

Watch a video presentation of this article
Watch the interview with the author
Abbreviations
- CO
cardiac output
- FDA
US Federal Drug Administration
- GMP
guanosine monophosphate
- mPAP
mean pulmonary artery pressure
- PAH
pulmonary arterial hypertension
- PAP
pulmonary artery pressure
- PCWP
pulmonary capillary wedge pressure
- PH
pulmonary hypertension
- POPH
portopulmonary hypertension
- PVR
pulmonary vascular resistance
- sGC
soluble guanylate cyclase
- TPG
transpulmonary gradient.
Background and Pathophysiology
Pulmonary hypertension (PH) is an umbrella term for elevated pulmonary pressures of any cause. In contrast, pulmonary arterial hypertension (PAH) is a specific disease of the small, precapillary pulmonary arteries. PAH is characterized by adventitial proliferation, smooth muscle proliferation, and growth of the endothelial cells into the vascular lumen, essentially “plugging up” the small artery. When approximately 30% of the pulmonary vasculature is involved, the pressure will start to rise in the pulmonary circulation. This pressure presents a higher workload for the right ventricle, and it eventually fails. Portopulmonary hypertension (POPH) is PAH that occurs in the setting of portal hypertension; much of the management for this condition relies on data from other types of PAH (Figs. 1 and 2).
Figure 1.
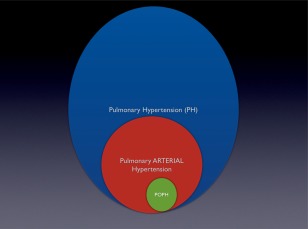
The relationship between pulmonary hypertension, pulmonary arterial hypertension, and portopulmonary hypertension.
Figure 2.

The classification of pulmonary arterial hypertension. Abbreviation: HIV, human immunodeficiency virus.
Epidemiology and Prognosis
POPH occurs in 2% to 6% of patients with decompensated liver disease and has a marked effect on prognosis. A modern cohort of treated and untreated POPH patients documented a 40% 5‐year survival.1 The severity of the liver disease does not appear to correlate with the presence of POPH, but it is more common among females and those with autoimmune hepatitis.2
Clinical Features and Diagnosis
Dyspnea and fatigue are the most common presenting complaints. As the disease progresses, the signs of right heart failure become evident. The best screening test for POPH is a resting echocardiogram. A screening protocol at the Mayo Clinic determined that an estimated PA systolic pressure of >50 mm Hg on echocardiogram or signs of significant right heart dysfunction predicted the presence of POPH 100% of the time.3 Therefore, if the estimated peak PA pressure on an echocardiogram in a patient with cirrhosis is >50 mm Hg, or the right heart is enlarged or hypokinetic, a workup for POPH should be pursued. Right heart catheterization is required to make the diagnosis, which must be done methodically because the cardiovascular derangements associated with cirrhosis complicate the hemodynamic evaluation.
The most common hemodynamic pattern in patients with decompensated cirrhosis is a peripherally vasodilated, high cardiac‐output state.3 The pattern is characterized by increased mean pulmonary artery pressure (mPAP), high cardiac output (CO), low pulmonary capillary wedge pressure (PCWP), and reduced pulmonary vascular resistance (PVR). In contrast, the pattern of POPH, or small vessel arteriopathy, is increased pressure resulting from high flow through the pulmonary circulation. A second abnormal but common hemodynamic profile is that of volume overload, or diastolic dysfunction. In these patients, pulmonary pressures may be elevated, but PCWPs are also increased, with a resulting low PVR that indicates pulmonary venous hypertension from elevated left heart‐filling pressures. Finally, and least commonly, are POPH patients. These patients have an elevated pulmonary artery pressure; a normal PCWP; a high PVR; and a high, normal, or low CO (depending on the degree of resultant right ventricular failure and liver disease) (Fig. 3). Not infrequently, patients may present with a combination of the above hemodynamic profiles, for example, POPH with volume overload. In this case, an increased transpulmonary gradient ([TPG] = mPAP – PCWP) can suggest the existence of combined volume overload and pulmonary arteriopathy. To characterize such complexity, many have suggested that the hemodynamic definition of POPH be expanded to include mPAP > 25, TPG > 12, and PVR > 3 Wood units.
Figure 3.
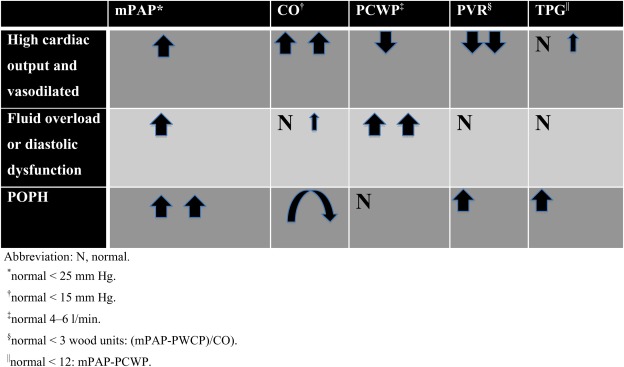
The hemodynamic profiles common in cirrhotics.
Medical Therapy
PAH therapy comes in four main classes: endothelin antagonists, phosphodiesterase type 5 inhibitors, soluble guanylate cyclase (sGC) stimulators, and prostacyclins.
Endothelin Antagonists
Endothelin‐1 is a pulmonary vasoconstrictor that is proproliferative. Its antagonism improves exercise capacity and hemodynamics and reduces clinical worsening in PAH patients. There are three available agents in this class: bosentan, ambrisentan, and macitentan. Bosentan is a dual endothelin receptor antagonist (ETA and ETB). POPH patients treated with this agent have shown improvements in exercise capacity and survival in small, single‐center uncontrolled trials.4, 5 The use of bosentan has been limited in POPH patients due to an approximately 10% incidence of hepatotoxicity. Ambrisentan is a selective ETA antagonist, also US Federal Drug Administration (FDA)‐approved for the treatment of PAH. Hepatotoxicity has not been reported with this agent. A single‐center uncontrolled observational trial of ambrisentan in POPH from the Mayo Clinic reported a significant improvement in hemodynamics and no hepatotoxic events.6 Macitentan is a recently approved dual‐receptor antagonist, also without known hepatotoxicity. There are no data with this agent in POPH.
Phosphodiesterase Type 5 Inhibitors
Two phosphodiesterase type 5 inhibitors, sildenafil and tadalafil, are currently FDA‐approved for the treatment of PAH. These agents act as vasodilators by inhibiting the breakdown of cyclic guanosine monophosphate (GMP), a product of the nitric oxide pathway. Small, uncontrolled observational trials reveal that these drugs increase 6‐minute walk test distance, lower N‐terminal prohormone of brain natriuretic peptide levels, and improve hemodynamics and patients with POPH.7, 8, 9
Soluble Guanylate Cyclase Stimulators
As a key enzyme in the nitric oxide signaling pathway, sGC stimulation catalyzes the creation of cyclic GMP, producing vasodilation. The recent PATENT‐1 trial demonstrated that riociguat improved exercise capacity and hemodynamics and reduced clinical worsening in patients with PAH. A small number of POPH patients were included in this pivotal trial.10
Prostacyclins
Prostacyclins are considered the agents of choice for the “sickest” patients with PAH. These drugs have traditionally been delivered in continuous subcutaneous, continuous intravenous, or intermittently inhaled forms. The current FDA‐approved prostacyclins for intravenous use are epoprostenol and treprostinil. Treprostinil is also available in subcutaneous infusion, inhaled form, and a recently approved oral formulation. In the United States, iloprost is available only in the inhaled form. Uncontrolled single‐center series have consistently demonstrated that prostacyclin infusions result in improvements in mean PAP, CO, and PVR in patients with POPH.11, 12, 13 Less data exist to support the use of inhalational prostacyclins for POPH, and there are no published data on the effects of oral treprostinil in POPH patients.14
Clinical Decision‐Making in the Modern Treatment Era
PH experts choose therapy by analyzing testing and performing a risk assessment for early mortality (Fig. 4). High‐risk patients are considered for intravenous prostacyclins, whereas those with low or moderate risk may be treated with oral agents or combination therapy. However, recent data suggest that PH practitioners may be “under‐treating” POPH patients. An analysis of prevalent cases in a large PH registry revealed that POPH patients had poorer survival and were less likely to be treated with PH‐specific therapy than were patients with other types of PAH.1
Figure 4.
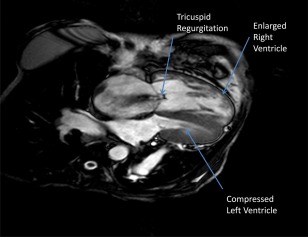
A cardiac MRI of a patient with POPH. This is a cardiac MRI image of a POPH patient with severe TR, an enlarged right atrium, a hypertrophied and dilated right ventricle, septal flattening, and a compressed left ventricle.
Summary
Providers should suspect POPH in patients with liver disease who present with dyspnea out of proportion to known cardiopulmonary disease. Treatment decisions for these patients are based on the severity of the disease and prognostic indicators. Severe POPH has a poor prognosis and early prostacyclin therapy should be considered.
Potential conflict of interest: Nothing to report.
References
- 1. Krowka MJ, Miller DP, Barst RJ, et al. Portopulmonary hypertension: a report from the US‐based REVEAL Registry. Chest 2012;141:906‐915. [DOI] [PubMed] [Google Scholar]
- 2. Kawut SM, Krowka MJ, Trotter JF, et al. Clinical risk factors for portopulmonary hypertension. Hepatology 2008;48:196‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krowka MJ, Swanson KL, Frantz RP, McGoon MD, Wiesner RH. Portopulmonary hypertension: results from a 10‐year screening algorithm. Hepatology 2006;44:1502‐1510. [DOI] [PubMed] [Google Scholar]
- 4. Hoeper MM, Seyfarth HJ, Hoeffken G, et al. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur Respir J 2007;30:1096‐1102. [DOI] [PubMed] [Google Scholar]
- 5. Hoeper MM, Halank M, Marx C, et al. Bosentan therapy for portopulmonary hypertension. Eur Respir J 2005;25:502‐508. [DOI] [PubMed] [Google Scholar]
- 6. Cartin‐Ceba R, Swanson K, Iyer V, Wiesner RH, Krowka MJ. Safety and efficacy of ambrisentan for the treatment of portopulmonary hypertension. Chest 2011;139:109‐114. [DOI] [PubMed] [Google Scholar]
- 7. Reichenberger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J 2006;28:563‐567. [DOI] [PubMed] [Google Scholar]
- 8. Deibert P, Bremer H, Roessle M, Kurz‐Schmieg AK, Kreisel W. PDE‐5 inhibitors lower portal and pulmonary pressure in portopulmonary hypertension. Eur Respir J 2007;29:220‐221. [DOI] [PubMed] [Google Scholar]
- 9. Gough MS, White RJ. Sildenafil therapy is associated with improved hemodynamics in liver transplantation candidates with pulmonary arterial hypertension. Liver Transpl 2009;15:30‐36. [DOI] [PubMed] [Google Scholar]
- 10. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369:330‐340. [DOI] [PubMed] [Google Scholar]
- 11. Kuo PC, Johnson LB, Plotkin JS, Howell CD, Bartlett ST, Rubin LJ. Continuous intravenous infusion of epoprostenol for the treatment of portopulmonary hypertension. Transplantation 1997;63:604‐606. [DOI] [PubMed] [Google Scholar]
- 12. Krowka MJ, Frantz RP, McGoon MD, Severson C, Plevak DJ, Wiesner RH. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): a study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology 1999;30:641‐648. [DOI] [PubMed] [Google Scholar]
- 13. Fix OK, Bass NM, De Marco T, Merriman RB. Long‐term follow‐up of portopulmonary hypertension: effect of treatment with epoprostenol. Liver Transpl 2007;13:875‐885. [DOI] [PubMed] [Google Scholar]
- 14. Melgosa MT, Ricci GL, Garcia‐Pagan JC, et al. Acute and long‐term effects of inhaled iloprost in portopulmonary hypertension. Liver Transpl 2010;16:348‐356. [DOI] [PubMed] [Google Scholar]