

SUMMARY
Neural crest (NC) cells undergo extensive migrations during development. Here, we couple in vivo live imaging at high resolution with custom software tools to reveal dynamic migratory behavior in chick embryos. Trunk NC cells migrate as individuals with both stochastic and biased features as they move dorsoventrally to form peripheral ganglia. Their leading edge displays a prominent fan-shaped lamellipodium that reorients upon cell-cell contact. Computational analysis reveals that when the lamellipodium of one cell touches the body of another, the two cells undergo “contact attraction,” often moving together and then separating via a pulling force exerted by lamellipodium. Targeted optical manipulation shows that cell interactions coupled with cell density generate a long-range biased random walk behavior, such that cells move from high to low density. In contrast to chain migration noted at other axial levels, the results show that individual trunk NC cells navigate the complex environment without tight coordination between neighbors.
Graphical Abstract
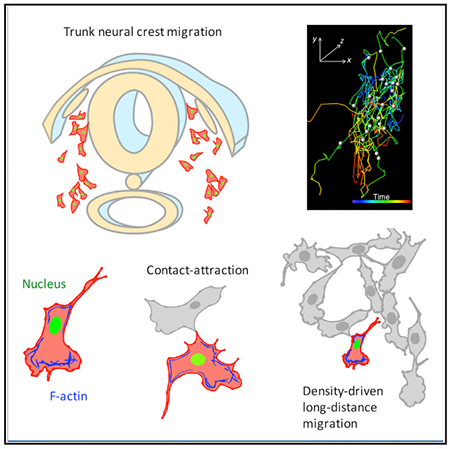
In Brief
Li et al. combine quantitative imaging with perturbation analysis to define the cellular dynamics driving trunk neural crest migration. Unlike chain migration at other axial levels, trunk neural crest cells move as individuals driven by the combined effect of lamellipodia mediated directionality, together with cell-cell contact and cell density.
INTRODUCTION
Cell migration is a critical aspect of normal development that abnormally recurs during cancer metastasis (Montell, 2006; Lecaudey and Gilmour, 2006; Friedl and Gilmour, 2009). The mechanisms underlying cell migration have been best described when cells collectively migrate as a group during events like tumor metastasis (Friedl and Gilmour, 2009), border cell migration in Drosophila (Prasad and Montell, 2007), and cranial neural crest migration in Xenopus (Carmona-Fontaine et al., 2008). In addition to collective migration, many vertebrate cells migrate as individuals, both during development and during cancer metastasis (De Pascalis and Etienne-Manneville, 2017). As these types of movements occur in a three-dimensional, often semi-opaque environment, clues to underlying mechanism typically have been gleaned by explanting individual or small groups of cells in tissue culture on two-dimensional substrates (Reig et al., 2014). In contrast, far less is known about how cells interact with each other within complex in vivo contexts and how this affects their speed, directionality, and pathfinding ability.
Studies based on static imaging indicate that neural crest cells in the trunk of amniote embryos undergo individual cell migration through a complex mesenchymal environment (Krull et al., 1995). These cells delaminate from the neural tube as single cells and approach the somites that are reiteratively arranged along the length of the trunk. Upon reaching the somitic milieu, they migrate ventrally to populate dorsal root ganglia, sympathetic ganglia, and the adrenal medulla (Le Douarin, 1982). However, trunk neural crest cells are constrained to the anterior half of each somitic sclerotome due to the presence of repulsive cues, most notably Semaphorin 3F and ephrins, in the posterior half of each somite (Gammill et al., 2006; Krull et al., 1997). Interestingly, both the migratory routes and modes of movement of individual trunk neural crest cells, as inferred from immunofluorescence (Krull et al., 1995), appear to be distinct from those of cranial neural crest cells in Xenopus that form collective sheets (Kuriyama et al., 2014; Theveneau et al., 2013). This is consistent with well-known differences in the gene regulatory networks governing cranial and trunk neural crest programs (Simoes-Costa and Bronner, 2016).
The molecular networks underlying the epithelial to mesenchymal transition (EMT) (Scarpa et al., 2015; Schiffmacher et al., 2016) and directing collective migration of neural crest cells of the head have been well described (Kuriyama et al., 2014; Theveneau et al., 2013). In contrast, the mechanisms acting at trunk levels remain to be determined. How do these cells migrate as individuals in developing embryos? Do they migrate autonomously and/or interact with their neighbors to arrive at the final destinations and differentiate into appropriate derivatives? Dynamic imaging, with longitudinal visualization and quantitative descriptions of migratory events in intact tissues (Megason and Fraser, 2007; Li et al., 2015), offers a unique opportunity to examine neural crest cell behavior. A major challenge is that neural crest cells become less accessible to optical microscopy as they move deep into tissue, rendering their complete trajectories difficult to follow. In addition, there is a trade-off between spatial resolution and field of view associated with microscope objectives. Consequently, previous studies have either used low magnification to capture multiple migration streams across the whole embryo (Kulesa and Fraser, 1998) or high magnification to distinguish cellular processes, such as cell division and cell volume changes, within a constrained context (Ahlstrom and Erickson, 2009; Ridenour et al., 2014), but obfuscating resolution of the relationship between cell morphological changes and cell migration. Moreover, limited quantitative tools are available to map the spatiotemporal activity of highly dynamic lamellipodia in an unbiased and statistically robust fashion.
Here, we tackle these challenges by examining migration of trunk neural crest cells in their in vivo environment. Using tissue slices through the trunk region of avian embryos, we visualize their extensive locomotion from the dorsal neural tube to the dorsal aorta with exquisite spatiotemporal resolution under normal and experimentally manipulated conditions. Combining optogenetics, laser ablation, and quantitative analysis, we reveal multiple factors that contribute to trunk neural crest migration, including lamellipodia mediated cell motion, cell-cell contact, cell density, and physical confinement. We further demonstrate the importance of F-actin flow and mechanical force in determining the direction of cell movement and cell-cell separation. These results offer insight into individual cell migration and point to distinctions between this mode of migration and collective cell migration noted in other regions of the embryo. Moreover, the software tools developed here can be used to distinguish different types of cell-cell contact and will be broadly applicable to analysis of diverse morphogenetic events.
RESULTS
Long-Term Time-Lapse Imaging of Trunk Neural Crest Migration
To follow the trajectories of neural crest cells moving deep inside the embryo, we adapted and optimized a method for using tissue slices through chick embryos (Shiau et al., 2011) for live imaging of the trunk region. To this end, a thick transverse slice (2 somite wide, ~500 μm in thickness) was made through the forelimb level of stage HH18-19 chick embryos (Figure 1A). For long-term imaging (13 h), a mold with a nylon grid was placed on the slice that only attached to structures ventral to the dorsal aortas, such that normal routes of cell migration through dorsal regions of interest was unperturbed. This method offers an excellent experimental system for studying neural crest migration as the tissue retains its morphology and cells continue to migrate and proliferate through their normal environment.
Figure 1. Live Imaging on Migrating Neural Crest Cells within Chick Embryonic Slices.
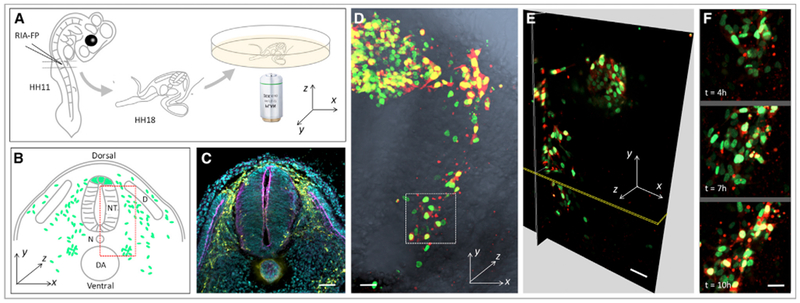
(A) Experimental setup. RIA virus encoding the fluorescent protein (FP) of interest was introduced into the chick neural tube at stage HH11. At stage HH18, a transverse slice at the forelimb level was transferred into the imaging dish for time-lapse imaging.
(B) Schematic diagram showing the general morphology of the sliced embryo and the coordinate axes used for quantitative analysis. The green dots and red dashed box depict migrating cells and the region imaged, respectively. NT, neural tube; D, dermomyotome; N, notochord; DA, dorsal aorta.
(C) Immunofluorescence of a frozen section to show the distribution of migrating trunk neural crest cells (HNK-1 positive, yellow) at stage HH18. The anatomical structure of the embryo was highlighted by phalloidin (magenta) and DAPI (cyan) staining to label F-actin and the nucleus, respectively. Scale bar, 100 μm.
(D–F) 4D (xyz and t) visualization of cell migration (n = 2 tissue slices).
(D) One selected time frame of a time-lapse video on a tissue slice expressing H2B-GFP (green) and mCherry (red), which was merged with phase-contrast image (gray) showing the in vivo context.
(E) Three orthogonal sections of the 3D-rendered image in (D).
(F) Representative snap shots of the selected region (white box in D).
Scale bars: (D and E) 25 μm; (F) 15 μm. The signal intensity of the images in (F) was adjusted from the corresponding video to present cell morphologies more clearly.
We employed replication-incompetent avian retrovirus (RIA) (Chen et al., 1999) to fluorescently tag neural crests with cytoplasmic mCherry and nuclear H2B-GFP. A high-titer virus (106–107 plaque-forming units [PFU]/mL) infects large numbers of premigratory cells, providing an opportunity to concomitantly track the movements of multiple cells (Figure 1D). In addition, a viral cDNA integrates into the host genome at a low copy number (Laufs et al., 2003) and thus labels individual cells with almost uniform fluorescent intensity (Li et al., 2018), enabling precise 3D (xyz) cell segmentation and 4D (xyz and t)trajectory mapping (Li et al.,2015).
To resolve neural crest migration at cellular resolution (10-μm scale) and capture complete cell trajectories as they move in a 240 × 100 × 80 μm3 region from dorsal neural tube to destinations adjacent to the dorsal aorta (Figure 1B), we performed time-lapse imaging using confocal microscopy (Figure 1A). Using a 20×/0.8 NA objective with large field of view, one lateral half of the slice was imaged (Figures 1B and 1C) at 8-min intervals (Figures 1D–1F; Video S1). By performing immunofluorescence on the post-imaged sample, we confirmed that all the infected cells expressed the HNK-1 epitope, a molecular marker of migrating neural crest cells (Figure S1), suggesting that the slice and imaging conditions reflect normal development.
Trunk Neural Crest Cells Undergo a Long-Range Biased Random Walk
Migrating trunk neural crest cell exhibited an array of cellular behaviors, ranging from persistent and directional migration from dorsal to ventral, to discontinuous and even backward (ventral to dorsal) motion (Video S1). The majority of cells appeared to move as individuals, although some were more clustered. This apparent heterogeneity in cell behavior necessitated quantitative analysis. To this end, we defined a coordinate system based on the geometry of the embryo (Figures 1A, 1B, 1D, and 2A). The dorsoventral (DV) axis was defined as the y axis with the dorsal neural tube and notochord as the proximal and distal ends. The mediolateral (ML) axis was defined as the x axis, with the z axis running anterioposteriorly (AP).
Figure 2. Neural Crest Cells Display a Biased Random Motion.
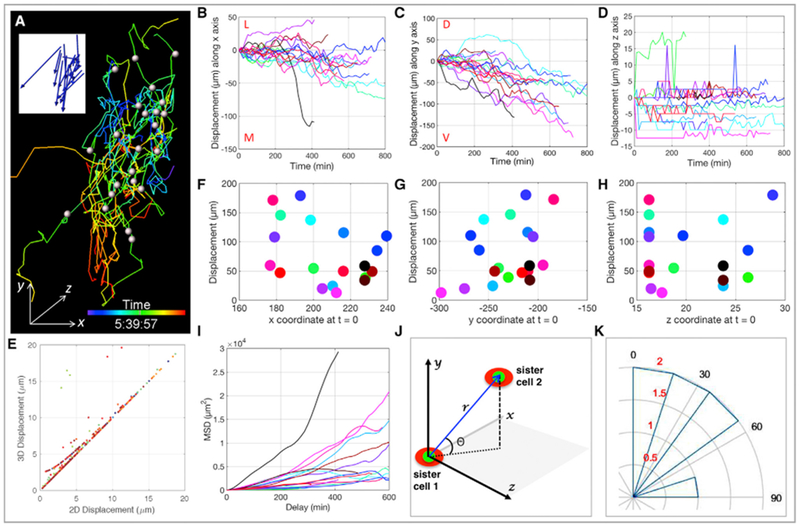
(A) Migrating cells were spot segmented with the white dots representing their centers at t = 0 and the corresponding cellular trajectories color-coded according to time. The blue arrows in the insert depict the final displacement directions of the cells.
(B–D) Track displacement lengths over time along x (B), y (C), and z (D) axes with each line representing the trajectory of one cell (n = 21). Maximal and minimal displacements were observed in the y (C) and z directions (D), respectively. M, L, D, and V stand for medial, lateral, dorsal, and ventral, respectively.
(E) Pearson correlation analysis shows the coordination between 2D (xy) and 3D (xyz) cell displacements.
(F–H) Total track displacement length as a function of cell initial position along x (F), y (G), and z (H) axes with each dot representing one cell. The random distribution (R2 = 0.1356) of the dots implies a lack of correlation.
(I) Mean square displacement (MSD) analysis of cell trajectories reveals all three modes of motion: directed (α > 1), free diffusion (α ≈ 1), and constrained (α < 1), highlighting the heterogeneous nature of cell migration.
(J and K) Neural crest cells did not divide along the DV axis of embryos.
(J) Coordinate system used to measure the orientation of cell division. r is the 3D distance between nucleus (green) of daughter cells (red), and θ is the angle between r relative to the xz plane.
(K) The orientation of mitosis was estimated based on the quadrant in which it was located. Angular histogram illustrates division angles, and the concentric rings are in correspondence to cell number colored in red (n = 7).
Dissecting cell trajectories along these axes revealed a spreading behavior (Figure 2A) with maximal cell displacement in the DV direction (Figures 2B–2D), as expected based on the anatomical locations of neural crest derivatives (Bronner and Simões-Costa, 2016). Intriguingly, this DV motion displayed a stable pattern: at any given time, cells had a similar chance of migrating toward the dorsal neural tube (dorsal) or dorsal aorta (ventral), with the probability of ventral-ward motion increasing overtime (Figure 2C). Along the ML axis, a similar cell spreading pattern was observed with more displacement toward the medial direction (Figure 2B). The lower axial resolution of one-photon imaging limits resolving finer details of cell displacement in the z direction, but cells generally exhibited a slightly zigzagged motion with negligible directional displacement along this axis (Figure 2D) compared with that of x and y axes. This is not surprising given that trunk neural crest cells are constrained to migrate through the anterior half of each somite (Gammill et al., 2006; Krull et al., 1997); as a result, this z axis motion did not significantly affect cell migration behavior in 3D (Figure 2E).
The combined long-term spreading and short-term oscillation of cell trajectories indicate that trunk neural crest migration is biased and stochastic in nature. Consistent with this, mean square displacement (MSD) analysis, a standard approach to evaluate the mode of movement of objects (Michalet, 2010), showed that population spreading is comprised of all three possible modes of movement, including free diffusion and directed and constrained movement (Figure 2I). On a global scale, there was little correlation between a cell’s original position and its total displacement length (R2 = 0.1356 in the y direction) (Figures 2F–2H), highlighting the likelihood that trunk neural crest cells do not move in a concerted fashion.
To label neural crest cells by an alternative approach that would enable more color combinations for cell tracking, embryos were co-electroporated with plasmids encoding Sox10E1-driven GFP (Betancur et al., 2010), NC2-driven cytoplasmic-mCherry (Simões-Costa et al., 2012) and nuclear-localized far-red fluorescent protein (NLS-iRFP). Subsequent imaging analysis on the multiplexed tagging samples confirmed the migratory patterns of trunk neural crest cells (Figures S2A–S2H; Video S2) observed above with RIA labeling.
During some types of cell morphogenetic events, such as zebra-fish gastrulation and Drosophila wing growth, contractile forces generated during oriented cell division drive cell displacement (Gong et al., 2004; Baena-López et al., 2005). To test whether this might be the case here, we identified cells undergoing mitosis (~33% of total) based on nuclear H2B-GFP expression. Using a defined coordinate system to calculate the angles between sister cells at the time of division (Figure 2J) (Li etal., 2017), we found that the division angles were shifted toward, rather than parallel to the DV direction (Figure 2K), ruling out the possibility that oriented cell division plays a major role in determining directionality.
Taken together, the results show that, despite the fact that the migratory behavior of individual trunk neural crest cells is not predictable, the cell population eventually move toward the dorsal aorta, consistent with the parabolic form of the mean square displacement curve of overall cell trajectories (Figure S2I). We refer to this behavior as a “biased random walk.” Such stochastic cell motion might lead to sister cells on distinct trajectories and final sites of localization. Indeed, we found that sister cells were frequently far apart by the end of imaging (Figure S2J), with siblings localized in distinct sites correlating with different fates (e.g., sensory versus sympathoadrenal). This supports the idea of stochastic motion and a random biased walk as the primary underlying mechanism.
Coordinated Relationships between Lamellipodia, Cell Morphology, and Cell Migration
Changes in cell shape and cell interactions are well known to be critical for cell movement (Köppen et al., 2006), but have been best studied in vitro or in explants by analyzing cell behavior on defined substrates (Reig et al., 2014). In contrast, much less is known about the influence of cell morphology on cell migration in vivo due to difficulties of capturing cells as they move deep into the complex environment of live embryos. To address trunk neural crest behavior at high temporal resolution, we increased the sampling frequency of our imaging analysis by collecting images every 2 min on cells expressing membrane-YFP, allowing us to resolve fine details of cell protrusions (Figure 3A; Video S3). Using a 20× objective, we could visualize multiple cellular events in the same tissue slice. In agreement with in vitro predictions, individual cells appeared to exhibit distinct broad lamellipodia followed by a more bulbous cell body and, finally, a thin tail (Figure 3D1; Video S3).
Figure 3. Neural Crest Migration Is Influenced by the Lamellipodium, Cell Morphology, and Cell Density.
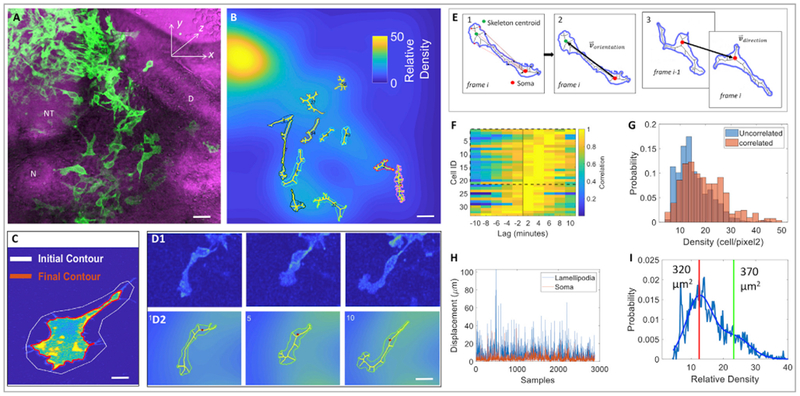
(A) A selected time frame of a time-lapse imaging on a tissue slice expressing membrane-YFP (green) (n = 2 tissue slices); it was merged with phase contrast image (magenta) showing neighboring tissues. NT, neural tube; D, dermomyotome; N, notochord. Scale bar, 15 μm.
(B) A selected 3D image of the same video in which segmented cells were filled with skeletons. Each cell was encoded with a distinct color for convenience of visual tracking of cell locomotion in the corresponding videos. The background of the image was further color-coded according to the relative cell density, defined as a Gaussian smoothed fluorescence intensity of membrane-YFP per pixel. Scale bar, 15 μm.
(C) Cell-surface segmentation in which the white line was the initial user defined contour, and the red line was the computer-generated contour of the cell. Scale bar, 3 μm.
(D) Representative snapshots of a migrating cell selected from (A) with pseudocolor based on fluorescence intensity (D1) and filled with skeleton (D2). Scale bar, 5 μm.
(E) Quantitative analyses of the orientation of lamellipodia and the direction of cell migration. (1) The contour was filled with a skeleton and the distance was calculated between the cell body/soma (centroid depicted as a big red dot) and all skeletal points (depicted as small red dots)that connect to cell protrusions. The three skeletal endpoints furthest from the cell body were identified to draw a polygon, which is the lamellipodium (its centroid is depicted as a green dot). (2) The orientation vector, depicting cell morphology, was generated by drawing a black line between the centroids of cell body and the lamellipodium. (3) The direction vector, depicting the direction of cell migration, was drawn between the cell body centroids one time frame apart. The change of the angle between the orientation vector and direction vector was used to assess the correlation between lamellipodial orientation and cell motion direction.
(F) Cell enlargement and displacement occurred in a sequential order. Cross-correlogram analysis of the area and cell body velocity over time revealed that the cell body and area only changed in a synchronized manner in a small fraction of cells (6% show symmetric cross-correlograms, Jarque-Bera test p < 0.05, n = 78). For the remaining cells with statistically significant cross-correlograms, 65% showed asymmetric cross-correlograms in which cell enlargement preceded cell movement (rank-sum test p < 0.05, n = 33).
(G) The lamellipodial orientation and direction of cell migration were positively correlated in a region with intermediate cell density. Cells whose direction and orientation vector were positively correlated (p < 0.05) were presented near the regions of relatively high density (average density when correlated 18 ± 9 vs. 14 ± 7 when not correlated, rank-sum test p < 10−21).
(H) The lamellipodium was more dynamic than the cell body. Over time, pixel shifts (the readout of displacement) of the lamellipodia were larger than cell bodies (median ± SD, soma: 2.7 ± 4.8 μm versus lamellipodia: 6.7 ± 6.7 μm, rank-sum test p < 10−280) and these changes were largely uncorrelated (0.08 correlation, p = 10−6).
(I) Cell size and regional density were negatively correlated. Cells in lower density areas (left of vertical red line) were significantly smaller than cells in higher density (left of red vertical line, median: 320 μm2 vs. 370 μm2; rank-sum test p = 10−14).
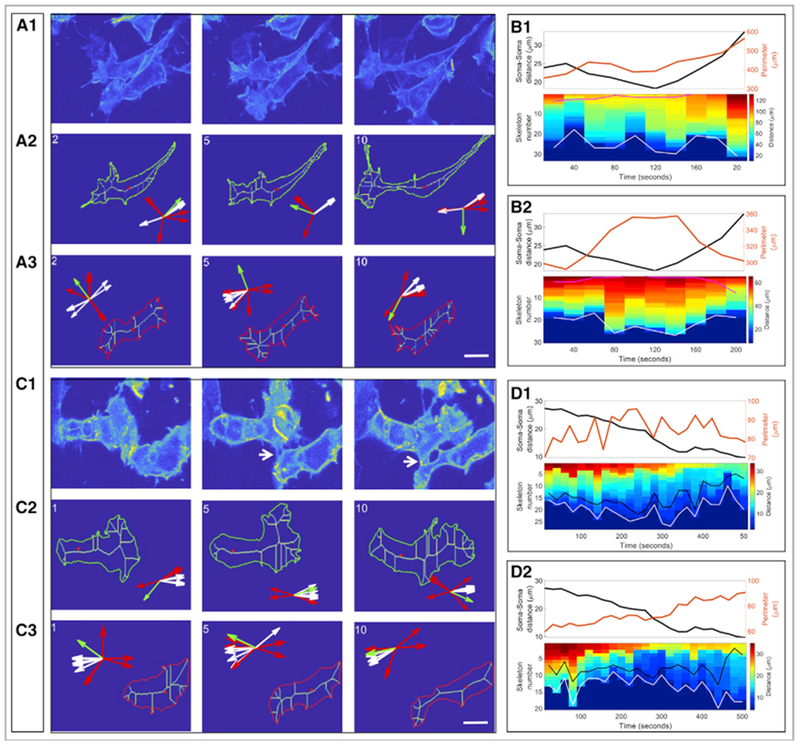
To quantitate cell shape changes in a statistically robust fashion, we performed surface segmentation of individual migrating cells by drawing a contour with an initial state boundary larger than the shape of the cell (Figure 3C) (Chan and Vese, 2001), thus minimizing shape bias introduced by the user. Further computational decomposition of cell morphology (Video S4) into a subset of quantifiable metrics revealed that cells increased their area prior to movement of the cell body (Figure 3F), suggesting that neural crest cells need to acquire a stretched configuration to carry out migration.
Next, we assessed the role of the lamellipodia during cell migration. Because of the highly dynamic nature of this subcellular structure, we extracted cellular contours using a minimalistic skeletonized representation which is subject to constraints in the image; in this case, the branches of the skeleton were connected to filopodia and changed accordingly (Figures 3D and 3E). Given that there are more filopodia on the lamellipodium than on the cell body/soma, this skeleton representation approach allowed us to decouple the changes of these two sub-cellular structures and further quantify their relative motion (Video S4). As cells exhibited variable behaviors in different regions of the tissue slice (Video S3), we generated a cell density map based on the intensity of membrane-YFP to further investigate the effect of crowding on cell motion (Figure 3B; Video S4).
Overlaying the cell skeleton map with the cell density map revealed that the orientation of lamellipodia extension and the direction of cell migration were coordinated in 42% of cells that could be cleanly segmented (Figure 3G). Compared with the cell body, the lamellipodium displayed more dynamic fluctuations (Figure 3H). Noticeably, the relationship between lamellipodial orientation and direction of cell movement was influenced by cell density, such that a statistically significant correlation was predominant in regions with optimal (intermediate) cell density (Figure 3G). This suggest that cells are likely to use contact as a means to maintain coordination between the lamellipodium and directional motion. Intriguingly, in the high-cell-density region with more frequent cell-cell contact events, cells became more compact (Figure 3I). The observed mixed heterogeneous cell morphological and motion changes are consistent with the spread and intermixture of cell trajectories (Figures 2A–2D).
In summary, our cross-correlation studies at a global scale suggest that neural crest cells display two primary morphologies: a stretched state during their migration with a broad fan-shaped lamellipodium at the leading edge, and a more compact state upon cell-cell contact that affects the direction of cell movement.
Lamellipodia Drive Short-Range Directional Cell Migration in a Neighbor-free Region
Next, we sought to determine the roles of lamellipodia and cell-cell contact at the single cell level. By performing both higher spatial (63×/1.4 NA objective) and temporal (collecting images every 20 s) resolution imaging analysis on individual migrating cells in a neighbor-free zone for ~5 min, we confirmed that lamellipodial turning was coordinated with the orientation of cell movement (Figures 4A–4C; Video S5). In support of a leading role for the lamellipodium, F-actin was highly expressed in the front side of migrating cells (Figure 4D; Video S5).
Figure 4. Neural Crest Migration Is Directed by the Lamellipodium in a Neighbor-free Zone.
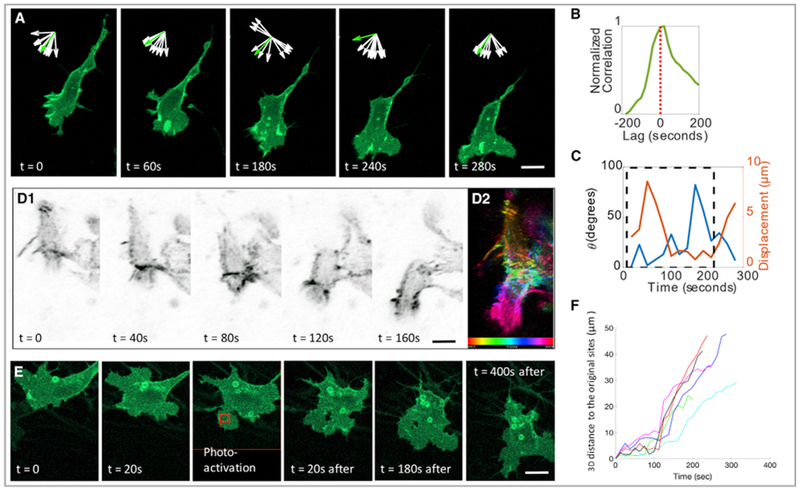
(A) Focusing on single cell at high resolution, the lamellipodium (membrane-YFP, green) was located on the leading edge of a migrating cell in a neighbor-free zone. Inset arrows show the normalized orientation of 10 skeleton endpoint (white) and the displacement vector (green) of cell body over time, suggesting the coordinated relationship between these two cellular factors. Scale bar, 5 μm.
(B and C) Quantitative analysis of the cell in (A) confirmed the coordinated relationship between lamellipodia and cell migration.
(B) Cross correlation between area change and soma displacement; a positive lag suggests that area increases prior to cell body displacement.
(C) Angle between the lamellipodium orientation and displacement vector (blue) and cell displacement (orange) were plotted. Maximal cell displacement occurred when both vectors were collinear (blue, θ close to 0). Dashed black rectangle highlights the frames during which the angle and displacement were inversely correlated (coefficient −0.62 and p < 0.05).
(D) Montage of a migrating cell with F-actin (utrophin-scarlet, gray) enriched in the front end of the cell (D1). F-actin distribution was color-coded according to time (D2). Scale bar, 5 μm.
(E and F) In one specific portion (red box) of a stationary cell expressing PA-Rac, photoactivation with a 458-nm laser induced the cell to migrate in the direction of laser illumination (E). Cell trajectory analysis with inflection points of the curves corresponded to the time points of photoactivation (F). (n = 6); two slices were imaged, and the cells were combined for analyses. Scale bar, 5 μm.
As a further test of their causal link, we asked whether activation of the Rac GTPase, known to regulate actin dynamics (Nobes and Hall, 1995), was sufficient to induce lamellipodial protrusion in a specific area of the cell and shift the direction of cell movement. To this end, we utilized a photoactivatable form of Rac (PA-Rac) (Wu et al., 2009) and activated it with light in a portion of the cell that previously lacked a lamellipodium; as a consequence, the cell elicited a lamellipodium in the activated region and subsequently changed orientation to move in the direction of the new lamellipodium (Figures 4E and 4F; Video S6). Thus, in developing embryos, the front-to-back cell polarity of neural crest cells is tightly associated with biased cell motility.
Lamellipodia and Cell-Cell Adhesion Cooperate to Control Cell-Cell Contact Attraction
Using the same high-resolution analyses, we observed several types of cell-cell contact. For example, when the lamellipodium of a dorsally moving cell came in contact with the cell body of a ventrally moving cell, the latter was pulled back in the direction of the former (Figure 5A; Video S7); we termed this behavior “contact attraction.” In the high cell density region, neural crest cells slid along each other (Figures 6A and 6B; Video S8) and appeared to use each other as substrates to crawl upon (Figures 6C and 6D; Video S8). In contrast, when the lamellipodia of two individual cells contacted each other, the lamellipodia collapsed and cells turned away from each other and dispersed, referred to here as contact repulsion (Figure S3A; Video S9); this phenomenon is similar to previously described contact inhibition of cell locomotion at the cranial level (Carmona-Fontaine et al., 2008; Mayor and Carmona-Fontaine., 2010), except that the trunk neural crest cells did not freeze at the site of contact but rather changed directions abruptly. These findings suggest that the cell body is more adhesive than the lamellipodium; in consistent with this, cross-correlation analyses at a global scale showed that cell density near the cell body was higher than near the lamellipodium (rank sum on values p = 10−6, n = 2,887).
Figure 5. Neural Crest Migration Is Controlled by Cell-Cell Contact Attraction.
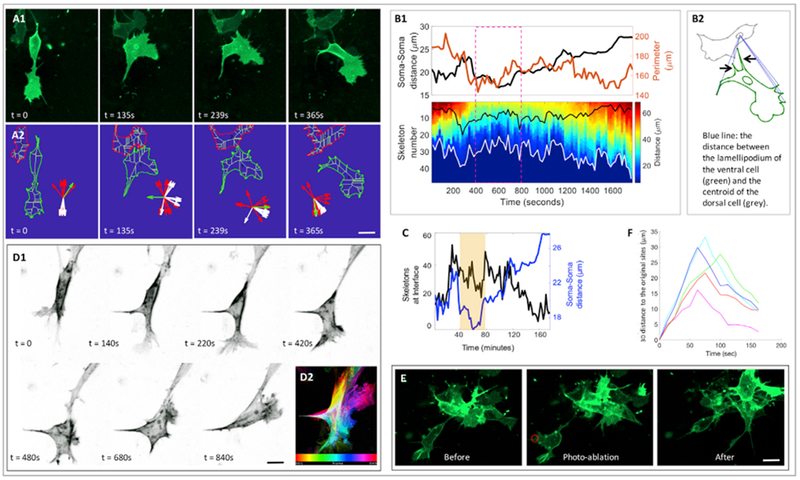
(A) Snap shots of contact attraction with both fluorescence (A1) and skeleton (A2) view: one cell (membrane-YFP, green) moving in the ventral direction with an elongated body was attracted by another cell on its dorsal side. The front (more ventral) cell became rounder and withdrew its lamellipodium. Subsequently, this front cell acquired its stretched shape and started to move forward. Inset (A2) showed the orientations of ten skeleton endpoints of the ventral cell (green) near to (red) and far from (white) its own cell body, which was further used for quantitative analyses in (B) and (C). Scale bar, 5 μm.
(B) Analyses of cell-cell distance change and cell morphological change of the cells in A. Data analyses (B1): the diagram showing the parameters used in (B1) (B2). Upper graph of (B1): the cell perimeter (orange line) of the ventral cell and its distance to the dorsal cell (black line) as a function of time. It confirmed that the ventral cell was more compacted when two cells were closer to each other (dashed magenta box). Lower graph of (B1) is the kymograph of the distance between the cell protrusions of the ventral cell and the centroid of the dorsal cell (white curve in the kymograph and the blue dashed straight lines in the diagram of B2). Focusing on the dashed magenta in the kymograph, the overall distance decreased as both cells were closer, consistent with the stationary state of the ventral cell upon its contacting with the neighbor. The black curve in the kymograph represented the number of cell protrusions of the ventral cell pointing toward the cell-cell interface (also illustrated in the diagram of B2 pointed by black arrows), which was termed inter-cellular protrusions. Prior to cell-cell association (250–400 s), inter-cellular protrusions increased, a sign of the reorientation of the lamellipodium; when the cells touched each other (400–1,300 s), these protrusions diffused around the cells and only a fraction was at the interface; after frame 1,300 s, the number of these protrusions decreased, consistent with the observation that the ventral cell moved away.
(C) Cell-cell distance change (blue curve) and the percentage of inter-cellular protrusions among total protrusions (black curve). The change of this percentage suggests that the ventral cell is more compact, and its protrusions are more uniformly distributed upon its associating with the neighbor.
(D) Montage of a front cell in which F-actin (utrophin-scarlet, gray) progressively moved to the other end of the cell (D1). F-actin distribution was color-coded according to time (D2). Scale bar, 5 μm.
(E and F) Lamellipodium generates a pulling force to break cell-cell adhesion. Focusing on a cell (membrane-YFP, green) leaving a cell cluster with a connecting tail (E), laser elimination of the lamellipodium (within the red circle) resulted in the backward motion of this cell. Cell trajectory analysis with the inflection points of the curves corresponded to the time points when laser ablation was applied (F). Two slices were imaged, and the cells were combined for analyses. Scale bar, 5 μm.
Figure 6. Cell Morphological Analysis of Cell Sliding and Crawling.
(A and B) Sell sliding.
(A) Representative snapshots showing two cells slide against each other with pseudo-color based on fluorescence-intensity (A1). Skeleton view of the upper (A2) and the lower (A3) cells.
(B) The same analyses in Figure 5B was applied and showed that both the upper (B1) and the lower (B2) cells displayed very few inter-cellular protrusions (the magenta lines in the kymographs), consistent with cell behaviors during sliding. Scale bar, 7.5 μm.
(C and D) Cell crawling. Representative snapshots showing two cells used each other as substrate to crawl (C1). Skeleton view of the upper (C2) and the lower (C3) cells. The majority inter-cellular protrusions in both cells fluctuated near the cell-cell interface (the black lines in the kymographs) (D1 and D2), a sign of cell crawl. Scale bar, 7.5 μm.
See also Video S8.
As contact attraction has not been previously described, we tested the underlying mechanism in the low cell density region in which we could perform more accurate cell-surface segmentation. Upon contact, the cell in front changed its morphology, becoming rounder, resulting in a pronounced increase in the number of lamellipodia at the cell-cell interface (Figures 5A–5C; Video S7). The F-actin within the lamellipodial fan appeared to progressively move to the other side of the front cell as the body of this cell stretched and acquired a polygonal shape (Figures 5A–5D; Video S7); concomitantly, the intercellular bridge became straight and eventually broke apart, allowing the cells to move forward. The stretched cell body, the straight tail during cell separation, and the directional F-actin flow are suggestive of pulling forces generated by the lamellipodium.
To test this, we performed targeted laser ablation focusing on the cells that were moving away from a cell cluster with the lamellipodium located in the front and the rear tail connected to the cluster. When the lamellipodium was partially eliminated, the targeted cell retracted (Figures 5E and 5F; Video S10), a sign that the lamellipodium exerted a pulling force. The backward motion of the cell after laser ablation was likely due to adhesion in the tail; as evidence for this, laser ablation of the tail caused rapid detachment of the cell from other cells (Figure S3B; Video S9). Thus, the lamellipodium and intercellular adhesion appear to play counteracting roles in separating cells following co-attraction.
Cell Density Promotes Biased Random Walk of Neural Crest Cells
Overlaying fluorescent images of migrating cells with phase-contrast images of whole tissue slices revealed that trunk neural crest cells were repelled by neighboring tissues like the neural tube, posterior sclerotome and notochord (Video S3), consistent with the findings of previous molecular perturbation studies (Krull et al., 1997; Gammill et al., 2006). In this constrained context, the lamellipodium tended to orient itself and drive cell locomotion toward the low cell density region (Figure 3B). Thus, we hypothesized that cell density may translate the random cell motion observed at short range into a more biased cell motion at long range.
To functionally test this possibility, we lowered the cell density by performing laser ablation experiments to remove a large number of migrating cells (n≈30) at a level adjacent to the ventral-most region of the neural tube (Figures 7A and 7B; Video S11). Strikingly, the remaining cells from the dorsal region aggregated into a chain and moved more rapidly with a higher directionality (Figures 7C–7G, 7K, and 7L). Accordingly, the cells’ original positions and their total displacement lengths were more coordinated along the dorsoventral body axis compared with unablated controls (Figures 7H–7J). All these cellular behaviors were similar to collective cell motion (Ilina and Friedl, 2009), supporting the view that trunk neural crest cells are subject to the combined effects of cell density and physical confinement, together promoting a biased random walk.
Figure 7. Cell Density Promotes Long-Range Directional Cell Migration.
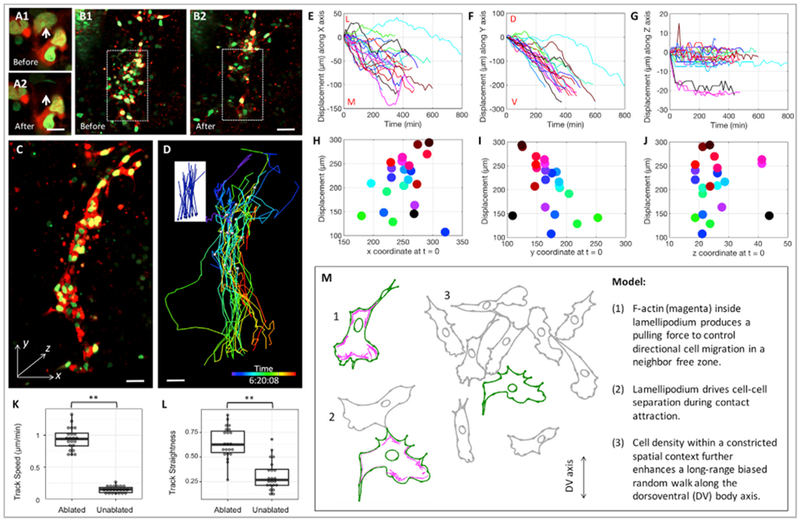
(A) Targeted laser illumination specifically ablated single cell in a tissue slice expressing H2B-GFP (green) and mCherry (red). The white arrows pointing to the cell before (A1) and after (A2) ablation. Scale bar, 10 μm.
(B) Around 30 cells in the ventral side of the tissue (enclosed within the white dashes box) were laser ablated. The tissue before (B1) and after (B2) laser ablation. Scale bar, 100 μm.
(C and D) A selected time frame of live imaging on the laser-ablated tissue (C) with the color-coded lines highlighting cellular trajectories (D) and net displacement vectors (insert, D). Scale bars, 20 μm.
(E–G) Cell displacement length over time along the x (E), y (F), and z (G) axes (n = 21). In the dorsolventral direction (F), cell trajectories appeared to be straighter compared with the ones in unablated tissues (Figure 2C).
(H–J) The relationship between total cell displacements and their original positions in the x (H), y (I), and z (J) directions. In the dorsolventral direction (I), these two factors appeared to be more coordinated compared with the ones in unablated tissues (Figure 2G).
(K) Statistical analysis of cell speed confirming faster cell movement in the ablated tissues compared with the un-ablated tissues (Figure 2) (n = 21 for each group). **p < 0.01 (rank-sum test).
(L) Statistical analysis of the straightness of cell trajectories showing higher directionality of cell movement in the ablated tissues compared with un-ablated tissues (Figure 2) (n = 21 for each group). **p < 0.01 (rank-sum test).
(M) Schematic diagram illustrating the migration mode of trunk neural crest cells.
See also Video S11.
DISCUSSION
Neural crest cells have the ability to migrate extensively to diverse sites throughout the embryo (Le Douarin, 1982). While gene expression analysis and perturbation studies have identified many molecules required for collective neural crest migration as occurs in the head region of Xenopus (Theveneau and Mayor, 2012), far less is known about their migration that occurs in the trunk of amniote embryos. In this study, we follow individual cells during long-distance migration and assess the roles of distinct cellular contributors (Figure 7M). The results uncover several features of trunk neural crest migration. First, these cells display a stereotypic morphology with F-actin and lamellipodium controlling short-distance cell migration in a neighbor-free zone. Second, cell movement is reoriented by multiple types of cell-cell interactions, including contact attraction when one cell contacts the lateral side of another, and contact repulsion, also called contact inhibition (Carmona-Fontaine et al., 2008; Mayor and Carmona-Fontaine, 2010), when two lamellipodium collide. Third, cell density and physical barriers of neighboring tissues influence biased cell motion.
High-resolution imaging reveals distinct geometric shapes of migrating trunk neural crest cell, likely reflecting changes in mechanical forces occurring during their movement (Heisenberg and Bellaïche, 2013). Notably, many cells tend to have a fan shaped lamellipodium with a narrow neck between the lamellipodium and cell body (Figure 5A, t = 0). One possible explanation for this characteristic shape is that rapid frontward extension of the lamellipodium precedes movement of the remaining cell body containing the rigid nucleus; as a result, a narrow region forms between the lamellipodium and the body. Our observations show that neural crest cells constantly touch each other, separate, and reorient themselves. It is not clear whether cells produce attraction forces at a distance, however, cells appear to touch each other randomly and display different behaviors based on the region of cell-cell contact. For example, during cell-cell contact-attraction, upon contact between the body of the front cell and the lamellipodium of the rear cell, the morphology of the front cell transforms from a stretched state to a more relaxed (rounder) state. At the same time, the F-actin and lamellipodium progressively flows to the other side and the cell body elongates, similar to an active cell sorting event. Accordingly, laser ablation experiments demonstrate the existence of pulling and adhesion forces mediated by the lamellipodium and intercellular bridge, respectively.
Despite the heterogeneous behaviors neural crest cells display, one common feature is that during all of these cell-cell interactions, two cells appear to establish an adherens junction and subsequently disassemble it, similar to what has been observed in other cell types including cranial neural crest cells in Xenopus (Davis et al., 2015; Scarpa et al., 2015). Junctions that form during contact attraction appear to be more stable than those forming during contact repulsion; this is likely due to differential concentrations of cadherin and myosin on the cell surface that, in turn, modulate both stiffness (tension) and viscosity (adhesion) properties of the cells (Amack and Manning, 2012). This possibility is supported by our quantitative analysis suggesting that cell body appears to be more adhesive than the lamellipodium (Figure 3H).
Compared with the concerted and directional chain migration displayed by cranial neural crest in Xenopus (Theveneau and Mayor, 2012) and neuronal precursor cells in mouse (Lois et al., 1996), our study shows that neural crest motion at the trunk level in chick exhibits more random motility. We speculate that this type of cell motion is more robust in response to genetic and environmental variations, as the number of cells moving to a specific destination for differentiation depends on multiple factors including cell-cell interactions, cell density, and adhesiveness to neighboring cells. In contrast, if cells move collectively, their final destinations may be predetermined by the spatiotemporal pattern of extracellular attractive/repulsive cues along the migration path. If irreversible damage occurs to the signaling source or the responding cells, the chances of generating specific progeny at the right place and time may dramatically decrease. However, in the case of a biased random walk, depletion of cells in a certain area might be compensated for by invasion of the remaining cells.
It has been proposed that neural crest migration could be driven by cell density (Kulesa and McLennan, 2015). Functional testing of this hypothesis in developing embryos requires artificial manipulation of cell distribution and/or number. Optical manipulation provides an accurate approach for achieving this type of targeted cell ablation. We show that one-photon laser ablation can eliminate individual neural crest cells without affecting adjacent ones in the xy (lateral) plane (Figure 7A). We cannot exclude the possibility that the laser may cause some damage in the z (axial) plane, but this should not affect our interpretation of the resultant collective cell migration, as cell displacements in the z direction is minimal in both unperturbed (Figure 2D) and perturbed (Figure 7G) samples.
One notable phenomenon from our laser ablation experiment is that reducing cell density in the ventral region of the embryos promotes cohesion of the remaining dorsal cells into a stream with rapid movement. One possibility is that the cells are channeled by undergoing a more classical contact inhibition of locomotion driven by the biased distribution of adhesion and repulsion force. For instance, when Xenopus cranial neural crests contact each other, cadherin mediated cell-cell interactions induce a repulsive response by promoting downstream GTPase activity (Scarpa et al., 2015). If the attraction force is larger than the repulsion force, cells can migrate as a cohesive group, similar to the movement observed in our laser ablation experiment in chick. In contrast, in the unperturbed chick embryos at the same stage, if the repulsion force plays a dominant role, it will trigger randomized cell dispersion. Thus, trunk neural crest cells appear to be able to switch between different migration modes according to the interplay between different types of physical forces.
As a solution to the challenge of cell morphological analysis (Megason and Fraser, 2007; Li et al., 2017), we developed a two-step segmentation method: the initial user defined a contour larger than the cell to minimize signal interference from neighboring cells, and an inner computer-generated contour based on the fluorescence intensity of the cell. Individual cell morphology is further analyzed with a skeleton representation whose endpoints connect to cell protrusions, enabling measurement of geometrical parameters of the cell. This segmentation approach permits automatic segregation of the spike-shaped filopodia, fan-shaped lamellipodium, or polygonal-shaped cell body, distinction between multiple types of cell-cell contact, and assessment of the link between cell morphogenesis and cell motion. With minor modifications, our toolkit could be applied to studying other cellular processes during both embryogenesis and organogenesis (Zhang et al., 2010).
In summary, by assessing the spatiotemporal correlation between cell morphology and cell locomotion within a single tissue preparation, we reveal that many dynamic events stochastically contribute to trunk neural crest migration at a given time, but in the long term, they are integrated to drive individual cell migration toward the ventral region of the embryos. Our work not only enriches the knowledge of cell migration mechanisms in higher vertebrates but also highlights the importance of studying cell biology in vivo through simultaneous quantitative imaging of multiple cellular events in conjugation with perturbation studies.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marianne E. Bronner (mbronner@caltech.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Chicken embryos (Gallus gallus) were obtained from AA Lab Eggs (Westminster, CA) and incubated at 37°C to reach the desired Hamburger-Hamilton stage (Hamburger and Hamilton, 1951). DF-1 fibroblasts were obtained from ATCC (ATCC, Manassas, VA; #CRL-12203, Lot number 62712171, Certificate of Analysis with negative mycoplasma testing available at ATCC website) and maintained at 37°C/5% CO2 in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin.
METHOD DETAILS
Molecular cloning, viral production and infection
RIA viral vector was modified by introducing unique AscI, NotI and ClaI digestion sites to facilitate cloning. H2B-GFP-T2A-mCherry, membrane-YFP, utrophin-scarlet, PA-Rac were cloned into the modified RIA vector. pCAG-CFP-2A-iRFP, pTK-Sox10E1-EGFP and pTK-NC2-mCherry were previously reported (Betancur et al., 2010; Simões-Costa et al., 2012). Recombinant RIA plasmids were co-transfected with Envelop A plasmid or VSV-G plasmid into chick DF1 cells in 15 cm culture dishes using standard transfection protocol (Li et al., 2015). When the cells were confluent, the cell culture medium was harvested once per day for three days, and was concentrated at 26,000 rpm for 1.5 hr. The pellet was dissolved in a minimal volume of DMEM.
Viral infection and plasmid electroporation
Fertilized chicken eggs were obtained from AA Lab Eggs (Westminster, CA) and incubated at 38°C for 44 hours or until embryos reached stages HH11+/12− (Hamburger and Hamilton, 1951). Eggs were windowed and injected with 5% Indian ink in Ringer’s underneath the embryo disc and Ringer’s solution was used to keep the embryos humid. To achieve efficient transfection of the neural tube and neural crest cells, concentrated virus (106-107 pfu/ml) were injected into the posterior neuropore, filling the entire tube (about 0.5 μl per embryo). For electroporation, injection of a solution containing 2 μg/μl pCAG(CFP-2A-iRFP), 2 μg/μl pTK(Sox10E1-EGFP) and 1.5 μg/μl pTK(NC2-mCherry) in 10 mM Tris, pH 8.5 was followed by application of 5 pulses of 18 V and 50-ms, with 100 ms intervals. Excess Ringer’s solution was removed and the egg shell opening was sealed with Blenderm tape (3M). Injected embryos were incubated at 38°C for 24 hours, collected with filter paper carriers and washed in Ringer’s solution.
Slice culture
Transverse cuts through posterior sclerotomes of the forelimb region were made every 2-somites with a Sharpoint Stab Knife, 22.5° Straight (Fine Science Tools). Next, the knife was used to cut through lateral tissue. Once freed of neighboring tissue, the slices were briefly washed in Ringer’s solution and immediately put into fluorodish (World Precision Instruments) containing prewarmed Neurobasal media (GIBCO) supplemented with glutamine, penicillin/streptomycin. The slices were positioned under nylon grids holding the limb buds or tissue ventral to the dorsal aorta and the whole fluorodish was transferred into the incubation chamber (37°C and 5% CO2) of a Zeiss LSM 800 microscope for time-lapse imaging.
In vivo live imaging
For imaging the tissue slices expressing H2B-GFP and mCherry, one-photon laser excitation was used with 0.5% and 0.8% relative power at wavelength of 488 and 561 nm, respectively. For imaging the samples expressing membrane-YFP and utrophin-scarlet, 1% and 1.3% relative power at wavelength of 488 and 561 nm, respectively, was used. Live tissue slices expressing electroporated Sox10E1-GFP, NC2-mCherry and PGK-NLS-iRFP plasmids were imaged using a Zeiss LSM 710 microscope (Zeiss). One-photon laser excitation was used with 6%, 10%, 12% relative power at wavelength of 488, 561 and 647 nm, respectively. In all the cases, optical sectioning was achieved at intervals of 2 μm. Phase-contrast imaging was performed using appropriate prisms.
For long-term imaging of the samples expressing H2B-GFP and mCherry, 20 × /0.8 NA objective was used and images were captured at 0.8 × magnification every 8 minute for 8-10 hours. For long-term imaging of the samples expressing membrane-YFP, 20 × /0.8 NA objective was used and images were captured at 0.8 × magnification every 2 minute for 8 hours. To resolve lamellipodial dynamics and cell morphological changes, 63 × /1.4 NA objective was used and images were captured at 1 × magnification every 20 s.
Optical manipulation
Laser ablation of the cells expressing H2B-GFP and mCherry was performed using a 63 × /1.4 NA objective and the laser tuned to 488 nm and set to 60% maximal power; the excitation was covered the whole nucleus. For laser ablation of membrane-YFP positive lamellipodia, the laser was tuned to 488 nm and set to 80% maximal power; the excitation covered one lamellipodium. In both cases, the laser ablation took about 30 s. Before and after ablation, 6 μm thick confocal images were acquired at 3 × magnification and captured every 20 s.
To photoactivate Rac, 63 × /1.4 NA objective was used and a one-photon laser tuned to 458 nm and set to 50% maximal power. The excitation was focused on a 1.3 × 1.3 μm2 region. Before and after activation, 6 μm thick confocal images were acquired at 3 × magnification and captured every 20 s.
Cell spot-segmentation, tracking and trajectory analysis
Live imaging data stored in ZEN software associated with LSM710 and 800 was imported into IMARIS 9.1.0 software for imaging processing. The centers of individual cell bodies were identified in each time frame were identified and connected to make a complete trajectory of the cell. The 4D coordinates (xyz and t) of cells were generated and further imported into MATLAB R2017a for mapping cell trajectory, calculating cell-cell coordination, cell speed, track straightness and division orientation.
Skeletal analysis of cell morphological changes
Images were imported and converted into an 8-bit grayscale temporal stack. Initial segmentation contours were drawn by hand and further refined by an unsupervised evolving contour using the activecontour MATLAB function. The coordinates of the soma for each cell were marked manually while skeletonized representations of the refined contour were calculated using the bwmorph function. The position of the leading lamellipodia was defined as the centroid position of 3 skeleton ends furthest from the soma position. A vector from the soma to the skeleton centroid was used to define the orientation of the cell. The displacement vector was defined as the vector between the position of the soma on the current frame and the next frame. The dot product between the orientation and direction vector was used to extract the angle between them. The correlation between the collinearity angle in frame i and the displacement of the soma center in a moving interval between frame i and i+10 was calculated. The cell density at the soma of each cell was obtained for each frame with statistically significant correlation (p < 0.05). The kymograph shown in Figure 5 was constructed by calculating all the skeleton end distances of one cell to the soma of the other cell, this distance is shown in sorted descending order on each column. The magenta line represents the number of skeleton ends whose distances is below the soma-soma distance, indicating these cellular protrusions lie at the cellular interface.
Immunofluorescence
A thick slice (2 somite and 500 μm in thickness) of stage HH18-19 chick embryos was made through the forelimb level. The slice was fixed in 4% PFA at 4°C for 30min. Frozen tissues sections were permeabilized with blocking buffer (1×PBS with: 10% vol/vol normal goat serum, 1% BSA, 0.1% vol/vol Triton X-100,), stained with primary antibody (1:10 dilution for mouse anti-HNK-1, DSHB) and then secondary antibody Goat anti-mouse IgG Alexa 488 (1:500 dilution, Thermo Fisher).
Quantification and Statistical Analysis
Statistical analyses were performed using MATLAB. Wilcoxon Rank-Sum test was used to examine the relationships between lamellipodia orientation, cell migration direction, cell morphological changes and cell density (Figures 3F–3I and 4C) and was used to examine the differences between cell migration behaviors in the ablated and unablated tissue slices (Figures 7K and 7L). P values are indicated in the text with significances are indicated as * (p < 0.05), ** (p < 0.01), and *** (p < 0.001).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse antibody against HNK1 | DSHB | 1C10; RRID: AB_10570406 |
| Goat anti-Mouse IgG, Secondary antibody, Alexa Fluor 488 | Thermo Fisher | A28175; RRID: AB_2536161 |
| Bacterial and Virus Strains | ||
| Replication-incompetent avian (RIA) retrovirus | Connie Cepko lab, Harvard | https://cepko.hms.harvard.edu |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Alexa Fluor 568 Phalloidin | Thermo Fisher | A12380 |
| DAPI | Thermo Fisher | D1306 |
| Neurobasal media | Thermo Fisher | LS21103049 |
| L-Glutamine | Sigma | G6392 |
| Experimental Models: Cell Lines | ||
| DF1 cells (Chicken embryonic fibroblast cells) | ATCC | CRL-12203 |
| Experimental Models: Organisms/Strains | ||
| Chick. Regular fertilized chicken eggs were obtained from farm and incubated at 37°C to reach the desired Hamburger–Hamilton stage. | AA Lab Eggs, CA, USA | http://www.balutpateros.com |
| Recombinant DNA | ||
| RIA-H2B-GFP-T2A-mCherry | This paper | N/A |
| RIA-membrane-YFP | This paper | N/A |
| RIA-Utrophin-Scarlet | This paper | N/A |
| RIA-PA-Rac-T2A-mCherry | This paper | N/A |
| pTK-NC2-mCherry | Simões-Costa et al., 2012 | https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1003142 |
| pTK-Sox10E1-EGFP | Betancur et al., 2010 | https://www.pnas.org/content/107/8/3570.long |
| ENV-A | Connie Cepko lab, Harvard | https://www.sciencedirect.com/science/article/pii/S0012160699994320?via%3Dihub |
| VSV-G | Connie Cepko lab, Harvard | https://www.sciencedirect.com/science/article/pii/S0012160699994320?via%3Dihub |
Highlights.
Trunk neural crest cells migrate individually and undergo a biased random walk
These cells show multiple types of cell-cell contacts, including contact attraction
Cell migration and cell-cell contact are actively influenced by mechanical forces
Manipulating cell density translates individual cell motion into chain migration
ACKNOWLEDGMENTS
We thank Dr. Vikas Trivedi (EMBL) for critically reading the manuscript and the Caltech Biological Imaging Center for sharing equipment. W.G.G. was supported by the Della Martin Foundation, the American Heart Association, and the Burroughs Wellcome Fund. This project was supported by DE024157 and DE027568 (to M.E.B.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures and eleven videos and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.01.039.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahlstrom JD, and Erickson CA (2009). The neural crest epithelial-mesenchymal transition in 4D: a ‘tail’ of multiple non-obligatory cellular mechanisms. Development 136, 1801–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amack JD, and Manning ML (2012). Knowing the boundaries: extending the differential adhesion hypothesis in embryonic cell sorting. Science 338, 212–215. [DOI] [PubMed] [Google Scholar]
- Baena-López LA, Baonza A, and García-Bellido A (2005). The orientation of cell divisions determines the shape of Drosophila organs. Curr. Biol 15, 1640–1644. [DOI] [PubMed] [Google Scholar]
- Betancur P, Bronner-Fraser M, and Sauka-Spengler T (2010). Genomic code for Sox10 activation reveals a key regulatory enhancer for cranial neural crest. Proc. Natl. Acad. Sci. USA 107, 3570–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner ME, and Simões-Costa M (2016). The neural crest migrating into the Twenty-First Century. Curr. Top. Dev. Biol 116, 115–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Matthews HK, Kuriyama S, Moreno M, Dunn GA, Parsons M, Stern CD, and Mayor R (2008). Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature 456, 957–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TF, and Vese LA (2001). Active contours without edges. IEEE Trans. Image Process. 10, 266–277. [DOI] [PubMed] [Google Scholar]
- Chen CMA, Smith DM, Peters MA, Samson MES, Zitz J, Tabin CJ, and Cepko CL (1999). Production and design of more effective avian replication-incompetent retroviral vectors. Dev. Biol 214, 370–384. [DOI] [PubMed] [Google Scholar]
- Davis JR, Luchici A, Mosis F, Thackery J, Salazar JA, Mao Y, Dunn GA, Betz T, Miodownik M, and Stramer BM (2015). Inter-cellular forces orchestrate contact inhibition of locomotion. Cell 161, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pascalis C, and Etienne-Manneville S (2017). Single and collective cell migration: the mechanics of adhesions. Mol. Biol. Cell 28, 1833–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, and Gilmour D (2009). Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol 10, 445–457. [DOI] [PubMed] [Google Scholar]
- Gammill LS, Gonzalez C, Gu C, and Bronner-Fraser M (2006). Guidance of trunk neural crest migration requires neuropilin 2/semaphorin 3F signaling. Development 133, 99–106. [DOI] [PubMed] [Google Scholar]
- Gong Y, Mo C, and Fraser SE (2004). Planar cell polarity signalling controls cell division orientation during zebrafish gastrulation. Nature 430, 689–693. [DOI] [PubMed] [Google Scholar]
- Hamburger V, and Hamilton H (1951). A Series of Normal Stages in the Development of the Chick Embryo. J. Morphol 88, 49–92. [PubMed] [Google Scholar]
- Heisenberg CP, and Bellaïche Y (2013). Forces in tissue morphogenesis and patterning. Cell 153, 948–962. [DOI] [PubMed] [Google Scholar]
- Ilina O, and Friedl P (2009). Mechanisms of collective cell migration at a glance. J. Cell Sci 122, 3203–3208. [DOI] [PubMed] [Google Scholar]
- Köppen M, Fernández BG, Carvalho L, Jacinto A, and Heisenberg CP (2006). Coordinated cell-shape changes control epithelial movement in zebra-fish and Drosophila. Development 133, 2671–2681. [DOI] [PubMed] [Google Scholar]
- Krull CE, Collazo A, Fraser SE, and Bronner-Fraser M (1995). Segmental migration of trunk neural crest: time-lapse analysis reveals a role for PNA-binding molecules. Development 121, 3733–3743. [DOI] [PubMed] [Google Scholar]
- Krull CE, Lansford R, Gale NW, Collazo A, Marcelle C, Yancopoulos GD, Fraser SE, and Bronner-Fraser M (1997). Interactions of Eph-related receptors and ligands confer rostrocaudal pattern to trunk neural crest migration. Curr. Biol 7, 571–580. [DOI] [PubMed] [Google Scholar]
- Kulesa PM, and Fraser SE (1998). Neural crest cell dynamics revealed by time-lapse video microscopy of whole embryo chick explant cultures. Dev. Biol 204, 327–344. [DOI] [PubMed] [Google Scholar]
- Kulesa PM, and McLennan R (2015). Neural crest migration: trailblazing ahead. F1000Prime Rep. 7, 02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama S, Theveneau E, Benedetto A, Parsons M,Tanaka M, Charras G, Kabla A, and Mayor R (2014). In vivo collective cell migration requires an LPAR2-dependent increase in tissue fluidity. J. Cell Biol 206, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufs S, Gentner B, Nagy KZ, Jauch A, Benner A, Naundorf S, Kuehlcke K, Schiedlmeier B, Ho AD, Zeller WJ, and Fruehauf S. (2003). Retroviral vector integration occurs in preferred genomic targets of human bone marrow-repopulating cells. Blood 101, 2191–2198. [DOI] [PubMed] [Google Scholar]
- Le Douarin N (1982). The Neural Crest (Cambridge University Press; ). [Google Scholar]
- Lecaudey V, and Gilmour D (2006). Organizing moving groups during morphogenesis. Curr. Opin. Cell Biol 18, 102–107. [DOI] [PubMed] [Google Scholar]
- Li Y, Trivedi V, Truong TV, Koos DS, Lansford R, Chuong CM, Warburton D, Moats RA, and Fraser SE (2015). Dynamic imaging of the growth plate cartilage reveals multiple contributors to skeletal morphogenesis. Nat. Commun 6, 6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li A, Junge J, and Bronner M. (2017). Planar cell polarity signaling coordinates oriented cell division and cell rearrangement in clonally expanding growth plate cartilage. eLife 6, 23279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Junge JA, Arnesano C, Gross GG, Miner JH, Moats R, Roberts RW, Arnold DB, and Fraser SE (2018). Discs large 1 controls daughter-cell polarity after cytokinesis in vertebrate morphogenesis. Proc. Natl. Acad. Sci. USA 115, E10859–E10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, García-Verdugo JM, and Alvarez-Buylla A (1996). Chain migration of neuronal precursors. Science 271, 978–981. [DOI] [PubMed] [Google Scholar]
- Mayor R, and Carmona-Fontaine C. (2010). Keeping in touch with contact inhibition of locomotion. Trends Cell Biol. 20, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megason SG, and Fraser SE (2007). Imaging in systems biology. Cell 130, 784–795. [DOI] [PubMed] [Google Scholar]
- Michalet X (2010). Mean square displacement analysis of single-particle trajectories with localization error: Brownian motion in an isotropic medium. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 82, 041914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell DJ (2006). The social lives of migrating cells in Drosophila. Curr. Opin. Genet. Dev 16, 374–383. [DOI] [PubMed] [Google Scholar]
- Nobes CD, and Hall A (1995). Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62. [DOI] [PubMed] [Google Scholar]
- Prasad M, and Montell DJ (2007). Cellular and molecular mechanisms of border cell migration analyzed using time-lapse live-cell imaging. Dev. Cell 12, 997–1005. [DOI] [PubMed] [Google Scholar]
- Reig G, Pulgar E, and Concha ML (2014). Cell migration: from tissue culture to embryos. Development 141, 1999–2013. [DOI] [PubMed] [Google Scholar]
- Ridenour DA, McLennan R, Teddy JM, Semerad CL, Haug JS, and Kulesa PM. (2014). The neural crest cell cycle is related to phases of migration in the head. Development 141, 1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa E, Szabó A, Bibonne A, Theveneau E, Parsons M, and Mayor R (2015). Cadherin switch during EMT in neural crest cells leads to contact inhibition of locomotion via repolarization of forces. Dev. Cell 34, 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmacher AT, Xie V, and Taneyhill LA (2016). Cadherin-6B proteolysis promotes the neural crest cell epithelial-to-mesenchymal transition through transcriptional regulation. J. Cell Biol. 215, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau CE, Das RM, and Storey KG (2011). An effective assay for high cellular resolution time-lapse imaging of sensory placode formation and morphogenesis. BMC Neurosci 12, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes-Costa M, and Bronner ME (2016). Reprogramming of avian neural crest axial identity and cell fate. Science 352, 1570–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões-Costa MS, McKeown SJ, Tan-Cabugao J., Sauka-Spengler T, and Bronner ME (2012). Dynamic and differential regulation of stem cell factor FoxD3 in the neural crest is Encrypted in the genome. PLoS Genet. 8, e1003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, and Mayor R. (2012). Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol 366, 34–54. [DOI] [PubMed] [Google Scholar]
- Theveneau E, Steventon B, Scarpa E, Garcia S, Trepat X, Streit A, and Mayor R (2013). Chase-and-run between adjacent cell populations promotes directional collective migration. Nat. Cell Biol 15, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, and Hahn KM (2009). A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 461, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gally C, and Labouesse M (2010). Tissue morphogenesis: how multiple cells cooperate to generate a tissue. Curr. Opin. Cell Biol 22, 575–582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.