

Watch a video presentation of this article
Watch the interview with the author
Abbreviations
- FD
the ferroportin disease
- FPN
ferroportin
- HC
hemochromatosis.
In 1999, just a few years after the discovery of HFE, the hemochromatosis (HC) gene, a non‐HFE hereditary iron overload condition was described in a large family from Italy.1 In contrast to classic HFE‐HC, the majority of affected individuals presented with marked iron overload, largely in reticuloendothelial cells, while transferrin saturation was normal. In the wake of the discovery of ferroportin, genome‐wide screening procedures confirmed that the gene SLC40A1, coding for ferroportin, on chromosome 2q32 was associated with the disease.2 All patients were heterozygous for a 230C→A substitution resulting in the replacement of alanine 77 with aspartate. This new disease entity was subsequently referred to as “the ferroportin disease” (FD).3 In 2001, a different ferroportin (FPN) mutation was also described (i.e., p.N144H) in patients with a similar form of autosomal dominant hereditary iron overload but presenting with parenchymal cell iron overload resembling classic HFE‐HC.4 This phenotype has been reported in a few other pedigrees with FPN mutations thereafter, and represents a variant of classic HC, since clinical presentation is almost identical to other HC syndromes, but substantially different from the FD described herein.
Epidemiology
Unlike HFE‐ and non–HFE‐HC, the pattern of inheritance of FD is autosomal dominant. Therefore, either parent carries the pathogenic mutation of FPN and presents with unexplained hyperferritinemia. Numerous mutations of the FPN gene have been identified so far in families with primary hyperferritinemia, with divergent findings with respect to the pattern of ferritin/transferrin dissociation in probands of French‐Canadian, Melanesian, Thai, Asian, and European heritage (reviewed in Pietrangelo et al.5). Yet a few more common FPN mutations have been reported independently in different countries (e.g., p.Val162del, p.Ala77Asp, p.Gly80Ser). Overall, based on the number of reported pedigrees worldwide, both in Caucasians and non‐Caucasians, FD is considered today the most common form of hereditary iron overload beyond classic HFE‐HC.
Pathogenesis
FPN, the main iron export protein in mammals, is expressed in cell types that play a key role in iron traffic, including placental syncytiotrophoblasts, duodenal enterocytes, hepatocytes, and reticuloendothelial macrophages. It is mainly regulated posttranslationally by hepcidin, a peptide hormone produced in the liver and secreted into the blood in response to iron and inflammation.6 Hepcidin binding to FPN at the cell surface results in its internalization, ultimately leading to degradation in lysosomes.
The disorder is due to a loss of iron export function of FPN: the resultant reduction in iron efflux causes a bottleneck in macrophages, which generate the largest iron flows, resulting in iron accumulation in Kupffer cells, spleen, and bone marrow macrophages, with high serum ferritin levels and low to normal transferrin saturation until late in the disease, when transferrin saturation also raises. The low‐normal transferrin saturation in spite of high serum ferritin is the biochemical hallmark of the disease and, along with the early and preferential accumulation of iron in hepatic Kupffer cells, is central in the diagnostic work‐up of the disorder (Figure 1). Although Kupffer cell iron load is an early feature of FD and is essential in the differential diagnosis of HFE‐HC, discrete hepatocytic iron deposits are also appreciable in classic FD, due to defective FPN activity in hepatocytes, even at early stages.1 The pathogenesis is different from the (hemochromatosis‐like) disease due to FPN mutations clinically characterized by high transferrin saturation and parenchymal iron overload, as alluded to above. Here, the FPN mutant localizes to the cell surface but is unable to respond to hepcidin degradation and therefore continuously export iron into the bloodstream from storage cells.
Figure 1.
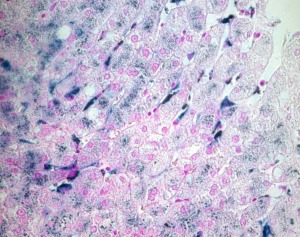
Liver biopsy specimen from a patient with FD (A77D FPN mutation). Perls' Prussian blue stain was used to detect the presence of iron. In addition to hepatocytes, and differently from classic HFE hemochromatosis, marked iron deposits are found in Kupffer cells throughout the liver lobule.
Clinical Aspects
Clinical presentation appears heterogeneous, but overall, expressivity is milder than classic HC, and the associated liver disease is usually not as severe. Hypochromic anemia is common in young female patients and may require iron supplementation, which may further exacerbate the iron overload. Although venesection is again the cornerstone of therapy, it may not be tolerated equally in all patients, and low transferrin saturation with anemia may be rapidly established despite serum ferritin still being elevated. If phlebotomy is discontinued, there is a rapid rise in the ferritin level, and both oral iron chelation and erythropoietin may be of some benefit.
Owing to the mild clinical expressivity reported in the literature, doubts have been raised on the penetrance of the genetic defect and the rationale for iron removal therapy in FD. However, the only published prospective study in FD has reported that the proband of the pedigree in which the disease was first described in 1999 developed a liver cancer (in the context of a concomitant occult hepatitis B virus infection), and two other family members with low adherence to phlebotomy showed progression of liver disease.7 This suggests that the FD may progress if not treated properly.
The disease must be suspected in any individual with unexplained hyperferritinemia and investigated with serum iron studies and genetic testing, if available, of the immediate family (Fig. 2). Abdominal magnetic resonance imaging (MRI) is a useful non‐invasive diagnostic tool to categorize and diagnose the disorder, as it can differentiate patients with the FD, characterized by liver, spleen and bone marrow iron retention (Fig. 3), from classic HFE‐HC and the rarer form of FPN‐related iron load (i.e., FPN‐HC), which is associated with liver iron overload but normal spleen and bone marrow iron content.8
Figure 2.
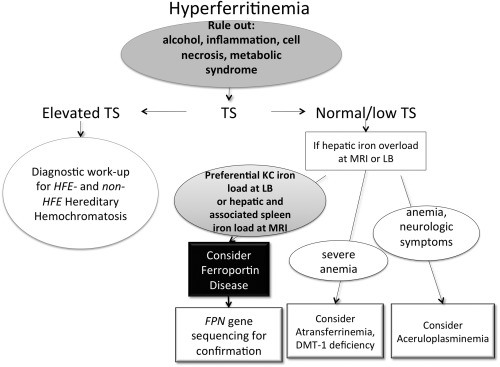
Diagnostic algorithm for FD. In patients with unexplained hyperferritinemia, regardless of the level of serum iron, cofactors and comorbidities associated with increased serum ferritin (e.g., chronic alcohol consumption, metabolic disturbances or obesity, inflammation, etc.) should be considered first. In the absence of these comorbidities, or if the iron abnormalities persist after these conditions have been treated effectively, if transferrin saturation (TS) is persistently elevated, HFE or non–HFE‐HC should be excluded. In patients with increased serum ferritin levels and normal to low transferrin saturation, in the absence of common causes of hyperferritinemia (see above), the work‐up should focus on documenting an iron overload state by liver biopsy (LB) or magnetic resonance imaging (MRI). If so, depending on the pattern of iron distribution (e.g., preferential Kupffer cells, Kupffer cells, iron overload) and/or accompanying symptoms (e.g., severe anemia and/or neurologic disorders), FD or other much rarer hereditary iron‐loading diseases should be considered.
Figure 3.
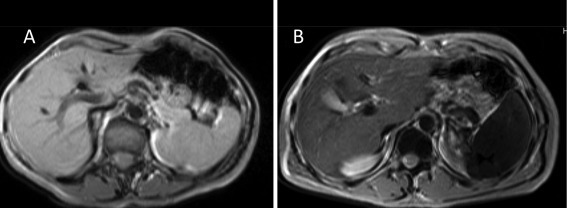
Magnetic resonance imaging in FD. T2‐weighted gradient echo sequences were used to detect iron accumulation. (A) Control subject. (B) Patient with the FPN A77D mutation. The patients show moderate iron overload in the liver but marked iron accumulation in the spleen and bone marrow due to macrophage iron retention.
Potential conflict of interest: Nothing to report.
References
- 1. Pietrangelo A, Montosi G, Totaro A, Garuti C, Conte D, Cassanelli S, et al. Hereditary hemochromatosis in adults without pathogenic mutations in the hemochromatosis gene [see comments]. N Engl J Med 1999;341:725–732. [DOI] [PubMed] [Google Scholar]
- 2. Montosi G, Donovan A, Totaro A, Garuti C, Pignatti E, Cassanelli S, et al. Autosomal‐dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 2001;108:619–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis 2004;32:131–138. [DOI] [PubMed] [Google Scholar]
- 4. Njajou OT, Vaessen N, Joosse M, Berghuis B, van Dongen JW, Breuning MH, et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet 2001;28:213–214. [DOI] [PubMed] [Google Scholar]
- 5. Pietrangelo A, Caleffi A, Corradini E. Non‐HFE hepatic iron overload. Semin Liver Dis 2011;31:302–318. [DOI] [PubMed] [Google Scholar]
- 6. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004;306:2090–2093. [DOI] [PubMed] [Google Scholar]
- 7. Corradini E, Ferrara F, Pollicino T, Vegetti A, Abbati GL, Losi L, et al. Disease progression and liver cancer in the ferroportin disease. Gut 2007;56:1030–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pietrangelo A, Corradini E, Ferrara F, Vegetti A, De Jong G, Luca Abbati G, et al. Magnetic resonance imaging to identify classic and nonclassic forms of ferroportin disease. Blood Cells Mol Dis 2006;37:192–196. [DOI] [PubMed] [Google Scholar]