

Watch a video presentation of this article
Alcoholic liver disease (ALD) has been recognized for centuries, but the growing epidemic of nonalcoholic fatty liver disease (NAFLD) makes it clear that ALD is a subset of the general class of fatty liver diseases. The histopathological findings of both ALD and NAFLD overlap significantly, and this is an important topic to which we will return at the end of this essay.
ALD has three categories of histopathological changes: steatosis, steatohepatitis, and steatofibrosis1, 2 (Table 1). All these changes begin and are usually most prominent in the centrilobular region of the hepatic lobule. The centrilobular hepatocytes are enzymatically prepared to metabolize alcohol through, for example, cytosolic alcohol dehydrogenase and the induction of cytochrome p450 enzymes.2 Such pathways produce the fatty acids accumulating in steatotic hepatocytes and create the toxic metabolites causing inflammation, hepatocyte injury, and fibrosis.
Table 1.
Histological Findings in ALD
| Steatosis | Alcoholic (Steato‐) Hepatitis | Steatofibrosis | Miscellaneous | |
|---|---|---|---|---|
| Classic findings | Small‐ and large‐droplet fat Beginning and/or most prominent in perivenular regions (acinus zone 3) and extending with increasing severity toward portal tracts | Hepatocyte ballooning with and without Mallory‐Denk bodies and/or neutrophil‐rich parenchymal inflammation | Perivenular and pericellular fibrosis (chicken‐wire fence pattern) Central‐central and/or central‐portal fibrous septa Micronodular (Laennec's cirrhosis) | Megamitochondria |
| Other possible findings | Diffuse microvesicular steatosis indicative of alcoholic foamy degeneration | Mixed inflammatory infiltrates | Features of fibrotic regression after sustained abstinence | Hepatocellular iron (whether it is related to alcohol use or concomitant hereditary hemochromatosis requires a clinical assessment) |
Of course the features described here may often be seen in autopsy and explant specimens because of the role of alcohol in death or the need for transplantation. However, they are most important in the biopsy setting. The publication “EASL Clinical Practical Guidelines: Management of Alcoholic Liver Disease” points to the lack of consensus regarding the role of liver biopsy in the assessment of ALD but suggests the following principles3:
Percutaneous biopsy may be performed in most patients, but transjugular biopsy is recommended for patients with a low platelet count and/or a prolonged prothrombin time.
Because of the potential mortality and morbidity of liver biopsy, it is not recommended for all patients with ALD.
Liver biopsy is recommended in “patients with aggressive forms of ALD such as severe steatohepatitis requiring specific therapies (e.g. corticosteroids and/or pentoxiphylline) and in patients with other cofactors suspected of contributing to liver disease.”
Liver biopsy for patients in clinical trials is recommended because liver biopsy “allows the risk of long‐term mortality prognosis of patients with ALD to be classified according to the severity of the histological lesions.”
Although the presence of acute steatohepatitis or acute‐on‐chronic hepatitis can be suspected on clinical and biochemical grounds, a definitive diagnosis of acute steatohepatitis requires liver biopsy.
Abbreviations.
ALD, alcoholic liver disease; H&E, hematoxylin and eosin; NAFLD, nonalcoholic fatty liver disease.
Steatosis
Hepatocellular steatosis progresses from small‐droplet fat (formerly called microvesicular steatosis) to large‐droplet fat, so the pattern is usually mixed (Fig. 1). As stated previously, steatosis begins in zone 3 and extends outward toward the portal tract with increasing severity. Steatosis may exist alone or in combination with the other changes of steatohepatitis or steatofibrosis.
Figure 1.
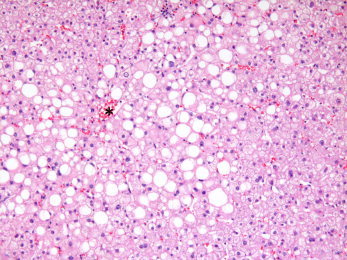
Moderate alcohol‐related steatosis. Steatosis, composed of small‐ and large‐droplet fat (as usual in fatty liver disease), is most prominent near the central vein (asterisk) and extends outward toward the portal tract with increasing severity. In this case, the fat extends into acinus zone 2 or the midzone of the hepatic lobule (H&E, ×4).
Alcoholic foamy degeneration is an uncommon form of ALD steatosis. The pathological appearance comprises microvesicular steatosis in virtually all hepatocytes, similar to that seen in diseases with mitochondrial dysfunction, such as Reye syndrome and fatty liver of pregnancy1, 4 (Fig. 2). It usually occurs without histological steatohepatitis or steatofibrosis despite chronic alcohol ingestion. Clinically, the serum gamma‐glutamyl transferase level is usually markedly elevated with or without transaminase elevations, and the patient may display severe acute hepatotoxicity, although the histologic change may also be present in an otherwise typical chronic alcohol user.4
Figure 2.
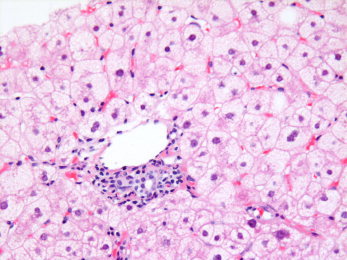
Acute alcoholic foamy degeneration. This condition shows diffuse microvesicular steatosis, an hepatic fatty change that indicates a very different process than other causes of fatty liver disease, usually indicating acute mitochondrial dysfunction. The small‐droplet fat is found in virtually all hepatocytes (here shown in the periportal region). This change is most often associated with toxins (e.g., Reye syndrome, valproate, and tetracycline), alcohol being another toxin that can sometimes show the same pattern (H&E, ×10).
Megamitochondria may be seen in association with steatosis, with or without other changes of ALD, further indicating the potential for alcohol to cause mitochondrial defects.1, 2
Steatohepatitis
Steatohepatitis includes all forms of hepatocyte injury and degeneration, whether or not there is associated inflammation. The most typical feature is hepatocyte ballooning with cell swelling, rarefaction of the cytoplasm, and disruption of the cytoskeleton, often with the formation of Mallory‐Denk bodies (Fig. 3). Apoptosis may also happen, with acidophil bodies sometimes still containing fat or Mallory‐Denk bodies. Again, these changes begin in the centrilobular region and extend toward portal tracts with increasing severity.
Figure 3.
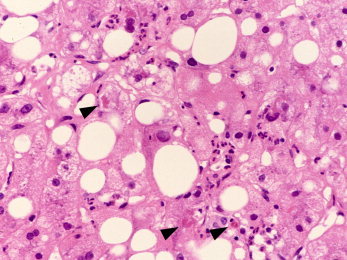
ALD with steatosis and steatohepatitis. Steatosis is present in the specimen, but so is hepatocyte ballooning with Mallory‐Denk bodies (arrows) and a diffuse inflammatory infiltrate rich in neutrophils. These features, together or separately, are classic hallmarks of alcoholic hepatitis (H&E, ×40; image courtesy of Wilson Tsui, Hong Kong, 2012).
Inflammation is variable and ranges from scant/mild, predominantly mononuclear infiltration of the portal tracts and hepatic parenchyma to more characteristic neutrophilic predominance, which either is focal around ballooned hepatocytes or courses diffusely through the lobule (Figs. 3 and 4).
Figure 4.
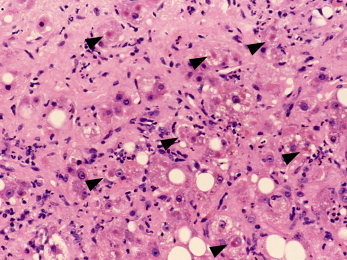
ALD with steatosis, steatohepatitis, and steatofibrosis. There can be seen diffuse steatosis, widespread steatohepatitis in the forms of hepatocyte ballooning (often with Mallory‐Denk bodies; arrows) and neutrophilia, and extensive pericellular steatofibrosis that, although more subtle with H&E stains versus trichrome stains, is clearly prominent and diffuse in this specimen (H&E, ×20; image courtesy of Wilson Tsui, Hong Kong, 2012).
Steatofibrosis and Progression to Late‐Stage Liver Disease
Steatofibrosis begins with perivenular fibrosis that extends outward along the sinusoids (Figs. 5 and 6). The accumulation initially is primarily in the space of Disse. Because this pericellular or perisinusoidal fibrosis extends outward, it shows a classic chicken‐wire fence pattern, sometimes all the way to the portal tract. In mild or early scarring, the changes may be virtually invisible on routine hematoxylin and eosin (H&E) stains; a trichrome or reticulin stain is essential for a sensitive assessment of the fibrotic stage of ALD.
Figure 5.
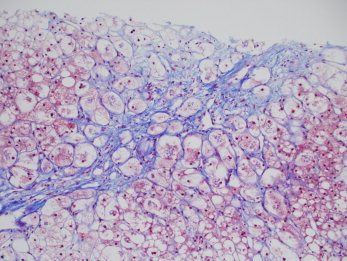
Early though prominent alcohol‐associated steatofibrosis. The trichrome stain highlights the perivenular and pericellular (or perisinusoidal) collagen deposition typical of ALD and other fatty liver diseases. This pattern is often called chicken‐wire fence fibrosis. The perisinusoidal fibers are often so thin and delicate that they may be greatly underestimated by H&E stains; therefore, collagen stains are important for the full evaluation of the stage of scarring in ALD. Note also that the central vein has been essentially obliterated by the scar; this event is often called sclerosing hyaline necrosis. Steatosis and steatohepatitis are also prominent in this specimen (chromotrope aniline blue, ×4; image courtesy of Linda Ferrell, University of California at San Francisco, 2012).
Figure 6.
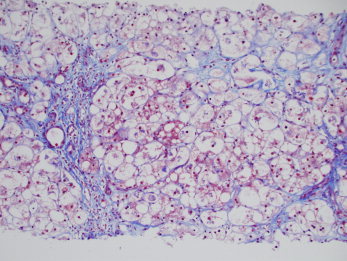
Later stage progression of alcohol‐associated steatofibrosis. The trichrome stain shows the linking of central veins to portal tracts. Over time and with continued alcohol use, areas with steatofibrosis develop hepatocyte atrophy, which leads to the collapse of such scars, whereas regeneration in areas with less scarring further compresses these regions. Eventually, these fibrous webs condense into compacted fibrous septa. However, with continued alcohol use, further steatofibrosis continues to subdivide regenerative nodules, and this leads to classic micronodular Laennec's cirrhosis of late‐ to end‐stage chronic alcohol use. Steatosis and steatohepatitis are also prominent in this specimen (chromotrope aniline blue, ×4; image courtesy of Linda Ferrell, University of California at San Francisco, 2012).
The progression of pericellular scarring to the end‐stage liver disease of typical micronodular Laennec's cirrhosis is complex (Fig. 6). Repeated cycles of hepatocellular regeneration within the fibrous webs lead to expansile hepatocyte nodules compressing adjacent hepatocytes and leading to their atrophy and the condensation of the fibrous scars into dense septa.2 With subsequent scarring of these newer hepatocytic nodules, the regenerated parenchyma continues to subdivide and becomes micronodular.
These cycles of injury and regeneration are parallel to parenchymal extinction resulting from double‐hit vascular injuries (to central veins and to one or both of the portal vessels).5 Subsequent vascular shunting leads to further fibrotic parenchymal collapse. Eventually, after years, hepatocytes undergo replicative senescence, at which point activation of the stem/progenitor cell compartment (i.e., a ductular reaction) becomes increasingly widespread.6 In a positive‐feedback loop, further vascular shunting follows these distortions and leads finally to end‐stage Laennec's cirrhosis.
Changes With Abstinence
With the cessation of drinking, steatosis rapidly disappears, probably within weeks, and this is followed by a subsidence of the inflammatory changes.7 Mallory‐Denk bodies may persist longer (up to 6 months).8 Finally, even some of the fibrotic changes can regress.8 Early lesions of pericellular fibrosis may disappear altogether with the cessation of alcohol use. A comparison of end‐stage cirrhotic livers from active drinkers and livers resected at transplant after more than 6 months of abstinence shows that later stage scarring and nodularity also remodel.9 The pattern becomes increasingly macronodular as fibrous scars regress: they first become densely compacted and then become fragmented with the coalescence of adjacent nodules. Whether alcoholic cirrhosis can regress altogether in rare patients remains an open question.
Distinguishing ALD From NAFLD
Two changes seen on biopsy may confidently be used to distinguish ALD from NAFLD. The first is marked cholestasis, which is usually indicative of acute decompensation (hepatocyte keratin 7 immunostaining becomes particularly prominent10), and the second is sclerosing hyaline necrosis, in which the central vein is essentially obliterated1, 2 (Fig. 5).
Other changes differ only in degree. Steatohepatitis may be far more prominent in ALD versus NAFLD in populations of affected patients—neutrophilic infiltrates may be more widespread, Mallory‐Denk bodies may be thicker and coarser, and mega‐mitochondria may be more plentiful—but there is substantial overlap in the findings between individual patients in the two disease categories. In the absence of a clinical history to indicate the nature of the patient's disease, the histology is not determinative.
On the other hand, pathologists and clinicians alike should remain aware that some patients with NAFLD may also be drinking. One should not rest contented with a diagnosis of metabolic syndrome as the sole etiology, particularly when the histological picture is very severe and, therefore, suggestive (but only suggestive) of ALD. Because of the potential social, psychological, and economic impacts of labeling a patient as alcoholic in a written part of the medical record, it is probably best for the diagnosing pathologist to make personal contact with the clinician to convey a heightened suspicion of alcohol use.
Acknowledgements
This article was completed by candlelight in the blackout zone of lower Manhattan in the aftermath of Hurricane Sandy only with the generous and profoundly appreciated support of members of the International Liver Pathology Study Group (the “Elves”) and the Laennec Hepatopathology Society (particularly Prithi Bhathal, James Crawford, Elizabeth Brunt, Linda Ferrell, Dale Snover, and Wilson Tsui).
Potential conflict of interest: Nothing to report.
References
- 1. Lefkowitch JH. Morphology of alcoholic liver disease. Clin Liver Dis 2005; 9: 37–53. [DOI] [PubMed] [Google Scholar]
- 2. Crawford JM. Histologic findings in alcoholic liver disease. Clin Liver Dis 2012; 16: 699–716. [DOI] [PubMed] [Google Scholar]
- 3. European Association for the Study of Liver . EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol 2012; 57: 399–420. [DOI] [PubMed] [Google Scholar]
- 4. Suri S, Mitros FA, Ahluwalia JP. Alcoholic foamy degeneration and a markedly elevated GGT: a case report and literature review. Dig Dis Sci 2003; 48: 1142–1146. [DOI] [PubMed] [Google Scholar]
- 5. Crawford JM, Burt AD. Anatomy, pathophysiology and basic mechanisms of disease In: Burt A, Portmann B, Ferrell L, eds. MacSween's Pathology of the Liver. 6th ed. Edinburgh, United Kingdom: Churchill Livingstone; 2012: 1–78. [Google Scholar]
- 6. Gouw AS, Clouston AD, Theise ND. Ductular reactions in human liver: diversity at the interface. Hepatology 2011; 54: 1853–1863. [DOI] [PubMed] [Google Scholar]
- 7. Brunt EM, Neuschwander‐Tetri BA, Burt AD. Fatty liver disease: alcoholic and non‐alcoholic In: Burt A, Portmann B, Ferrell L, eds. MacSween's Pathology of the Liver. 6th ed. Edinburgh, United Kingdom: Churchill Livingstone; 2012: 293–360. [Google Scholar]
- 8. Galambos JT. Natural history of alcoholic hepatitis. 3. Histological changes. Gastroenterology 1972; 63: 1026–1035. [PubMed] [Google Scholar]
- 9. Zhu CC, Li P, Zhu L, Sidhu G, John D, Teperman L, et al. Evidence for regression of alcoholic cirrhosis with abstinence. Arch Pathol Lab Med 2004; 2: 183. [Google Scholar]
- 10. Sancho‐Bru P, Altamirano J, Rodrigo‐Torres D, Coll M, Millán C, José Lozano J, et al. Liver progenitor cell markers correlate with liver damage and predict short‐term mortality in patients with alcoholic hepatitis. Hepatology 2012; 55: 1931–1941. [DOI] [PubMed] [Google Scholar]