

Watch a video presentation of this article
Watch the interview with the author
Abbreviations
- ALIX
ALG‐2 interacting protein X
- ALT
alanine aminotransferase
- Anti‐HAV
antibody to hepatitis A virus
- CD4/CD8
cluster of differentiation 4 and 8 T cells
- eHAV
hepatitis A virus encased in host‐derived membranes
- ESCRT‐III
endosomal sorting complex required for transport III
- GI
gastrointestinal
- HAV
hepatitis A virus
- HAVcr1/TIM‐1
hepatitis A virus cellular receptor 1/T cell transmembrane, immunoglobulin and mucin gene family
- HAV 3C
hepatitis A virus nonstructural 3C protease
- Huh7.5
human hepatoma cell line
- IgM/IgG
immunoglobulin G and M
- IM
intramuscular
- NK
natural killer cell
- RNA
ribonucleic acid
- VPS4B
vacuolar protein sorting 4 homolog B
Hepatitis A has been around for centuries, perhaps dating back to the 8th century A.D. or earlier according to Chinese literature. It is one of the most common causes of infectious jaundice in the world and is often associated with epidemics. The natural history of this infection (Fig. 1) begins with a relevant exposure to hepatitis A virus (HAV) in a susceptible person, typically through the fecal–oral route, but may include blood‐borne transmission in some situations. Following ingestion of HAV from material contaminated with feces, trafficking to the liver occurs via the gastrointestinal (GI) tract, ultimately resulting in hepatitis after replication of the virus in hepatocytes (reviewed by Hollinger and Martin1). The question of whether an independent GI replication occurs has not been resolved, although detection of genomic material and antigens at various levels of the GI tract following infection of owl monkeys and a rebound of fecal HAV RNA shedding during relapsing hepatitis in HAV‐infected chimpanzees without a concomitant rise in HAV RNA in the serum or liver suggests this possibility. Regardless, the primary site of replication resides in the liver and occurs within hours or days after infection. Attachment and entry is felt to be in a calcium‐dependent manner, with binding to a cellular receptor such as HAVcr1/TIM‐1 followed by slow uncoating and release of viral RNA into the cytoplasm within 4 hours.2 The receptor comprises an N‐terminal immunoglobulin‐like domain and a mucin‐like domain.3 The former domain is sufficient for binding, but both regions are necessary for uncoating. Another possible mechanism of entry involves immune complexes of HAV and virus‐specific immunoglobulin A antibody, present in significant amounts during infection, that lead to receptor‐mediated endocytosis through hepatic asialoglycoprotein receptors.4
Figure 1.
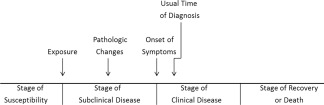
Natural history timeline. Modified with permission from Centers for Disease Control and Prevention. Principles of Epidemiology. 2nd ed. Atlanta, GA: US Department of Health and Human Services; 1992. Copyright 1992, Centers for Disease Control and Prevention.
Following genome translation, polyprotein processing, and replication, HAV is released into bile canaliculi and is transported to the intestine, where it is detected in the stool at concentrations that can range up to 109 infectious virions per gram of stool.1 This occurs during the incubation period or subclinical stage of the disease before alanine aminotransferase (ALT) levels become elevated and before clinical symptoms or jaundice ensue (Fig. 2). Communicability is highest during this stage but is severely curtailed following the onset of symptoms or jaundice, requiring larger concentrations of fecal material or serum to cause an infection (Fig. 3).5 Higher concentrations of virions are observed in the stools of patients with jaundice than in those without jaundice or clinical disease. Concurrently, virus is observed in blood (viremia) at levels that are at least 1000 times lower than those found in the stool. It precedes the appearance of clinical and laboratory evidence of hepatitis by at least 2 weeks, but diminishes dramatically when the ALT begins to rise. An early replicative event also has been affirmed in the oropharynx and salivary glands of orally inoculated chimpanzees and marmosets shortly after the appearance of virus in the blood.6, 7 Infectious virus also has been detected in low concentrations in the saliva of patients during acute disease, but its presence may be due to contamination by serous crevicular fluid released from the root–capillary interface during mastication.
Figure 2.
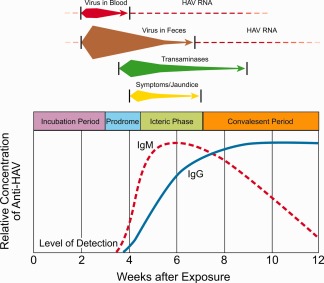
Immunologic and clinically relevant biologic events associated with HAV infection in humans based on stages of disease. Reprinted with permission from Fields Virology.1 Copyright 2013, Lippincott Williams & Wilkins.
Figure 3.
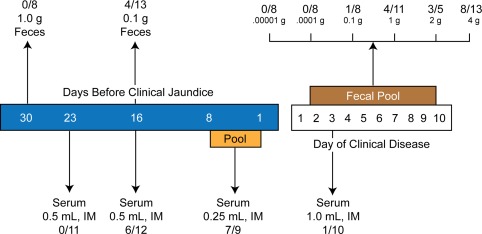
Communicability of HAV from the feces and serum of infected humans before and after jaundice or clinical disease (the number of subjects infected is followed by the number tested per quantity of sample). The infectivity rate may be underestimated because susceptibility of the subjects was not known.5 Reprinted with permission from Fields Virology.1 Copyright 2013, Lippincott Williams & Wilkins.
In contradistinction to the transient infection seen in humans, cell culture–adapted virus often leads to noncytolytic changes and long‐term persistence in vitro. Recently, there has been a resurgence of interest in the possibility that HAV also may persist in the blood and stool of patients and that this might have clinical implications. This had its origins in a number of earlier studies in which fecal shedding of presumably encapsidated HAV RNA occurred for up to 5 months after hepatitis developed, mostly in infants or preterm neonates, but also in the 3%‐20% of patients who develop recrudescence of their disease, often designated relapsing hepatitis.1 This clinical condition is usually less severe than the original episode and occurs 4–15 weeks after the initial symptoms have resolved. In some patients, only biochemical changes are observed. Immunoglobulin M anti‐HAV either reappears or increases in titer, and HAV genomic material may be detected in feces and serum. More than one relapse can occur, and enzyme elevations can persist for 5–12 months, although chronic sequelae are not observed. In addition, HAV RNA and antigen have been found to persist in the liver of infected chimpanzees more than 30 weeks after ALT levels have returned to normal and hepatic inflammation has resolved.8
In conjunction with these observations, HAV also has been found to circulate in the blood of some infected patients and chimpanzees for >10 weeks surrounded by lipid‐associated membrane fragments at concentrations several orders of magnitude less than in acute‐phase samples and always accompanied by HAV‐specific antibody. In a recent series of innovative experiments, Feng et al.9 showed that HAV released from Huh7.5 cells is encased in host‐derived membranes, which they term eHAV, protecting the virion from in vitro antibody‐mediated neutralization. The enveloped particles appear to be fully infectious and have been found to circulate in the blood of infected humans and chimpanzees. In contrast, virus in feces was nonenveloped. They found that the endosomal sorting complex required for transport (ESCRT‐III)‐associated proteins (VPS4B and ALIX), which are known to facilitate the budding of many enveloped viruses, were apparently used by HAV for egress of both eHAV and nonenveloped HAV from the cell. The authors postulate that this cloaking of the HAV particle allows eHAV to evade neutralizing antibodies and could explain how virus and anti‐HAV might coexist in serum. Unfortunately, these studies apparently did not evaluate eHAV in sera collected from patients in the late acute or convalescent phase of the disease, when antibody was present, to determine whether the low buoyant density eHAV particles were still present. Despite these novel experiments, the clinical significance of this observation remains uncertain, because transmission or infectivity of blood components or fecal matter containing HAV genomic material in the late acute or convalescent stages of the disease has never been documented. Similarly, if this phenomenon could be generalized to potential blood donors who donate in the late convalescent phase of their infection, a number of transfusion‐transmitted HAV infections should have been detected, and this has not occurred. Correspondingly, it is reassuring to know that the current recipient risk of acquiring HAV infection following the administration of blood components from asymptomatic donors who donate in the seronegative window period is very low, based on the fact that the HAV nucleic acid–reactive yield during this interval ranges from 1 positive donation in 120,000 to 1,805,500 donations (S. Stramer, personal communication, 2011).
The outcome of hepatitis A infection varies from inapparent or subclinical hepatitis (neither symptoms nor jaundice) that is usually observed in children, to anicteric hepatitis (symptoms without jaundice) or icteric hepatitis that occur primarily in adults leading occasionally to fulminant hepatitis and death, especially in patients older than 50 years (Table 1). More than 70% of adult clinical cases are jaundiced. The molecular mechanisms and immune responses responsible for these outcomes are being addressed and can be summarized as follows. Clinical studies in humans and chimpanzees have shown intrahepatic disruption of the two major cellular interferon‐activating pathways by HAV 3C protease precursors that are formed during the initial stages of genome translation and polyprotein processing.1, 10, 11 Thus, HAV may have evolved strategies to evade protective host innate immune responses typically induced by double‐stranded RNA replicating forms and intermediates, thereby facilitating HAV replication during the clinically silent early stages of the infection.8 Subsequently, hepatocellular injury and viral clearance ensue, but the mechanisms responsible for these events have only partially been characterized. A direct cytopathic effect can be excluded due to the presence of large quantities of virus in the liver and stools prior to the onset of hepatitis, as well as the fact that wild‐type HAV is not cytopathic in cell culture, nor does it lead to alterations in cellular metabolism.
Table 1.
Predicted Outcome After HAV Infection
| Parameter | Predicted Outcome | |
|---|---|---|
| Children (<5 yr) | Adults | |
| Inapparent infection | 80%‐95% | 10%‐25% |
| Anicteric or icteric disease | 5%‐20% | 75%‐90%a |
| Complete recovery | ≥99% | ≥98% |
| Chronic disease | None | None |
| Mortality rate (hospitalized cases) | ||
| <30 yr | 0.23% | |
| 30‐49 yr | 0.3%‐0.6% | |
| >49 yr | 1.8%‐2.1% | |
| Fuliminant hepatitis | <1% | |
| Relapsing hepatitis | 3%‐20% | |
| Prolonged cholestasis | 5% | |
More than 70% of adult clinical cases are icteric.
In contrast, clinical hepatitis and recovery appears to coincide with the emergence of robust humoral and cellular immune responses (reviewed by Hollinger and Martin1). For example, nonspecific immune mechanisms involving natural killer (NK) cells have been postulated to play a role in hepatocellular damage, since NK cells isolated from acute hepatitis A patients are capable of lysing cultured HAV‐infected cells. In addition, human leukocyte antigen–restricted, HAV‐specific, CD8+ cytotoxic T cells, which are capable of inducing an interferon‐γ response following exposure to HAV‐infected cultured cells, have been isolated from the liver and blood of patients with acute hepatitis A. The possible role of host characteristics or specific subtypes of HAV and their relationship to outcome has been explored. Accordingly, the occurrence of liver failure from acute HAV infection in first‐degree relatives and the presence of HAV subgenotype IB in many cases of fulminant hepatitis A are noteworthy.12 In terms of viral clearance, a recent study in chimpanzees suggests that CD4+ T cells may play a significant role, because multiple cytokines are expressed by these cells in association with viral clearance or transient relapses, whereas no similar relationship was seen with CD8+ T cells.13 The authors postulate that failure to maintain a CD4+ T cell response may increase the risk of relapsing hepatitis A. Additionally, the presence of HAV‐specific immunoglobulin A antibodies produced by the GI mucosa‐associated lymphoid tissue may allow for enterohepatic cycling of HAV causing endogenous reinfection of the liver in the presence of neutralizing immunoglobulin G antibodies contributing to relapsing disease. Finally, humoral immunity unequivocally plays a major role in viral clearance, as viremia promptly declines following the appearance of neutralizing antibodies.14
Long‐term immunity through universal immunization is becoming a reality and will eventually lead to a decline in morbidity and mortality.15 Two types of monovalent hepatitis A vaccines are available globally: formaldehyde‐inactivated, alum‐ or influenza‐virosome adjuvanted vaccines (Havrix®, Vaqta®, Avaxim®, Healive®, Epaxal®) and live, attenuated, freeze‐dried vaccines (H2 and LA‐1) that have been developed in China. In addition, there are combination vaccines that combine the hepatitis A antigenic component with other vaccines such as the hepatitis B vaccine (Twinrix®, Twinrix Junior®, Ambirix®) and typhoid vaccines (ViATIM® or Vivaxim®, Hepatyrix®). All of the inactivated vaccines, except Epaxal, are free of thimerosol as a preservative and are safe and comparably efficacious. Herd immunity is often observed in the nonvaccinated adult population following large‐scale childhood vaccination programs. Reversion of the live, attenuated vaccines to a more virulent strain has not been observed, and single‐dose injections are as effective as two doses of the inactivated vaccines.
Potential conflict of interest: Nothing to report.
Supported in part by the Eugene B. Casey Foundation and the William and Sonya Carpenter Fund, Baylor College of Medicine (F.B.H.).
REFERENCES
- 1. Hollinger FB, Martin A. Hepatitis A virus In: Knipe DM, Howley PM, eds. Fields Virology. 6th ed. Philadelphia: Lippincott Williams & Wilkins; pp. 550–581, 2013. [Google Scholar]
- 2. Bishop NE, Anderson DA. Uncoating kinetics of hepatitis A virus virions and provirions. J Virol 2000;74:3423‐3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thompson P, Lu J, Kaplan GG. The Cys‐rich region of hepatitis A virus cellular receptor 1 is required for binding of hepatitis A virus and protective monoclonal antibody 190/4. J Virol 1998;72:3751‐3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dotzauer A, Gebhardt U, Bieback K, Göttke U, Kracke A, Mages J, et al. Hepatitis A virus‐specific immunoglobulin A mediates infection of hepatocytes with hepatitis A virus via the asialoglycoprotein receptor. J Virol 2000;74:10950‐10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krugman S, Ward R, Giles JP, Bodansky O, Jacobs AM. Infectious hepatitis: detection of virus during the incubation period and in clinically inapparent infection. N Eng J Med 1959;261:729‐734. [DOI] [PubMed] [Google Scholar]
- 6. Cohen JI, Feinstone S, Purcell RH. Hepatitis A virus infection in a chimpanzee: duration of viremia and detection of virus in saliva and throat swabs. J Infect Dis 1989;160:887‐890. [DOI] [PubMed] [Google Scholar]
- 7. Pinto MA, Marchevsky RS, Baptista ML, de Lima MA, Pelajo‐Machado M, Vitral CL, et al. Experimental hepatitis A virus (HAV) infection in Callithrix jacchus: early detection of HAV antigen and viral fate. Exp Toxicol Pathol 2002;53:413‐420. [DOI] [PubMed] [Google Scholar]
- 8. Lanford RE, Feng Z, Chavez D, Guerra B, Brasky KM, Zhou Y, et al. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc Natl Acad Sci U S A 2011;108:11223‐11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, et al. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013;496:367‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qu L, Feng Z, Yamane D, Lanford RE, Li K, Lemon SM. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease‐polymerase processing intermediate, 3CD. PLoS Pathog 2011;7:e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fensterl V, Grotheer D, Berk I Schlemminger S, Vallbracht A, Dotzauer A. Hepatitis A virus suppresses RIG‐I‐mediated IRF‐3 activation to block induction of beta interferon. J Virol 2005;79:10968‐10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ajmera V, Xia G, Vaughan G, Forbi JC, Ganova‐Raeva LM, Khudyakov Y, et al. What factors determine the severity of hepatitis A‐related acute liver failure? J Viral Hepat 2011;18:e167‐e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou Y, Callendret B, Xu D, Brasky KM, Feng Z, Hensley LL, et al. Dominance of the CD4(+) T helper cell response during acute resolving hepatitis A virus infection. J Exp Med 2012;209:1481‐1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lemon SM, Murphy PC, Provost PJ, Chalikonda I, Davide JP, Schofield TL, et al. Immunoprecipitation and virus neutralization assays demonstrate qualitative differences between protective antibody responses to inactivated hepatitis A vaccine and passive immunization with immune globulin. J Infect Dis 1997;176:9‐19. [DOI] [PubMed] [Google Scholar]
- 15. Shouval D. Module 18: hepatitis A. In: The Immunological Basis for Immunization Series. Geneva, Switzerland: World Health Organization; 2011:1‐39. [Google Scholar]