

Abstract
Cas12a (Cpf1) is a bacterial RNA-guided nuclease used widely for genome editing and diagnostic applications. In bacteria, Cas12a enzymes can be inhibited by bacteriophage-derived proteins, anti-CRISPRs (Acrs), to thwart clustered regularly interspaced short palindromic repeat (CRISPR) adaptive immune systems. How these inhibitors disable Cas12a by preventing programmed DNA cleavage is unknown. We show that three inhibitors (AcrVA1, AcrVA4 and AcrVA5) block Cas12a activity using functionally distinct mechanisms, including a previously unobserved enzymatic strategy. AcrVA4 and AcrVA5 inhibit double-stranded DNA (dsDNA) recognition with AcrVA4 driving Cas12a dimerization. In contrast, AcrVA1 is a multiple-turnover inhibitor that triggers cleavage of the target recognition sequence of the Cas12a-bound guide RNA to irreversibly inactivate the Cas12a complex. These distinct mechanisms equip bacteriophage with tools to evade CRISPR-Cas12a and support biotechnological applications where multiple-turnover enzymatic inhibition of Cas12a are desirable.
Bacteria and archaea can protect themselves against mobile genetic elements and viruses, including bacteriophage, using CRISPR-Cas adaptive immunity1. When challenged by a mobile genetic element, bacteria deploy CRISPR-associated (Cas) nucleases guided by an RNA2,3 to base-pair with the target and mediate target interference to provide immunity against reinfection3–5. While bacteriophage can undergo rapid mutation and selection to prevent Cas-effector targeting, genetic variation alone is insufficient to escape the potent programmability of bacterial CRISPR-Cas adaptive immunity6. To effectively evade CRISPR systems, bacteriophage have evolved protein-based inhibitors – anti-CRISPRs (Acrs)7–9 – that inactivate RNA-guided Cas nucleases10,11 and enable phage replication12,13. In the case of CRISPR-Cas914, such inhibitory Acrs can prevent DNA cutting by blocking dsDNA binding15–17, promoting Cas9 dimerization17, or preventing target DNA cleavage17. Recently, Acrs were discovered that inhibit the activities of the type V-A DNA-targeting CRISPR-Cas12a system18,19. These inhibitors targeting Cas12a might be expected to differ in mechanism given that Cas12a has a distinct structure20,21 and DNA cleavage pathway relative to Cas922. After expression of the CRISPR array and Cas proteins, Cas12a catalyzes precursor CRISPR-RNA (pre-crRNA) processing to form a Cas12a-crRNA complex (or ribonucleoprotein, RNP)23,24. Unlike the commonly used SpCas9, which utilizes two nuclease domains (HNH and RuvC) to cut dsDNA with single-turnover kinetics25,26, Cas12a possesses a single nuclease domain (RuvC) that is activated upon a crRNA targeting sequence (or spacer) binding to a complementary single-stranded DNA (ssDNA) or dsDNA target molecule23,27. Furthermore, the Cas12a RuvC domain catalyzes both single-turnover target DNA cutting (cis-cleavage) and multiple turnover non-target ssDNA cutting (trans-cleavage)27.
To determine the mechanistic basis for Cas12a inhibition, we biochemically assayed inhibition by AcrVA1, AcrVA4, and AcrVA5 on a panel of Cas12a orthologs. While biochemical experiments revealed that no AcrVA was capable of competitively inhibiting the RuvC-nuclease, each AcrVA was able to robustly inhibit dsDNA-targeting and to some extent ssDNA-targeting. Each AcrVA blocked dsDNA binding, but only AcrVA4 dimerized Cas12a and at high concentrations outcompeted dsDNA bound to dLbCas12a. Finally, we show that AcrVA1 triggers multiple turnover endoribonucleolytic cleavage of a Cas12a-bound crRNA to truncate the spacer sequence and permanently inactivate the complex. Together, these data provide the first insights into the mechanisms of AcrVAs, shedding light on the vulnerabilities in Cas12a and the evolutionary arms race between bacteriophage and their host bacteria.
RESULTS
AcrVAs do not inhibit all modes of DNA targeting by Cas12a.
Cas12a has a kinetically distinct DNA-cleavage pathway (Fig 1a). To determine how AcrVA proteins inhibit Cas12a, we first tested whether AcrVAs competitively inhibit the conserved RuvC nuclease domain. To do this, we determined the Michaelis-Menten constants, Vmax and Km, for the Cas12a RuvC nuclease in the presence of each inhibitor. AcrVA1, AcrVA4 and AcrVA5 each reduced the maximum velocity of trans-ssDNA cutting by Lachnospiraceae bacterium (Lb) Cas12a27 without dramatically affecting the Michaelis-Menten constant for the activated RuvC (Fig. 1b; Supplementary Fig. 1a-d). These data indicated that AcrVAs were not competitive inhibitors of the RuvC nuclease, but instead somehow depleted the pool of active Cas12a enzyme. We next tested whether AcrVAs could inhibit cis-DNA cleavage by Cas12a (Fig. 1a) using radiolabeled DNA substrates and three recombinantly purified Cas12a orthologs that are phylogenetically divergent and/or have been used for genome editing (Fig. 1c; Supplementary Fig. 2a). AcrVA1 blocked dsDNA cleavage by all three Cas12a orthologs, whereas AcrVA4 and AcrVA5 were effective against Moraxella bovoculi (Mb) Cas12a and LbCas12a but not Acidaminococcus sp. (As) Cas12a (Fig. 1c), consistent with plasmid cleavage data18. The pattern of inhibition was generally the same for Cas12a-mediated ssDNA cleavage but activity was not completely abolished by any inhibitor (Fig. 1d; Supplementary Fig. 3a-c). Collectively, these data indicated that ArcVA1 was a broad-spectrum inhibitor of Cas12a-catalyzed cis-DNA cleavage, whereas AcrVA4 and AcrVA5 inhibited cis-DNA cleavage catalyzed by MbCas12a and LbCas12a.
Figure 1 – AcrVAs do not inhibit all modes of DNA targeting by Cas12a.

a) Schematic representation of the steps in Cas12a target interference, b) Michaelis-Menten fit for 0.1 nM effective LbCas12a holoenzyme in the absence (black) or presence of AcrVA1 (blue), AcrVA4 (red), or AcrVA5 (yellow). The mean initial velocity (V0) is plotted against increasing DNase-Alert substrate concentrations (µM), where n = 3 replicates. The Vmax and Km for wild-type LbCas12a RuvC are indicated with dashed lines. c) Radiolabeled kinetic dsDNA cleavage assays for (left to right) MbCas12a, LbCas12a, and AsCas12a complexes with or without AcrVAs. Time courses represent 1’, 2’, 5’, 15’, and 60’. The uncleaved and cis-cleaved fractions are indicated with black triangles, d) Quantified percentage ssDNA cleaved for LbCas12a in the presence or absence of AcrVAs (mean ± sd, n = 3 independent experiments). Experimental fits are shown as solid lines and the calculated pseudo-first-order rate constants (kobs) (mean ± sd) are 2.6 ± 0.3 min−1, 0.15 ± 0.01 min−1, 0.7 ± 0.06 min−1, and 1.2 ± 0.09 min−1 for LbCas12a, LbCas12a+AcrVA1, LbCas12a+AcrVA4, and LbCas12a+AcrVA5 respectively. Source data for panels b and d are available with the paper online. The uncropped gel images are available in Supplementary Data Set 1.
AcrVAs block dsDNA binding and AcrVA4 dimerizes Cas12a.
We next tested whether AcrVAs affect DNA binding to Cas12a, the rate limiting step of Cas12a-targeting activity28 (Fig. 1a). To test this, we assayed 5’-radiolabelled DNA binding to catalytically dead (dLbCas12a) by electrophoretic mobility shift assays (EMSAs). The dLbCas12a-crRNA complex was formed before the separate addition of each AcrVA and incubation with dsDNA, revealing that AcrVAs abolished dsDNA binding (Fig. 2a), while ssDNA binding was perturbed to a lesser extent (Supplementary Fig. 4a, b). Notably, we observed a slow-mobility species representing the DNA-bound dLbCas12a complex in the presence of AcrVA4 (Supplementary Fig. 4c), hinting at a possible multimeric assembly reminiscent of inhibitor-induced N. meningitidis (Nme) Cas9 dimerization17. To test this possibility, we assessed the solution oligomeric state of each AcrVA and when mixed with LbCas12a-crRNA (Fig. 2b; Supplementary Fig; 5a, b). While AcrVA1 and AcrVA5 appeared monomeric (Supplementary Fig. 5a, b), AcrVA4 appeared dimeric prior to complexing with Cas12a (Fig. 2b). Although neither AcrVA1 nor AcrVA5 triggered a substantial change in estimated molecular weight when complexed with LbCas12a-crRNA (Supplementary Fig. 5a, b), mixing AcrVA4 with LbCas12a-crRNA produced two higher molecular weight species (Fig. 2b). Using light scattering, we estimated the mass of these species to be 349 kDa and 214 kDa respectively, consistent with a dimeric LbCas12a protein-crRNA-AcrVA4 complex and a monomeric LbCas12a-crRNA bound to a dimer of AcrVA4. To directly visualize the dimerization of LbCas12a-crRNA with AcrVA4, we analyzed gel filtration purified fractions by negative stain electron microscopy, revealing a distribution of particles including a symmetrical complex of LbCas12a-crRNA dimers (Fig. 2c; Supplementary Fig. 5c). Taken together, these results demonstrate that AcrVAs block dsDNA binding to Cas12a and that the mechanism for AcrVA4 involves dimerization of the LbCas12a-crRNA complex.
Figure 2 – AcrVAs block dsDNA binding and AcrVA4 dimerizes Cas12a.
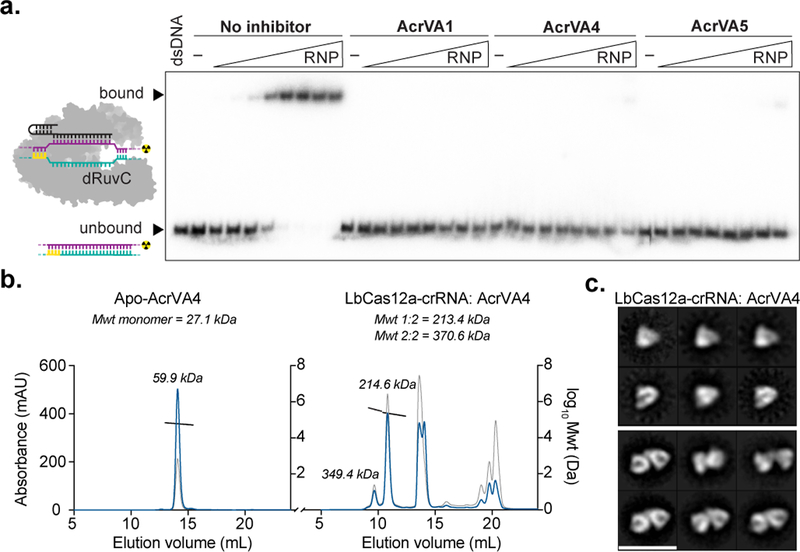
a) Radiolabeled dsDNA electrophoretic mobility shift assay of increasing concentrations of dLbCas12a-crRNA complexed with or without AcrVA before association with dsDNA. The bound and unbound fractions are indicated with black triangles, b) Size exclusion chromatography coupled light scattering traces for (left) AcrVA4 alone and (right) LbCas12a-crRNA complexed with AcrVA4. The absorbance at 280 nm (blue) and 260 nm (grey) are shown (left axis) with the linear region for the mass estimate corresponding to the relevant peaks (black lines, central and right axis). The predicted molecular weights for each sample are shown above the graph and the calculated molecular weights are indicated adjacent to the relevant peak c) 2D-class averages of LbCas12a-crRNA monomers (top) and LbCas12a-crRNA dimers bound to AcrVA4 (bottom). The scale bar represents 28 nm. The uncropped gel images are available in Supplementary Data Set 1.
AcrVA4 can dislodge dsDNA bound to dCas12a.
We next wondered if any AcrVA was capable of disrupting dsDNA-bound complexes of Cas12a-crRNA, a mechanism that may have evolved to disable an activated and trans-cleaving Cas12a (Fig. 1a). To test this possibility, we formed a ternary complex of dLbCas12a-crRNA bound to radiolabeled dsDNA to which was added a titration series of excess AcrVA1, AcrVA4, AcrVA5, or unlabeled dsDNA and visualized by EMSA. At high concentrations, AcrVA4 triggered the release of dsDNA bound to Cas12a, whereas little dsDNA release occurred in the presence of AcrVA1, AcrVA5, or unlabeled dsDNA competitor (Fig. 3a; Supplementary Fig. 6a). In contrast, a stoichiometric excess of any AcrVA or ssDNA competitor had no effect on dLbCas12a-crRNA bound to radiolabeled ssDNA (Supplementary Fig. 6b). These data suggest that at high concentrations AcrVA4 can dislodge dsDNA after it has formed an R-loop interaction with Cas12a. Depletion or addition of ATP had no effect on dsDNA displacement from LbCas12a-crRNA complexes by AcrVA4, suggesting an ATP-independent process (Supplementary Fig. 6c). AcrVA4 did not trigger release of ssDNA bound to dLbCas12a-crRNA, suggesting that the non-target strand (NTS) of the DNA (the strand not base-paired to the crRNA) might be required to drive re-annealing with the target strand (TS). In support of this possibility, addition of the NTS ssDNA molecule to the dLbCas12a-crRNA-TS DNA complex led to TS DNA displacement in the presence of AcrVA4; a non-complementary ssDNA used in a similar experiment had no effect (Supplementary Fig. 7a). Consistent with TS DNA release requiring base pairing to a complementary NTS strand, AcrVA4 was unable to drive DNA release from dLbCas12a-crRNA bound to a dsDNA substrate containing mismatched nucleotides along all or some of the 20-nt NTS (Supplementary Fig. 7b, c).
Figure 3 – AcrVA4 displaces dsDNA bound to dCas12a but not wild-type Cas12a.
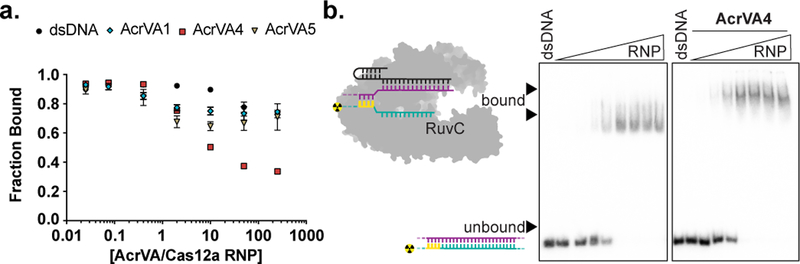
a) Quantified fraction dsDNA bound by dLbCas12a-crRNA after complexing with dsDNA before the addition of increasing concentrations of AcrVA or dsDNA competitor determined by EMSA (mean ± sd, n = 3 independent experiments), Source data are available with the paper online. b) Radiolabeled dsDNA electrophoretic mobility shift assay of increasing concentrations of LbCas12a-crRNA first complexed with dsDNA before addition of AcrVA. The bound and unbound fractions are indicated with black triangles. The uncropped gel images are available in Supplementary Data Set 1.
The preceding experiments were conducted using catalytically inactive Cas12a, which prevents cutting of bound DNA and hence remains associated with an intact dsDNA molecule. Given that catalytically active Cas12a would cut and release the PAM distal dsDNA fragment after the formation of an R-loop interaction29, we reasoned that this release might prevent AcrVA4 from displacing the PAM proximal dsDNA bound to the crRNA (Fig. 1a). To test this possibility, we incubated wild-type LbCas12a-crRNA with a dsDNA substrate, followed by addition of AcrVA4 and analysis of the resulting samples by EMSA (Fig. 3b). In contrast to dLbCas12a (Fig. 3a; Supplementary Fig. 7d), AcrVA4 had no effect on DNA bound by wild-type LbCas12a (Fig. 3b). These data demonstrate that AcrVA4 can dislodge dsDNA bound to catalytically dead but not active Cas12a, presumably due to a shift in binding equilibrium that favors DNA strand re-annealing.
AcrVA1 triggers endoribonucleolytic truncation of a Cas12a-bound crRNA
We next explored whether any AcrVA might prevent target DNA binding by disruption of the Cas12a-crRNA complex (Fig. 1a). To test this possibility, we incubated Cas12a with each AcrVA individually before adding radiolabeled RNA to probe crRNA integrity, the efficacy of pre-crRNA processing, and the affinity of Cas12a for mature crRNA. We were surprised to observe that AcrVA1 induced rapid 3’-end truncation of both mature and pre-crRNA in the presence of Cas12a (Fig. 4a; Supplementary Fig. 8a). In these experiments, neither binding to mature crRNA or pre-crRNA processing were affected (Fig. 4b; Supplementary Fig. 8a). Notably, AcrVA1 had no effect on the integrity of mature or pre-crRNA in the absence of Cas12a, and neither AcrVA4 nor AcrVA5 had any effect on crRNA in the absence or the presence of Cas12a (Fig. 4a, b; Supplementary Fig. 8a, b). Pre-assembly of Cas12a and crRNA forms the Cas12a-crRNA complex which was also susceptible to AcrVA1-mediated crRNA 3’-truncation (Supplementary Fig. 8b). However, pre-assembly of an activated Cas12a-crRNA complex with the addition of complementary ssDNA or dsDNA prevented AcrVA1-mediated crRNA truncation (Supplementary Fig. 8c). Furthermore, AcrVA1-mediated crRNA truncation was specific for a crRNA bound by Cas12a regardless of spacer sequence (Supplementary Fig. 9a) or lengths that support Cas12a DNA-targeting23,27 (Supplementary Fig. 9b), and without any detectable non-specific ribonuclease activity (Supplementary Fig. 9c). Taken together, the above data indicate that AcrVA1 triggers crRNA truncation on an assembled Cas12a-crRNA complex.
Figure 4 – AcrVA1-triggered endoribonuclease activity truncates a Cas12a-bound crRNA.
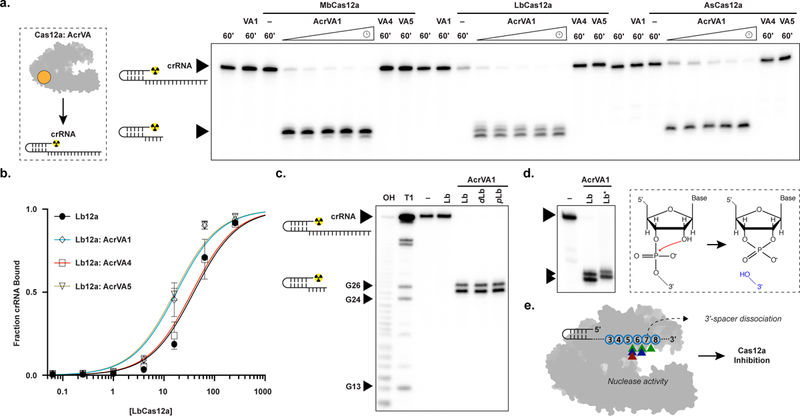
a) Radiolabeled kinetic crRNA cleavage assays for (left to right) MbCas12a, LbCas12a, and AsCas12a complexed with or without AcrVAs. Time courses represent 1, 2, 5, 15, and 60 min. Black triangles indicate full-length and truncated crRNA, b) Quantified fraction crRNA bound by LbCas12a in the presence or absence of AcrVAs determined by EMSA (mean ± sd, n = 3 independent experiments). Measured dissociation constants (Kd) are 38.9 nM ± 4.7, 17.6 ± 2.4, 35.8 ± 4.4, and 16.4 ± 2.1 in absence of inhibitor or in the presence of AcrVA1, AcrVA4, or AcrVA5, respectively, Source data are available with the paper online. c) Radiolabeled crRNA cleavage assay with LbCas12a-crRNA complexed without or with AcrVA1. Treatments in the absence of AcrVA1 are (left to right) crRNA hydrolysis ladder (OH), crRNA RNase T1 digestion (T1), untreated crRNA (−), and crRNA incubated with LbCas12a (Lb). Treatments in the presence of AcrVA1 are (left to right) wild-type LbCas12a (Lb), dLbCas12a (D832A, dLb), and processing dead Cas12a (K785A, pLb). A large black triangle indicates the full-length crRNA, smaller triangles indicate RNase T1 mapped G-nucleotides, d) Radiolabeled crRNA cleavage assay using LbCas12a-crRNA complexed with AcrVA1 that is either untreated (Lb) or treated with PNK (Lb*), e) Schematic representation of AcrVA1-triggered crRNA spacer cleavage activity on Cas12a. Cleavage sites for Mb (blue), Lb (green), and AsCas12a (red) are indicated with triangles. The uncropped gel images are available in Supplementary Data Set 1.
Interestingly, AcrVA1 is not predicted to be a nuclease18,19, nor does it have detectable RNA cleavage activity in the absence of a Cas12a-crRNA complex (Fig. 4a; Supplementary Fig. 9a-c). To assess the mechanism of AcrVA1-mediated RNase activity, we mapped the scissile phosphates at positions five to eight within the crRNA spacer, with some plasticity in position dependent on the Cas12a ortholog (Fig. 4c; Supplementary Fig. 9d). The activity is that of an endoribonuclease where catalysis generates an intact 3’-fragment of the crRNA that is released by Cas12a after AcrVA1-triggered truncation (Supplementary Fig. 9e-f). To identify the nuclease center responsible for crRNA truncation we targeted Cas12a’s nucleases for mutagenesis, the RuvC or pre-crRNA processing nuclease, and observed that mutation of either did not prevent AcrVA1-triggered spacer truncation (Fig. 4c; Supplementary Fig. 9d). We next assayed the metal-dependency of the nuclease by supplementing with MgCl2 or EDTA and observed that AcrVA1-triggered spacer truncation was not dependent on divalent cations (Supplementary Fig. 9g). Consistent with this, we determined the end-group chemistry of the 5’-radiolabeled crRNA fragment with T4 polynucleotide kinase (PNK) treatment which resulted in an upward shift in polyacrylamide gel migration, indicating that the 5’-fragment generated had a 3’-phosphate at its terminus (Fig. 4d). Taken together, these data demonstrate that AcrVA1 triggers metal-ion independent endoribonucleolytic cleavage of the targeting portion of the crRNA which then dissociates to render the complex rudderless with respect to DNA targeting (Fig. 4e).
AcrVA1 is a multiple-turnover inhibitor and competes with AcrVA5
AcrVA1 stands out among the known Cas12a and Cas9 inhibitors as a highly effective and broad-spectrum inhibitor of RNA-guided dsDNA targeting by Cas12a18. Given its unique enzymatic activity, we wondered if the potency of AcrVA1 inhibition might be attributed to multiple-turnover kinetics. To test this, we incubated a range of AcrVA1 concentrations with Cas12a-crRNA complexes and in all cases observed ~95% of crRNAs truncated, even at sub-stoichiometric concentrations of AcrVA1 (Fig. 5a). Thus, AcrVA1 activity is multiple-turnover where cleavage of a crRNA will permanently inactivate Cas12a-crRNA complexes through a mode of inhibition not previously observed for any anti-CRISPR protein. However, we earlier demonstrated that AcrVA1 was not a robust inhibitor of ssDNA targeting by Cas12a (Fig. 1d; Supplementary Fig. 3) which is at odds with the observed nuclease activity on the crRNA (Fig. 4a; Fig. 5a). We wondered if the 5’- or 3’-fragments of the crRNA, together or separately, might still be sufficient for ssDNA-targeting. To test this, we prepared RNA fragments that mimic products of AcrVA1 activity and assayed ssDNA-targeting by LbCas12a. Remarkably, LbCas12a cleaved ssDNA in the presence of both the 5’- and 3’-fragments (Supplementary Fig. 10c), suggesting that the ssDNA-targets can be recruited to Cas12a with a two-component crRNA. Taken together, our data demonstrate that AcrVA1 triggers crRNA truncation and release of the 3’-fragment from Cas12a which can hybridize with a target ssDNA to activate cis and trans-ssDNA cleavage by Cas12a.
Figure 5 – AcrVA1-triggered endoribonuclease activity is multiple turnover, requires the PID, and competes with AcrVA5.
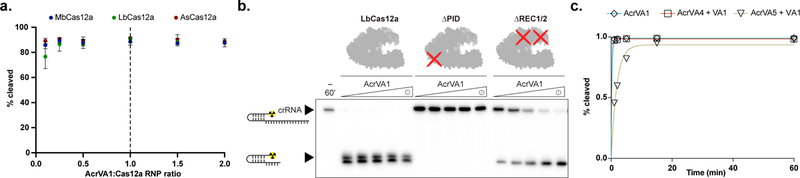
a) Percentage crRNA spacers truncated after 1 hr at different ratios of AcrVA1:Cas12a-crRNA (mean ± sd, n = 3 independent experiments). b) Radiolabeled kinetic crRNA cleavage assays for (left to right) LbCas12a, ∆PID LbCas12a, and ∆REC1/2 LbCas12a in the presence of AcrVA1. Time courses represent 1, 2, 5, 15, and 60 min. Black triangles indicate the full-length and truncated crRNA. The approximate position of the truncated domains is shown with a red cross. c) Quantified time-course of percentage AcrVA1-triggered spacers truncated in the presence AcrVA4 or AcrVA5 (n = 3 independent experiments). Experimental fits are shown as solid lines and the calculated pseudo-first-order rate constants (kobs) (mean ± sd) are 4.8 ± 0.2 min−1, 4.4 ± 0.3 min−1, and 0.5 ± 0.04 min−1 for AcrVA1, AcrVA1 + AcrVA4, and AcrVA1 + AcrVA5, respectively. Source data for panels a and c are available with the paper online. The uncropped gel images are available in Supplementary Data Set 1.
The ability of AcrVA1 to inhibit diverse Cas12a orthologs (Fig. 1c) suggested that it might exploit an evolutionarily conserved domain of Cas12a. To determine domains required for AcrVA1-triggered spacer truncation, we generated truncations that still allowed for crRNA binding and pre-crRNA processing. Removal of either the PAM-interacting domain (PID) or both recognition (REC) domains generated stable constructs that maintained near wild-type mature crRNA binding affinity or pre-crRNA processing (Supplementary Fig. 10b, c). However, only in the absence of the PID was AcrVA1-triggered crRNA truncation prevented (Fig. 5b). Finally, we wondered if either AcrVA4 or AcrVA5 might compete with the AcrVA1-triggered spacer truncation activity. To test this, we first incubated an LbCas12a RNP with either AcrVA4 or AcrVA5 before adding AcrVA1 and found that AcrVA5 reduced the rate of AcrVA1-triggered crRNA truncation (Fig. 5c), suggesting that AcrVA5 does compete with the spacer truncation activity of AcrVA1.
DISCUSSION
CRISPR-Cas12a are RNA-guided DNA-targeting nucleases with robust cis-cleavage and ssDNA trans-cleavage, activities that have led to their rapid implementation as tools for genome engineering and diagnostics30. In this work, we present mechanistic insights into type V-A bacteriophage-derived anti-CRISPRs elucidating the distinct modes leveraged to inactivate Cas12a (Fig. 6). We found that AcrVA1, AcrVA4, and AcrVA5 robustly inhibited Cas12a dsDNA targeting, not unlike inhibitors that evolved to target Cas98.
Figure 6 – Three distinct modes of CRISPR-Cas12a inactivation.
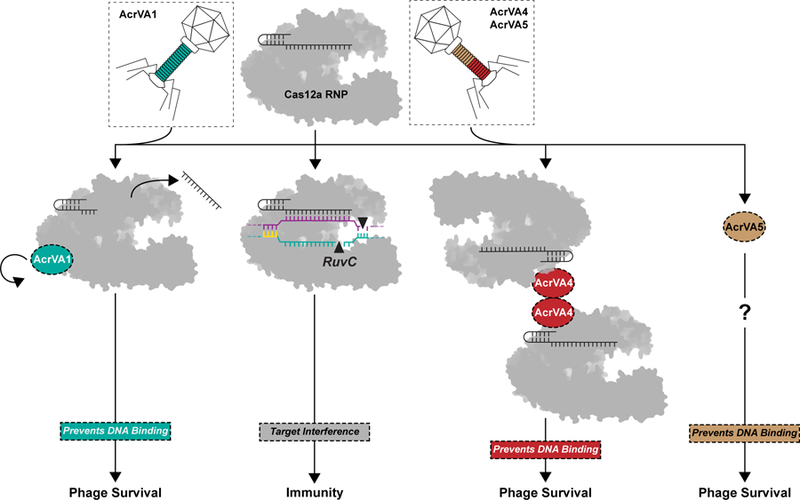
Model for AcrVA1, AcrVA4, and AcrVA5 inhibition of Cas12a. Cas12a assembles with its crRNA to form a surveillance complex (top). In the absence of inhibitors, Cas12a recognizes a complementary target DNA activating the RuvC leading to target interference and immunity. Phage-encoded AcrVA1 (teal) associates with Cas12a triggering crRNA spacer truncation preventing DNA binding. Phage-encoded AcrVA4 dimerizes (red) Cas12a and blocks dsDNA binding. Phage-encoded AcrVA5 (brown) blocks Cas12a dsDNA binding via an unknown mechanism.
AcrVA1 provides a uniquely potent mechanism for evading CRISPR adaptive immunity by triggering crRNA truncation with multiple turnover kinetics to rapidly and permanently inactivate the Cas12a surveillance complex. We demonstrated that the nuclease activity is entirely dependent on the presence of a Cas12a-crRNA complex and AcrVA1, but our data do not describe the identity of the component bearing the catalytic center for the observed nuclease activity. It is likely that AcrVA1 is an RNase, however we could not detect any RNase activity on free crRNA or trans-ssRNA substrates, suggesting that its activity is allosterically activated by binding to a Cas12a-crRNA complex or that Cas12a harbors the nuclease domain or a part thereof. AcrVA1 has a broad spectrum of inhibition, disabling divergent Cas12a nucleases in vitro and in mammalian cell editing18 potentially exploiting the broadly conserved PAM interacting domain for direct access to the pre-ordered seed of the crRNA31. Interestingly, AcrVA1 displayed less robust inhibition of Cas12a ssDNA-targeting, a potential artefact of working in vitro as the cleaved 3’-crRNA fragments can readily associate with ssDNA and be recruited back to Cas12a for activation. In the bacterial host, it is likely that cleavage of the crRNA creates an ineffective two-component system. However, further experiments are required to determine if AcrVA1 provides a selective advantage to ssDNA plasmids or ssDNA phage. It was recently shown that bacteriophage cooperate to immunocompromise bacterial hosts, delivering Acrs iteratively to gradually overcome CRISPR-Cas immunity12,13. In light of these observations, bacteriophage encoding AcrVA1 may be the most effective in supporting populations of phage lacking Acrs given its multiple turnover kinetics even with the recent data suggesting that Cas12a endonuclease activity can be reset32. Furthermore, Cas effectors are universally steered by programmable RNA guides raising the possibility that all CRISPR-Cas systems are susceptible to this mode of inhibition. The unique mechanism for AcrVA1-mediated CRISPR-Cas12a inhibition may lend itself to potent control of Cas12a in gene editing applications where it is desirable to block DNA targeting or limit unintended editing events.
We also found that AcrVA4 blocks dsDNA binding in addition to driving dimerization of Cas12a-crRNA complexes. This mechanism has also been described for AcrIIC3 which targets NmeCas917. While mechanistically and structurally divergent, Cas9 and Cas12a are susceptible to a convergent mechanism of inhibition suggesting that higher order assembly of Cas nucleases and the associated inhibitors offers an as yet unclear benefit to bacteriophage. AcrVA4 was also able to disrupt a dLbCas12a-crRNA complex stably associated with dsDNA, an activity that required high concentrations of the inhibitor. The disruption of dLbCas12a dsDNA bound states may have applicability in dLbCas12a-mediated transcriptional control applications, however further experiments are required to establish the off-rate for dsDNA in the presence of AcrVA4. Furthermore, while it is interesting to consider that AcrVA4 may shift the equilibrium in favor of dsDNA dissociation, this mode of action is unlikely to have biological significance given that wild-type Cas12a rapidly catalyzes DNA-cleavage once an R-loop is formed29,30.
Finally, we demonstrated that AcrVA5 robustly inhibited Cas12a dsDNA-targeting activity by preventing dsDNA binding. Given that AcrVA5 competed with AcrVA1 and that AcrVA1 activity is dependent on the PID, we speculate that AcrVA5 may directly exploit the PID to block PAM recognition on dsDNA substrates. If true, this raises the possibility that AcrVA5 might be leveraged as a tool to block in vivo dsDNA targeting by Cas12a in order to exclusively select for ssDNA targeting. Furthermore, this panel of AcrVA inhibitors are more potent inhibitors of dsDNA-targeting than ssDNA-targeting by Cas12a which may reflect an evolutionary pressure from dsDNA phage in the hosts microbial community. Taken together, these mechanistic insights reveal vulnerabilities in the modes of Cas12a targeting, providing scope for greater control of a rapidly expanding landscape of Cas12a33 and Acr34 applications.
METHODS
Phylogenetic analysis
A multiple sequence alignment of the Cas12 proteins23,35,36 was generated using MAFFT L-INS-i37 and a maximum-likelihood phylogenetic tree was constructed using RAxML38 with PROTGAMMALG as the substitution model and 100 bootstrap samplings. The tree was visualized using iTOL v339 to highlight the phylogeny of Cas12a with Cas12b-e as a collapsed outgroup.
Protein expression and purification
Plasmids encoding Moraxella bovoculi (33362) Cas12a, Lachnospiraceae bacterium (ND2006) Cas12a, Acidaminococcus sp. (BV3L6) Cas12a, AcrVA1, AcrVA4, and AcrVA5 were generated from a custom pET-based expression vector as described previously18. Cas12a point mutations and truncations were introduced by either around-the-horn PCR or Gibson Assembly verified by DNA sequencing. Proteins were purified as described previously18. Briefly, E. coli Rosetta 2 (DE3) containing Cas12a or AcrVA expression plasmids were grown in Lysogeny Broth overnight with ampicillin (100 µg mL−1). Overnight cultures were sub-cultured in Terrific Broth to an OD600 of 0.6–0.8, after which the cultures were cooled on ice for 15 min before induction with 0.5 mM IPTG and incubated overnight at 16°C for 16 hrs. Cells were harvested by centrifugation and resuspended in wash buffer (20 mM Tris-Cl, (pH 7.5), 500 mM NaCl, 1 mM TCEP, 5% (v/v) glycerol) supplemented with 0.5 mM PMSF and cOmplete protease inhibitor (Roche), lysed by sonication, and purified over Ni-NTA Superflow resin (Qiagen) in wash buffer supplemented with either 10 mM imidazole (wash) or 300 mM imidazole (elution). Eluted proteins were digested overnight with TEV protease at 4°C in a Slide-A-Lyzer (10 kDa MWCO, Thermofisher) against dialysis buffer (20 mM Tris-Cl (pH 7.5), 125 mM NaCl, 1 mM TCEP, 5% (v/v) glycerol). Digested proteins were loaded onto an MBP-Trap (GE Healthcare) upstream of a Heparin Hi-Trap (GE Healthcare, Cas12a) or a Hi-Trap Q (GE Healthcare, AcrVA) and eluted over a salt gradient (20 mM Tris-Cl, (pH 7.5), 1 mM TCEP, 5% (v/v) glycerol, 125 mM – 1 M KCl). The eluted protein was concentrated before injection to a Superdex 200 10/300 Increase (GE Healthcare) developed in 20 mM HEPES-K (pH 7.5), 200 mM KCl, 1 mM TCEP, 5% (v/v) glycerol). Purified proteins were concentrated and snap frozen in LN2 for storage at −80°C. The purity and integrity of proteins used in this study were assessed by SDS-PAGE (Coomassie blue staining) (Supplementary Fig. 2b).
RNA and DNA preparation
RNA used in this study were ordered from Integrated DNA Technologies (IDT) (Supplementary Table 1). RNA substrates were purified by gel extraction from 12% (v/v) urea-denaturing PAGE (0.5X TBE) and ethanol precipitation as described previously40. All DNA substrates were synthesized by IDT and purified as described above. Radiolabeled RNA substrates were prepared by 5’-end-labeling with T4 PNK (NEB) in the presence of gamma 32P-ATP. For 3’-end-labeled substrates, the crRNA was labeled with T4 RNA Ligase 1 (NEB) in the presence of 32P-PcP. Radiolabeled DNA substrates were prepared by 5’-end-labeling with T4 PNK (NEB) in the presence of gamma 32P-ATP. For dsDNA substrates, non-target strand or target-strand was first 5’-end-labeled before annealing a 1.2-fold molar excess of the complementary strand at 95°C for 3 min in 1X hybridization buffer (20 mM Tris-Cl, pH 7.5, 150 mM KCl, 5 mM MgCl2, 1 mM DTT) followed by slow-cooling to room temperature.
DNase-Alert trans-cleavage assays
For Michaelis-Menten kinetics in the presence or absence of AcrVA, 0.1 nM of pre-assembled and activated LbCas12a-crRNA-activator holoenzyme was prepared by complexing 5 nM Cas12a and 6.25 nM crRNA (15 min at 37°C) with 25–50 nM AcrVA (30 min at 37°C) and 0.1 nM DNA activator (15 min at 37°C) in 1X trans-cleavage buffer (20 mM HEPES-K (pH 7.5), 100 mM KCl, 5 mM MgCl2, 5% (v/v) glycerol, 1 mM DTT, 50 µg/mL heparin). Reactions were initiated with 0.001, 0.01, 0.1, 0.25, 0.5, 1, or 2 µM of DNase-Alert™ substrate (IDT). Reactions were incubated in a fluorescence plate reader (BioTek) for 30 min at 37°C with fluorescence measurements taken every 20 sec (λex: 535 nm; λem: 595 nm). The initial velocity (V0) was calculated by fitting to a linear regression and plotted against the substrate concentration to determine the Michaelis-Menten constants (Prism7, GraphPad), according to Y = (Vmax x X)/(Km + X), where X is the substrate concentration and Y is the initial velocity (n = 3 independent experiments).
Radiolabeled DNA cleavage assays
Cas12a-mediated DNA-cleavage assays were carried out in 1X cleavage buffer (20 mM Tris-Cl pH (7.8), 150 mM KCl, 5 mM MgCl2, 1% (v/v) glycerol, and 2 mM DTT). Radiolabeled DNA-cleavage assays consisted of Cas12a, crRNA, and 32P-labeled DNA substrates in the presence or absence of AcrVA at 30 nM, 36 nM, 1 nM, and 300 nM, respectively. The RNP was formed at 37°C for 15 minutes before addition of AcrVA (unless otherwise indicated) and incubated at 37°C for 30 minutes. Reactions were initiated with the addition of target DNA at 37°C and timepoints (1, 2, 5, 15, 60 min) quenched in 1.5X formamide loading buffer (final concentration 45% (v/v) formamide, 15 mM EDTA, 0.1% (w/v) SDS, 200 µg mL−1 Heparin, and 0.25% (w/v) bromophenol blue) for 3 min at 95°C. Samples were resolved by 12% (v/v) urea-denaturing PAGE (0.5X TBE) and visualized by phosphoroimaging (Amersham Typhoon, GE Healthcare). The percentage cleavage was calculated as a ratio of the intensity of the product band relative to the total intensity of both the product and uncleaved DNA normalized to the background within each measured substrate in ImageQuant TL Software (GE Healthcare). Apparent rates were calculated by a fit to a single exponential decay (Prism7, GraphPad). The rates with their associated standard deviations are included in the figure legends (n = 3 independent experiments).
Radiolabeled crRNA cleavage assays
crRNA-cleavage assays were carried out in 1X cleavage buffer. Radiolabeled crRNA-cleavage assays consisted of Cas12a, and 32P-labeled RNA substrates in the presence or absence of AcrVA at 50 nM, 1 nM, and 500 nM, respectively. Complexing was carried out by incubating Cas12a and AcrVA or 32P-labeled crRNA substrates at 37°C for 30 min before initiating the reaction with the addition of AcrVA or 32P-labeled RNA substrates. Reaction timepoints (1, 2, 5, 15, 60 min) were quenched in 1.5X formamide loading buffer for 3 min at 95°C. Samples were resolved by 12% (v/v) urea-denaturing PAGE (0.5X TBE), visualized by phosphoroimaging (Amersham Typhoon, GE Healthcare), and quantified with ImageQuant TL Software (GE Healthcare). Where appropriate, apparent rates were calculated by a fit to a single exponential decay (Prism7, GraphPad), and the calculated rates with their associated standard deviations are included in the figure legends (n = 3 independent experiments). For substrate turnover experiments, 120 nM of Cas12a was complexed with 100 nM of mature crRNA and incubated with 10, 25, 50, 100, 150, or 200 nM AcrVA1 at 37°C for 60 min before quenching in 1.5X formamide loading buffer for 3 min at 90°C. Samples were resolved by 12% (v/v) urea-denaturing PAGE (0.5X TBE), visualized with SYBR Gold (Invitrogen) post-staining, imaged, and quantified with a ChemiDoc (BioRad). The percentage of crRNA spacers cleaved was calculated as a ratio of the intensity of the product band relative to the total intensity of both the product and uncleaved crRNA normalized to background.
Product size mapping and 3’ end chemistry identification
Cleavage product length was determined biochemically by comparing the gel migration of AcrVA1-triggered cleavage products with alkaline hydrolysis and RNase T1 digestion ladders of the matched untreated crRNA. Hydrolysis ladders were generated by incubating 15 nM 5’-radiolabeled crRNA at 95°C for 10 min in 1X alkaline hydrolysis buffer (Ambion). Reactions were quenched in 1.5X formamide loading buffer and immediately loaded to a urea-denaturing PAGE (0.5X TBE) gel. For RNase T1 digestion ladders, 15 nM 5’-radiolabeled crRNA were unfolded in 1X RNA sequencing buffer (Ambion) at 65°C for 5 min and cooled to ambient temperature before the addition of 1 U of RNase T1 (Ambion). After incubating at ambient temperature for 15 min, reactions were extracted in phenol-chloroform (pH 8.0) and stored in 1.5X formamide loading buffer before loading to a urea-denaturing PAGE (0.5X TBE) gel. For 3’ end chemistry identification, products from AcrVA1-triggered crRNA truncation reactions were extracted in phenol-chloroform (pH 8.0) before incubation with 10 U of T4 polynucleotide kinase (NEB) in 1X T4 polynucleotide kinase buffer (NEB) for 30 min at 37°C. Reactions were quenched with 1.5X formamide loading buffer for 3 min at 95°C and resolved on a 15% (v/v) urea-denaturing PAGE (0.5X TBE) gel and visualized by phosphoroimaging (Amersham Typhoon, GE Healthcare).
RNA electrophoretic mobility-shift assays
All experiments were equilibrated in 1X binding buffer (20 mM Tris-Cl (pH 7.5), 150 mM KCl, 5 mM MgCl2, 1mM DTT, 5% (v/v) glycerol, 50 µg mL−1 heparin, 50 µg mL−1 BSA, and 0.01% (v/v) IGEPAL CA-630). Cas12a 32P-crRNA EMSAs were prepared by a titration of Cas12a (0 nM, 0.063 nM, 0.25 nM, 1 nM, 4 nM, 16 nM, 64 nM, 256 nM) in the presence or absence of an excess of AcrVA (5 µM) incubated for 30 min at 37°C before the addition of 0.2 nM 32P-crRNA and incubation for 30 min at 37°C.
DNA electrophoretic mobility-shift assays
All experiments were equilibrated in 1X binding buffer. To avoid the dissociation of the dLbCas12a-crRNA or LbCas12a-crRNA complex during DNA binding experiments, Cas12a 32P-DNA EMSAs were prepared as an excess of Cas12a (960 nM) incubated with a titration of crRNA (0 pM, 2.4 pM, 9.7 pM, 39 pM, 0.156 nM, 0.625 nM, 2.5 nM, 10 nM, 40 nM) to pre-form the RNP for 30 min at 37°C, unless otherwise indicated. For experiments testing if AcrVA block ssDNA or dsDNA binding, the RNP was first incubated with an excess of AcrVA (10 µM) for 30 min at 37°C before incubation with 32P-DNA substrate (0.1 nM) for 30 min at 37°C, unless indicated otherwise. For experiments testing if AcrVA could disrupt a dLbCas12a-ssDNA bound complex, RNP was prepared as described above except 32P-ssDNA addition preceded the addition of AcrVA (10 µM), cold non-target strand, and/or cold ssDNA competitor. For experiments testing if AcrVA could disrupt a dLbCas12a-dsDNA or LbCas12a-crRNA bound complex, RNP was prepared at 40 nM effective concentration as described above before pre-forming a DNA bound state with 32P-dsDNA (0.1 nM) for 30 min at 37°C. This was followed by the addition of AcrVA or cold DNA competitor (0 nM, 1 nM, 3 nM, 16 nM, 80 nM, 400 nM, 2µM, 10 µM) for another 30 min at 37°C. To test ATP dependence, an excess of Cas12a (960 nM) was incubated with a titration of crRNA (0 pM, 2.4 pM, 9.7 pM, 39 pM, 0.156 nM, 0.625 nM, 2.5 nM, 10 nM, 40 nM, 160 nM, 640 nM) to pre-form the RNP for 30 min at 37°C before the addition of 32P-dsDNA (0.1 nM) with or without 1 µM apyrase (NEB) or 2 µM ATP (NEB) and further incubation for 30 min at 37°C. AcrVA4 (10 uM) was then introduced for a final incubation at 37°C for 30 min. For all EMSAs, the resulting complexes were resolved by 6% (v/v) native PAGE (0.5X TBE supplemented with 5 mM MgCl2), visualized by phosphoroimaging (Amersham Typhoon, GE Healthcare), and quantified with ImageQuant (GE Healthcare). The fraction bound was determined as the ratio of the bound band intensity relative to the total intensity of both the unbound and bound intensity normalized to background and fit to a binding isotherm (Prism7, GraphPad) to calculate the dissociation constants (n = 3 independent experiments). Affinities and their associated standard deviations are reported in the figure legends.
Size-exclusion chromatography and coupled di-angle light scattering
All experiments were run in 20 mM HEPES.K (pH 7.5), 200 mM KCl, 1 mM TECP, 1 mM MgCl2) on a Superdex 10/300 Increase column (GE Healthcare) at 0.5 mL min−1 using the Infinity 1260 Bio-SEC with light scattering module (Agilent). Light scattering was collected at 15° and 90° using a 658 nm laser. The system was calibrated using a 2 mg/mL BSA and dn/dc of 0.18541. Calibration constants were determined as: 280 nm UV = 567.9, LS 90° = 39111.5 and LS 15° =29921.9. LS 15° data were not used in our calculations. Cas12a and AcrVA concentrations were determined via nanodrop before combination with nucleic acid substrates and used as manual inputs for the mass calculation and a dn/dc of 0.185. All masses were determined using a first degree fit over the linear region of mass estimates for each peak using the Bio-SEC software V A.02.01 (Agilent).
Single particle negative stain electron microscopy
Purified LbCas12a RNP bound to AcrA4 was prepared at ~50 nM and negatively stained in 2% (w/v) uranyl acetate (Electron Microscopy Sciences) solution following the standard deep-stain procedure on holey carbon-coated EM copper grids covered with a thin layer of continuous carbon. Negative stained specimens were mounted on a transmission electron microscope holder and examined by a Tecnai Spirit electron microscope operated at 120-kV acceleration voltage. Magnified digital micrographs of the specimen were taken at a nominal magnification of 51,000 on a Gatan Ultrascan4000 CCD camera with a pixel size of 2.18-Å at the specimen level by Leginon42. The defocus values ranged from −0.9 μm to −1.5 μm, and the total accumulated dose at the specimen was about 60 electrons per Å2. Image analysis was performed in Appion43.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Ye and K. Zhou for technical assistance; A. Lapinaite, B. Cress, and members of the Doudna laboratory for critical discussions. G.J.K is a recipient of an Australian American Association Fellowship from the Australian American Association. B.A.S is supported by the National Science Foundation Graduate Research Fellowship (DGE 1752814). The authors acknowledge financial support from the Defense Advanced Research Projects Agency (DARPA) (award HR0011-17-2-0043 to J.A.D.), the Paul G. Allen Frontiers Group, and the National Science Foundation (MCB-1244557 to J.A.D.). J.A.D. is an investigator of the Howard Hughes Medical Institute (HHMI), and this study was supported in part by HHMI; J.A.D is also a Paul Allen Distinguished Investigator.
Footnotes
COMPETING INTERESTS
The Regents of the University of California have patents pending for CRISPR technologies on which the authors are inventors. J.A.D. is a co-founder of Caribou Biosciences, Editas Medicine, Intellia Therapeutics, Scribe Therapeutics, and Mammoth Biosciences. J.A.D. is a scientific advisory board member of Caribous Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Synthego, Metagenomi, Mammoth Biosciences, and Inari. J.A.D. is a Director at Johnson & Johnson and has sponsored research projects by Pfizer, Roche Biopharma, and Biogen.
DATA AVAILABILITY
Source data for Figures 1b, 1d, 3a, 4b, 5a, and 5c are available with the paper online. Source data for all other biochemical experiments that support the findings of this study are available from the corresponding author upon reasonable request. The uncropped images for the main text or supplementary figures are available in Supplementary Data Set 1.
REFERENCES
- 1.Wright AV, Nuñez JK & Doudna JA Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 164, 29–44 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Brouns SJJ et al. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science (80-. ) 321, 960–964 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marraffini LA & Sontheimer EJ CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science (80-. ) 322, 1843–1845 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrangou R et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science (80-. ) 315, 1709–1712 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Garneau JE et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71 (2010). [DOI] [PubMed] [Google Scholar]
- 6.van Houte S et al. The diversity-generating benefits of a prokaryotic adaptive immune system. Nature 532, 385–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pawluk A, Davidson AR & Maxwell KL Anti-CRISPR: Discovery, mechanism and function. Nature Reviews Microbiology 16, 12–17 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Bondy-Denomy J Protein Inhibitors of CRISPR-Cas9. ACS Chem. Biol 13, 417–423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koonin EV & Makarova KS Anti-CRISPRs on the march. Science (80-. ) 362, 156–157 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Bondy-Denomy J, Pawluk A, Maxwell KL & Davidson AR Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pawluk A, Bondy-Denomy J, Cheung VHW, Maxwell KL & Davidson AR A New Group of Phage Anti-CRISPR Genes Inhibits the Type I-E CRISPR-Cas System of Pseudomonas aeruginosa. MBio 5, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landsberger M et al. Anti-CRISPR Phages Cooperate to Overcome CRISPR-Cas Immunity. Cell 174, 908–916.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borges AL et al. Bacteriophage Cooperation Suppresses CRISPR-Cas3 and Cas9 Immunity. Cell 174, 917–925.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pawluk A et al. Naturally Occurring Off-Switches for CRISPR-Cas9. Cell 167, 1829–1838.e9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong D et al. Structural basis of CRISPR–SpyCas9 inhibition by an anti-CRISPR protein. Nature 546, 436–439 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Shin J et al. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci. Adv 3, e1701620 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington LB et al. A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell 170, 1224–1233.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watters KE, Fellmann C, Bai HB, Ren SM & Doudna JA Systematic discovery of natural CRISPR-Cas12a inhibitors. Science (80-. ) eaau5138 (2018). doi: 10.1126/science.aau5138 [DOI] [PMC free article] [PubMed]
- 19.Marino ND et al. Discovery of widespread Type I and Type V CRISPR-Cas inhibitors. Science (80-. ) eaau5174 (2018). doi: 10.1126/science.aau5174 [DOI] [PMC free article] [PubMed]
- 20.Yamano T et al. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell 165, 949–962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong D et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 532, 522–526 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Swarts DC & Jinek M Cas9 versus Cas12a/Cpf1: Structure-function comparisons and implications for genome editing. Wiley Interdiscip. Rev. RNA 9, e1481 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Zetsche B et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 163, 759–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fonfara I, Richter H, Bratovič M, Le Rhun A & Charpentier E The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Savell KE & Day JJ Applications of CRISPR/CAS9 in the mammalian central nervous system. Yale J. Biol. Med 90, 567–581 (2017). [PMC free article] [PubMed] [Google Scholar]
- 26.Sternberg SH, Redding S, Jinek M, Greene EC & Doudna JA DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen JS et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science (80-. ) 360, 436–439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strohkendl I, Saifuddin FA, Rybarski JR, Finkelstein IJ & Russell R Kinetic Basis for DNA Target Specificity of CRISPR-Cas12a. Mol. Cell 71, 816–824.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swarts DC & Jinek M Mechanistic Insights into the cis- and trans-Acting DNase Activities of Cas12a. Mol. Cell (2019). doi: 10.1016/j.molcel.2018.11.021 [DOI] [PMC free article] [PubMed]
- 30.Knott GJ & Doudna JA CRISPR-Cas guides the future of genetic engineering. Science (80-. ) 361, 866–869 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swarts DC, van der Oost J & Jinek M Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stella S et al. Conformational Activation Promotes CRISPR-Cas12a Catalysis and Resetting of the Endonuclease Activity. Cell 175, 1856–1871.e21 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Kleinstiver BP et al. Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol (2019). doi: 10.1038/s41587-018-0011-0 [DOI] [PMC free article] [PubMed]
- 34.Nakamura M et al. Anti-CRISPR-mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat. Commun 10, 194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burstein D et al. New CRISPR-Cas systems from uncultivated microbes. Nature 542, 237–241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shmakov S et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 60, 385–397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh K & Standley DM MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stamatakis A RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Letunic I & Bork P Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44, W242–W245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.East-Seletsky A et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao H, Brown PH & Schuck P On the Distribution of Protein Refractive Index Increments. Biophys. J 100, 2309–2317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suloway C et al. Automated molecular microscopy: The new Leginon system. J. Struct. Biol 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Lander GC et al. Appion: An integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.