

Abstract
The mitochondrial permeability transition pore (mPTP) has been considered a key contributor to cell death, inducing the process in several major neurodegenerative diseases. To date, the molecular nature of the mPTP remains confounding but its significance is universally acknowledged. Several targets have been screened and inhibition of mPTP has emerged as an attractive field for researchers. Nowadays, in silico-directed studies help to explore new small molecules targeting the mPTP to improve their drug-like properties and bioactivity. Here, we briefly summarize the role of mPTP in neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson disease (PD), and Huntington’s disease (HD), and discusses current and future potential therapeutic targets.
Introduction
Mitochondria, the power house of the cell, have an essential role in cell survival by providing energy in the form of ATP, regulating intracellular calcium homeostasis, and maintaining redox stress inside the cell. Mitochondria are also important in regulating ion channels, securing cellular signal transduction, and in causing pathophysiological events [1]. In the central nervous system (CNS), mitochondria are crucial for neuronal survival and death by regulating cellular survival and various apoptotic pathways [2,3]. Structurally, mitochondria are rod-shaped (0.75–3 mm), double membrane-bound organelles. The two membranes of mitochondria, outer and inner, comprise phospholipids and proteins. Compared with the outer membrane of mitochondria (OUM), which is smooth and permeable to a large number of nutrients, ions, and energy molecules (ATP and ADP), the inner membrane of mitochondria (IMM) is more complex, intensively folded, and relatively impermeable to large molecules. The impermeable nature of the IMM is crucial in maintaining the homeostasis of the inner mitochondrial environment as well as integrity of the mitochondria [4]. Different ion channels and transporters in IMM help in communications between mitochondria and the external environment. For instance, calcium (Ca2+) is taken into mitochondria from cytoplasm by energy-driven uniporters, while the efflux of Ca2+ from the mitochondria to the cytoplasm is regulated by ion gradient-driven antiporters. Increased oxidative stress and impaired energy metabolism compromise Ca2+ homeostasis, causing further calcium overload and mishandling by the mitochondria. This abnormal condition result in the quick efflux of calcium through a nonselective pathway that eventually leads to the formation of the mPTP [4,5].
Mitochondrial permeability transition pore
After the discovery of the permeability transition pore by Hunter et al. [6], the permeability transition (PT) was later explained as an increase in the permeability of IMM to ions and solutes with molecular masses up to 1.5 kDa, leading to mitochondrial matrix swelling and cellular death through apoptosis (programmed cell death) and necrosis (nonprogrammed, autolysis) [7]. Furthermore, Hunter and Haworth characterized mPTP as a nonselective channel with a peak conductance of ~1.3 nS [8]. Although the concept of mPTP was considered as in vitro artefact of uncertain pathophysiological significance, investigators later supported the essence of mPTP formation in disease manifestation [9]. Until recently, the structure of mPTP was controversial, but research by Szabó and Zoratti [10], Halestrap [11], and others suggested that the mPTP comprised a voltage-dependent anion channel (VDAC) in the OMM [12], adenine nucleotide translocator (ANT) in the IMM [13], and cyclophilin D (CypD) in the mitochondrial matrix [4,14,15]. VDACs contribute to outer membrane permeability and are associated with the mitochondrial benzodiazepine receptor; thus, they regulate extramitochondrial cholesterol transfer to the intermembrane space [16]. ANT allows the influx of phosphorylated and nonphosphorylated derivatives of adenine nucleotides through the IMM, and CypD helps in protein folding because it exhibits peptidyl propyl isomerase activity [5]. CypD is also a key component of mitochondrial permeability transition and is involved in neurodegenerative diseases.
Under normal physiological conditions, all components of mPTP are disaggregated and, upon apoptotic or stressing stimuli, unite to form pores of 1.0–1.3 nm that trigger the nonselective flux of molecules [17]. Given the mechanistic basis of mPTP formation, researchers proposed that mPTP formation is initiated by the binding of ANT with CypD in IMM, where CypD translocates from the mitochondrial matrix to the IMM. This binding facilitates the formation of a tunnel-like structure comprising ANT and VDAC and connects the mitochondrial matrix to the cytosol by crossing both mitochondrial membranes [4,12,13,17]. The formation of mPTP is not only a key event in cell death, but is also suggested to be important for mitochondrial function and cell survival [18,19]. Using a calcium-overload model, assembly of the C-subunit of ATPase (C-subunit), inorganic polyphosphate (polyP), and polyhydroxybutyrate (PHB) were found to be required for mPTP opening [20].
In addition to the components discussed above, F1F0 ATP synthase is also involved in mPTP opening [21,22]. Using genetic approaches, Bonora et al. suggested that the dissociation of F1F0 ATP synthase dimers and involvement of the C-ring are crucial in mPTP induction [23]. Importantly, blocking the interaction of F1F0 ATP synapse with CypD alleviated mitochondrial and synaptic defects and improved cognitive function in diabetes milieu [24]. Under the influence of oxidative stress, the translocation of the tumor suppressor p53 was also reported in mitochondria, which elicits mPTP formation after interacting with CypD [25]. Using light scattering, fluorescence, and electron microscopy techniques, the induction of calcium-induced PTP was found to be significantly inhibited in low osmotic-strength solutions [26]. Thus, the effect is linked to changes in the curvature of the IMM. Leung et al. proposed the participation of the phosphate carrier (PiC) in pore formation [27]. PiC is present in IMM and forms mPTP either by itself or by binding to ANT [27]. In rat brain cortex mitochondria, the calcium uniporter has a role in regulating mitochondrial permeability [28]. Additionally, using genetic silencing approaches, the involvement of soluble matrix peptidyl prolyl isomerase F cyclophilin D (PPIF) was confirmed in Ca2 +-induced mPTP opening [15,29,30]. Likewise, using an RNAi-based screening approach, Shanmughapriya et al. identified the necessary and conserved role of spastic paraplegia 7 (SPG7) in Ca2+ and ROS-induced PTP opening [31]. The authors proposed that SPG7 is a conserved core component that constitutes the PTP complex and regulates its function [31]. A mouse model of experimental autoimmune encephalomyelitis (EAE) lacking CyPD was found to be partially rescued from EAE by Forte et al., suggesting CypD and mPTP as a potential target for neuroprotective therapy [32]. These studies suggest additional and important molecular components that can facilitate mPTP formation given the contrasting reports for the involvement of VDAC and ANT in mPTP formation [33,34]. Although there is disagreement over the involvement of VDAC and ANT as core components for mPTP configuration, CypD has been considered to be crucial in mPTP formation, at least in Ca2+-dependent mechanisms [9] (Fig. 1).
FIGURE 1.
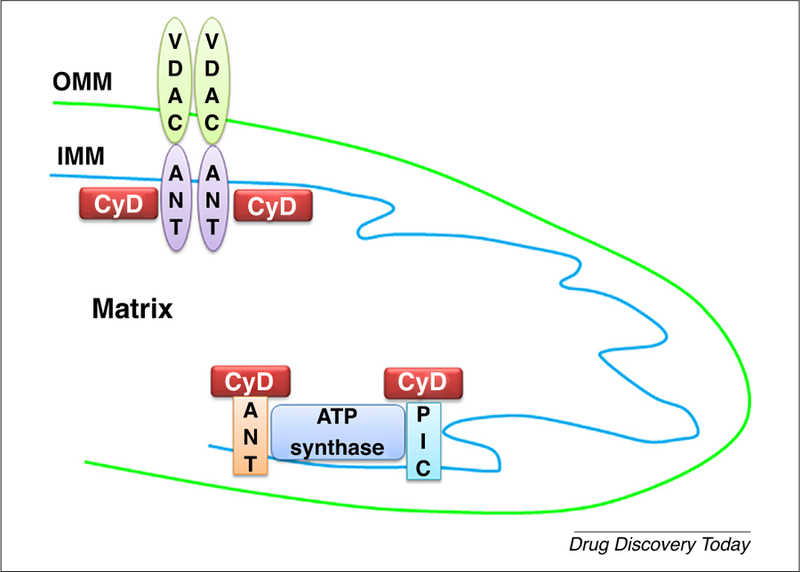
Components of the mitochondrial permeability transition pore (mPTP) according to the published literature. (a) Historical mPTP model that contains voltage-dependent anion channels (VDAC), adenine nucleotide translocators (ANT), and cyclophilin D (CypD). (b) Components of more recent models of mPTP formation. Abbreviations: IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; PiC, phosphate carrier.
mPTP opening is implicated in various neurodegenerative diseases, including AD, PD, prion disease, motor neuron disease (MND), HD, spinal muscular atrophy (SMA), and amyotrophic lateral sclerosis (ALS) [35–37]. The opening of mPTP is characterized by decoupling of electrochemical potential across the IMM and alterations of mitochondrial osmotic disturbances that accompanies Ca2+ mishandling and reduced ATP generation in the mitochondria [38]. Cellular death via the apoptotic pathway occurs when mPTP opening occurs, resulting in reduced mechanisms of oxidative phosphorylation and ATP hydrolysis [39]. In the process, the presence of sufficient ATP levels and transient opening of the mPTP results in cytochrome c release through the mitochondria, which activates caspase cascades and mitochondria-driven apoptosis [40]. In contrast to apoptosis, sustained and extensive mPTP opening limits ATP synthesis by mediating the uncoupling of oxidative phosphorylation [33]. With the onset of a high energy demand and compromised ATP production, the cell mass loses its structural and functional integrity, resulting in irreversible damage and narcotic cell death [40,41] (Fig. 2).
FIGURE 2.
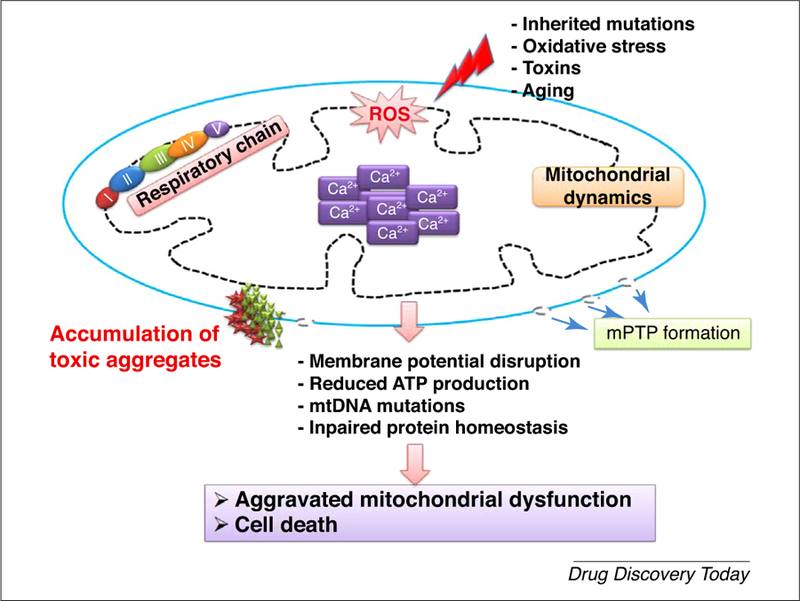
The cell death cascade resulting from mitochondrial permeability transition pore (mPTP) formation and mitochondrial dysfunction in neurodegenerative diseases. In neurodegenerative diseases, oxidative stress resulting from toxins induces reactive oxygen species (ROS) generation, which promotes mitochondrial calcium mishandling and abrupt mitochondrial dynamics. Altered redox dynamics aggravate mitochondrial dysfunction by promoting mPTP formation, which promotes cell death.
Role of mPTP in different neurodegenerative diseases
Alzheimer’s disease and mPTP
AD is a progressive neurodegenerative disease that impairs memory and other important mental functions. The AD brain is characterized pathologically by the accumulation of toxic protein amyloid beta (Aβ) and neurofibrillary tangles (NFTs), formed by pathological forms of tau protein [42]. The deposition of neurotoxic aggregates is mediated by overproduction of toxic peptides and failure of clearance mechanisms. Studies have shown that these aggregates induce neuronal dysfunction, leading to cognitive and memory impairment [42,43]. The neurotoxicity effects of aggregates have been reported to be facilitated through free radical generation and calcium dysregulation [36,44]. The presence of Aβ has also been confirmed in mitochondria of human AD brain [37] and transgenic (Tg) animal models of AD [45]. It is unclear whether Aβ in mitochondria is generated in situ or imported. However, researchers have suggested that Aβ is derived from extracellular and intracellular pools and then internalized in the mitochondria [46]. For example, a study in isolated rat mitochondria by Hanson Petersen et al. showed the co-localization of extracellularly applied Aβ and mitochondrial markers in IMM, suggesting that Aβ is taken by the cells [47]. Similarly, studies using confocal microscopy confirmed the presence of Aβ42 fragments co-localized with complex II of the respiratory chain in IMM and OMM, and with the chaperon matrix protein HSP60 [48,49]. Studies in patients with AD and Tg-AβPP mice suggested formation of the complex after the binding of Aβ with CypD in cortical mitochondria [37,45]. The interaction between Aβ and CypD causes the formation of mPTP, resulting in reduced mitochondrial potential, decreased mitochondrial respiratory function, and increased formation of free radicals, leading to oxidative stress and the release of pro-apoptotic proteins [4,5].
Potential drugs designed to interfere with the Aβ-CypD complex could help relieve the neurotoxic effect of Aβ. Given that the pathophysiological cascades of AD start with mPTP formation, therapeutic interventions against mPTP could be helpful in reversing disease pathology [4]. Hence, mPTP has been studied widely as a potential drug target for neurodegenerative diseases [4,35]. The specific binding of Aβ has also been shown with other mitochondrial proteins that cause cell physiology defects and efficient respiratory halts in AD [50,51]. Lustbader et al. elucidated the binding of Aβ with Aβ-binding alcohol dehydrogenase (ABAD), also called 17-β-dehydrogenase type-10 (HSD-10), an intracellular enzyme in the mitochondrial matrix, which resulted in increased cellular oxidative stress, DNA fragmentation, and reactive oxygen species (ROS) generation [45,46,51]. Increased expression of HSD-10 was associated with increased mitochondrial Aβ in the hippocampus region of the AD brain compared with healthy brain [52]. Studies in transgenic AD mice suggested that inhibition of the ABAD/HSD-10-Aβ interaction significantly diminished mitochondrial Aβ accumulation, thus protecting neuromitochondrial and learning and/or memory functions in AD mice [45]. Another study in Tg mAPP/ABAD mice showed that ABAD-Aβ interactions affected the expression levels of endophilin-1 (EP-1), a cytoplasmic SH3 domain-containing protein, and Prdx-2, an antioxidant protein [53], which mediate neuronal death by activating JNK (c-jun-N terminal kinase) signaling [53,54]. In addition, compromised oxidative phosphorylation during ADpathologyhasbeen widelystudiedand Aβ wasfoundtoaffect ATP synthase, cytochrome c oxidase, cytochrome c reductase (complex III), complex I, and complex IV activities [55,56]. Furthermore, Aβ has also been implicated in disease progression by inducing lipid peroxidation and superoxide radical production, and reducing cellular antioxidant levels [56]. Indeed, suppression of mitochondrial ROS by eliminating damaged mitochondria significantly protected against Aβ-mediated mitochondrial and synaptic dysfunction [57]. Recent studies demonstrated that enhancing the clearance of dysfunctional mitochondria by augmenting PTEN-induced putative kinase 1 (PINK1) function reduced cerebral and mitochondrial Aβ, and improved mitochondrial and synaptic function as well as cognition [57]. Furthermore, suppressing excessive mitochondrial fission or ROS generation significantly reversed AD-derived mitochondrial defects and axonal mitochondrial transport in human transmitochondrial cybrid cells [58–61].
Parkinson’s disease and mPTP
PD currently affects around 6.3 million people worldwide, and is expected to affect 8 million–9 people by 2030 [62]. It mainly affects motor neurons through the loss of dopaminergic neurons in the substantia nigra pars compacta (SNPc) and the presence of excess α-synuclein protein in presynaptic neuronal cells. The presence of clumps of α-synuclein protein, also called Lewy bodies, is the hallmark of PD. Clinical symptoms associated with the disease are body stiffness, bradykinesia, frequent tremors, postural instability, and balance disorders. An impaired cellular redox state, defective metabolism, and dysfunctional mitochondria are also reported in PD [36,62]. The sporadic form of the PD is reflected through the loss of Ca2+ homeostasis and α-synuclein aggregates [62,63]. Defects in mitochondrial respiratory complex I and mitochondrial autophagy (mitophagy) are also associated with PD [62]. Similar to AD, mPTP formation in PD through mitochondrial dysfunction occurs in the same manner and includes a series of events, including IMM depolarization, impaired oxidative phosphorylation, increased ROS production, mitochondria matrix swelling, IMM cristae unfolding, loss of Ca2+ homeostasis, and the release of apoptogenic proteins through the OMM, which eventually leads to cell death. mPTP opening has been linked to neuronal death by various mechanisms, including excitotoxicity, neurotoxicity, apoptosis, and necrosis [64].
Huntington’s disease and mPTP
HD, also known as Huntington’s chorea, is an inherited disorder caused by mutations in the gene encoding Huntington (Htt), located on Chr 4 [65]. Abnormal elongation of cytosine-adenine-guanine (CAG) triplet repeats in Htt results in pathological elongation of polyglutamine in the Htt protein, which gradually damages brain cells. The disease mainly affects basal ganglia and causes uncontrolled movements, mood disorders, psychiatric problems, mental disabilities, and dementia. Metabolic deficiencies have been reported in patients with HD, suggesting the relevance of mitochondrial dysfunction in its pathogenesis. Additionally, mitochondrial abnormalities, such as mPTP formation, Ca2+ mishandling, and redox stress, have been implicated in in vivo and in vitro models of HD [66,67].
mPTP as a potential drug target
Irreversible mPTP formation has been linked to various neurodegenerative diseases, and pharmacological, genetic interventions have been identified and formulated to inhibit mPTP formation [33,68]. Of all the components thought to be involved in mPTP formation, CypD is the most important.
Mitochondrial dysfunction is an early pathological feature of AD. AD-derived mitochondria exhibit failure in respiratory function, energy metabolism, and mitochondrial morphological alterations [57–60,69–76]. Mitochondria isolated from cortical and hippocampal brain regions of patients with AD showed significant increased expression of CypD compared with non-AD brain mitochondria [37]. Increased CypD expression is also positively associated with increased Aβ expression [4]. In addition, mouse models of AD that express mutant forms of APP also showed age-dependent increases in CypD in cortical and hippocampus regions of the brain compared with their littermate controls [4,45]. CypD ablation has also been studied for preventing mPTP formation, given that CypD is considered a crucial molecule for mPTP opening [4,5]. Research using a mAPP model has established various events that occur during mPTP formation [4,5], including increased expression of CypD in cortical mitochondria that translocate to the IMM, altered Ca2+ homeostasis, and mitochondrial swelling [4,5,77]. Interestingly, ablating CypD with cyclosporine A (CsA), a CypD inhibitor, mitigated these detrimental effects [37,45]. The binding affinity of CypD with Aβ has been proven experimentally, and was found to be sequence specific [37,45]. Mitochondria from a CypD-deficient mAPP mouse model (mAPP/Ppif−/−) showed reduced mitochondrial swelling and permeability transition following Ca2+ induction compared with mAPP mice [4,37,45]. However, introduction of CsA in mAPP mitochondria relieved mitochondrial dysfunction in response to Ca2+ [4,37,45]. Given the role of CypD in ameliorating spatial learning and memory function, a radial arm water-maze test was performed and mAPP/Ppif−/− mice showed considerable improvements in the test compared with mAPP mice [4,37,45]. Different selective approaches have been designed and modified to target lipophilic cations to mitochondria. Smith et al. showed that mitochondria-targeted antioxidants comprising hydrophobic delocalized cations linked to antioxidant quinone moieties had beneficial impacts resulting from a reduction in oxidative effects [78]. Similarly, Warne et al. designed and synthesized JW47, a CypD inhibitor, using a quinolinium cation tethered to cyclosporine. In the EAE disease model, JW47 showed significant protection of axons and improved motor assessments with minimal immunosuppression [79]. The use of multiple pharmacological approaches will facilitate the development of new drugs by increasing the drug-like properties and bioactivities of candidate molecules, such as enhancing mitochondrial biogenesis, and suppressing excessive mitochondrial ROS production and mPTP opening.
An acute model of PD prepared in Ppif−/− mice, using neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), showed cytoprotection compared with control mice [80]. CypD-deficient mice also displayed a considerable reduction in brain infarct size after acute middle cerebral artery occlusion and reperfusion. These results strongly supported a vital role of CypD in an ischemic injury model in which calcium overload and oxidative stress are affected [30]. Likewise, in an ex vivo study by Li et al., the essential role of CypD in mitochondrial permeability transition (CyD-mPT) was elucidated in glutamate-triggered delayed calcium deregulation (DCD) and excitotoxic cell death [81]. The study results suggested that, compared with cortical neurons isolated from control mice, cortical neurons from Ppif−/− mice were more resistant to DCD and cell death when induced with moderate but pathologically relevant glutamate concentrations. These studies highlight CypD as a strong candidate to prevent progression of chronic neurodegenerative diseases.
However, to establish the role of mPTP formation in the progression of neurodegenerative diseases, clinical studies have mostly used cyclosporine, a CypD inhibitor. Unfortunately, cyclosporine has certain limitations that restrict its use as a tool to look at the clinical potential of CypD inhibition and mitochondrial dysfunction. The limitations include toxicity and immunosuppressive effects [82]. Other limitations, such as reduced tissue penetration and reduced ability to cross the blood–brain barrier (BBB), are other examples of issues that restrict the role of cyclosporine as a future potential candidate [83]. Using a fragment-based drug discovery approach based on nucleic magnetic resonance (NMR) and X-ray crystallography, Ahmed-Belkacem et al. generated a new family of nonpeptidic, small-molecule cyclophilin inhibitors, unrelated to CsA, with potent in vitro PPIase inhibitory activity [84].
Using molecular docking and virtual screening methods, Valasani et al. described the design, synthesis, diastereomeric crystallization, docking 2D quantitative structure–activity relationship (QSAR) studies, and pharmacophore modelling for a series of acetylcholinesterase inhibitors [86] and CypD-selective inhibitors [85,87] for AD treatment. Guo et al. elucidated quinoxaline derivatives that also bind CypD and inhibit mitochondrial swelling in response to Ca2+ [36,88]. Elkamhawy et al. reported novel quinazoline urea analogs that bind selectively to CypD, and showed neuroprotective effects against Aβ-induced toxicity in neuronal cells [89]. Azzolin et al. reported the use of antamanide, a natural cyclic peptide, to inhibit mPTP formation in a manner similar to CsA [90].
Using pharmacokinetics data, authors have determined that pharmacological tools can be developed against neurodegenerative pathologies [90–97]. In addition, according to the published literature, ~122 natural compounds derived from traditional plant sources are in use in modern medicine, of which 80% are used in forms similar to the naturally occurring molecules [98]. Natural products with antioxidant and anti-inflammatory activities have been found to be beneficial against mPTP formation [99–102]. Russo et al. described the potential of compounds from Galanthus spp., Crocus sativus, Ginkgo biloba, Salvia spp., and Huperzia serrata in ameliorating dementia or AD [103]. Modifications of plant products are also under way to further improve their efficacy and bioavailability [103]. Since 1929, compounds isolated or expressed in microbial systems have been used as important regulators of disease. Bryostatins are a group of macrolide lactones isolated from the bryozoan, Bugula neritina. Using bryostatin-1, a Phase 2 clinical trial (NCT00606164) was approved for the treatment of AD [104]. Their main pharmacological mechanism of action is modulation of protein kinase C (PKC) activity [105]. In an APP/PS1 transgenic mouse model of AD, oral administration of bryostatin-1 showed improvements in learning and enhanced long-acting memory, thus, highlighting this as a future potential candidate for treating AD [106]. Furthermore, natural redox-active compounds, such as polyphenols, which act as antioxidants, were also shown to be valuable [107,108].
To target mitochondrial mPTP anomalies, candidate drug molecules can be selected by virtual screening of best-fi synthetic, semisynthetic, and/or microbial molecules and their chemical transformation into novel active analogs using in silico QSAR modeling, molecular docking, and absorption, distribution, metabolism, excretion and toxicity (ADME/T) studies. It will be necessary to design compounds with increased bioavailability, reduced toxicity and other properties that can overcome the BBB. This step will involve a large number of compounds and could be used to screen the most important novel formulations before wet lab testing. Figure 3 depicts ways in which to develop lead or drug-like molecules. Furthermore, virtually active compounds or series of compounds could be synthesized by using suitable schemes followed by chromatographic purification and spectroscopic characterization. The lead molecules isolated would then be tested against neurodegenerative diseases in vitro, ex vivo and in vivo. Directing advanced studies using genomics and structural biology can create new frontiers for bioactive drugs with reduced or no adverse effects. Moreover, formulations with improved solubility and stability would also be useful for treating AD.
FIGURE 3.
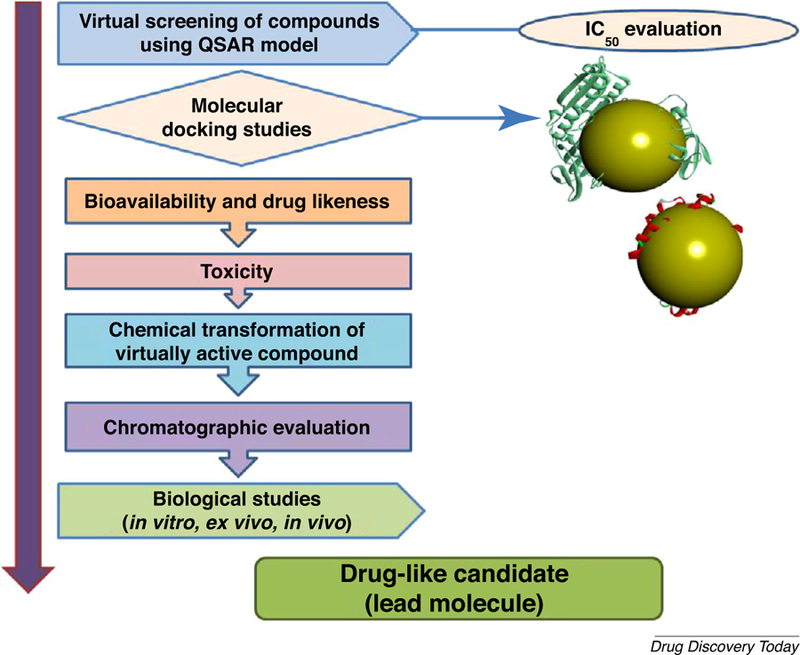
Lead optimization using in silico quantitative structure–activity relationship (QSAR) modeling, molecular docking, bioavailability, drug likeness, and toxicity parameters followed by wet lab synthesis and biological validation.
Concluding remarks
Here, we discussed the role of mitochondrial dysfunction and mPTP formation in mediating neurodegenerative diseases. Although the structural and mechanistic basis of mPTP formation associated with neurodegenerative diseases has not been clearly elucidated, the results obtained with mPTP inhibition suggest its promising role as a potential candidate in the treatment of various neurodegenerative diseases. Once the therapeutic aspects of mPTP inhibition through potential compounds have been elucidated further, more avenues for future research might become clear. Hence, exploring therapeutic interventions through potential therapeutic compounds can potentiate their use against different neurodegenerative diseases. The use of QSAR and molecular docking-guided lead optimization can help in screening virtual analogs and their drug-like potentials. Furthermore, wet lab preparations of selected candidate molecules and in vivo and in vitro testing can further translate the therapeutics for use in the clinic.
Acknowledgments
This study was supported by NIH/NIA (R37AG037319, R01AG044793, R01AG053041, and R21AG058985), and NIH/NINDS (R01NS089116).
Reference
- 1.Duchen MR (2004) Roles of mitochondria in health and disease. Diabetes 53 (Suppl. 1), S96–S102 [DOI] [PubMed] [Google Scholar]
- 2.Yeo W and Gautier J (2004) Early neural cell death: dying to become neurons. Dev. Biol 274, 233–244 [DOI] [PubMed] [Google Scholar]
- 3.Kole AJ et al. (2013) Mature neurons: equipped for survival. Cell Death Dis 4, e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du H and Yan SS (2010) Mitochondrial permeability transition pore in Alzheimer’s disease: cyclophilin D and amyloid beta. Biochim. Biophys. Acta 1802, 198–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rao VK et al. (2014) Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 1842, 1267–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunter DR et al. (1976) Relationship between configuration, function, and permeability in calcium-treated mitochondria. J. Biol. Chem 251, 5069–5077 [PubMed] [Google Scholar]
- 7.Lemasters JJ et al. (2009) Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 1787, 1395–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haworth RA and Hunter DR (1979) The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys 195, 460–467 [DOI] [PubMed] [Google Scholar]
- 9.Bernardi P (2013) The mitochondrial permeability transition pore: a mystery solved? Front. Physiol 4, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zoratti M and Szabo I (1995) The mitochondrial permeability transition. Biochim. Biophys. Acta 1241, 139–176 [DOI] [PubMed] [Google Scholar]
- 11.Halestrap AP (1999) The mitochondrial permeability transition: its molecular mechanism and role in reperfusion injury. Biochem. Soc. Symp 66, 181–203 [DOI] [PubMed] [Google Scholar]
- 12.Szabo I et al. (1993) The mitochondrial permeability transition pore may comprise VDAC molecules. II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett 330, 206–210 [DOI] [PubMed] [Google Scholar]
- 13.Halestrap AP et al. (1997) Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J. Biol. Chem 272, 3346–3354 [DOI] [PubMed] [Google Scholar]
- 14.Biasutto L et al. (2016) The mitochondrial permeability transition pore in AD 2016: an update. Biochim. Biophys. Acta 1863, 2515–2530 [DOI] [PubMed] [Google Scholar]
- 15.Baines CP et al. (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 [DOI] [PubMed] [Google Scholar]
- 16.Chipuk JE et al. (2006) Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ 13, 1396–1402 [DOI] [PubMed] [Google Scholar]
- 17.Crompton M (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem. J 341, 233–249 [PMC free article] [PubMed] [Google Scholar]
- 18.Morciano G et al. (2015) Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol 78, 142–153 [DOI] [PubMed] [Google Scholar]
- 19.Perez MJ and Quintanilla RA (2017) Development or disease: duality of the mitochondrial permeability transition pore. Dev. Biol 426, 1–7 [DOI] [PubMed] [Google Scholar]
- 20.Elustondo PA et al. (2016) Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov 2, 16070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alavian KN et al. (2014) An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. U. S. A 111, 10580–10585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonora M et al. (2013) Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12, 674–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonora M et al. (2017) Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep 18, 1077–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan S et al. (2016) F1F0 ATP synthase-cyclophilin D interaction contributes to diabetes-induced synaptic dysfunction and cognitive decline. Diabetes 65, 3482–3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaseva AV et al. (2012) p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baev AY et al. (2017) Osmotic regulation of the mitochondrial permeability transition pore investigated by light scattering, fluorescence and electron microscopy techniques. Anal. Biochem 552, 38–44 [DOI] [PubMed] [Google Scholar]
- 27.Leung AW et al. (2008) The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem 283, 26312–26323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu N et al. (2012) The calcium uniporter regulates the permeability transition pore in isolated cortical mitochondria. Neural Regen. Res 7, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakagawa T et al. (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658 [DOI] [PubMed] [Google Scholar]
- 30.Schinzel AC et al. (2005) Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. U. S. A 102, 12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shanmughapriya S et al. (2015) SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol. Cell 60, 47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forte M et al. (2007) Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. U. S. A 104, 7558–7563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Javadov S et al. (2009) Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J. Pharmacol. Exp. Ther 330, 670–678 [DOI] [PubMed] [Google Scholar]
- 34.Kokoszka JE et al. (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du H and Yan SS (2010) Mitochondrial medicine for neurodegenerative diseases. Int. J. Biochem. Cell. Biol 42, 560–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo C et al. (2013) Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res 8, 2003–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du H et al. (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med 14, 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernardi P (1999) Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev 79, 1127–1155 [DOI] [PubMed] [Google Scholar]
- 39.Waterhouse NJ et al. (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J. Cell Biol 153, 319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol 35, 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vanden Berghe T et al. (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol 15, 135–147 [DOI] [PubMed] [Google Scholar]
- 42.Serrano-Pozo A et al. (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Irvine GB et al. (2008) Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med 14, 451–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohandas E et al. (2009) Neurobiology of Alzheimer’s disease. Indian J. Psychiatry 51, 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao J et al. (2011) Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci 31, 2313–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Picone P et al. (2014) Mitochondrial dysfunction: different routes to Alzheimer’s disease therapy. Oxid. Med. Cell Longev 2014, 780179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansson Petersen CA et al. (2008) The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U. S. A 105, 13145–13150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tillement L et al. (2006) The spirostenol (22R, 25R)-20alpha-spirost-5-en-3beta-yl hexanoate blocks mitochondrial uptake of Abeta in neuronal cells and prevents Abeta-induced impairment of mitochondrial function. Steroids 71, 725–735 [DOI] [PubMed] [Google Scholar]
- 49.Walls KC et al. (2012) Swedish Alzheimer mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein (APP) and beta-amyloid. J. Biol. Chem 287, 30317–30327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takuma K et al. (2005) ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J 19, 597–598 [DOI] [PubMed] [Google Scholar]
- 51.Lustbader JW et al. (2004) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452 [DOI] [PubMed] [Google Scholar]
- 52.Chen JX and Yan SD (2007) Amyloid-beta-induced mitochondrial dysfunction. J. Alzheimers Dis 12, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ren Y et al. (2008) Endophilin I expression is increased in the brains of Alzheimer disease patients. J. Biol. Chem 283, 5685–5691 [DOI] [PubMed] [Google Scholar]
- 54.Ramjaun AR et al. (2001) Endophilin regulates JNK activation through its interaction with the germinal center kinase-like kinase. J. Biol. Chem 276, 28913–28919 [DOI] [PubMed] [Google Scholar]
- 55.Rhein V et al. (2009) Amyloid-beta leads to impaired cellular respiration, energy production and mitochondrial electron chain complex activities in human neuroblastoma cells. Cell. Mol. Neurobiol 29, 1063–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bobba A et al. (2013) Mitochondrial respiratory chain Complexes I and IV are impaired by beta-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 13, 298–311 [DOI] [PubMed] [Google Scholar]
- 57.Du F et al. (2017) PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 140, 3233–3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu Q et al. (2017) Mitochondrial dysfunction triggers synaptic deficits via activation of p38 MAP kinase signaling in differentiated Alzheimer’s disease trans-mitochondrial cybrid cells. J. Alzheimers Dis 59, 223–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu Q et al. (2016) Antioxidants rescue mitochondrial transport in differentiated Alzheimer’s disease trans-mitochondrial cybrid cells. J. Alzheimers Dis 54, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fang D et al. (2016) Increased electron paramagnetic resonance signal correlates with mitochondrial dysfunction and oxidative stress in an Alzheimer’s disease mouse brain. J. Alzheimers Dis 51, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gan X et al. (2015) Blockade of Drp1 rescues oxidative stress-induced osteoblast dysfunction. Biochem. Biophys. Res. Commun 468, 719–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rasheed MZ et al. (2017) Mitochondrial permeability transition pore: a promising target for the treatment of Parkinson’s disease. Protoplasma 254, 33–42 [DOI] [PubMed] [Google Scholar]
- 63.Stefanis L (2012) alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med 2, a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berman SB and Hastings TG (1999) Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J. Neurochem 73, 1127–1137 [DOI] [PubMed] [Google Scholar]
- 65.(1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983 [DOI] [PubMed] [Google Scholar]
- 66.Brustovetsky N et al. (2003) Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J. Neurosci 23, 4858–4867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Milakovic T et al. (2006) Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: functional consequences. J. Biol. Chem 281, 34785–34795 [DOI] [PubMed] [Google Scholar]
- 68.Bouchier-Hayes L et al. (2005) Mitochondria: pharmacological manipulation of cell death. J. Clin. Invest 115, 2640–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Akhter F et al. (2017) Mitochondrial perturbation in Alzheimer’s disease and diabetes. Prog. Mol. Biol. Transl. Sci 146, 341–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Z et al. (2015) NR2B-dependent cyclophilin D translocation suppresses the recovery of synaptic transmission after oxygen-glucose deprivation. Biochim. Biophys. Acta 1852, 2225–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fang D et al. (2015) Increased neuronal PreP activity reduces Abeta accumulation, attenuates neuroinflammation and improves mitochondrial and synaptic function in Alzheimer disease’s mouse model. Hum. Mol. Genet 24, 5198–5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gan X et al. (2014) Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim. Biophys. Acta 1842, 220–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guo L et al. (2013) Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons. PLoS One 8, e54914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Du H et al. (2014) Cyclophilin D deficiency rescues Abeta-impaired PKA/CREB signaling and alleviates synaptic degeneration. Biochim. Biophys. Acta 1842, 2517–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gan X et al. (2014) Oxidative stress-mediated activation of extracellular signal-regulated kinase contributes to mild cognitive impairment-related mitochondrial dysfunction. Free Radic. Biol. Med 75, 230–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Du H et al. (2010) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. U. S. A 107, 18670–18675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hung CH et al. (2010) Modulation of mitochondrial calcium as a pharmacological target for Alzheimer’s disease. Ageing Res. Rev 9, 447–456 [DOI] [PubMed] [Google Scholar]
- 78.Smith RA and Murphy MP (2011) Mitochondria-targeted antioxidants as therapies. Discov. Med 11, 106–114 [PubMed] [Google Scholar]
- 79.Warne J et al. (2016) Selective inhibition of the mitochondrial permeability transition pore protects against neurodegeneration in experimental multiple sclerosis. J. Biol. Chem 291, 4356–4373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thomas B et al. (2012) Mitochondrial permeability transition pore component cyclophilin D distinguishes nigrostriatal dopaminergic death paradigms in the MPTP mouse model of Parkinson’s disease. Antioxid. Redox Signal 16, 855–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li V et al. (2009) Role of cyclophilin D-dependent mitochondrial permeability transition in glutamate-induced calcium deregulation and excitotoxic neuronal death. Exp. Neurol 218, 171–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Graham RM (1994) Cyclosporine: mechanisms of action and toxicity. Cleve. Clin. J. Med 61, 308–313 [DOI] [PubMed] [Google Scholar]
- 83.Atkinson K et al. (1984) Distribution and concentration of cyclosporin in human blood. J. Clin. Pathol 37, 1167–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ahmed-Belkacem A et al. (2016) Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities. Nat. Commun 7, 12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Valasani KR et al. (2014) Structure based design, synthesis, pharmacophore modeling, virtual screening, and molecular docking studies for identification of novel cyclophilin D inhibitors. J. Chem. Inf. Model 54, 902–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Valasani KR et al. (2013) Acetylcholinesterase inhibitors: structure based design, synthesis, pharmacophore modeling, and virtual screening. J. Chem. Inf. Model 53, 2033–2046 [DOI] [PubMed] [Google Scholar]
- 87.Valasani KR et al. (2016) Identification of a small molecule cyclophilin D inhibitor for rescuing abeta-mediated mitochondrial dysfunction. ACS Med. Chem. Lett 7, 294–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo HX et al. (2005) Novel cyclophilin D inhibitors derived from quinoxaline exhibit highly inhibitory activity against rat mitochondrial swelling and Ca2+ uptake/release. Acta Pharmacol. Sin 26, 1201–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Elkamhawy A et al. (2014) Novel quinazoline-urea analogues as modulators for Abeta-induced mitochondrial dysfunction: design, synthesis, and molecular docking study. Eur. J. Med. Chem 84, 466–475 [DOI] [PubMed] [Google Scholar]
- 90.Nunez-Figueredo Y et al. (2014) JM-20, a novel benzodiazepine-dihydropyridine hybrid molecule, protects mitochondria and prevents ischemic insult-mediated neural cell death in vitro. Eur. J. Pharmacol 726, 57–65 [DOI] [PubMed] [Google Scholar]
- 91.Nunez-Figueredo Y et al. (2014) Antioxidant effects of JM-20 on rat brain mitochondria and synaptosomes: mitoprotection against Ca2+-induced mitochondrial impairment. Brain Res. Bull 109, 68–76 [DOI] [PubMed] [Google Scholar]
- 92.Martin LJ et al. (2014) GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci 8, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shevtsova EF et al. (2014) Dimebon attenuates the Abeta-induced mitochondrial permeabilization. Curr. Alzheimer Res 11, 422–429 [DOI] [PubMed] [Google Scholar]
- 94.Wang H et al. (2007) Nortriptyline delays disease onset in models of chronic neurodegeneration. Eur. J. Neurosci 26, 633–641 [DOI] [PubMed] [Google Scholar]
- 95.Neganova ME et al. (2016) Neuroprotective effects of the securinine-analogues: identification of allomargaritarine as a lead compound. CNS Neurol. Disord. Drug Targets 15, 102–107 [DOI] [PubMed] [Google Scholar]
- 96.Malouitre S et al. (2009) Mitochondrial targeting of cyclosporin A enables selective inhibition of cyclophilin-D and enhanced cytoprotection after glucose and oxygen deprivation. Biochem. J 425, 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dube H et al. (2012) A mitochondrial-targeted cyclosporin A with high binding affinity for cyclophilin D yields improved cytoprotection of cardiomyocytes. Biochem. J 441, 901–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fabricant DS and Farnsworth NR (2001) The value of plants used in traditional medicine for drug discovery. Environ. Health Perspect 109 (Suppl. 1), 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kumar A et al. (2011) Protective effect of curcumin (Curcuma longa) against D-galactose-induced senescence in mice. J. Asian Nat. Prod. Res 13, 42–55 [DOI] [PubMed] [Google Scholar]
- 100.Zhou JS et al. (2014) Ginsenoside Rd attenuates mitochondrial permeability transition and cytochrome C release in isolated spinal cord mitochondria: involvement of kinase-mediated pathways. Int. J. Mol. Sci 15, 9859–9877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rastogi M et al. (2008) Curcuminoids modulates oxidative damage and mitochondrial dysfunction in diabetic rat brain. Free Radic. Res 42, 999–1005 [DOI] [PubMed] [Google Scholar]
- 102.Abdel-Kader R et al. (2007) Stabilization of mitochondrial function by Ginkgo biloba extract (EGb 761). Pharmacol. Res 56, 493–502 [DOI] [PubMed] [Google Scholar]
- 103.Russo P et al. (2013) Multitarget drugs of plants origin acting on Alzheimer’s disease. Curr. Med. Chem 20, 1686–1693 [DOI] [PubMed] [Google Scholar]
- 104.Cragg GM and Newman DJ (2013) Natural products: a continuing source of novel drug leads. Biochim. Biophys. Acta 1830, 3670–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pettit GR et al. (1982) Isolation and structures of bryostatin-1. J. Am. Chem. Soc 104, 6846–6848 [Google Scholar]
- 106.Schrott LM et al. (2015) Acute oral Bryostatin-1 administration improves learning deficits in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res 12, 22–31 [DOI] [PubMed] [Google Scholar]
- 107.Cao G et al. (1997) Antioxidant and prooxidant behavior of flavonoids: structure-activity relationships. Free Radic. Biol. Med 22, 749–760 [DOI] [PubMed] [Google Scholar]
- 108.Galati G et al. (1999) Glutathione-dependent generation of reactive oxygen species by the peroxidase-catalysed redox cycling of flavonoids. Chem. Res. Toxicol 12, 521–525 [DOI] [PubMed] [Google Scholar]