

Abstract
The strong hemolytic toxicity of pulsatilla saponin D (1, HD50 6.3 μM) has hampered its clinical development as an injectable anticancer agent. To combat this challenge, 17 new derivatives of 1 with ring C, C-28, or C-3 modifications were synthesized and evaluated for cytotoxicity against several selected human tumor lines, as well as for hemolytic toxicity against rabbit erythrocytes. Structure−activity relationship (SAR) and structure−toxicity relationship (STR) correlations were also elucidated. Compared to the lead compound 1, the hemolytic activity of all 17 derivatives dropped dramatically. Notably, compound 14 exhibited significant cytotoxicity toward A549 human lung cancer cells (IC50 2.8 μM) in a dose-dependent manner without hemolytic toxicity (HD50 > 500 μM). Molecular studies indicated that 14 induced typical G1 cell cycle arrest and apoptosis in A549 cells, and Western blot assays suggested that both intrinsic and extrinsic apoptosis pathways were activated by 14. Collectively, compound 14 may merit further development as a potential anti-lung cancer agent.
Graphical Abstract
Cancerous tumors are a major cause of human death worldwide. Therefore, new antitumor agents with novel mechanisms of action are continually and urgently needed. One effective strategy is to generate new antitumor drugs from natural products. Compared to synthetic compounds, natural products generally have higher molecular weights, fewer nitrogen and halogen atoms, and more oxygen and bridgehead atoms. Moreover, natural products typically have more chiral centers and more rings in their structures. The importance of natural products in anticancer drug discovery could go beyond molecular diversity and structural novelty. The discovery of novel natural structures with significant biological relevance and new mechanisms of action could also pioneer innovative research.1
At least 150 types of natural saponins from more than 100 plant families possess significant antitumor properties.2 Unfortunately, some saponins can also cause hemolysis of red blood cells, which can induce toxicity in most animals, including humans. This effect is a major drawback for the clinical development of saponins as injectable antitumor agents. Although the exact mechanism for saponin-related hemolysis is not clearly understood, this effect is often thought to be associated with the amphiphilic properties of saponins. However, Winter proposed a different mechanism in which the interaction of saponins with the water channel aquaporin resulted in increased water transport inside the cells, inducing erythrocyte hemolysis.3 Furthermore, in other studies, no correlation was evident between the hemolytic toxicity and cytotoxicity of saponins.4 That is, toxicity toward erythrocytes and cytotoxicity against cancer cells may be two separate biological characteristics of saponins. Therefore, it may be possible to identity a type of saponin that can be used as an antitumor agent, but is devoid of toxicity toward erythrocytes.
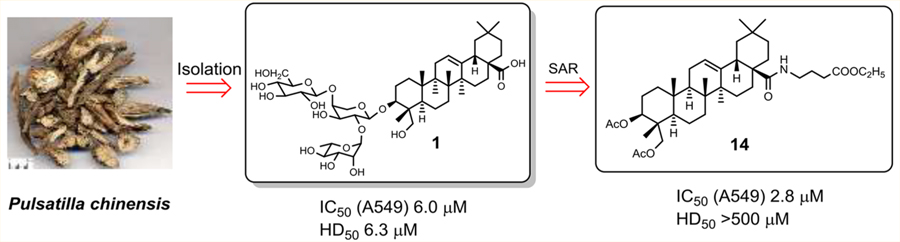
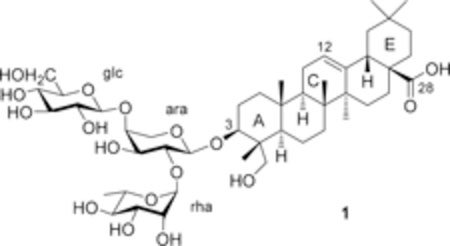
Pulsatilla saponin D (1, hederagenin 3-O-α-L-rhamnopyr-anosyl-(1→2)-[β-D-glucopyranosyl-(1→4)]-α-L-arabinopyrano-side), also named hederacolchiside A1 or SB365 in the literature, can be isolated from Pulsatilla chinensis (Bunge) Regel (Ranunculaceae) and other related plants. This compound exhibited potent in vivo antitumor activity, with even more potent effects than paclitaxel and doxorubicin, in mice bearing Lewis lung carcinoma.5,6 Compound 1 also induced apoptosis, inhibited cell growth, and reduced angiogenesis through modulation of the PI3K/Akt/mTOR pathway in human hepatocellular carcinoma (HCC) with strong suppression of the Huh-7 cell xenograft in nude mice.7 Moreover, compound 1 effectively suppressed the proliferation of gefitinib-resistant HCC827GR non-small-cell lung cancer (NSCLC) cells with Met amplification, suppressed the anchorage-independent growth, migration, and invasion of HCC827GR cells, and induced apoptosis. Compound 1 may be a good candidate as a natural product for use in the treatment of Met-amplified NSCLCs.8 However, compound 1 also exhibited severe hemolytic toxicity (HD50 = 6.3 μM),9 which is a major obstacle to its clinical development as an injectable antitumor agent. Consequently, resolution of the hemolytic toxicity of 1 is urgent for continued drug development.
Considering the potent antitumor value and low natural yield of 1, a practical synthesis in 17.4% overall yield from L-arabinose in eight steps was reported in 2013.10 Novel analogues of 1, including some with various sugar moieties linked at the C-3 position of hederagenin, have been synthesized, but did not show improved cytotoxicity.11 In recent years, our group has reported new triterpenoid saponins modified at the C-28 COOH substituent with amide, ester, or nitric oxide (NO) donating groups. Moreover, some derivatives exhibited dramatically decreased hemolytic toxicity accompanied by potent tumor growth inhibition against murine H22 hepatocellular cells in vivo.12–15 In light of the above results,11–15 new structure−activity relationship (SAR) correlations and structure−toxicity relationship (STR) correlations of 1 were explored in this report. First, new derivatives of 1 with a modified C ring were designed and synthesized (Scheme 1). Second, derivatives were generated without a sugar chain at the C-3 position (Scheme 2) to evaluate its role on the activities of 1. The determination of the effects of such modifications on cytotoxicity and hemolytic toxicity should help to resolve the hemolysis issue of pentacyclic triterpenoid saponins.
Scheme 1.

Synthesis of 2−11 from 1a, aReagents and conditions: (a) Ac2O, py, rt, 4 h, 95%; (b) K2CrO4, glacial acetic acid, reflux, 6 h, 80%; (c) KOH, MeOH/THF/H2O, rt, overnight; (d) m-CPBA, CHCl3, dark, 2 days; (e) BrBn, K2CO3, DMF, rt, 8 h, 95%; (f) (COCl)2, CH2Cl2, rt, 6 h; (g) NH2CH2COOCH2CH3 HCl, CH2Cl2, Et3N, rt, overnight; (h) H2, 10% Pd/C, THF, 6 h; (i) DDQ, anhydrous benzene, 90 °C, 2 days.
Scheme 2.
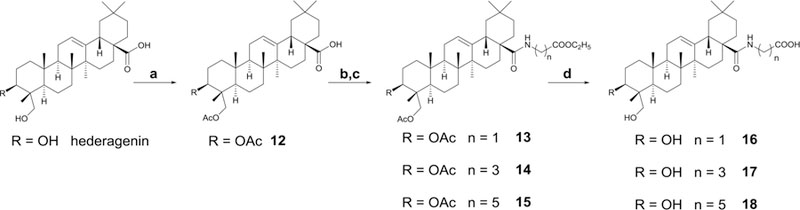
Synthesis of 12−18 from Hederagenina, aReagents and conditions: (a) Ac2O, py, rt, 4 h; (b) (COCl)2, CH2Cl2, rt, 6 h; (c) aminoalkanoic acid ethyl ester hydrochloride, Et3N, CH2Cl2, rt, overnight; (d) KOH, MeOH/THF/H2O, rt, overnight.
RESULTS AND DISCUSSION
Scheme 1 outlines the general synthetic procedure for compounds 2−11. Compound 1 was treated with acetic anhydride in pyridine to afford 2, which was oxidized by potassium chromate in glacial acetic acid to give 3,18 followed by hydrolysis of the acetate under basic catalysis to yield 4 (11-oxo derivative of Pulsatilla Saponin D (PSD)). Compound 3 was also reacted with oxalyl chloride in dichloromethane, followed by amination with glycine and hydrolysis to yield 7.12 Furthermore, compound 2 was treated with 3-chloroperoxybenzoic acid (m-CPBA) in chloroform to afford 5,19 followed by hydrolysis of the acetate to give 11 with a OH-12 substituent and a 28,13-lactone unit. Interestingly, when the C-28 COOH group of 2 was protected as a benzyl ester to afford 6, oxidation with m-CPBA and benzyl deprotection with Pd/H2 yielded the key intermediate 8 with a C-12 oxo moiety. Finally, hydrolysis of 8 gave 9, which has C-12 oxo and C-28 COOH groups rather than a 28,13-lactone. Oxidation of 8 with DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) in anhydrous benzene,20 followed by hydrolysis, provided 10 with a C-12 oxo group and 28,13-lactone. The synthetic methods used to obtain compounds 12−18 are illustrated in Scheme 2. With hederagenin as starting material, 12 was synthesized by acetylation with acetic anhydride in pyridine. Subsequent reaction of 12 with oxalyl chloride in CH2Cl2, followed by amination with three different ω-aminoalkanoic acid ethyl esters, yielded 13−15. Compounds 16−18, with a free terminal COOH group, were obtained by hydrolysis of 13−15, respectively, in MeOH/THF/H2O.15
Except for intermediates 3 and 8, the newly synthesized derivatives were evaluated for cytotoxic effects against different tumor cell lines using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) or sulforhodamine (SRB) assay, according to methods previously described.16 Cisplatin or paclitaxel served as the positive control, and the cultured cells were exposed to the test compounds for 48 or 72 h. The hemolytic toxicities of the new derivatives of 1 against rabbit red blood cells were determined according to the reported method21 with 1 as the positive control. Table 1 lists the IC50 and HD50 values.
Table 1.
In Vitro Cytotoxicity and Hemolytic Activity Data of Pulsatilla Saponin D (1) Derivatives
| IC50a (μM) |
||||||
|---|---|---|---|---|---|---|
| compound | A549b | MDA-MB-231c | KBd | KB-VINe | MCF-7f | HD50g (μM) |
| 1i | 6.0 ± 0.65 | ndh | ndh | ndh | ndh | 6.3 ± 0.58 |
| 4i | >10 | ndh | ndh | ndh | ndh | 239 ± 22.2 |
| 7i | >10 | ndh | ndh | ndh | ndh | 290 ± 27.0 |
| 9i | >10 | ndh | ndh | ndh | ndh | 73.1 ± 9.0 |
| 10i | >10 | ndh | ndh | ndh | ndh | 290 ± 25.5 |
| 11i | >10 | ndh | ndh | ndh | ndh | 308 ± 24.2 |
| hederageninj | >10 | >10 | >10 | >10 | >10 | >200 |
| 12j | 6.2 ± 0.040 | >10 | 7.4 ± 0.315 | 8.5 ± 0.148 | >10 | >500 |
| 13j | 4.6 ± 0.450 | 5.5 ± 0.290 | 4.2 ± 0.215 | 4.9 ± 0.159 | 7.7 ± 1.74 | >500 |
| 14j | 2.8 ± 0.548 | 3.7 ± 0.572 | 3.6 ± 0.265 | 5.1 ± 0.070 | 8.6 ± 1.01 | >500 |
| 15j | 5.1 ± 0.107 | 6.8 ± 0.099 | 6.9 ± 0.092 | >10 | >10 | >500 |
| 16j | >10 | >10 | >10 | >10 | >10 | >500 |
| 17j | >10 | >10 | >10 | >10 | >10 | >500 |
| 18j | >10 | >10 | >10 | >10 | >10 | >500 |
| cisplatini | 49.4 ± 06.32 | ndh | ndh | ndh | ndh | ndh |
| paclitaxelj (nM) | 6.2 ± 0.266 | 9.4 ± 0.502 | 6.6 ± 0.330 | 1860 ± 65.4 | 11.4 ± 0.381 | ndh |
Concentration inhibiting 50% of cell growth for a 72 h exposure period of test samples.
Data represent mean values ± standard deviation for three independent experiments.
A549, human lung cancer cell line.
MDA-MB-231, human breast adenocarcinoma cell line.
KB, human epidermoid carcinoma of the nasopharynx cell line.
KB-VIN, MDR cell line overexpressing P-glycoprotein.
MCF-7, human breast cancer.
HD50 is the concentration inducing 50% of erythrocyte hemolysis.
Not determined.
MTT assay with cisplatin as a positive control.
SRB assay with paclitaxel as a positive control.
Compound 1 exhibited cytotoxicity against human A549 (lung cancer), SMMC-771 (hepatocellular carcinoma), and BGC-823 (gastric carcinoma) cell lines with IC50 values of 6.0, 4.4, and 7.2 μM, respectively. However, compounds 2, 4−7, and 9−11, with a sugar chain at C-3 but modification in the C ring, were inactive (IC50 > 10 μM) against the same three cancer cell lines.
At the same time, the hemolytic toxicity was decreased. For example, compounds 10 (HD50 290 μM) and 11 (HD50 308 μM), which have 12-oxo and −OH groups, respectively, as well as a 28,13-lactone, exhibited almost 50-fold lower hemolytic toxicity than 1 (HD50 6.3 μM), which has a double bond between C-12 and C-13. These data are consistent with prior findings15 and suggest that the C-ring, especially the 12,13-double bond, is very important for mediation of the cytotoxicity and hemolytic toxicity of compounds related to 1. Although compound 9 (HD50 73.1 μM) showed greater hemolytic toxicity than the remaining new derivatives in Table 1, it was still at least 10-fold weaker than 1. Compound 9 has a C-12 oxo group rather than the C-12,13-double bond found in PSD. Interestingly, compound 4 (HD50 239 μM) exhibited much less hemolytic toxicity than 1, even though it retains a 12,13-double bond and varies structurally from 1 only by the addition of an 11-oxo group in the C-ring. Thus, even this slight change in the C-ring could affect hemolytic toxicity, as well as cytotoxicity.
Overall, derivatives 7, 10, and 11 displayed the lowest hemolytic toxicity (HD50 = 290−308 μM) among the compounds with a sugar chain at C-3. Interestingly, in all three compounds, the C-28 COOH group is involved in either an amide (7) or lactone (10 and 11) moiety. Thus, it is likely that the free C-28 COOH group of 1 is another key group connected with its hemolytic toxicity.
As shown in Table 1, hederagenin, the aglycone of 1, also displayed low hemolytic toxicity, but was inactive toward A549 cancer cells. To explore the effect of the sugar chain at C-3 as well as the above observation concerning the C-28 COOH group, this group of hederagenin was linked with different aminoalkanoic acids to give 13−18. These six compounds exhibited no hemolytic toxicity (HD50 > 500 μM), which indicates that the sugar chain and the C-28 COOH group may affect the hemolytic toxicity of compounds related to 1. Interestingly, compounds 13−15, with terminal ethyl esters, displayed more potent cytotoxicity than their corresponding hydrolyzed products 16−18, with terminal carboxylic acids, which suggested that greater lipophilicity may increase the cytotoxicity of hederagenin. The length of the 28-aminoalkanoic acid in 13−15 also influenced the cytotoxicity. Compound 14, the 28-amide with 4-aminobutanoic acid, displayed cytotoxicity against all five tested human cancer cell lines (IC50 2.8−8.6 μM) in a dose-dependent manner, without hemolytic toxicity (HD50 > 500 μM) (Table 1). Compounds 13 and 15, with shorter and longer chains, respectively, were less cytotoxic. Compound 14 was most potent toward A549 cells (IC50 2.8 μM) and also quite cytotoxic toward KB-VIN cells (IC50 5.1 μM), which is an MDR cell line overexpressing P-glycoprotein.
Next, according to the previous method,22 it was investigated whether compound 14 could induce A549 cell cycle arrest. A549 cells were treated with 2, 4, and 8 μM 14 for 24 h, with combretastatin A-4 (20 nM) used as the positive control. Treated cells were fixed with 70% EtOH, stained with propidium iodide (PI), and then analyzed by FCS Express 6 Flow Research Edition to determine the cell cycle distribution of treated cells. As shown in Figure 1, the DNA cell cycle analysis revealed typical G1 arrest in response to treatment with 14. After treatment with 2, 4, and 8 μM 14 for 24 h, the populations of G1 cells were 53.4%, 61.1%, and 68.4%, respectively, whereas that of the control was 46.6%.
Figure 1.

Compound 14 induces G1 cycle arrest in A549 cells. A549 cells were treated with 2, 4, and 8 μM 14 for 24 h. Distribution in the cell cycle was monitored by flow cytometry. Representative histograms for cell cycle distribution in A549 cells are shown.
Subsequently, annexinV/PI staining was used to detect the apoptosis induction of 14. As shown in Figure 2, after being treated with 2, 4, and 8 μM 14 for 24 h, the early and median apoptotic cells (right lower section of fluorocytogram) were determined to be 14.8%, 26.5%, and 30.3%, respectively, whereas the control was only 1.43%. Meanwhile, the late apoptotic and necrotic cells (right upper section of fluorocytogram) were determined to be 3.80%, 4.40%, and 4.87%, while the control was 2.97%. The results showed that compound 14 induced apoptosis in A549 cells.
Figure 2.

Compound 14 induces apoptosis in A549 cells. An annexinV/PI assay was used to detect the apoptosis, after A549 cells were treated with 14 (2, 4, and 8 μM) for 24 h. The data are expressed as the means ± SEM of the data obtained from three separate experiments (*p < 0.05; **p < 0.01).
Finally, a Western blot assay was used to detect the expressions of apoptotic-related proteins. The results showed that, as the concentration of 14 increased, the expression of PARP decreased, while the expression of cleaved PARP increased (Figure 3). The involvement of caspases was examined in 14-mediated apoptosis. Compared to the controls, the expressions of pro-caspase-3, pro-caspase-9, and pro-caspase-8 were decreased after treatment with 14 for 24 h. These results suggested that both intrinsic and extrinsic apoptosis pathways were activated by 14.
Figure 3.
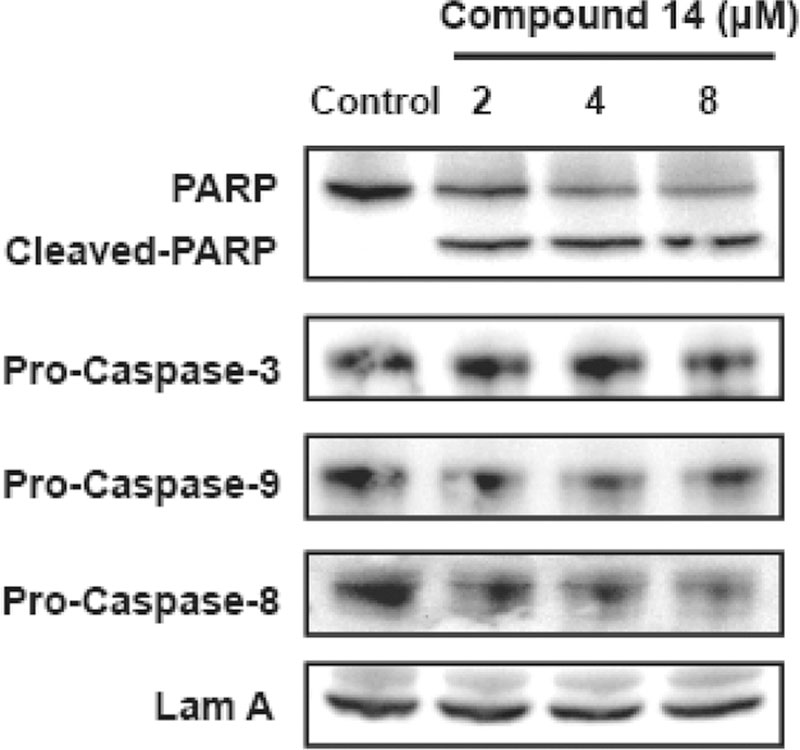
Expression of apoptosis-related protein PARP, pro-caspase-3, pro-caspase-9, and pro-caspase-8 levels as assessed by Western blot analysis.
In summary, 17 new derivatives of 1 were synthesized from either 1 or its aglycone hederagenin as starting material. The new compounds were evaluated for cytotoxic effects against selected cancer cell lines and also for hemolytic toxicity against rabbit red blood cells. The overall conclusions from this current and prior studies15 are shown in Figure 4. With significant cytotoxicity but no hemolytic toxicity, 3β,23-diacetoxyolean-12-en-N-[4-aminobutanoic acid ethyl ester]-28-formamide (14) was identified as a potential antitumor drug candidate.
Figure 4.
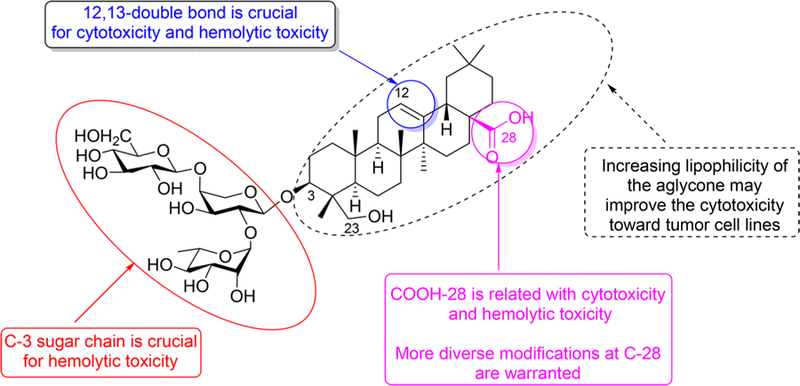
Graphical depiction of the general SAR for cancer cell cytotoxicity and hemolytic toxicity of 1 and its derivatives.
EXPERIMENTAL SECTION
General Experimental Procedures.
All commercially available reagents and solvents were used without further purification. Melting points were measured on the Fisher-Johns melting point apparatus (Fisher Scientific Company, USA). 1H NMR and 13C NMR spectra were measured in CDCl3 or pyridine-d5 on a Bruker AV500 spectrometer (Bruker Biospin Company, Karlsruhe, Germany). Chemical shifts are reported in ppm using tetramethylsilane (TMS) as the internal standard. HRESIMS was determined on a Q-TOF2 spectrometer (Waters Corporation, Milford, MA, USA).
Plant Material.
The roots of Pulsatilla chinensis (Bunge) Regel (Ranunculaceae) were purchased from Suzhou, Jiangsu, People’s Republic of China, in September 2014 and were authenticated by one of the authors (Z.C.). A voucher specimen (ZC-PC-2014–09) is deposited in the Herbarium of Materia Medica, Department of Traditional Chinese Medicine, College of Pharmaceutical Science, Soochow University, Suzhou, People’s Republic of China.
Extraction and Isolation.
Dried Pulsatilla chinensis (10 kg) was extracted three times with 70% EtOH (3 × 30 L, 3 h each) under reflux. The solvent was subsequently removed under reduced pressure to yield a residue (1.2 kg), which was dissolved in distilled water and then passed through a D101 macroporous resin column (26 cm × 150 cm) and eluted with a stepwise EtOH gradient (0%, 30%, 60%, and 90%; 30 L each). The eluate (280 g) from 60% EtOH was dissolved in 10 L of 1 M NaOH and hydrolyzed under reflux for 8 h. The hydrolysate solution was cooled to room temperature (rt), and its pH was adjusted to 7.0 with 1 M HCl. The solution was passed through a D101 column, eluted with a stepwise EtOH gradient (0%, 40%, 70%, and 90%; 50 L each). The eluate (150 g) from 70% EtOH was separated by dynamic axial compression chromatography with ODS as the stationary phase and eluted with a stepwise gradient solvent of MeOH/H2O (76:24, 78:22, 80:20, and 90:10; 10 L each) at a flow rate of 300 mL/min. Pulsatella saponin D (1) (105 g) was obtained in the fraction eluted with 80:20 MeOH/H2O. The isolate exhibited physical and spectroscopic data consistent with literature values.16,17 The purity of 1 isolated from this protocol was 96.1%, as determined by analytical high-performance liquid chromatography with photodiode array detection.
Chemical Procedures for Preparation of New Derivatives 2−12.
3β-Hydroxy-23-acetoxyolean-12-en-28-oic acid-3-O-2,3,4-tri-O-acetyl-α-L-O-rhamnopyranosyl-(1→2)-[2,3,4,6-tetra-O-acetyl-β-D-O-glucopyranosyl-(1→4)]-(3-O-acetyl)-α-L-arabinopyranoside (2).
To a solution of 1 (3.40 g, 6.66 mmol) in dry pyridine (25 mL) was added acetic anhydride (8.0 mL, 83.2 mmol), and the solution was stirred at rt for 4 h. Then, ethyl acetate (EtOAc, 100 mL) was added, and the pH of the solution was adjusted to 4−5 with 10% HCl. The organic layer was washed with brine (50 mL × 3) and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated, and the residue was dissolved in EtOAc and purified on a silica gel column (100 g, petroleum/EtOAc, 1:1) to give 2 (4.52 g, yield 94%): white solid; mp 172−173 °C; 1H NMR (400 MHz, CDCl3) δ 5.26 (1H, d, J = 7.1 Hz, H-1″), 5.12−5.22 (1H, m, H-12), 5.05 (1H, s, H-1′), 4.96 (1H, d, J = 8.5 Hz, H-1‴), 4.53 (2H, dd, J = 16.1, 7.5 Hz), 4.25 (2H, d, J = 13.7 Hz), 4.12−4.16 (3H, m), 3.87−4.02 (3H, m), 3.68−3.84 (3H, m), 3.58 (1H, s), 3.43 (1H, d, J = 8.0 Hz), 2.80−2.83 (1H, m), 2.14 (3H, s, CH3CO), 2.08 (3H, s, CH3CO), 2.08 (3H, s, CH3CO), 2.04 (9H, s, CH3CO), 2.01 (3H, s, CH3CO), 1.99 (3H, s, CH3CO), 1.97 (3H, s, CH3CO), 1.25 (3H, s), 1.18 (3H, d, J = 5.3 Hz), 1.10 (3H, s), 0.93 (3H, s), 0.92 (3H, s), 0.89 (3H, s), 0.74 (3H, s); 13C NMR (100 MHz, CDCl3) δ 183.5 (C-28), 170.8 (CH3CO), 170.7 (CH3CO), 170.6 (CH3CO), 170.3 (CH3CO), 170.2 (CH3CO), 170.1 (CH3CO), 170.0 (CH3CO), 169.6 (CH3CO), 169.5 (CH3CO), 143.7 (C-13), 122.6 (C-12), 101.5 (C-1‴), 101.3 (C-1′), 98.2 (C-1″), 82.0 (C-3), 77.4, 74.7, 72.7, 71.9, 71.1, 71.0, 69.6, 69.0, 68.4, 67.3, 65.4, 61.9, 60.5, 48.0, 47.9, 46.6, 46.0, 45.9, 42.0, 41.8, 41.7, 41.1, 39.4, 38.3, 36.7, 33.2, 32.5, 30.8, 29.8, 25.9, 23.7, 21.2, 21.1, 21.1, 21.0, 21.0, 20.9, 20.9, 20.9, 20.8, 20.7, 20.7, 17.4, 17.0, 15.9, 14.3, 12.7; HRESIMS m/z 1289.5975 [M − H]− (calcd for C65H93O26, 1289.5961).
3β-Hydroxy-23-acetoxy-11-oxo-olean-12-en-28-oic acid-3-O-2,3,4-tri-O-acetyl-α-L-O-rhamnopyranosyl-(1→2)-[2,3,4,6-tetra-O-acetyl-β-D-O-glucopyranosyl-(1→4)]-(3-O-acetyl)-α-L-arabinopyra-noside (3).
To a solution of 2 (680 mg, 0.52 mmol) in glacial acetic acid (20 mL) was added potassium dichromate (305 mg, 1.04 mmol), and the solution was refluxed for 6 h. The solution was cooled to rt and filtered. Most of the glacial acetic acid was removed under a vacuum, and the residue was diluted with EtOAc (50 mL). Then, the organic layer was washed with brine (30 mL × 3) and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated, and the residue was dissolved in EtOAc and purified on a silica gel column (70 g, petroleum/EtOAc, 2:1) to give 3 (537 mg, yield 78%): yellow solid; mp 168−169 °C; 1H NMR (600 MHz, CDCl3) δ 5.58 (1H, s, H-1″), 5.18 (1H, d, J = 13.2 Hz, H-12), 5.15−5.09 (1H, m, H-1′), 4.99 (2H, d, J = 10.6 Hz), 4.89−4.95 (1H, m, H-1‴), 4.87 (1H, s), 4.50 (1H, d, J = 7.4 Hz), 4.46 (1H, s), 4.20 (1H, d, J = 9.2 Hz), 4.11 (1H, d, J = 11.6 Hz), 4.06 (1H, d, J = 6.7 Hz), 3.98 (2H, d), 3.91 (1H, s), 3.80 (1H, d, J = 11.2 Hz), 3.71 (1H, s), 3.67 (1H, d, J = 6.8 Hz), 3.52 (1H, d, J = 5.0 Hz), 3.37 (1H, d, J = 7.0 Hz), 2.91 (1H, d, J = 11.2 Hz), 2.75 (1H, d, J = 12.0 Hz), 2.09 (3H, s), 2.04 (3H, s), 2.04 (3H, s), 2.01 (3H, s), 1.99 (3H, s), 1.98 (3H, s), 1.96 (3H, s), 1.94 (3H, s), 1.92 (3H, s), 1.28 (3H, s), 1.14 (3H, s), 1.09 (3H, s), 0.88 (3H, s), 0.86 (3H, s), 0.72 (3H, s); 13C NMR (150 MHz, CDCl3) δ 200.1 (C-11), 182.3 (COOH), 170.7 (CH3CO), 170.6 (CH3CO), 170.4 (CH3CO), 170.2 (CH3CO), 170.1 (CH3CO), 170.0 (CH3CO), 169.9 (CH3CO), 169.4 (CH3CO), 169.4 (CH3CO), 168.4 (C-12), 127.9 (C-13), 101.4 (C-1‴), 101.2 (C-1′), 98.2 (C-1″), 81.6, 74.7, 72.8, 72.6, 71.8, 71.0, 70.8, 69.5, 68.9, 68.3, 67.2, 65.3, 61.8, 61.7, 47.7, 45.9, 45.0, 44.2, 43.4, 42.3, 41.4, 38.8, 36.8, 33.7, 32.9, 32.6, 31.6, 30.7, 29.7, 27.6, 25.0, 23.4, 23.4, 22.7, 21.0, 20.9, 20.8, 20.8, 20.8, 20.7, 20.6, 20.6, 19.1, 17.3, 17.1, 16.7, 14.2, 12.6; HRESIMS m/z 1305.5871 [M + H]+ (calcd for C65H93O27, 1305.5904).
3β,23-Dihydroxy-11-oxo-olean-12-en-28-oic acid-3-O-α-L-rhamnopyranosyl-(1→2)-[β-D-gluco-pyranosyl-(1→4)]-α-L-arabinopyranoside (4).
To a solution of 3 (237 mg, 0.18 mmol) in a mixture of MeOH/THF/H2O (2:1:1, 20 mL) was added potassium hydroxide (182 mg, 3.24 mmol), and then the solution was stirred at rt overnight. The organic solvent was removed under vacuum, and the residue was diluted with n-butyl alcohol (50 mL). The pH of the solution was adjusted to 4−5 with 10% HCl. The organic layer was washed with brine (30 mL × 3) and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated, and the residue was dissolved in MeOH and purified on a silica gel column (30 g, CH2Cl2/MeOH, 8:1) to give 4 (159 mg, yield 95%): yellow solid; mp 223−224 °C; 1H NMR (600 MHz, pyridine-d5) δ 6.26 (1H, s, H-1″), 5.97 (1H, s, H-12), 5.11 (1H, d, J = 7.9 Hz, H-1′), 4.96 (1H, d, J = 6.7 Hz, H-1‴), 4.71 (1H, s), 4.63 (1H, d, J = 8.5 Hz), 4.03 (1H, d, J = 7.9 Hz), 3.88 (1H, s), 3.74 (1H, d, J = 10.4 Hz), 3.28 (1H, dd, J = 12.8, 6.0 Hz), 2.62 (1H, s, H-10), 1.64 (3H, d, J = 5.5 Hz, H-6″), 1.37 (3H, s), 1.35 (3H, s), 1.27 (3H, s), 1.18 (3H, s), 1.12 (3H, s), 0.91 (3H, s); 13C NMR (150 MHz, pyridine-d5) δ 200.4 (C-11), 180.0 (COOH), 170.0 (C-12), 128.5 (C-13), 107.1 (C-1‴), 104.8 (C-1′), 102.0 (C-1″), 81.4 (C-3), 80.8, 80.0, 79.2, 78.9, 76.6, 75.8, 75.5, 74.5, 72.8, 72.6, 71.5, 70.0, 67.5, 65.8, 64.2, 62.8, 62.7, 50.0, 48.1, 46.5, 45.7, 44.9, 44.4, 44.3, 42.7, 40.2, 37.8, 34.3, 34.2, 33.4, 33.2, 32.6, 32.5, 32.2, 31.2, 30.0, 28.7, 26.8, 26.0, 24.0, 23.8, 23.3, 21.5, 19.8, 19.0, 17.6, 17.5; HRESIMS m/z 949.4767 [M + Na]+ (calcd for C47H74O18Na, 949.4773).
3β,12α-Dihydroxy-23-acetoxyolean-28,13β-olide 3-O-[2,3,4-tri-O-acetyl]-α-L-rhamnopyranosyl-(1→2)-[(3-O-acetyl)-β-D-glucopyranosyl-(1→4)]-[2,3,4,6-tetra-O-acetyl]-α-L-arabinopyranoside (5).
To a solution of 2 (290 mg, 0.22 mmol) in anhydrous CHCl3 (15 mL) was added 75% m-CPBA (127 mg, 0.55 mmol), and the solution was stirred in the dark at rt for 48 h. The solvent was removed under vacuum, and the residue was diluted with EtOAc (50 mL). Then the organic layer was washed with 5% sodium hydroxide solution (20 mL) and brine (30 mL × 2) and dried over anhydrous Na2SO4. After the mixture was filtered, the filtrate was concentrated and the residue was dissolved in EtOAc and purified on a silica gel column (60 g, petroleum/EtOAc, 3:1) to give 5 (243 mg, yield 80%): white solid; mp 155−156 °C; 1H NMR (400 MHz, CDCl3) δ 5.23 (1H, s, H-1″), 5.21 (1H, s, H-12), 5.16 (1H, d, J = 9.5 Hz, H-1′), 4.99−5.08 (3H, m), 4.90−4.99 (3H, m), 4.54 (1H, d, J = 7.9 Hz), 4.50 (1H, d, J = 3.4 Hz), 4.25 (1H, dd, J = 12.3, 4.6 Hz), 4.12−4.16 (3H, m), 4.02 (1H, t, J = 6.3 Hz), 3.82−3.91 (1H, m), 3.68−3.78 (2H, m), 3.55 (1H, q, J = 6.9 Hz), 3.43 (1H, dd, J = 11.7, 4.4 Hz), 2.14 (3H, s), 2.08 (3H, s), 2.08 (3H, s) 2.04 (3H, s), 2.03 (3H, s), 2.03 (3H, s), 2.01 (3H, s), 1.98 (3H, s), 1.97 (3H, s), 1.28 (3H, s), 1.25 (3H, s), 1.13 (3H, s), 0.96 (3H, s), 0.91 (3H, s), 0.89 (3H, s), 0.75 (s,3H); 13C NMR (100 MHz, CDCl3) δ 180.0 (C-28), 170.8 (CH3CO), 170.7 (CH3CO), 170.5 (CH3CO), 170.3 (CH3CO), 170.2 (CH3CO), 170.1 (CH3CO), 170.1 (CH3CO), 169.6 (CH3CO), 169.6 (CH3CO), 101.6 (C-1‴), 101.4 (C-1′), 98.2 (C-1″), 90.6 (C-13), 81.8 (C-3), 77.4, 76.4, 74.6, 73.0, 72.7, 71.9, 71.1, 71.0, 69.6, 69.0, 68.4, 67.3, 65.3, 61.9, 60.5, 51.2, 47.9, 44.9, 44.8, 42.4, 42.1, 42.1, 39.5, 38.6, 36.1, 34.2, 33.8, 33.4, 31.7, 29.8, 29.0, 28.1, 27.6, 25.2, 24.0, 21.3, 21.2, 21.0, 21.0, 21.0, 20.9, 20.9, 20.8, 20.7, 20.7, 18.7, 17.6, 17.4, 16.9, 14.3, 12.5; HRESIMS m/z 1329.5896 [M + Na]+ (calcd for C65H94O27Na, 1329.5881).
Benzyl 3β-hydroxy-23-acetoxyolean-12-en-28-oic acid-3-O-2,3,4-tri-O-acetyl-α-L-O-rhamnopyranosyl-(1→2)-[2,3,4,6-tetra-O-acetyl-β-D-O-glucopyranosyl-(1→4)]-(3-O-acetyl)-α-L-arabinopyranoside (6).
To a solution of 2 (1.03 g, 0.79 mmol) in dimethylformamide (DMF) (20 mL) was added anhydrous potassium carbonate (328 mg, 2.38 mmol), and benzyl bromide was added dropwise. The solution was stirred at rt for 8 h. The mixture was diluted with distilled water (100 mL). Then the mixture was extracted with EtOAc (50 mL × 3), and the organic layer was combined, washed with brine (30 mL × 3), and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated, and the residue was dissolved in EtOAc and purified on a silica gel column (80 g, petroleum/EtOAc, 4:1) to give 6 (1.02 g, yield 95%): white solid; mp 164−165 °C; 1H NMR (600 MHz, CDCl3) δ 7.28−7.37 (5H, m, Ar−H), 5.28 (1H, t, J = 3.4 Hz, H-1″), 5.18 (1H, brs, H-12), 4.92 (1H, s, H-1′), 4.55 (1H, d, J = 7.9 Hz, H-1‴), 4.52 (1H, d, J = 3.4 Hz), 4.26 (1H, dd, J = 12.3, 4.7 Hz), 4.15 (1H, dd, J = 12.3, 2.2 Hz), 4.01−4.04 (3H, m), 3.96 (1H, dt, J = 16.1, 6.2 Hz), 3.81 (1H, d, J = 11.5 Hz), 3.76 (1H, dd, J = 5.6, 3.5 Hz), 3.72 (1H, d, J = 4.6 Hz), 3.57 (1H, d, J = 7.2 Hz), 3.42 (1H, dd, J = 11.8, 4.6 Hz), 2.90 (1H, dd, J = 13.6, 3.8 Hz), 2.14 (3H, s), 2.09 (3H, s), 2.08 (1H, s), 2.05 (3H, s), 2.04 (3H, s), 2.03 (3H, s), 2.02 (3H, s), 1.99 (3H, s), 1.97 (3H, s), 1.19 (3H, d, J = 6.2 Hz), 1.09 (3H, s), 0.91 (6H, s), 0.89 (3H, s), 0.75 (3H, s), 0.59 (3H, s); 13C NMR (150 MHz, CDCl3) δ 177.5 (C-28), 170.8 (CH3CO), 170.7 (CH3CO), 170.6 (CH3CO), 170.3 (CH3CO), 170.2 (CH3CO), 170.1 (CH3CO), 170.0 (CH3CO), 169.6 (CH3CO), 169.6 (CH3CO), 143.9 (C-13), 136.6 (Ar−C), 128.5 (Ar−C), 128.1 (Ar−C), 128.1 (Ar−C), 122.6 (C-12), 101.6 (C-1‴), 101.3 (C-1′), 98.2 (C-1″), 82.1 (C-3), 74.7, 73.0, 72.7, 71.9, 71.1, 71.0, 69.6, 69.0, 68.5, 67.3, 66.1, 65.4, 62.0, 48.0, 47.9, 46.9, 46.0, 42.1, 41.7, 41.5, 39.4, 38.3, 36.7, 34.0, 33.3, 32.5, 32.5, 30.8, 29.8, 27.7, 25.9, 25.2, 23.8, 23.6, 23.2, 21.1, 21.0, 21.0, 21.0, 20.9, 20.9, 20.8, 20.7, 18.8, 17.4, 17.0, 15.9, 12.8; HRESIMS m/z 1403.6385 [M + Na]+ (calcd for C72H100O26Na, 1403.6401).
3β,23-Dihydroxy-11-oxo-olean-12-en-28-(glycine)oic acid-3-O-α-L-rhamnopyranosyl-(1→2)-[β-D-glucopyranosyl-(1→4)]-α-L-arabinopyranoside (7).
To a solution of 3 (300 mg, 0.23 mmol) in anhydrous CH2Cl2 (15 mL) was added oxalyl chloride (0.1 mL, 1.17 mmol), and the solution was stirred at rt for 6 h. Then the solvent and remaining oxalyl chloride were removed under vacuum to give the acyl chloride intermediate. The intermediate was dissolved in anhydrous CH2Cl2 (10 mL) with glycine ethyl ester (35 mg, 0.57 mmol) and triethylamine (87 mg, 0.86 mmol). This solution was stirred at rt overnight and then diluted with additional CH2Cl2 (30 mL), washed with brine (20 mL × 3), and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated, the residue was dissolved in a mixture of MeOH/THF/H2O (2:1:1, 15 mL), potassium hydroxide (232 mg, 4.14 mmol) was added, and the mixture was stirred at rt overnight. The organic solvent was removed under vacuum, and the residue was diluted with n-butyl alcohol (30 mL). Then the pH of the solution was adjusted to 4−5 with 10% HCl. Then the organic layer was washed by brine (20 mL × 3) and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated and purified on a silica gel column (30 g, CH2Cl2/MeOH, 10:1) to give 7 (102 mg, overall yield 45%): white solid; mp 253−254 °C; 1H NMR (600 MHz, pyridine-d5) δ 8.36 (1H, s, NHCH2), 6.25 (1H, s, H-1″), 5.99 (1H, s, H-12), 5.11 (1H, d, J = 7.8 Hz, H-1′), 4.97 (1H, d, J = 6.6 Hz, H-1‴), 4.64 (1H, dd, J = 9.2, 3.0 Hz), 4.44−4.55 (2H, m), 3.87−3.89 (1H, m), 3.75 (1H, d, J = 10.5 Hz), 3.29 (2H, dd, J = 12.9, 6.0 Hz), 2.79 (1H, t, J = 7.4 Hz), 2.63 (1H, d, J = 11.6 Hz), 1.65 (3H, d, J = 6.1 Hz), 1.37 (3H, s), 1.36 (3H, s), 1.21 (3H, s), 1.12 (3H, s), 0.88 (3H, s), 0.85 (3H, s); 13C NMR (150 MHz, pyridine-d5) δ 200.6 (C-11), 177.6 (COOH), 174.0 (C-28), 170.1 (C-12), 128.5 (C-13), 107.0 (C-1″), 104.8 (C-1′), 102.0 (C-1‴), 81.4 (C-3), 80.6, 79.1, 78.8, 76.6, 75.8, 75.2, 74.4, 72.8, 72.5, 71.5, 70.0, 65.7, 64.2, 62.8, 61.7, 50.0, 48.0, 46.2, 45.8, 45.1, 44.3, 44.3, 43.1, 42.4, 40.2, 37.7, 34.5, 33.3, 33.2, 31.1, 28.2, 26.7, 24.0, 23.9, 19.9, 18.9, 17.6, 17.5, 14.3; HRESIMS m/z 982.5034 [M − H]− (calcd for C49H76NO19, 982.5012).
3β-Hydroxy-23-acetoxy-12-oxo-olean-28-oic acid-3-O-2,3,4-tri-O-acetyl-α-L-O-rhamnopyranosyl-(1→2)-[2,3,4,6-tetra-O-acetyl-β-D-O-glucopyranosyl-(1→4)]-(3-O-acetyl)-α-L-arabinopyranoside (8).
To a solution of 6 (1.00 g, 0.72 mmol) in anhydrous CHCl3 (20 mL) was added 75% m-CPBA (420 mg, 1.82 mmol), and the solution was stirred in the dark at rt for 48 h. The solvent was removed under vacuum, and the residue was diluted with EtOAc (50 mL). Then, the organic layer was washed by 5% sodium hydroxide solution (20 mL) and brine (30 mL × 2) and dried with anhydrous Na2SO4. After filtration, the filtrate was concentrated. The residue was dissolved in anhydrous THF (15 mL); then 10% Pd/C (15 mg) was carefully added under nitrogen gas. The solution was stirred at rt for 6 h. The Pd/C was filtered, and the residue was concentrated under vacuum, dissolved in EtOAc, and purified on a silica gel column (60 g, petroleum/EtOAc, 4:1) to give 8 (626 mg, overall yield 66%): white solid; mp 176−177 °C; 1H NMR (400 MHz; CDCl3) δ 5.23 (1H, d, J = 1.0 Hz, H-1″), 5.15 (1H, d, J = 9.6 Hz, H-1′), 4.89−5.08 (6H, m), 4.85 (1H, d, J = 9.1 Hz), 4.53 (1H, d, J = 7.9 Hz), 4.48 (1H, d, J = 2.8 Hz), 3.55 (1H, d, J = 8.3 Hz), 3.42 (1H, s), 2.75 (1H, d, J = 13.7 Hz), 2.67 (1H, d, J = 3.9 Hz), 2.14 (3H, s), 2.08 (3H, s), 2.08 (3H, s), 2.05 (3H, s), 2.04 (3H, s), 2.02 (3H, s), 2.01 (3H, s), 1.98 (3H, s), 1.97 (3H, s), 1.24 (3H, s), 0.97 (3H, s), 0.96 (3H, s), 0.91 (3H, s), 0.89 (3H, s), 0.89 (3H, s), 0.75 (3H, s); 13C NMR (100 MHz, CDCl3) δ 211.4 (C-12), 183.2 (COOH), 170.8 (CH3CO), 170.7 (CH3CO), 170.5 (CH3CO), 170.3 (CH3CO), 170.2 (CH3CO), 170.1 (CH3CO), 170.0 (CH3CO), 169.5 (CH3CO), 169.5 (CH3CO), 101.6 (C-1‴), 101.4 (C-1′), 98.3 (C-1″), 81.6 (C-3), 74.8, 73.0, 72.7, 71.9, 71.1, 71.0, 69.6, 69.0, 68.4, 67.3, 65.3, 61.9, 60.5, 52.0, 50.0, 47.9, 47.2, 42.1, 42.0, 41.4, 38.6, 37.8, 36.7, 36.3, 34.6, 33.5, 33.2, 32.0, 31.7, 31.6, 30.7, 30.3, 29.8, 27.6, 25.1, 23.2, 22.7, 21.2, 21.1, 21.0, 20.9, 20.9, 20.9, 20.8, 20.7, 20.7, 20.7, 18.1, 17.5, 16.3, 15.8, 14.3, 12.6; HRESIMS m/z 1329.5868 [M + Na]+ (calcd for C65H94O27Na, 1329.5881).
3β,23-Dihydroxy-12-oxo-olean-28-oic acid-3-O-a-L-rhamnopyranosyl-(1→2)-[β-D-glucopyranosyl-(1→4)]-a-L-arabinopyranoside (9).
To a solution of 8 (210 mg, 0.16 mmol) in a mixture of MeOH/THF/H2O (2:1:1, 15 mL) was added potassium hydroxide (162 mg, 2.89 mmol), and the solution was stirred at rt overnight. The organic solvent was removed under vacuum, and the residue was diluted with n-butyl alcohol (20 mL). Then the pH of the solution was adjusted to 4−5 with 10% HCl. Then the organic layer was washed with brine (15 mL × 3) and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated, and the residue was dissolved in MeOH and purified on a silica gel column (30 g, CH2 Cl2 /MeOH, 8:1) to give 9 (133 mg, yield 95%): white solid; mp 197−198 °C; 1H NMR (600 MHz, pyridine-d5) δ 6.28 (1H, s, H-1″), 5.13 (1H, d, J = 7.4 Hz, H-1′), 4.95 (1H, d, J = 6.4 Hz, H-1‴), 4.63 (2H, d, J = 8.7 Hz), 4.47−4.55 (2H, m), 4.24−4.44 (5H, m), 4.11−4.23 (3H, m), 4.04 (2H, brs), 3.90 (1H, s), 3.72 (2H, d, J = 10.3 Hz), 3.33 (1H, d, J = 12.8 Hz), 3.20 (1H, s), 1.66 (3H, d, J = 5.2 Hz, H-6″), 1.12 (3H, s), 1.07 (3H, s), 1.04 (3H, s), 1.04 (3H, s), 0.94 (3H, s), 0.82 (3H, s); 13C NMR (150 MHz, pyridine-d5) δ 211.8 (C-12), 181.0 (COOH), 107.1 (C-1‴), 104.8 (C-1′), 102.1 (C-1″), 81.2 (C-3), 80.9, 79.2, 78.9, 76.6, 75.9, 75.5, 74.5, 72.8, 72.6, 71.6, 70.0, 65.9, 64.1, 62.8, 52.6, 50.6, 50.0, 48.1, 47.7, 43.9, 42.6, 41.9, 39.2, 38.7, 37.2, 37.0, 35.2, 34.1, 34.0, 33.2, 32.5, 32.3, 32.2, 31.4, 30.3, 28.5, 26.6, 23.9, 23.8, 21.6, 21.0, 19.0, 18.5, 16.7, 16.2, 14.1; HRESIMS m/z 951.4912 [M + Na]+ (calcd for C47H76O18Na, 951.4930).
3β,23-Dihydroxy-12-oxo-olean-28-oic acid-28,13β-lactone-3-O-α-L-rhamnopyranosyl-(1→2)-[β-D-glucopyranosyl-(1→4)]-α-L-arabinopyranoside (10).
To a mixture of 8 (415 mg, 0.32 mmol) and DDQ (142 mg, 0.64 mmol) was added anhydrous benzene (20 mL). The solution was refluxed at 90 °C for 48 h and then cooled. The solvent was removed under vacuum, and the residue was dissolved in CH2Cl2 (20 mL), washed with saturated sodium bicarbonate solution (20 mL × 3), and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated to provide a residue, which was dissolved in a mixture of MeOH/THF/H2O (2:1:1, 15 mL). Potassium hydroxide (167 mg, 2.90 mmol) was added, and the solution was stirred at rt overnight. The organic solvent was removed under vacuum, and the residue was diluted with n-butyl alcohol (20 mL). After the pH of the solution was adjusted to 4−5 with 10% HCl, the organic layer was washed with brine (15 mL × 3) and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated, and the residue was dissolved in EtOAc and purified on a silica gel column (30 g, CH2Cl2/MeOH, 8:1) to give 10 (131 mg, overall yield 45%): white solid; mp 198−199 °C; 1H NMR (600 MHz, pyridine-d5) δ 6.26 (1H, s, H-1″), 5.13 (1H, d, J = 7.7 Hz, H-1′), 4.96 (1H, d, J = 6.9 Hz, H-1‴), 4.63 (2H, dd, J = 9.1, 3.3 Hz), 4.50 (2H, dd, J = 15.2, 8.5 Hz), 4.33−4.41 (2H, m), 4.30 (1H, t, J = 9.3 Hz), 4.25 (1H, t, J = 9.1 Hz), 4.18 (1H, dd, J = 17.0, 10.0 Hz), 4.03 (1H, t, J = 16.3 Hz), 3.77 (1H, d, J = 10.7 Hz), 3.67 (1H, d, J = 12.4 Hz), 2.30 (2H, t, J = 12.2 Hz), 1.66 (3H, d, J = 6.0 Hz), 1.56 (3H, s), 1.34 (3H, s), 1.11 (3H, s), 0.94 (3H, s), 0.88 (3H, s), 0.81 (3H, s); 13C NMR (150 MHz, pyridine-d5) δ 206.8 (C-12), 180.1 (C-28), 107.1 (C-1‴), 104.8 (C-1′), 102.0 (C-1″), 91.8 (C-13), 81.4 (C-3), 80.8, 79.1, 78.9, 76.6, 76.1, 75.8, 75.4, 74.5, 72.8, 72.6, 71.6, 70.0, 65.8, 64.2, 62.9, 52.0, 50.0, 48.3, 45.4, 45.3, 44.0, 43.1, 43.0, 39.8, 39.5, 39.0, 37.3, 37.3, 37.2, 36.7, 34.8, 34.5, 33.6, 33.4, 32.0, 29.7, 28.8, 28.6, 26.7, 26.5, 24.2, 22.1, 19.4, 19.1, 19.0, 18.4, 18.0, 17.6, 16.8, 14.1; HRESIMS m/z 925.4786 [M − H]− (calcd for C47H73O18, 925.4797).
33β,12α,23-Trihydroxyolean-28-oic acid 28,13β-lactone-3-O-α-3L3-rhamnopyranosyl-(1→32)-[β-3D3-glucopyranosyl-(1→34)]-α-3L3-arabinopyranoside (11).
To a solution of 5 (243 mg, 0.18 mmol) in a mixture of MeOH/THF/H2O (2:1:1, 20 mL) was added potassium hydroxide (187 mg, 3.34 mmol), and the solution was stirred at rt overnight. The organic solvent was removed under vacuum, and the residue was diluted with n-butyl alcohol (50 mL). Then, the pH of the solution was adjusted to 4−5 with 10% HCl. The organic layer was washed with brine (30 mL × 3) and dried over anhydrous Na2SO4. After the mixture was filtered, the filtrate was concentrated, and the residue was dissolved in EtOAc and purified on a silica gel column (30 g, CH2Cl2/MeOH, 10:1) to give 11 (152 mg, yield 88%): white solid; mp 198−199 °C; 1H NMR (400 MHz, CD3OD) δ 5.15 (1H, s, H-1″), 4.46 (1H, d, J = 4.0 Hz, H-1′), 4.41 (1H, d, J = 4.0 Hz, H-1‴), 4.08 (1H, dd, J = 12.1, 3.5 Hz), 1.29 (3H, s), 1.18 (3H, d, J = 6.0 Hz, H-6″), 1.05 (3H, s), 0.91(3H, s), 0.86 (3H, s), 0.86 (3H, s), 0.63 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 182.6 (C-28),105.9 (C-1‴), 104.5 (C-1′), 101.8 (C-1″), 93.3 (C-13), 82.2 (C-3), 79.5 (C-12), 77.9, 77.7, 76.8, 76.5, 75.2, 73.8, 71.9, 71.9, 71.2, 70.1, 64.9, 64.5, 62.5, 52.4, 46.1, 45.6, 44.0, 43.3, 43.2, 40.2, 39.7, 37.0, 35.1, 34.5, 33.7, 32.3, 29.5, 28.9, 28.6, 26.6, 24.2, 22.3, 20.9, 19.2, 19.0, 18.2, 18.0, 17.3, 13.6; HRESIMS m/z 927.4945 [M − H]− (calcd for C47H75O18, 927.4953).
3β,23-Diacetoxyolean-12-en-28-oic acid (12).
The synthesis of 12 was similar to that of 2 but with hederagenin as starting material (980 mg, yield 96%): white solid; mp 163−164 °C; 1H NMR (400 MHz, CDCl3) δ 5.28 (brs, 1H, H-12), 4.79 (1H, dd, J = 8.0, 4.0 Hz, H-3), 3.88(d, J = 8.0 Hz, 1H, H-23), 3.70 (1H, d, J = 8.0 Hz, H-23), 2.07 (3H, s, CH3CO), 2.02 (3H, s,CH3CO), 1.12, 0.97, 0.93, 0.91, 0.83, 0.75 (each 3H, s, CH3 × 6); 13C NMR (100 MHz, CDCl3) δ 184.2, 171.1, 170.8, 143.7, 122.6, 74.6, 65.6, 48.0, 47.8, 46.7, 45.9, 41.6, 41.1, 40.7, 39.4, 37.8, 37.0, 33.9, 33.2, 32.6, 32.4, 30.8, 27.8, 25.9, 23.7, 23.5, 23.1, 23.0, 21.4, 21.1, 18.1, 17.3, 16.0, 13.2; HRESIMS m/z 555.3682 [M − H]− (calcd for C34H51O6, 555.3691).
General Procedure for Synthesis of Compounds 13−15.
To a solution of 12 (556 mg, 1 mmol) in anhydrous CH2Cl2 (15 mL) was added oxalyl chloride (0.42 mL, 5 mmol), and the solution was stirred at rt for 6 h. The solvent and remaining oxalyl chloride were removed under vacuum to give the intermediate, which was dissolved in anhydrous CH2Cl2 (15 mL) with the aminoalkanoic acid ethyl ester hydrochloride (1.2 mmol) and triethylamine (0.17 mL, 1.2 mmol). The solution was stirred at rt overnight, diluted with CH2Cl2 (50 mL), washed with brine (30 mL × 3), and dried over anhydrous Na2SO4. After being filtered, the filtrate was concentrated and purified on a silica gel column (30 g, hexanes−EtOAc) to give compounds 13−15.
3β,23-Diacetoxyolean-12-en-N-[glycine ethyl ester]-28-formamide (13):
546 mg (85%); white solid; mp 140−141 °C; 1H NMR (600 MHz,CDCl3) δ 6.51 (1H, brs, CONH), 5.45 (1H, s, H-12), 4.78 (1H, dd, J = 12.0, 6.0 Hz, H-3), 4.21 (2H, q, J = 6.0 Hz, OCH2CH3), 4.11 (1H, dd, J = 12.0, 6.0 Hz), 3.86(1H, d, J = 12.0 Hz, H-23), 3.80 (1H, dd, J = 12.0, 6.0 Hz), 3.70 (1H, d, J = 12.0 Hz, H-23), 2.06 (3H, s, CH3CO), 2.02 (3H, s, CH3CO), 1.28 (3H, t, J = 6.0 Hz, OCH2CH3), 1.15 (3H, s, CH3), 0.95 (3H, s, CH3), 0.92 (3H, s, CH3), 0.91 (3H, s, CH3), 0.83 (3H, s, CH3), 0.71 (3H, s, CH3); 13C NMR (150 MHz, CDCl3) δ 178.3, 171.1, 170.8, 170.2, 144.5, 123.2, 74.6, 65.5, 61.6, 47.9, 47.8, 46.7, 46.4, 42.3, 42.0, 41.8, 40.5, 39.5, 37.9, 36.8, 34.2, 33.1, 32.5, 32.2, 30.9, 27.4, 25.8, 23.9, 23.7, 23.7, 23.1, 21.4, 21.1, 18.1, 16.6, 16.0, 14.3, 13.2; HRESIMS m/z 642.4332 [M + H]+ (calcd for C38H60NO7, 642.4370).
3β,23-Diacetoxyolean-12-en-N-[4-aminobutanoic acid ethyl ester]-28-formamide (14):
556 mg (83%); white solid; mp 135−136 °C; 1H NMR (600 MHz, CDCl3) δ 6.04 (1H, brs, CONH), 5.38 (1H, s, H-12), 4.77 (1H, dd, J = 11.5, 4.5 Hz, H-3), 4.13 (2H, q, J = 7.1 Hz, OCH2CH3), 3.86 (1H, d, J = 11.6 Hz, H-23), 3.70 (1H, d, J = 11.6 Hz, H-23), 3.40 (1H, td, J = 13.7, 6.8 Hz), 2.98−3.08 (1H. m), 2.50−2.56 (1H, m), 2.33 (2H, td, J = 7.2, 2.3 Hz), 2.05 (3H, s, CH3CO), 2.02 (3H, s, CH3CO), 1.25 (3H, t, J = 7.1 Hz, OCH2CH3), 1.14 (3H, s, CH3), 0.97 (3H, s, CH3), 0.90 (3H, s, CH3), 0.90 (3H, s, CH3), 0.83 (3H, s, CH3), 0.75 (3H, s, CH3); 13C NMR (150 MHz, CDCl3) δ 178.4, 173.5, 171.1, 170.8, 145.1, 122.7, 74.6, 65.5, 60.7, 47.9, 47.8, 46.8, 46.4, 42.3, 42.1, 40.7, 39.5, 39.1, 37.9, 36.8, 34.3, 33.1, 32.8, 32.2, 32.0, 30.9, 27.4, 25.8, 24.6, 23.8, 23.7, 23.6, 23.1, 21.4, 21.1, 18.1, 17.1, 16.0, 14.4, 13.2; HRESIMS m/z 670.4677 [M + H]+ (calcd for C40H64NO7, 670.4683).
3β,23-Diacetoxyolean-12-en-N-[6-aminocaproic acid ethyl ester]-28-formamide (15):
565 mg (81%); white solid; mp 125−126 °C; 1H NMR (600 MHz, CDCl3) δ 6.04 (1H, brs, CONH), 5.37 (1H, s, H-12), 4.77 (1H, dd, J = 11.4, 4.7 Hz, H-3), 4.11 (2H, q, J = 7.1 Hz, OCH2CH3), 3.86 (1H, d, J = 11.6 Hz, H-23), 3.70 (1H, d, J = 11.7 Hz, H-23), 3.33−3.35 (1H, m), 2.98−3.03 (1H, m), 2.51 (1H, d, J = 12.0 Hz), 2.29 (2H, t, J = 7.4 Hz), 2.05 (3H, s, CH3CO), 2.02 (3H, s, CH3CO), 1.25 (3H, t, J = 7.1 Hz, OCH2CH3), 1.14 (3H, s, CH3), 0.97 (3H, s, CH3), 0.90 (3H, s, CH3), 0.90 (3H, s, CH3), 0.83 (3H, s, CH3), 0.76 (3H, s, CH3); 13C NMR (150 MHz, CDCl3) δ 178.2, 173.7, 171.1, 170.8, 145.3, 122.6, 74.6, 65.5, 60.4, 47.9, 47.8, 46.9, 46.4, 42.4, 42.2, 40.7, 39.5, 39.4, 37.9, 36.8, 34.3, 34.3, 33.1, 32.7, 32.2, 30.9, 29.3, 27.4, 26.7, 25.8, 24.7, 23.9, 23.7 (CH3CO), 23.7, 23.1 (CH3CO), 21.4, 21.1, 18.1, 17.1, 16.0, 14.4 (CH2CH3), 13.2; HRESIMS m/z 698.4990 [M + H]+ (calcd for C42H68NO7, 698.4996).
General Procedure for Synthesis of Compounds 16−18.
Compound 13, 14, or 15 (0.5 mmol) was dissolved in a mixture of MeOH/THF/H2O (2:1:1, 15 mL), and then potassium hydroxide (128 mg, 2.25 mmol) was added. This mixture was stirred at rt overnight. The organic solvent was removed under vacuum, and the residue was diluted with EtOAc (50 mL). Then, the pH of the solution was adjusted to 4−5 with 10% HCl. The organic layer was washed with brine (30 mL × 3) and dried over anhydrous Na2SO4. After being filtered, the filtrate was concentrated and purified on a silica gel column (30 g, hexanes/EtOAc) to give the pure 16−18, respectively.
3β,23-Dihydroxyolean-12-en-28-(glycine)oic acid amide (16):
251 mg (95%); white solid; mp 172−173 °C; 1H NMR (600 MHz, pyridine-d5) δ 7.93 (1H, brs, CONH), 5.55 (1H, s, H-12), 4.60 (1H, dd, J = 17.7, 5.7 Hz), 4.35 (1H, dd, J = 17.7, 4.5 Hz), 4.18−4.25 (2H, m), 3.74 (1H, d, J = 10.4 Hz), 3.17 (1H, d, J = 3.8 Hz), 1.24, 1.08, 1.04, 1.01, 0.94, 0.91 (each 3H, s, 6 × CH3); 13C NMR (150 MHz, pyridine-d5) δ 178.3, 173.6, 145.1, 123.5, 73.7, 68.2, 48.9, 48.5, 47.2, 46.8, 43.3, 42.8, 42.6, 42.5, 40.2, 39.2, 37.6, 34.8, 33.9, 33.5, 33.1, 31.3, 28.3, 28.0, 26.5, 24.3, 24.1, 18.9, 17.6, 16.4, 13.5; HRESIMS m/z 528.3694 [M − H]− (calcd for C32H50NO5, 528.3689).
3β,23-Dihydroxyolean-12-en-28-(4-aminobutanoic acid)oic acid amide (17):
256 mg (92%); white solid; mp 169−170 °C; 1H NMR (600 MHz, pyridine-d5) δ 7.52 (1H, s, CONH), 5.50 (1H, brs, H-12), 4.21 (2H, dd, J = 16.4, 7.7 Hz), 3.64−3.77 (2H, m), 3.55 (1H, dd, J = 12.9, 6.2 Hz), 3.12 (1H, dd, J = 13.1, 3.5 Hz), 2.65 (2H, t, J = 7.2 Hz), 1.22, 1.08, 1.05, 1.03, 0.94, 0.91 (each 3H, s, 6 × CH3); 13C NMR (150 MHz, pyridine-d5) δ 178.0, 176.3, 145.3, 123.3, 73.8, 68.3, 48.9, 48.5, 47.2, 46.7, 43.3, 42.6, 42.3, 40.2, 40.1, 39.2, 37.6, 34.8, 34.2, 33.6, 33.2, 33.1, 31.3, 28.3, 28.1, 26.5, 26.1, 24.3, 24.2, 24.1, 19.0, 18.0, 16.4, 13.5; HRESIMS m/z 556.4007 [M − H]− (calcd for C34H54NO5, 556.4002).
3β,23-Dihydroxyolean-12-en-28-(6-aminocaproic acid)oic acid amide (18):
263 mg (90%); white solid; mp 165−166 °C; 1H NMR (600 MHz, CDCl3) δ 5.36 (1H, brs, H-12), 4.11−4.12 (1H, m), 3.43 (1H, d, J = 10.2 Hz), 3.25−3.39 (3H, m), 2.98−3.30 (2H, m), 2.49 (1H, dd, J = 12.8, 3.4 Hz), 2.26−2.37 (4H, m), 1.14, 0.95, 0.89, 0.89, 0.88, 0.75 (3H, each s, 6 × CH3); 13C NMR (150 MHz, CDCl3) δ 178.4, 174.1, 145.2, 122.8, 76.8, 72.1, 51.7, 49.8, 47.7, 46.9, 46.4, 42.5, 42.3, 41.9, 39.6, 39.6, 39.5, 39.4, 38.3, 37.0, 34.3, 34.0, 34.0, 33.7, 33.1, 32.6, 32.3, 30.9, 29.2, 29.0, 29.0, 27.4, 26.7, 26.4, 26.3, 25.9, 24.7, 24.6, 24.4, 23.9, 23.7, 23.7, 18.6, 17.1, 15.9, 11.6; HRESIMS m/z 586.4456 [M + H]+ (calcd for C36H60NO5, 586.4471).
Cell Culture.
A549 human lung cancer, MDA-MB-231 and MCF-7 human breast cancer, KB human epidermoid cancer, and KB-VIN cells were obtained from ATCC. SMMC-7721 human hepatocellular carcinoma cells and BGC-823 human gastric carcinoma cells were obtained from the Cell Center of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, People’s Republic of China. A549, MDA-MB-231, MCF-7, KB, KB-VIN, SMMC-7721, and BGC-823 cells were cultured in RPMI-1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco/Invitrogen, Carlsbad, CA, USA), 100 u/mL benzyl penicillin, and 100 u/mL streptomycin in a humidified environment with 5% CO2 at 37 °C.
MTT Assay.
After diluting to 5 × 104 cells mL−1 with the complete medium, 100 μL of the cell suspension obtained was added to each well of 96-well culture plates. The subsequent incubation was performed at 37 °C, 5% CO2 atmosphere for 24 h before the cytotoxicity assessments. Tested samples at preset concentrations were added to each well, and the cells were incubated for 48 h. The MTT solution (100 μL, 0.5 mg/mL) was added to each well, and the cells were incubated for another 4 h. The formazan crystals were dissolved in 150 μL of DMSO. Cell viability was assessed by measuring the absorbance at a 490 nm wavelength using a Thermo Multiskan FC microplate photometer (Thermo Fisher Scientific Instruments Co., Ltd., Shanghai, People’s Republic of China).
The inhibition was calculated with the formula
where ODc is the absorbance of negative control and ODt is the absorbance of tested drug.
SRB Assay.
In brief, the cells (3−5 × 103 cells/well) were seeded in 96-well plates filled with RPMI-1640 medium supplemented with 10% fetal bovine serum containing various concentrations of samples and incubated for 72 h. At the end of the exposure period, the attached cells were fixed with cold 50% trichloroacetic acid for 30 min followed by staining with 0.04% SRB (Sigma Chemical Co.) for 30 min. The bound SRB was solubilized in 10 mM Tris-base, and the absorbance was measured at 515 nm on an ELx800 microplate reader (Bio-Tek Instruments, Winooski, VT, USA) with Gen5 software. All results were representative of three or more experiments.
The inhibition was calculated with the formula
Hemolytic Assay.
Nonheparinized blood was collected from healthy New Zealand rabbits (Experimental Animal Center of Soochow University; approved protocol number SUAEC-2016–028). The erythrocytes were washed three times in PBS (phosphate-buffered saline: NaCl, 8 g/L; KCl, 0.2 g/L; Na2HPO4, 1.44 g/L; and KH2PO4, 0.24 g/L; pH 7.4) and then diluted with PBS to obtain a 10% suspension. All tested compounds were dissolved in DMSO at a concentration of 10 mM, and then PBS was added to dilute the solution to the testing concentrations ranging from 5 to 500 μM. The final volume of the derivative solution was 1.0 mL. The erythrocyte suspension (100 μL) was added to 1 mL of the suspension to be tested, and the samples were rapidly stirred and incubated at 37 °C with periodic stirring during a 60 min incubation period. The solution was then centrifuged at 3000 rpm for 5 min. Absorbance of the supernatant was measured at 540 nm using a Thermo Multiskan FC microplate photometer (Thermo Fisher Scientific Instruments Co., Ltd., Shanghai, People’s Republic of China), and the hemolytic % was calculated by comparison with the 100% hemolytic activity caused by distilled water as maximal hemolytic control. The hemolytic % developed by the PBS control was subtracted from all groups. Each experiment was performed in triplicate at all concentrations used.
The hemolytic % was calculated with the formula
where ODt is the absorbance of tested drug, ODpc is the absorbance of positive control, and ODnc is the absorbance of negative control. The concentration inducing 50% of the maximum hemolytic activity is the HD50.
Cell Cycle Analysis.
Cells treated for different periods were trypsinized, washed with PBS, and fixed in 100% EtOH for 12 h at −20 °C. Then, they were washed with PBS and stained with PI containing 0.05% RNase before the DNA content was determined. The DNA content and cell cycle parameters were determined using a Becton-Dickinson (BD) LSR-II Fortessa flow cytometer. The data were analyzed using the software FCS Express 6 Flow Research Edition.
AnnexinV/PI Staining.
Apoptosis-mediated cell death of tumor cells was examined using a FITC-labeled annexinV/PI apoptosis detection kit (Invitrogen Corporation, NY, USA) according to the manufacturer’s instructions. In brief, 1 × 106 cells were harvested and washed with PBS. They were resuspended in 500 μL of binding buffer, and then 5 μL of annexin-V-FITC and 1 μL of PI were added. Flow cytometric analysis was performed immediately after supravital staining. Data acquisition and analysis were performed with a BD LSR-II Fortessa flow cytometer using the software FCS Express 6 Flow Research Edition. The cells in early stages of apoptosis were annexin V positive and PI negative, whereas the cells in the late stage of apoptosis were both annexin V and PI positive.
Western Blot Analysis.
Cells were collected and lysed in lysis buffer (100 mM Tris-Cl, pH 6.8, 4% SDS, 20% glycerol, 200 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride, and 1 g/mL aprotinin). The lysates were then centrifuged at 12000g for 15 min at 4 °C. The supernatant was collected, and the concentrations of the total proteins were determined using the bicinchoninic acid assay by Varioskan spectrofluorometer and spectrophotometer (Thermo, Waltham, MA, USA). Then protein samples were separated with 10−15% SDS-PAGE gel and transferred onto the PVDF membranes (Millipore, Billerica, MA, USA). Immune complexes were formed by incubation of proteins with primary antibodies overnight at 4 °C followed by horseradish peroxidase-labeled secondary antibodies. Immunoreactive proteins were detected by using enhanced chem-iluminescence reagent.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by the National Natural Science Foundation of China (Nos. 81302660, 81673513) and the Provincial Universities of Jiangsu Province Natural Science Foundation (13KJB350006). Partial support was also provided by NIH grant CA177584 from the National Cancer Institute awarded to K.H.L.
Footnotes
DEDICATION
Dedicated to Dr. Susan Band Horwitz, of Albert Einstein College of Medicine, Bronx, NY, for her pioneering work on bioactive natural products.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnat-prod.7b00578.
1H and 13C NMR spectra of 2−18 (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Xiao ZY; Morris-Natschke SL; Lee KH Med. Res. Rev 2016, 36, 32–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Man SL; Gao WY; Zhang YJ; Huang LQ; Liu CX Fitoterapia 2010, 81, 703–714. [DOI] [PubMed] [Google Scholar]
- (3).Winter WP Blood 1994, 84, Suppl 1–10, Abstr. 445.7949116 [Google Scholar]
- (4). (a)Wang Y; Zhang Y; Zhu Z; Zhu S; Li Y; Li M; Yu B Bioorg. Med. Chem 2007, 157, 2528–2532. [DOI] [PubMed] [Google Scholar]; (b) Gauthier C; Legault J; Karl GL; Mshvildadze V; Pichette A Bioorg. Med. Chem 2009, 17, 2002–2008. [DOI] [PubMed] [Google Scholar]; (c) Chwalek M; Lalun N; Bobichon H; Ple K; Nazabadioko LV Biochim. Biophys. Acta, Gen. Subj 2006, 1760, 1418–1427. [DOI] [PubMed] [Google Scholar]; (d) Oda K; Matsuda H; Murakami T; Katayama S; Ohgitani T; Yoshikawa M Biol. Chem 2000, 381, 67–74. [DOI] [PubMed] [Google Scholar]
- (5).Kim Y; Bang SC; Lee JH; Ahn BZ Arch. Pharmacal Res 2004, 27, 915–918. [DOI] [PubMed] [Google Scholar]
- (6).Bang SC; Lee JH; Song GY; Kim DH; Yoon MY; Ahn BZ Chem. Pharm. Bull 2005, 53, 1451–1454. [DOI] [PubMed] [Google Scholar]
- (7).Hong SW; Jung KH; Lee HS; Choi MJ; Son MK; Zheng HM; Hong SS Cancer Sci 2012, 103, 1929–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jang WJ; Park B; Jeong GS; Hong SS; Jeong CH Oncol. Rep 2014, 32, 2612–2618. [DOI] [PubMed] [Google Scholar]
- (9).Gauthier C; Legault J; Karl GL; Mshvildadze V; Pichette A Bioorg. Med. Chem 2009, 17, 2002–2008. [DOI] [PubMed] [Google Scholar]
- (10).Kim M; Lim E; Jung M Tetrahedron 2013, 69, 5481–5486. [Google Scholar]
- (11).Kim M; Park Y; Chung WY; Park KK; Jung M Chem. Pharm. Bull 2015, 63, 669–677. [DOI] [PubMed] [Google Scholar]
- (12).Chen Z; Duan HQ; Wang ML; Han L; Liu YL; Zhu YM; Yang SL Bioorg. Med. Chem. Lett 2015, 25, 2550–2554. [DOI] [PubMed] [Google Scholar]
- (13).Fang YY; Yang ZH; Ouyang H; Wang RK; Li J; Huang HS; Jin Y; Feng YL; Yang SY Bioorg. Med. Chem. Lett 2016, 26, 4576–4579. [DOI] [PubMed] [Google Scholar]
- (14).Fang YY; Wang RK; He MZ; Huang HS; Wang Q; Yang ZH; Li Y; Yang SL; Jin Y Bioorg. Med. Chem. Lett 2017, 27, 98–101. [DOI] [PubMed] [Google Scholar]
- (15).Tong XH; Han L; Duan HQ; Cui YR; Feng YL; Zhu YM; Chen Z; Yang SL Eur. J. Med. Chem 2017, 129, 325–336. [DOI] [PubMed] [Google Scholar]
- (16).Xu QM; Shu Z; He WJ; Chen LY; Yang SL; Yang G; Liu YL; Li XR Phytomedicine 2012, 19, 293–300. [DOI] [PubMed] [Google Scholar]
- (17).Shi BJ; Li Q; Zhang XQ; Wang Y; Ye WC; Yao XS Acta Pharm. Sin 2007, 42, 862–866. [PubMed] [Google Scholar]
- (18).Siewert B; Wiemann J; Köwitsch A; Csuk R Eur. J. Med. Chem 2014, 72, 84–101. [DOI] [PubMed] [Google Scholar]
- (19).Qian S; Li HJ; Chen Y; Zhang WY; Yang SY; Wu YJ Nat. Prod 2010, 73, 1743–1750. [DOI] [PubMed] [Google Scholar]
- (20).Ding Y; Huang ZJ; Yin J; Lai YS; Zhang SB; Zhang ZG; Fang L; Peng SX; Zhang YH Chem. Commun 2011, 47, 9495–9497. [DOI] [PubMed] [Google Scholar]
- (21).Gong W; Jiang ZH; Sun P; Li L; Jin YS; Shao LC; Zhang W; Liu BS; Zhang HW; Tang H; Chen YF; Yi YH; Zhang DZ Chem. Biodiversity 2011, 8, 1833–1852. [DOI] [PubMed] [Google Scholar]
- (22).Chen YF; Lin YC; Huang PK; Chan HC; Kuo SC; Lee KH; Huang LJ Bioorg. Med. Chem 2013, 21, 5064–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.