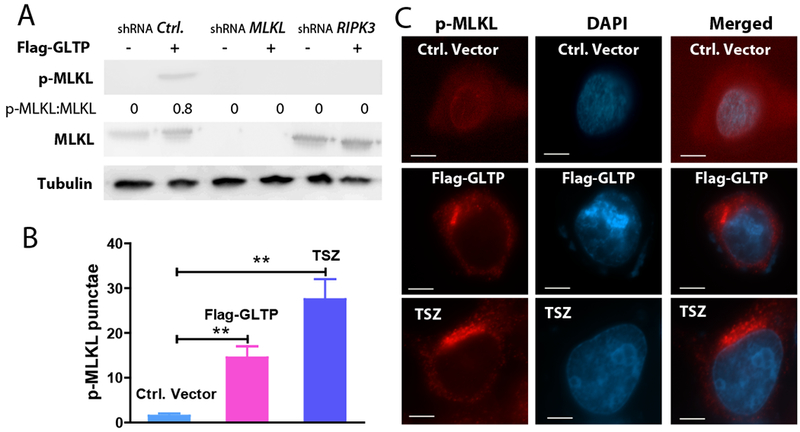
(A) Depletion of RIPK3 and MLKL using gene specific shRNA vectors in HT-29 cells abolishes p-MLKL expression in cells overexpressing GLTP. Quantitative insights are provided by ratiometric comparisons with tubulin (loading control). (B) Quantification analyses of images in (C) involved 20 cells per group from 3 independent experiments. Values were means ± sem. *P<0.05, **P<0.01, **P<0.001 using Student t-test. (C) HT-29 cells, transfected with empty control vector, GLTP overexpressing plasmid (Flag-GLTP) or treated with TSZ (TNFα, SMAC mimetic, and zVAD-FMK combination), were immunolabeled with primary antibody to p-MLKL followed by Alexa Fluor 595 secondary antibody. In GLTP overexpressing cells and TSZ-treated positive controls, the red channel shows increased pMLKL punctae otherwise minimally present in control cells. Scale bars: 10μm.
3.5. Sphingolipid changes induced by GLTP overexpression in colon cancer cells
To determine if the sphingolipid metabolic changes triggered by GLTP overexpression are consistent with the induction of cell death, sphingolipidomic analyses were performed. Sphingolipid mass levels [ceramide, monohexosylceramides (MHCer), sphingomyelin, ceramide-1-phosphate (C1P), sphingosine, and sphingosine-1-phosphate (S1P)] in wild-type HCT-116 and HT-29 cells as well as in these same cells overexpressing GLTP are shown in Fig. 7 and Supplementary Fig. S6. Of particular interest are the GLTP-induced alterations in S1P and Cer levels due to the opposing effects of these sphingolipids on cell survival [53,54].
Fig. 7. Changes in ceramides and S1P induced by GLTP overexpression.
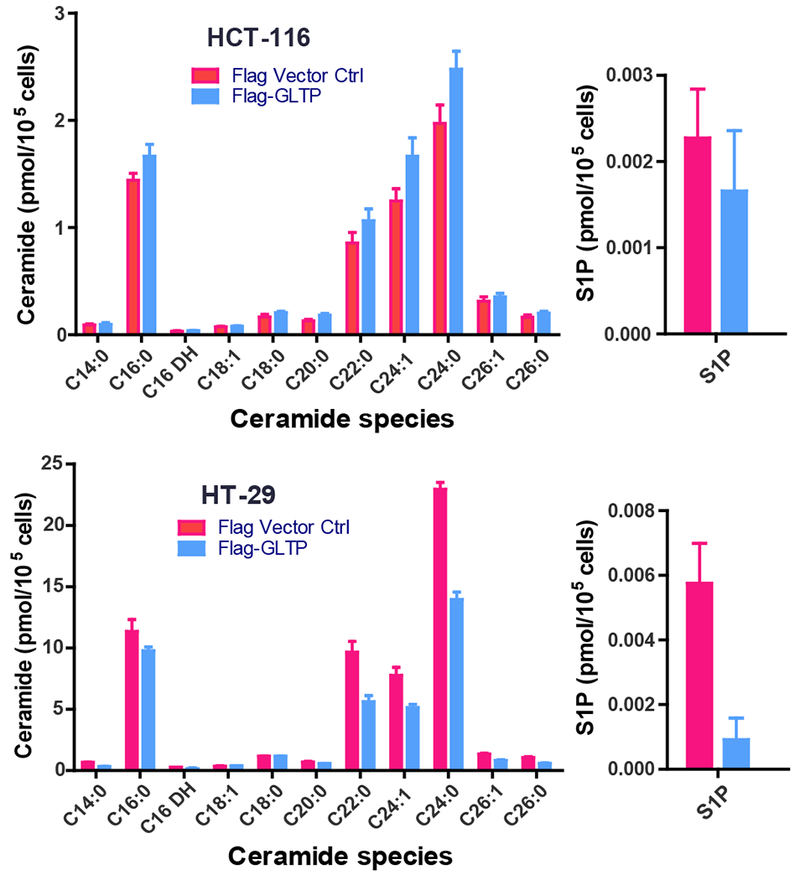
Sphingolipidomics analyses were carried out as described in the Materials and Methods. (Upper panel) HCT-116 cells and (Lower Panel) HT-29 cells (see Supplemental Fig. S6 for analyses of additional sphingolipids)
Notably (Fig. 7), in HT-29 cells, 16:0-ceramide remained nearly unchanged while S1P levels dropped substantially (~5-fold), consistent with tipping the S1P/16:0-Cer ratio towards cell death. Notably, no significant change in S1P and only slightly increased 16:0-Cer were observed in HCT-116 cells overexpressing GLTP (Fig. 7) yielding a relatively unaltered ‘sphingolipid rheostat’ (S1P/16:0-Cer ratio). Also, in wild-type HCT-116 cells, the ceramide, MHCer, and S1P mass levels are 6- to 10-fold lower than in HT-29 cells but sphingomyelin levels are similar (Supplementary Fig. S6). Altogether, the findings provide evidence that GLTP-induced necroptosis in HT-29 cells requires viable levels of RIPK3 and p-MLKL and that a decreased S1P to 16:0-Cer ratio contributes to cell death. The failure of GLTP upregulation to induce necroptosis in HCT-116 cells and other cancer cell lines (e.g. HeLa cells) [19] correlates with their poor expression of endogenous RIPK-3 [55] and minimal effect on the ‘sphingolipid rheostat’ (S1P/16:0-Cer ratio).
Discussion
The present study reveals that overexpression of human GLTP induces cell death via necroptosis in certain colon cancer cells (HT-29), but not in others (HCT-116) or in normal colonic cells. Despite tremendous advances in early detection, cases of colon cancer remain high and new pathways and targets to improve colon cancer treatment outcomes are desirable. While much is known about the structure/function and genomic aspects of GLTP and related GLTP homologs [14,15,17,18,41,56–58], the in vivo function(s) of these cytosolic amphitropic SL transfer proteins have remained unclear and a subject of debate. Mammalian GLTPs have been proposed to function as molecular transporters of certain GSLs to the plasma membrane [59–61] as well as regulatory sensors of GSL levels at various sites in the cell [13,15,42 ,56,62]. The latter function could be especially pertinent to programmed cell death processes because of the known involvement of GSLs and related metabolites in regulating surface adhesion, neurodegeneration, and apoptosis.
Our findings of upregulated Kip1/p27 and Cip1/p21 along with decreased CDK2, CDK4, cyclin E and cyclin D1 levels in response to GLTP overexpression are consistent with the observed growth arrest at the G0/G1 cell cycle checkpoint. CDKIs of the Kip/Cip family are established regulators of the activities of CDK–cyclin complexes known to negatively modulate cell cycle exit from the G1-phase [43,44,63]. CDKIs inhibit the kinase activities by binding with active CDK–cyclin complexes to regulate the cell cycle progression [44,63]. Not surprisingly, Kip1/p27 shows high expression levels in quiescent cells [64,65]. Kip1/p27 also has been shown to be a prognostic indicator for various cancers including colon cancer [64,66,67]. Cip1/p21 is a universal inhibitor of CDKs whose expression is mainly regulated by p53 tumor suppressor protein [43,67]. Thus, the increased levels of Kip1/p27 and Cip1/p21 induced by GLTP overexpression are consistent with the observed G1/S cell cycle arrest via inhibition of CDK2 and CDK4.
Another way that CDK activity can be regulated is by cyclin binding proteins. Without bound cyclins, CDKs exhibit very low kinase activity. Thus, only the cyclin-CDK complex is an active kinase [43,44,67]. Cyclin D-dependent CDK4 and cyclin E-dependent CDK2 are largely involved in controlling G1/S restriction point [43,44]. Persistent mitogenic signaling, which occurs in the majority of cancer cells, is required for the induction, synthesis, and assembly of cyclin D1 with CDK4 [63]. Our finding that GLTP overexpression decreases the protein expression of CDK4 and cyclin D1 in HT-29 cells is consistent with the dramatically increased levels of G1/G0-phase and correspondingly decreased levels of S-phase cells. Moreover, the finding that GLTP overexpression also decreases the protein expression of CDK2 and cyclin E reveals an additional way of inducing G1/G0-phase cell cycle arrest. Thus, a ‘multi-faceted’ regulatory mechanism for arresting HT-29 cells at the G1-S transition emerges for GLTP overexpression. The mechanism involves not only diminished levels of the CDK4-cylin D1 and CDK2-cyclin E complexes, but also increased levels of the Kip1/p27 and Cip1/p21 inhibitory proteins to ensure arrest at the G1/S and strong reduction of S-phase cell levels.
The disruption of the cell cycle by GLTP overexpression results in HT-29 cell death for HT-29 cells but a quiescent state for HCT-116 cells. The HT-29 cell death is non-apoptotic in nature as revealed by the lack of cleaved caspase-3 derived p17 and PARP derived p89 fragments. Also, the GLTP-induced cell death is not abrogated by pretreatment of the cells with pan caspase inhibitor zVAD. The PS externalization observed in HT-29 cells overexpressing GLTP supports necroptosis because PS externalization is not limited to apoptosis but also occurs during necroptosis As pointed out earlier, PS exposure occurs in necroptosis via shedding of damaged plasma membrane, i.e. vesicle ‘bubbling’ [48–50]. The ‘coup de grâce’ indicating necroptotic cell death is the finding of upregulated phosphorylation of MLKL via RIPK-3, a hallmark event in necrosome formation and necroptosis induction. Indeed, high endogenous RIPK-3 expression is a key indicator for necroptosis susceptibility that is missing in normal colonic cells (CCD-18Co) and HCT-116 colon cancer cells but not in HT-29 colon cancer cells [68,69]. Not surprisingly, HT-29 cells overexpressing GLTP also contain elevated cytosolic Ca2+ levels derived from internal stores, a condition linked to oligomerization of pMLKL that permeabilizes the plasma membrane and drives necroptotic cell death.
Cell death via apoptosis is known to be regulated by sphingolipids [7,12,53,54]. Ceramide is a well-established inducer of apoptosis; whereas S1P exerts pro-mitogenic and pro-survival cell growth. The ratio between S1P and ceramide, i.e. the ‘sphingolipid rheostat’, correlates with cell survival versus programmed cell death induction [53,54]. Most previous studies of ceramide-induced apoptosis focused on 16:0-ceramide and 18:0-ceramide with fewer and varying insights into the impact of long-chain ceramide species [7,12]. Recently, 24:0-ceramide has been reported to reduce the potency of 16:0-ceramide and associated channel formation in mitochondria during apoptosis [70]. Compared to apoptosis, much less is currently known about the potential modulatory roles of ceramide and S1P in necroptosis. Our data indicate that necroptosis induced by GLTP overexpression in HT-29 cells is characterized by dramatically lowered S1P (~85%) accompanied by only slightly decreased 16:0-ceramide (~10%) thus tipping the ‘sphingolipid rheostat’ towards cell death. In contrast, no significant change in S1P and only slightly increased 16:0-Cer were observed in HCT-116 cells overexpressing GLTP. In a recent study of TNFα-induced necroptosis in HT-29 cells [71], significant changes were observed in four ceramide species including 16:0-ceramide but S1P levels were not reported thus preventing assessment of the sphingolipid rheostat. In human lung epithelial and endothelial cells undergoing necroptosis induced by exposure to cigarette smoke, 16:0-ceramide levels were increased, but again S1P levels were not assessed [72]. In other studies, increased ceramide levels have been detected during necroptosis [35,73]. Also ceramide nanoliposomes are an effective necroptosis-inducing chemotherapeutic reagent in ovarian cancer [74].
Another sphingolipid of potential interest in the HT-29 cell death response is globotriaosylceramide (Gb3), a highly expressed GSL that has been linked to the enhanced metastatic transformation and invasiveness of HT-29 cells [75]. A GlcCer synthase inhibition study in various cancer cells has confirmed elevated Gb3 levels in HT-29 cells but not in HCT-116 cells [76]. Yet, direct links between Gb3 expression and cell death remain poorly understood. The only other sphingolipidomics analysis of GLTP expression involved HeLa cells [42]. Despite increases of 16:0-ceramide (~20%), MHceramides (~10%) and Gb3 (~20%) (but no S1P levels reported) [42], GLTP overexpression fails to induce cell death in adherent HeLa cells [19]. Yet, Fabry’s cells, with genetically defective α-galactosidase A that results in massive Gb3 accumulation undergo cell death by the apoptotic instrinsic pathway [77]. On the other hand, Gaucher’s cells which contain massively accumulated GlcCer due to genetically defective glucocerebrosidase, have upregulated RIPK-3 [78,79]. Thus, the impact of various upregulated GSLs on cell death induction and the specific cell death pathway are complex and in need of additional future studies.
Another complicating factor in need of future study is the expressional relationship between GLTP and FAPP2 and the associated impact on sphingolipid metabolism. Whereas, a parallel downregulation of GLTP occurs upon depletion of GlcCer synthase [80], the impact of GLTP overexpression on endogenous FAPP2 levels has not been reported. The GLTP homology domain in FAPP2 is responsible for intracellular transfer of newly synthesized GlcCer during higher GSL synthesis that includes Gb3 [81,82]. Although FAPP2 expression regulates both GlcCer and Gb3 cellular levels, it remains unclear whether changes in FAPP2 expression impact 16:0-ceramide and S1P levels and whether GLTP overexpression can exert a ‘dominant negative effect’ over FAPP2. What is known is that FAPP2 and GLTP can exert partial compensatory effects on GlcCer transfer when one or the other is depleted [61].
Other members of the GLTP superfamily, i.e. ceramide-1 -phosphate transfer protein (CPTP) have recently been shown to regulate stress-related processes such as autophagy [38]. RNAi-induced downregulation of CPTP in HeLa, HEK-293, A549, or THP-1 cells was found to induce autophagy but not apoptosis. In the case of the macrophage-like THP-1 surveillance cells, autophagy-dependent inflammasome assembly was also triggered resulting in increased release of the IL-1β and IL-18 pro-inflammatory cytokines and eventual cell death by pyroptosis. Altogether, the emerging importance of GLTP superfamily members in the regulation of non-apoptotic cell death and stress-related pathways is becoming increasingly clear.
Supplementary Material
Highlights.
GLTP inhibits colon carcinoma cell growth but spares normal colonic cells
GLTP upregulation induces necroptosis in HT-29 cells, but not in HCT 116 cells
In HT-29 cells, GLTP upregulation tips the sphingolipid rheostat towards death
GLTP-induced necroptosis requires RIPK-3 and a high 16:0-ceramide/S1P ratio
Acknowledgements
Research support provided by NIH HL-125353 (CEC & REB), GM-45928 (REB), HD087198 (CEC), RR031535 (CEC), the Veteran’s Administration (VA Merit Review, I BX001792 (CEC)), Research Career Scientist Award 13F-RCS-002 (CEC), Southern Minnesota Paint-the-Town Pink Cancer Research Funds (REB) and the Hormel Foundation (REB) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Portions of this work were presented in preliminary form at the 2017 AACR Annual Meeting (Washington, DC) and appeared in Cancer Res 2017; 77 (13 Suppl): Abstr. 1123. doi: 10.1158/1538-7445.AM2017-1123
Disclosure Statement: The authors report no conflict of interest and have no relevant financial or nonfinancial relationships to disclose.
References
- [1].Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, Jemal A, Colorectal cancer statistics, 2017. CA Cancer J. Clin 67 (2017) 177–193. [DOI] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A, Cancer Statistics, 2017. CA Cancer J Clin 67 (2017) 7–30. [DOI] [PubMed] [Google Scholar]
- [3].Matarrese P, Malorni W, The role of sphingolipids and lipid rafts in determining cell fate. Apoptosis 20 (2015) 581–583. [DOI] [PubMed] [Google Scholar]
- [4].Maceyka M, Spiegel S, Sphingolipid metabolites in inflammatory disease. Nature 510 (2014) 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Garcia-Barros M, Coant N, Truman JP, Snider AJ, Hannun YA, Sphingolipids in colon cancer. Biochim. Biophy. Acta 1841 (2014) 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Astudillo L, Therville N, Colacios C, Segui B, Andrieu-Abadie N, Levade T, Glucosylceramidases and malignancies in mammals. Biochimie 125 (2016) 267–80. [DOI] [PubMed] [Google Scholar]
- [7].Hannun YA, Obeid LM, Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol 19 (2018) 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mohammad RM, Muqbil I, Lowe L, Yedjou C, Hsu HY, Lin LT. Siegelin MD, Fimognari C, Kumar NB, Dou QP, Yang H, Samadi AK, Russo GL, Spagnuolo C, Ray SK, Chakrabarti M, Morre JD, Coley HM, Honoki K, Fujii H, Georgakilas AG, Amedei A, Niccolai E, Amin A, Ashraf SS, Helferich WG, Yang X, Boosani CS, Guha G, Bhakta D, Ciriolo MR, Aquilano K, Chen S, Mohammed SI, Keith WN, Bilsland A, Halicka D, Nowsheen S, Azmi AS, Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol 35 (2015) Suppl, S78–S103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Camp ER, Patterson LD, Kester M, Voelkel-Johnson C, Therapeutic implications of bioactive sphingolipids: A focus on colorectal cancer. Cancer Biol. Ther 18 (2017) 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nagahashi M, Tsuchida J, Moro K, Hasegawa M, Tatsuda K, Woelfel IA, Takabe K, Wakai T, High levels of sphingolipids in human breast cancer. J Surg Res 204 (2016) 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Giussani P, Tringali C, Riboni L, Viani P, Venerando B, Sphingolipids: key regulators of apoptosis and pivotal players in cancer drug resistance. Intl. J. Mol. Sci 15 (2014) 4356–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ogretmen B, Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 18 (2018) 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Brown RE, Mattjus P, Glycolipid transfer proteins. Biochim. Biophys. Acta 1771 (2007) 746–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zou X, Chung T, Lin X, Malakhova ML, Pike HM, Brown RE, Human glycolipid transfer protein (GLTP) genes: organization, transcriptional status and evolution. BMC Genomics 9 (2008) 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Malinina L, Simanshu DK, Zhai X, Samygina VR, Kamlekar R, Kenoth R, Ochoa-Lizarralde B, Malakhova ML, Molotkovsky JG, Patel DJ, Brown RE, Sphingolipid transfer proteins defined by the GLTP-fold. Q. Rev. Biophys 48 (2015) 281–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Garcia-Ruiz C, Morales A, Fernandez-Checa JC, Glycosphingolipids and cell death: one aim, many ways. Apoptosis 20 (2015) 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Malinina L, Patel DJ, Brown RE, How alpha-helical Motifs form functionally diverse lipid-binding compartments. Annu. Rev. Biochem 86 (2017) 609–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Malinina L, Malakhova ML, Teplov A, Brown RE,Patel DJ, Structural basis for glycosphingolipid transfer specificity. Nature 430 (2004) 1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gao Y, Chung T, Zou X, Pike HM, Brown RE, Human glycolipid transfer protein (GLTP) expression modulates cell shape. PLoS One 6 (2011) e19990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Su Z, Yang Z, Xie L, DeWitt JP, Chen Y, Cancer therapy in the necroptosis era. Cell Death Differ 23 (2016) 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang T, Jin Y, Yang W, Zhang L, Jin X, Liu X, He Y, Li X, Necroptosis in cancer: An angel or a demon? Tumour Biol 39 (2017) 1010428317711539. [DOI] [PubMed] [Google Scholar]
- [22].Shan B, Pan H, Najafov A, Yuan J, Necroptosis in development and diseases. Genes Dev 32 (2018) 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G, Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol 11 (2010) 700–714. [DOI] [PubMed] [Google Scholar]
- [24].Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, Han J, Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24 (2014) 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P, Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol 15 (2014) 135–147. [DOI] [PubMed] [Google Scholar]
- [26].Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG, Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nature Cell Biol 16 (2014) 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Linkermann A, Green DR , Necroptosis. N. Engl. J. Med 370 (2014) 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nomura M, Ueno A, Saga K, Fukuzawa M, Kaneda Y, Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res 74 (2014) 1056. [DOI] [PubMed] [Google Scholar]
- [29].Stenovec M, Trkov S, Kreft M, Zorec R, Alterations of calcium homoeostasis in cultured rat astrocytes evoked by bioactive sphingolipids. Acta Physiol. (Oxf) 212 (2014) 49–61. [DOI] [PubMed] [Google Scholar]
- [30].Carpio LC, Shiau H, Dziak R, Changes in sphingolipid levels induced by epidermal growth factor in osteoblastic cells. Effects of these metabolites on cytosolic calcium levels. Prostaglandins Leukot. Essent. Fatty Acids 62 (2000), 225–232. [DOI] [PubMed] [Google Scholar]
- [31].Voccoli V, Tonazzini I, Signore G, Caleo M, Cecchini M, Role of extracellular calcium and mitochondrial oxygen species in psychosine-induced oligodendrocyte cell death. Cell Death Dis 5 (2014), e1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sundaram K, Mather AR, Marimuthu S, Shah PP, Snider AJ, Obeid LM, Hannun YA, Beverly LJ, Siskind LJ, Loss of neutral ceramidase protects cells from nutrient- and energy - deprivation-induced cell death. Biochem. J 473 (2016), 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, Bielawski J, Szulc ZM, Thomas RJ, Selvam SP, Senkal CE, Garrett-Mayer E, De Palma RM, Fedarovich D, Liu A, Habib AA, Stahelin RV, Perrotti D, Ogretmen B, Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med 5 (2013) 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pacheco FJ, Almaguel FG, Evans W, Rios-Colon L, Filippov V, Leoh LS, Rook-Arena E, Mediavilla-Varela M, De Leon M, Casiano CA, Docosahexanoic acid antagonizes TNF-alpha-induced necroptosis by attenuating oxidative stress, ceramide production, lysosomal dysfunction, and autophagic features. Inflamm. Res 63 (2014) 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sawai H, Ogiso H, Okazaki T Differential changes in sphingolipids between TNF-induced necroptosis and apoptosis in U937 cells and necroptosis-resistant sublines. Leuk. Res 39 (2015) 964–970. [DOI] [PubMed] [Google Scholar]
- [36].Pimentel AA, Benaim G Ca2+ and sphingolipids as modulators for apoptosis and cancer. Invest. Clin 53 (2012) 84–110. [PubMed] [Google Scholar]
- [37].Chan JP, Sieburth D, Localized sphingolipid signaling at presynaptic terminals is regulated by calcium influx and promotes recruitment of priming factors. J. Neurosci 32 (2012) 17909–17920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mishra SK, Gao YG, Deng Y, Chalfant CE, Hinchcliffe EH, Brown RE, CPTP: A sphingolipid transfer protein that regulates autophagy and inflammasome activation. Autophagy 14 (2018) 862–879; doi: 10.1080/15548627.2017.1393129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wijesinghe DS, Allegood JC, Gentile LB, Fox TE, Kester M, Chalfant CE, Use of high performance liquid chromatography-electrospray ionization-tandem mass spectrometry for the analysis of ceramide-1-phosphate levels. J. Lipid Res 51 (2010) 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Contaifer D, Carl DE, Warncke UO, Martin EJ, Mohammed BM, Van Tassell B, Brophy DF, Chalfant CE, Wijesinghe DS, Unsupervised analysis of combined lipid and coagulation data reveals coagulopathy subtypes among dialysis patients. J. Lipid Res 58 (2017) 586–599; doi: 10.1194/jlr.P068833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Simanshu DK, Kamlekar RK, Wijesinghe DS, Zou X, Zhai X, Mishra SK, Molotkovsky JG, Malinina L, Hinchcliffe EH, Chalfant CE, Brown RE, Patel DJ,Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature 500 (2013) 463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kjellberg M, Backman A, Ohvo-Rekilä H, Mattjus P, Alternation in the Glycolipid Transfer Protein Expression Causes Changes in the Cellular Lipidome. PLoS ONE 9 (2014) e97263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Grana X, Reddy EP, Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 11 (1995) 211–219. [PubMed] [Google Scholar]
- [44].Morgan DO, Principles of CDK regulation. Nature 374 (1995) 131–134. [DOI] [PubMed] [Google Scholar]
- [45].Wiman KG, Zhivotovsky B, Understanding cell cycle and cell death regulation provides novel weapons against human diseases. J. Intern. Med 281 (2017) 483–495. [DOI] [PubMed] [Google Scholar]
- [46].Clarke PR, Allan LA, Cell-cycle control in the face of damage--a matter of life or death. Trends Cell Biol 19 (2009) 89–98. [DOI] [PubMed] [Google Scholar]
- [47].Zebell SG, Dong X, Cell-cycle regulators and cell death in immunity. Cell Host Microbe 18 (2015) 402–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, Regev-Rudzki N, Edry-Botzer L, Gerlic M, Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol 15 (2017), e2002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR, ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169 (2017) 286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Krysko O, Aaes TL, Kagan VE, D’Herde K, Bachert C, Leybaert L, Vandenabeele P, Krysko DV, Necroptotic cell death in anti-cancer therapy. Immunol. Rev 280 (2017), 207–219. [DOI] [PubMed] [Google Scholar]
- [51].Sun W, Wu X, Gao H, Yu J, Zhao W, Lu JJ, Wang J, Du G, Chen X, Cytosolic calcium mediates RIP1/RIP3 complex-dependent necroptosis through JNK activation and mitochondrial ROS production in human colon cancer cells. Free Radic. Biol. Med 108 (2017) 433–444. [DOI] [PubMed] [Google Scholar]
- [52].Bootman MD, Collins TJ, Peppiatt CM, Prothero LS, MacKenzie L, De Smet P, Travers M, Tovey SC, Seo JT, Berridge MJ, Ciccolini F,Lipp P, Calcium signalling—an overview. Semin. Cell Devel. Biol 12 (2001) 3–10. [DOI] [PubMed] [Google Scholar]
- [53].Newton J Lima S, Maceyka M, Spiegel S, Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exptl. Cell Res 333 (2015) 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Espaillat MP, Shamseddine AA, Adada MM, Hannun YA Obeid LM, Ceramide and sphingosine-1-phosphate in cancer, two faces of the sphinx. Transl. Cancer Res 4 (2015) 484–499. [Google Scholar]
- [55].He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X, Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137 (2009, 1100–1111. [DOI] [PubMed] [Google Scholar]
- [56].Lin X, Mattjus P, Pike HM, Windebank AJ, Brown RE, Cloning and expression of glycolipid transfer protein from bovine and porcine brain. J. Biol. Chem 275 (2000) 5104–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Malinina L, Malakhova ML, Kanack AT, Lu M, Abagyan R, Brown RE, Patel DJ, The liganding of glycolipid transfer protein is controlled by glycolipid acyl structure. PLoS Biol 4 (2006) e362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Simanshu DK, Zhai X, Munch D, Hofius D, Markham JE, Bielawski J, Bielawska A, Malinina L, Molotkovsky JG, Mundy JW, Patel DJ, Brown RE, Arabidopsis accelerated cell death 11, ACD11, is a ceramide-1-phosphate transfer protein and intermediary regulator of phytoceramide levels. Cell Reports 6 (2014) 388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sasaki T, Glycolipid transfer protein and intracellular traffic of glucosylceramide. Experientia 46 (1990), 611–616. [DOI] [PubMed] [Google Scholar]
- [60].Warnock DE, Lutz MS, Blackburn WA, Young WW Jr., Baenziger JU, Transport of newly synthesized glucosylceramide to the plasma membrane by a non-Golgi pathway. Proc. Natl. Acad. Sci. USA 91 (1994) 2708–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Halter D, Neumann S, van Dijk SM, Wolthoorn J, de Mazière AM, Vieira OV, Mattjus P, Klumperman J, van Meer G, Sprong H, Pre- and post-Golgi translocation of glucosylceramide in glycosphingolipid synthesis. J. Cell Biol 179 (2007) 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Malakhova ML, Malinina L, Pike HM, Kanack AT, Patel DJ, Brown RE, Point mutational analysis of the liganding site in human glycolipid transfer protein. Functionality of the complex. J. Biol. Chem 280 (2005) 26312–26320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hunter T, Pines J, Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell 79 (1994) 573–82. [DOI] [PubMed] [Google Scholar]
- [64].Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massague J, Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78 (1994) 59–66. [DOI] [PubMed] [Google Scholar]
- [65].Mori M, Mimori K, Shiraishi T, Tanaka S, Ueo H, Sugimachi K, Akiyoshi T, p27 expression and gastric carcinoma. Nat. Med 3 (1997) 593. [DOI] [PubMed] [Google Scholar]
- [66].Malumbres M, Therapeutic opportunities to control tumor cell cycles. Clin. Transl. Oncol 8 (2006) 399–408. [DOI] [PubMed] [Google Scholar]
- [67].Otto T, Sicinski, Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17 (2017) 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FKM, Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis 6 (2015) e1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Morgan MJ, Kim Y-S, The serine threonine kinase RIP3: lost and found. BMB Reports 48 (2015) 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Stiban J, Perera M, Very long chain ceramides interfere with C16-ceramide-induced channel formation: A plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim. Biophys. Acta 1848 (2015) 561–567; 10.1016/j.bbamem.2014.11.018 [DOI] [PubMed] [Google Scholar]
- [71].Parisi LR, Li N, Atilla-Gokcumen GE, Very long chain fatty acids are functionally involved in necroptosis. Chem. Biol 24 (2017) 1445–1454; 10.1016/j.chembiol.2017.08.026 [DOI] [PubMed] [Google Scholar]
- [72].Mizumura K, Justice MJ, Schweitzer KS, Krishnan S, Bronova I, Berdyshev EV, Hubbard WC, Pewzner-Jung Y, Futerman AH, Choi AMK, Petrache I, Sphingolipid regulation of lung epithelial cell mitophagy and necroptosis during cigarette smoke exposure. FASEB J 32 (2018) 1880–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Thon L, Möhlig H, Mathieu S, Lange A, Bulanova E, Winoto-Morbach S, Schütze S, Bulfone-Paus S, Adam D, Ceramide mediates caspase-independent programmed cell death. FASEB J 19 (2005) 1945–1956. [DOI] [PubMed] [Google Scholar]
- [74].Zhang X, Kitatani K, Toyoshima M, Ishibashi M, Usui T, Minato J, Egiz M, Shigeta S, Fox T, Deering T, Kester M, Yaegashi N, Ceramide Nanoliposomes as a MLKL-dependent, necroptosis-inducing, chemotherapeutic reagent in ovarian cancer. Mol. Cancer Ther 17 (2018) 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kovbasnjuk O, Mourtazina R, Baibakov B, Wang T, Elowsky C, Choti MA, Kane A, Donowitz M, The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc. Natl. Acad. Sci. USA 102 (2005) 19087–19092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Alam S, Fedier A, Kohler RS, Jacob F, Glucosylceramide synthase inhibitors differentially affect expression of glycosphingolipids. Glycobiology 25 (2015) 351–356. [DOI] [PubMed] [Google Scholar]
- [77].De Francesco PN, Mucci JM, Ceci R, Fossati CA, Rozenfeld PA, Higher apoptotic state in Fabry disease peripheral blood mononuclear cells.: effect of globotriaosylceramide. Mol. Genet. Metab 104 (2011) 319–324. [DOI] [PubMed] [Google Scholar]
- [78].Zhang S, Tang MB, Luo HY, Shi CH, Xu YM, Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Dis 8 (2017) e2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Vitner EB, Salomon R, Farfel-Becker T, Meshcheriakova A, Ali M, Klein AD, Platt FM, Cox TM, Futerman AH, RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat.Med 20 (2014) 204–208. [DOI] [PubMed] [Google Scholar]
- [80].Kjellberg MA, Mattjus P, Glycolipid transfer protein expression is affected by glycosphingolipid synthesis. PLoS ONE 8 (2013) e70283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].D’Angelo G, Polishchuk E, Di Tullio G, Santoro M, Di Campli A, Godi A, West G, Bielawski J, Chuang CC, van der Spoel AC, Platt FM, Hannun YA, Polishchuk R, Mattjus P, De Matteis MA, Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature 449 (2007) 62–67. [DOI] [PubMed] [Google Scholar]
- [82].D’Angelo G, Uemura T, Chuang CC, Polishchuk E, Santoro M, Ohvo-Rekilä H, Sato T, Di Tullio G, Varriale A, D’Auria S, Daniele T, Capuani F, Johannes L, Mattjus P, Monti M, Pucci P, Williams RL, Burke JE, Platt FM, Harada A, De Matteis MA, Vesicular and non-vesicular transport feed distinct glycosylation pathways in the Golgi. Nature 501 (2013) 116–120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.