

Watch a video presentation of this article
Watch the interview with the author
Autoimmune liver diseases are rare and chronic and exert a significant burden on quality and quantity of life. The failure to understand the true etiopathogenesis of disease leaves clinicians diagnosing patients based on collective positive and negative clinical findings, such that three particular autoimmune diseases are apparent (Table 1). In the clinical setting, patients frequently exhibit features that overlap between these entities, a common concern for clinicians.1, 2 The fact that patients have overlapping features of autoimmune liver injury should, however, not be used to define new syndromes, nor should it be used as evidence that therapy must be applied; overlap is the inherent result of syndromic diagnosis based on collective serologic, histologic, and radiologic findings. It is also a reflection of the inadequacy of present diagnostic labels: it is not difficult to see that patients with primary biliary cirrhosis (PBC), primary sclerosing cholangitis (PSC), and autoimmune hepatitis (AIH) are much more varied in their presentation and outcome, than three disease labels does credit for.
Table 1.
Features of AIH, PBC, and PSC
| Feature | AIH | PBC | PSC |
|---|---|---|---|
| Sex | Females: 60%‐75% | Females: >90% | Females: 30%‐35% |
| Age | All age groups; median age ∼45 years | Typically 30‐65 years; not diagnosed in children | Typically 30‐50 years, but all age groups |
| Aminotransferases | Markedly elevated, often 3‐ to 10‐fold, but may be normal or only minimally elevated | Normal or slightly elevated | Normal or slightly elevated |
| ALP | Elevated levels may be seen | Moderately to markedly elevated | Moderately to markedly elevated (typically at least 3× ULN; but variable levels, may even be normal) |
| Bilirubin | Variable increase | Variable increase, but normal in majority at diagnosis | Variable increase, but normal in majority at diagnosis |
| Immunoglobulins | Hyper‐gammaglobulinemia, especially elevated IgG (generally elevated 1.2‐3.0× ULN) | IgM increased in most patients | IgG increased in up to 61%; IgM increased in up to 45% |
| Autoantibodies | |||
| ANA, SMA | Significant titers (≥1:40) of ANA and/or SMA in 70%‐80% | ANA in >30% (anti‐gp210 and anti‐Sp100 highly specific); SMA may be present | ANA in 8%‐77%; SMA in 0%‐83% |
| Anti‐LKM | Anti‐LKM in 3%‐4% | ||
| Anti‐SLA/LP | Anti‐SLA/LP in 10%‐30% | Anti‐SLA/LP may be detected | Anti‐SLA/LP may be detected |
| pANCA | pANCA in 50%‐96% (often atypical, pANNA); conventional autoantibodies not detected in up to 10% | pANCA in 26%‐94% | |
| AMA | AMA in low titer occasionally seen (AMA anti–PDC‐E2 pattern rarely detected) | AMA in 90%‐95% (AMA anti–PDC‐E2 pattern highly specific) | AMA occasionally positive |
| Liver biopsy | |||
| Interface hepatitis | Typical findinga | In a proportion of casesb | In a variable number of casesc |
| Portal inflammation | Portal plasma cell infiltrate | Portal lymphocytic infiltrate | Portal lymphocytic infiltrate |
| Biliary changes | In a proportion of cases | Typical | Typical |
| Granulomas | Atypical | Suggestive of PBC, but invariably present | Atypical, but may be observed |
| Cholangiography | Normal or signs of liver cirrhosis | Normal or signs of liver cirrhosis | Characteristic findings, diagnostic of PSC; normal cholangiography in small duct PSC |
| Irritable bowel disease | Rarely associated with AIH; PSC should be excluded | Rarely associated with PBC | Present in up to 80% |
Abbreviations: IgG, immunoglobulin G; IgM, immunoglobulin M; pANCA, perinuclear anti‐neutrophil cytoplasmic antibodies; pANNA, anti‐neutrophil nuclear antibody; PDC‐E2, pyruvate dehydrogenase complex‐E2; LKM, Liver Kidney Microsomal; SLA/LP, soluble liver antigen/liver‐pancreas.
A diagnosis of definite AIH should not be concluded without a liver biopsy.
A liver biopsy is not required in AMA‐positive cases. In early disease, characteristic features are uncommon.
A liver biopsy is not necessary for the diagnosis of large duct PSC but is required for the diagnosis of small duct PSC. Based on Trivedi and Hirschfield.2
Adapted from Reference 2.
The often slow natural history of autoimmune liver disease, the absence of disease‐specific markers, and the invasive nature and sampling error of liver biopsy result in a clinical reliance on surrogates of disease activity. The way in which one applies surrogates of treatment efficacy must be critically appraised when faced with overlap presentations. The ability of changes to alkaline phosphatase levels in PBC to predict outcome3 is not comparable in PSC, where the same treatment has been trialed,4 and similarly the efficacy of corticosteroids in AIH5 does not imply that steroid treatment for a significant transaminitis associated with interface hepatitis in PBC is equally beneficial to patients.6
Underlying Concepts
Overlap autoimmune liver syndromes are best not considered as distinct entities, but more the reflection of an inherent distribution of clinical features across patient populations presenting with autoimmune liver disease. The more extreme the distribution, the more distinct the apparent overlap, and the greater the likelihood that treatment based on the classic autoimmune liver disease distinctions will be of value. Clinically, overlap should be considered in the differential when a patient deviates from the normal clinical course and expected response to therapy, but it is not necessary to overdiagnose, nor is it necessary at presentation of a predominant disease process to consider overlap a means of justifying nonstandard therapy. The prevalence of overlap features is difficult to ascertain because of publication bias, challenges in definitions, and limitations in test interpretation, particularly those that are qualitative and/or subjective. Overlap designations, therefore, tend to be arbitrary and imprecise, and the clinical phenotypes of patients with the same overlap designation exhibit considerable heterogeneity. Patient management should focus on careful evaluation of immune markers of liver injury and therapy tailored to the individual, with close attention to avoidance of side effects and inappropriate application of surrogates of treatment efficacy. The timing of overlap features in the course of the patient illness is also of relevance: greater benefit from treatment is likely when overlaps are very distinct in time, as the early phases of disease presentation in autoimmune liver disease can commonly have indistinct features, in which case patients are best re‐evaluated after treatment of the predominant process.
In autoimmune liver disease, it is recognized that if you have abnormal tests on or off therapy, then disease progression is likely. Clinicians also recognize in PBC, for example, that the degree of interface hepatitis associates with disease progression to cirrhosis; where they don't agree is the mechanism of the interface hepatitis (Fig. 1). In this context, when clinicians broadly apply the term “overlap syndrome” for instances in which (concurrently or consecutively) there exists a coexistence of immune‐mediated hepatitis, as well as features of either PBC or PSC, they are in essence reflecting their unease that a patient is at risk of poor outcome. However, for these diseases, clinicians need to develop the confidence to accept some uncertainty for their patients, particularly as long as we lack true disease mechanisms.
Figure 1.
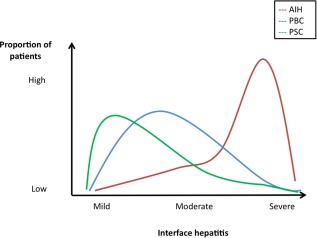
The inherent distribution of features of autoimmune liver disease. Interface hepatitis is a good example of a facet of all autoimmune liver disease that varies in its prominence. In AIH, the majority of patients will have a significant degree of interface hepatitis; however, a degree of interface hepatitis can be also observed in a proportion of patients with PBC and PSC. Approximately 10% of patients with AIH may also have histologic features of bile duct injury. A similar graph can be adopted for other parameters such as autoantibody titer, serum immunoglobulins, and liver biochemistry (e.g. 5%‐20% of patients with AIH have anti‐mitochondrial antibodies (AMAs), 15% have increased serum levels of immunoglobulin M, and 19% have a disproportionate elevation of serum ALP). Adapted with permission from Alimentary Pharmacology & Therapeutics.2 Copyright 2012, Wiley‐Blackwell.
Patient presentation must be kept in context, particularly with recognition of liver disease epidemiology and outcomes:
Autoimmune hepatitis is a syndrome of immune‐mediated hepatitis that is especially sensitive to corticosteroids, with usual normalization of liver biochemistry, and for the majority of patients a nonspecific immunoserologic reactivity (i.e., non–disease‐specific antinuclear antibody [ANA] reactivity): interface hepatitis on histology cannot be taken to equal classic steroid responsive autoimmune hepatitis, and there is a clear need for a more detailed systematic comparison of the features of interface hepatitis in classic PBC compared with classic AIH, for example.
PBC is an immune‐mediated cholangitis in which treatment response is associated with age at presentation (50% of those presenting under the age of 50 will fail treatment with ursodeoxycholic acid [UDCA]7), disease progression is associated with not only the degree of ductopenia but also the degree of interface hepatitis, and patients can have evidence of serologic reactivity beyond antimitochondrial antibodies (AMAs) (specific ANA reactivity as characterized by gp210 or sp100 positivity)—in other words, one should not (a) mistake a nonresponse to UDCA as overlap, (b) forget to look for specific ANA reactivity in patients who are AMA‐negative, or (c) assume that interface activity in PBC has the same significance, pathophysiology, or treatment response, as it does in AIH.
The biliary inflammation associated with PSC will classically give a cholestatic hepatitic profile with elevation of both alkaline phosphatase (ALP) and alanine aminotransferase (ALT), and recognizing pediatric perspectives highlights how features can overlap: young patients with AIH will have concurrent cholangiopathy 50% of the time8 (i.e., the younger the patient, the more hepatitic the features of PSC may be).
The Spectrum of Overlap
Presentations that raise the spectrum of overlap span:
immunoserologic overlap (e.g., positive ANA/anti–smooth muscle antibody [SMA] titers and elevated immunoglobulin G in conjunction with AMA‐positive PBC or AMA positivity in AIH);
biochemical overlap (aspartate aminotransferase/ALT >5× the upper limit of normal [ULN] in patients with PBC or PSC or ALP >3× ULN in patients with AIH [or gamma‐glutamyltransferase >5× ULN in children]);
radiologic overlap: clinical features of AIH with cholangiographic abnormalities indicative of inflammatory cholangiopathy;
histologic overlap: lymphoplasmacytic infiltrate and interface hepatitis on liver biopsy with bile duct lesions indicative of either PBC or PSC; and
varying combinations of the above, including temporally (i.e., consecutive versus sequential presentations).
Appraisal must be performed longitudinally rather than at a single point in time, and in particular the stratification of disease by time and response to initial monotherapy is very helpful: hence defining patients as having true overlap syndromes at presentation seems inevitably flawed and a root cause of much confusion (Fig. 2). If there is evidence at all for true overlaps, it occurs in those very rare patients that have distinct changes of presentation over prolonged follow‐up; in this setting, one either defines them as overlap or the coincidental occurrence of two diseases in one patient over time.
Figure 2.
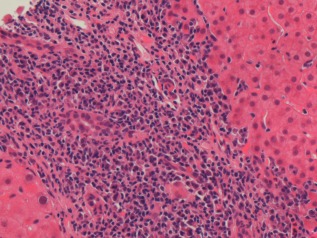
Interface hepatitis in PBC. At presentation, this patient was AMA‐positive with a cholestatic liver chemical profile. Because of an ALT that was >5× ULN alongside an immunoglobulin G level of 20 g/L and SMA reactivity, liver biopsy was performed. This demonstrated significant interface hepatitis. However, treatment with UDCA alone led to a complete normalization of liver chemistry and immunoglobulins at 6 months. Notably, when patients in a clinical trial of PBC were identified as having potential AIH overlap retrospectively, it was clear that the clinical course on UDCA treatment was not distinct compared with classical PBC patients, nor was outcome.13
Diagnostic Criteria?
It is of course possible to conceive diagnostic criteria for overlap presentations (Table 2), but one must evaluate any such approach critically: In the development of criteria for classifying an autoimmune liver disease, what was the purpose of the scoring system? Was it to create a homogenous population of patients that are easily compared, to disprove that overlap exists, or to champion the concept of overlap and a different approach to therapy? Any such criteria must also be critically appraised: Is the presence of an autoantibody of equal significance to histologic observations? In the absence of rules that work, clinicians must individualize their approach at times for autoimmune liver disease and maintain a balanced outlook (e.g., a nonjaundiced patient rarely has a need for emergency intervention). Historical definitions and descriptors of an overlap syndromes should perhaps give way to a culture by which an individual's disease will be defined by the history, simultaneous and sequential appropriate investigations, and response to therapy.
Table 2.
Paris Criteria for AIH/PBC Overlap11
| AIH (2 out of 3 criteria) |
| 1. ALT levels >5× ULN |
| 2. Serum immunoglobulin G levels >2× ULN or a positive test for SMAs |
| 3. Liver biopsy showing moderate or severe periportal or periseptal lymphocytic piecemeal necrosis |
| PBC (2 out of 3 criteria) |
| 1. ALP levels >2× or gamma‐glutamyltranspeptidase levels >5× ULN |
| 2. Positive test for AMAs |
| 3. Liver biopsy specimen showing florid bile duct lesions |
The present simplified International Autoimmune Hepatitis Group criteria facilitate making the diagnosis of pure AIH with a specificity and sensitivity of ∼90%.9 Although widely applied to diagnose overlap syndromes in patients with an existing diagnosis of PBC or PSC, the International Autoimmune Hepatitis Group scoring criteria was never intended for such use,10 nor was the simplified score. The classification proposed by Chazouillères et al.11 remains the most commonly used tool for diagnosing AIH/PBC overlap, yet the reported prevalence in the literature of AIH occurring in the context of PBC (depending on the criteria used) varies between 2.8% and 19%. Up to 25% of these patients are ASMA‐positive. The diagnostic criteria described for the overlap syndrome of PSC with AIH are more uniform compared with the overlap syndrome of PBC with AIH. AIH/PSC is a descriptor for overt cholangiographic or histologic findings typical of PSC, alongside robust histologic features of AIH concurrently or historically (Fig. 3).
Figure 3.

Duct lesions of PSC in AIH. Liver histology demonstrating classic sclerosing bile duct lesions in a man with an established diagnosis of type 1 AIH. Biopsy was arranged because of an unexpected rise in aminotransferases despite compliant treatment.
How to Practice in the Real World
An overlap classification at presentation is not recommended because there is no opportunity to see the response to primary therapy with UDCA for PBC and corticosteroids for AIH; it is much easer to convince a patient who has true stepwise change to take a different approach to treatment. Practically, clinicians should appraise the origin of the evidence for overlap (serology, histology, imaging) and the confidence of the observers and the observations. Additionally, they should consider why the tests were performed and what the patient was expected to have. An incidental AMA‐positive result can be discarded if the patient is a young male who is cholestatic with an abnormal cholangiogram and colitis; however, a patient with anti‐centromere antibodies and gp210 reactivity has PBC, even if their biochemistry is a transaminitis and not a classic cholestasis.
It has been established that in patients with PBC, UDCA (13–15 mg/kg/day) leads to slowed progression of fibrosis and liver failure, particularly in patients who demonstrate an adequate biochemical response to therapy.12 The use of corticosteroids, in particular, in patients with AIH/PBC is understandably considered; however, no randomized data exist to support this intervention. Similarly for AIH/PSC, the combination of UDCA and immunosuppressive therapy may improve liver biochemistry, but it must be emphasized that no double‐blind, randomized‐controlled trials have yet evaluated the efficacy of such a strategy. Furthermore, although immunosuppressive therapy benefits the hepatitic component of AIH, no robust survival benefit of UDCA in PSC has been demonstrated. Treatments should thus be individualized based on liver biochemistry, autoantibodies, immunoglobulin titers, cholangiography, and histologic findings. If one therefore feels the need to intervene, because features of distinct diseases are so pronounced, a stepwise approach is generally appropriate wherein the first and predominant disease is addressed before additional therapy started. We therefore do the following:
In the setting of a new diagnosis of autoimmune liver disease, the predominant process is treated first and time given (3–6 months) for a full treatment response before evaluation for added therapy (e.g., patients with likely PBC but overlap features of hepatitis are given UDCA first and are only considered for steroids after 3–6 months). Similarly, a young patient with colitis who has a hepatitic presentation would be treated initially for AIH, even if long‐term concerns existed that the “true” autoimmune liver disease was an evolving PSC syndrome.
In the setting of established autoimmune liver disease, wherein a stepwise, otherwise unexplained (e.g., drug injury) change in presentation occurs, then therapy is augmented with either UDCA or corticosteroids in accordance with a new diagnosis of PBC or AIH, respectively.
Finally however in the context of an era where clinical trials for PBC and PSC are likely, and in which trials may exclude patients on immunosuppression, great care is needed to ensure this is recognized and that clinicians avoid treating test results rather than individuals.
Abbreviations
- AIH
autoimmune hepatitis
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- ANA
antinuclear antibody
- AMA
antimitochondrial antibody
- SMA
smooth muscle antibody
- PBC
primary biliary cirrhosis
- PSC
primary sclerosing cholangitis
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
Potential conflict of interest: Nothing to report.
References
- 1. Boberg KM, Chapman RW, Hirschfield GM, Lohse AW, Manns MP, Schrumpf E; International Autoimmune Hepatitis Group. Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. J Hepatol 2011;54:374‐385. [DOI] [PubMed] [Google Scholar]
- 2. Trivedi PJ, Hirschfield GM. Review article: overlap syndromes and autoimmune liver disease. Aliment Pharmacol Ther 2012;36:517‐533. [DOI] [PubMed] [Google Scholar]
- 3. Corpechot C, Abenavoli L, Rabahi N, Chrétien Y, Andréani T, Johanet C, et al. Biochemical response to ursodeoxycholic acid and long‐term prognosis in primary biliary cirrhosis. Hepatology 2008;48:871‐877. [DOI] [PubMed] [Google Scholar]
- 4. Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, et al. High‐dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009;50:808‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cook GC, Mulligan R, Sherlock S. Controlled prospective trial of corticosteroid therapy in active chronic hepatitis. Q J Med 1971;40:159‐185. [DOI] [PubMed] [Google Scholar]
- 6. Rautiainen H, Kärkkäinen P, Karvonen AL, Nurmi H, Pikkarainen P, Nuutinen H, et al. Budesonide combined with UDCA to improve liver histology in primary biliary cirrhosis: a three‐year randomized trial. Hepatology 2005;41:747‐752. [DOI] [PubMed] [Google Scholar]
- 7. Carbone M, Mells GF, Pells G, Dawwas MF, Newton JL, Heneghan MA, et al. Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology 2013;144:560‐569 e7; quiz e13‐e14. [DOI] [PubMed] [Google Scholar]
- 8. Gregorio GV, Portmann B, Karani J, Harrison P, Donaldson PT, Vergani D, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16‐year prospective study. Hepatology 2001;33:544‐553. [DOI] [PubMed] [Google Scholar]
- 9. Hennes EM, Zeniya M, Czaja AJ, Parés A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008;48:169‐176. [DOI] [PubMed] [Google Scholar]
- 10. Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999;31:929‐938. [DOI] [PubMed] [Google Scholar]
- 11. Chazouillè res O, Wendum D, Serfaty L, Montembault S, Rosmorduc O, Poupon R. Primary biliary cirrhosis‐autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology 1998;28:296‐301. [DOI] [PubMed] [Google Scholar]
- 12. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology 2009;50:291‐308. [DOI] [PubMed] [Google Scholar]
- 13. Joshi S, Cauch‐Dudek K, Wanless IR, Lindor KD, Jorgensen R, Batts K, et al. Primary biliary cirrhosis with additional features of autoimmune hepatitis: response to therapy with ursodeoxycholic acid. Hepatology 2002;35:409‐413. [DOI] [PubMed] [Google Scholar]