

Abstract
Background:
Human microbiota have many functions that could contribute to cancer initiation and/or progression at local sites, yet the relation of the lung microbiota to lung cancer prognosis has not been studied.
Methods:
In a pilot study, 16S rRNA gene sequencing was performed on paired lung tumor and remote normal samples from the same lobe/segment in 19 non-small cell lung cancer patients. We explored associations of tumor or normal tissue microbiome diversity and composition with recurrence-free and disease-free survival, and compared microbiome diversity and composition between paired tumor and normal samples.
Results:
Higher richness and diversity in normal tissue were associated with reduced recurrence-free survival (richness p=0.08, Shannon index p=0.03) and disease-free survival (richness p=0.03, Shannon index p=0.02), as was normal tissue overall microbiome composition (Bray-Curtis p=0.09 for recurrence-free and p=0.02 for disease-free survival). In normal tissue, greater abundance of family Koribacteraceae was associated with increased recurrence-free and disease-free survival, while greater abundance of families Bacteroidaceae, Lachnospiraceae, and Ruminococcaceae were associated with reduced recurrence-free or disease-free survival (p<0.05). Tumor tissue diversity and overall composition were not associated with recurrence-free or disease-free survival. Tumor tissue had lower richness and diversity (p≤0.0001) than paired normal tissue, though overall microbiome composition did not differ between paired samples.
Conclusions:
We demonstrate, for the first time, a potential relationship between the normal lung microbiota and lung cancer prognosis, which requires confirmation in a larger study.
Impact:
Definition of bacterial biomarkers of prognosis may lead to improved survival outcomes for lung cancer patients.
Keywords: non-small cell, microbiome, recurrence, disease-free survival
INTRODUCTION
Lung cancer is the most common cancer, excluding non-melanoma skin cancer, and the most common cause of cancer death in the world, with approximately 1.8 million diagnoses and 1.6 million deaths per year (1). While incidence rates for lung cancer have been declining in the U.S. due to reductions in smoking, challenges in early detection have left lung cancer as the leading cause of U.S. cancer death (5-year survival rate 18% on average in the U.S.) (2). Non-small cell lung cancer (NSCLC), the most common form of lung cancer, is typically treated at the early stages with surgical resection, with or without chemotherapy or chemoradiotherapy (3); these early stage cancers have better 5-year survival rates (50–90%), however a substantial proportion of patients still die of disease recurrence (4). Improvements in early detection with low-dose computed tomography (CT) (5) will inevitably increase the identification of early stage lung cancers and offer more opportunities for curative resection, making it extremely timely to investigate factors contributing to long-term disease-free survival following resection. Better identification of early stage patients at high risk of recurrence can improve survival by indicating which patients may benefit from increased surveillance and/or adjuvant therapy.
The healthy human lung is host to a unique and dynamic bacterial community, determined by bi-directional movement of non-sterile air and mucus in and out of the airways (6). In lung disease, regional changes in the lung environment create permissive niches for bacterial growth, resulting in significant differences in community composition between healthy and diseased lungs (7). Studies have explored the oral or airway microbiome in lung cancer cases compared with controls (8–12), noting lower microbial diversity and altered abundance of specific bacterial groups in cases. However, few studies have characterized the microbiome in lung tumor tissue (13,14), and no studies have explored the relationship between the microbiome of resected lung tissue and lung cancer prognosis. Bacteria have many functions that could contribute to cancer initiation and/or progression at local sites, including genotoxic pathways, bacterial metabolite signaling, and induction of host inflammatory pathways (15). Investigation of potential bacterial involvement in lung cancer prognosis may lead to new biomarkers and therapies to improve survival outcomes for lung cancer patients.
We conducted a pilot study of paired tumor and remote normal lung tissue samples from 19 NSCLC patients at NYU Langone Health, 17 of them with prospective follow up. Using 16S rRNA gene sequencing, we explored whether the tumor or normal lung microbiome was associated with recurrence-free and disease-free survival, and compared the lung microbiome of paired tumor and normal samples.
MATERIALS AND METHODS
Patients and sample collection.
Samples were selected from the NYU Thoracic Surgery Archives (NTSA). Established in 2006, the NTSA has prospectively collected serum, plasma, buffy coat, peripheral blood mononuclear cells, along with lung cancer and matching normal lung specimens under the IRB approved 8896 protocol. Patients identified on pre-operative workup as having a pulmonary nodule suspicious for lung cancer were consented for collection of blood and snap frozen tissues (tumor and remote lung from the same lobe/segment) in the operating room at the time of their resection. Lung and matching tumor are sterilely cut at the operating room table, transferred to pre-labeled nunc vials and immediately snap frozen in liquid nitrogen within 10 minutes of resection. Samples are de-identified for storage at −80°C until use. Since these specimens remain sterile and are immediately frozen, they are ideal for microbiome analysis, as immediate freezing does not impact microbiome composition (16). Less is known regarding long-term (i.e. years) storage at −80°C, which may impact certain aspects of the microbiome (17,18); however, length of storage time in our samples was not associated with overall microbiome diversity and composition (α- and β-diversity).
Clinical and pathologic demographics are recorded in an encrypted Research Electronic Data Capture (REDCap) spreadsheet. Patients are seen at 3 month intervals for 2 years, and then at 6 month intervals for 1 year, and then annually, with CT scans performed for surveillance in order to document any systemic and loco-regional recurrences, or the development of a second primary tumor. The 19 patients’ samples included in this study were originally chosen as pilot samples to test whether sufficient material was available for DNA extraction from tumor and matching normal lung. The samples were also chosen to represent patients with different stages of NSCLC and patients with recurrence, to explore the lung microbiome in relation to these factors.
Definitions.
Endpoints were defined according to the consensus agreement in Punt et al. (19). Disease-free survival (DFS) includes recurrences (loco-regional and systemic), new primaries (same or other cancer), and death from any cause as events. Recurrence-free survival (RFS) includes recurrences (loco-regional and systemic) and death from any cause as events, ignoring new primaries as events. For both endpoints, person time is defined as time from surgery to event or loss to follow-up (censored).
Microbiome assay.
Lung tissue samples underwent 16S rRNA gene sequencing at the Environmental Sample Preparation and Sequencing Facility at Argonne National Laboratory. DNA extraction and amplification steps occurred in two batches (batch 1: 10 samples and batch 2: 28 samples; tumor-normal pairs from same patient kept together), but all samples were sequenced in the same batch. DNA was extracted from tissue using the Mo Bio PowerSoil DNA isolation kit, following the manufacturer’s protocol. This protocol uses mechanical bead beating and chemical methods to achieve sample homogenization and cell lysis, ensuring that sample features do not interfere with DNA extraction. The V4 region of the 16S rRNA gene was PCR amplified with the 515F/806R primer pair, which included sequencer adapter sequences used in the Illumina flowcell and sample-specific barcodes (20,21). Each 25 µL PCR reaction contained 9.5 µL of Mo Bio PCR Water (Certified DNA-Free), 12.5 µL of QuantaBio’s AccuStart II PCR ToughMix (2x concentration, 1x final), 1 µL Golay barcode tagged Forward Primer (5 µM concentration, 200 pM final), 1 µL Reverse Primer (5 µM concentration, 200 pM final), and 1 µL of template DNA. The conditions for PCR were as follows: 94 °C for 3 minutes to denature the DNA, with 35 cycles at 94 °C for 45 s, 50 °C for 60 s, and 72 °C for 90 s; with a final extension of 10 min at 72 °C. PCR products were quantified using PicoGreen (Invitrogen) and a plate reader (Infinite 200 PRO, Tecan). Each batch included 2 extraction blanks and 10 amplification blanks, all of which did not amplify. Additionally, amplification levels for samples were in the same range for both batches. Sample PCR products were then pooled in equimolar amounts, purified using AMPure XP Beads (Beckman Coulter), and then quantified using a fluorometer (Qubit, Invitrogen). Molarity was then diluted to 2 nM, denatured, and then diluted to a final concentration of 6.75 pM with a 10% PhiX spike for sequencing on the Illumina MiSeq. Amplicons were sequenced on a 151bp x 12bp x 151bp MiSeq run (21).
Sequence read processing.
Sequence reads were processed using QIIME 2 (22). Briefly, sequence reads were demultiplexed and paired-end reads were joined, followed by quality filtering as described in Bokulich et al. (23). Next the Deblur workflow was applied, which uses sequence error profiles to obtain putative error-free sequences, referred to as “sub” operational taxonomic units (s-OTU) (24). s-OTUs were assigned taxonomy using a naïve Bayes classifier pre-trained on the Greengenes (25) 13_8 99% OTUs, where the sequences have been trimmed to only include 250 bases from the 16S V4 region, bound by the 515F/806R primer pair. A phylogenetic tree was constructed via sequence alignment with MAFFT (26), filtering the alignment, and applying FastTree (27) to generate the tree. One tumor sample without detectable s-OTUs was dropped, leaving 37 samples (19 normal, 18 tumor) from 19 patients for final analysis. The number of sequence reads per sample prior to the Deblur workflow was similar in tumor compared to normal tissue samples (Wilcoxon signed-rank p = 0.61), and marginally higher in batch 1 compared to the batch 2 (Wilcoxon rank-sum p = 0.08) (Supplementary Figure 1). Due to amplification of human mitochondrial DNA in these tissue samples, the majority of sequence reads belonged to human mitochondria and were dropped when not matching to the bacterial 16S database during Deblur. The number of sequence reads per sample after the Deblur workflow was marginally lower in tumor compared to normal tissue samples (Wilcoxon signed-rank p = 0.09), and higher in batch 1 compared to batch 2 (Wilcoxon rank-sum p = 0.001) (Supplementary Table 1, Supplementary Figure 1).
α-diversity.
α-diversity (within-sample microbiome diversity) was assessed using richness (number of s-OTUs) and the Shannon diversity index, calculated in 100 iterations for rarefied s-OTU tables (63 sequence reads per sample [lowest sequencing depth among samples]) using the QIIME 2 diversity plugin. Rarefaction curves suggested that this depth reflected the general ranking of community richness and diversity of the samples (Supplementary Figure 2). We used Cox proportional hazards models to determine whether α-diversity was associated with recurrence-free and disease-free survival. We examined whether α-diversity differed between paired tumor and normal samples using the Wilcoxon signed-rank test.
β-diversity.
β-diversity (between-sample microbiome diversity) was assessed using unweighted and weighted UniFrac distances (28), the Bray-Curtis dissimilarity, and the Jaccard index. Principal coordinate analysis (PCoA) (29) was used for visualization. The community-level test of association between the microbiota and survival times (MiRKAT-S) (30) and the optimal microbiome-based survival analysis test (OMiSA) (31) were used to test the association of overall bacterial composition with recurrence-free and disease-free survival. We also assigned samples to clusters by applying Ward’s Hierarchical Agglomerative Clustering method (32) to the distance matrices, and then tested whether these clusters were related recurrence-free and disease-free survival using log-rank tests. Permutational multivariate analysis of variance (PERMANOVA) (33) was used to examine statistically whether overall bacterial composition differed between paired tumor and normal samples, using patient ID as strata. We also compared between-pair distances in overall bacterial composition for tumor and normal tissue sample pairs with distances for all possible pairings of tumor and normal samples from different subjects (i.e. true pairs vs. not true pairs) using the Wilcoxon rank-sum test, to determine whether true sample pairs are more similar to each other than random pairings. These analyses were performed with and without rarefying s-OTU tables to an even depth (63 sequence reads per sample), as β-diversity can be sensitive to sequencing depth (34).
Differential abundance.
Relative abundance of s-OTUs (total sum scaling) was calculated, and s-OTUs were agglomerated to phylum, class, order, family, genus, and species levels. We filtered taxa to include in analysis only those present in 25% of the samples. We used Cox proportional hazards models to assess whether taxa centered log ratio (clr) transformed (35,36) abundance or carriage was associated with recurrence-free and disease-free survival. We used the Wilcoxon signed-rank test and McNemar’s test to assess differences in taxon relative abundance and carriage, respectively, between paired tumor and normal samples. P-values were adjusted for the false discovery rate (FDR).
Sensitivity analyses.
We checked whether results for overall α-diversity and β-diversity were consistent when restricting to adenocarcinoma cases only, restricting to patients in the larger extraction batch (batch 2), excluding stage 3–4 cases, excluding current smokers, and excluding samples with low sequencing depths (≤124 reads/sample). We did not perform analyses within other histology groups or the smaller extraction batch due to small sample size (n=4 squamous cell carcinoma, n=1 sarcomatoid carcinoma, n=5 in batch 1).
RESULTS
Patient characteristics.
Demographic and clinical characteristics of the 19 patients are presented in Table 1. The average patient age was 71.6 years old, and 37% were male, 100% were white, and 95% formerly or currently smoked. The majority of patients had lung adenocarcinomas (74%), while a minority had other histologic types (squamous cell carcinoma 21%; sarcomatoid carcinoma 5%). Two patients with no follow up due to postoperative death were excluded from survival analysis; of the remaining 17 patients, 3 had new primaries and 9 had recurrences (loco-regional or systemic) during follow up (follow up times ranged from 1–12 years).
Table 1.
Characteristics of 19 lung cancer patients.
| Characteristic | All (n=19) |
Disease-free (n=5) |
Recurrence (n=9) |
New primary (n=3) |
|---|---|---|---|---|
| Age, mean ± SD | 71.6 ± 6.7 | 73.6 ± 6.3 | 73.6 ± 6.3 | 64.3 ± 6.3 |
| Male, n (%) | 7 (36.8) | 3 (60.0) | 2 (22.2) | 0 (0) |
| White, n (%) | 19 (100.0) | 5 (100.0) | 9 (100.0) | 3 (100.0) |
| Smoking status, n (%) | ||||
| Never | 1 (5.3) | 1 (20.0) | 0 (0) | 0 (0) |
| Former | 16 (84.2) | 4 (80.0) | 8 (88.9) | 2 (66.7) |
| Current | 2 (10.5) | 0 (0) | 1 (11.1) | 1 (33.3) |
| Histology, n (%) | ||||
| Adenocarcinoma | 14 (73.7) | 4 (80.0) | 9 (100.0) | 1 (33.3) |
| Squamous cell carcinoma | 4 (21.1) | 0 (0) | 0 (0) | 2 (66.7) |
| Sarcomatoid carcinoma | 1 (5.3) | 1 (20.0) | 0 (0) | 0 (0) |
| Stage, n (%) | ||||
| I | 10 (52.6) | 3 (60.0) | 4 (44.4) | 2 (66.7) |
| II | 5 (26.3) | 2 (40.0) | 2 (22.2) | 1 (33.3) |
| III | 2 (10.5) | 0 (0) | 1 (11.1) | 0 (0) |
| IV | 2 (10.5) | 0 (0) | 2 (22.2) | 0 (0) |
Normal lung tissue microbiome diversity and composition is associated with recurrence-free and disease-free survival.
Patients with recurrence or a new primary during follow up had greater bacterial richness (p = 0.01) and diversity (p = 0.06), in their normal lung tissue, than disease-free patients, at the evenly rarefied depth of 63 sequences per sample (Figure 1; Supplementary Table 2). Consistently, higher richness and diversity in normal tissue were significantly associated with reduced recurrence-free and disease-free survival in Cox proportional hazards models (RFS: p = 0.08 for richness, p = 0.03 for Shannon index; DFS: p = 0.03 for richness, p = 0.02 for Shannon index; Supplementary Table 2). Results remained largely consistent in the sensitivity analyses (Supplementary Table 3).
Figure 1. α-diversity in normal lung tissue and survival.
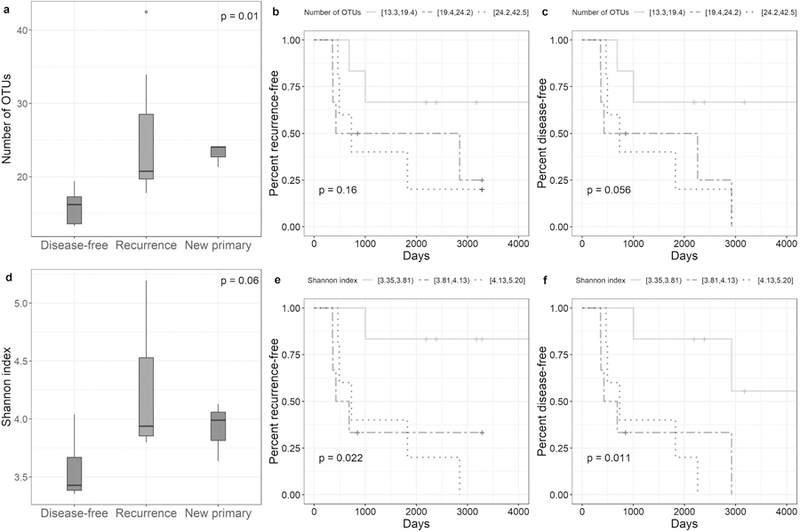
(a) Distribution of number of OTUs at an even depth of 63 sequence reads per sample in normal lung tissue by recurrence status of patients (p-values are from Kruskal-Wallis tests). (b-c) Recurrence-free and disease-free survival curves for patients grouped in tertiles of number of OTUs at an even depth of 63 sequence reads per sample in normal lung tissue (p-values are from log-rank tests for trend). (d) Distribution of the Shannon index at an even depth of 63 sequence reads per sample in normal lung tissue by recurrence status of patients (p-values are from Kruskal-Wallis tests). (e-f) Recurrence-free and disease-free survival curves for patients grouped in tertiles of the Shannon index at an even depth of 63 sequence reads per sample in normal lung tissue (p-values are from log-rank tests for trend).
Overall microbiome composition in normal lung tissue was associated with recurrence-free and disease-free survival according to several distance measures with the MiRKAT-S test (RFS p≤0.09 and DFS p≤0.04 for unweighted and weighted UniFrac distances, Bray-Curtis dissimilarity, and Jaccard index; Supplementary Table 4), though not with the OMiSA test (RFS p = 0.20, DFS p = 0.12). Results were similar in the sensitivity analyses (Supplementary Table 3). Results were also similar when rarefying to an even depth for the UniFrac distances, but somewhat attenuated for the Bray-Curtis dissimilarity and Jaccard index (Supplementary Table 4). Principal coordinate analysis of the Bray-Curtis dissimilarity from normal tissue revealed clustering of patients by recurrence status (Figure 2a–b); results were similar for the unweighted and weighted UniFrac distances and the Jaccard index (Supplementary Figure 3). We grouped patients into four discrete clusters based on the Bray-Curtis dissimilarity in normal tissue (Figure 2c), and observed that these clusters were significantly related to recurrence-free and disease-free survival as well (RFS p = 0.03, DFS p = 0.015; Figure 2d–e).
Figure 2. β-diversity in normal lung tissue and survival.
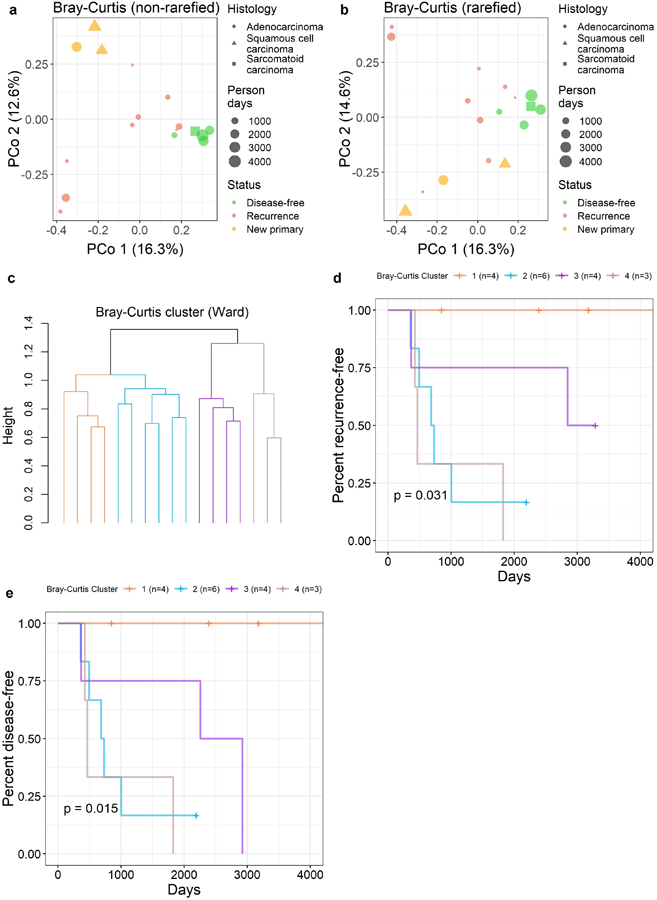
Principal coordinate analysis of the Bray-Curtis dissimilarity, with samples annotated according to recurrence status, histology, and person days: (a) non-rarefied, (b) rarefied to an even depth of 63 sequence reads per sample. (c) Unsupervised clustering (ward.D2 method) of the Bray-Curtis dissimilarity grouped patients into four clusters. These clusters were significantly related to (d) recurrence-free survival (log-rank p = 0.031) and (e) disease-free survival (log-rank p = 0.015).
We observed several taxa in normal tissue for which relative abundance and/or carriage were associated with both recurrence-free and disease-free survival in Cox proportional hazards models at p<0.05 (Supplementary Table 5; Figure 3); these taxa were not significant after FDR adjustment. Greater abundance of family Koribacteraceae in normal tissue was associated with increased recurrence-free and disease-free survival, while greater abundance of family Lachnospiraceae, and genera Faecalibacterium and Ruminococcus (from Ruminococcaceae family), and Roseburia and [Ruminococcus] (from Lachnospiraceae family) were associated with reduced recurrence-free and disease-free survival. Taxa associated only with recurrence-free survival (p<0.05) included family S24–7 (increased recurrence-free survival), and family Bacteroidaceae and genus Bacteroides (reduced recurrence-free survival). Taxa associated only with disease-free survival (p<0.05) included family Sphingomonadaceae and genus Sphingomonas (increased disease-free survival), and family Ruminococcaceae (reduced disease-free survival). A heatmap of these 12 taxa in normal tissue clustered patients somewhat by recurrence status (Figure 3).
Figure 3. Taxa in normal lung tissue associated with recurrence-free or disease-free survival.
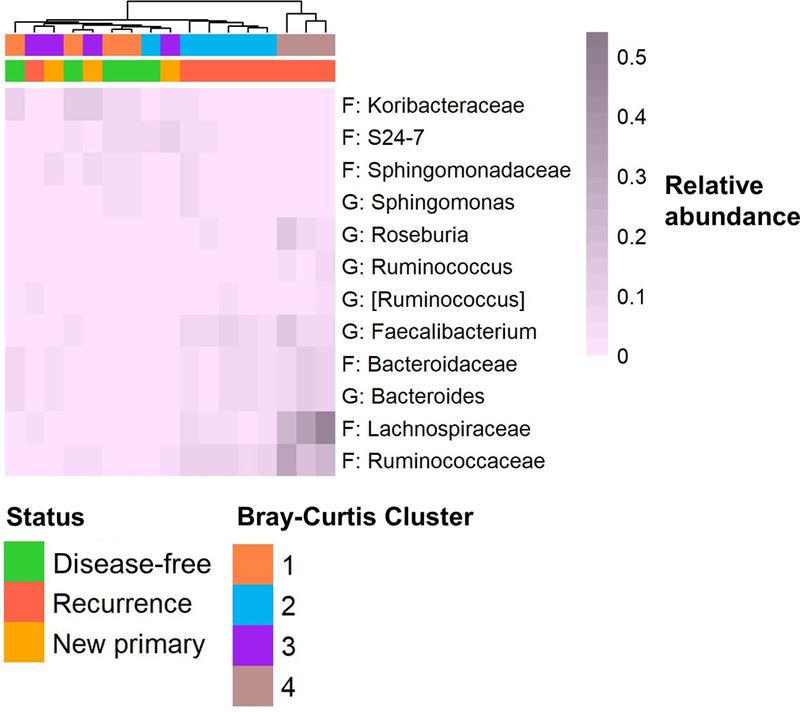
Heatmap shows relative abundance of families and genera (F: family, G: genus) with p≤0.05 from Cox proportional hazards models of clr-transformed abundance or carriage (Supplementary Table 4). Heatmap was generated with average linkage clustering, and the Manhattan distance method; samples are annotated with recurrence status and Bray-Curtis cluster (from Figure 2).
Lung tumor tissue microbiome is not associated with survival.
Tumor tissue richness and diversity were not associated with recurrence status or with recurrence-free and disease-free survival (Supplementary Figure 4; Supplementary Table 2), and this was consistent in the sensitivity analyses (Supplementary Table 3). Additionally, tumor overall microbiome composition was not associated with recurrence-free or disease-free survival (Supplementary Table 4; Supplementary Figure 5), and this was consistent when rarefying to an even depth and in the sensitivity analyses (Supplementary Table 3; Supplementary Table 4). In tumor tissue, only families Koribacteraceae and Lachnospiraceae were associated with reduced recurrence-free and disease-free survival (p<0.05; Supplementary Table 5).
Lung tumor tissue microbiome is less diverse than, but compositionally similar to, paired normal tissue microbiome.
Tumor tissue samples had significantly lower bacterial richness (observed OTUs; p = 0.0001) and diversity (Shannon index; p < 0.0001) than paired normal tissue samples at the evenly rarefied depth of 63 sequences per sample (Figure 4; Supplementary Table 6). Significance remained at higher sequencing depths despite dropped samples with lower depths (15 tumor/normal pairs at 124 sequence reads per sample: p = 0.02 for number of OTUs, p < 0.0001 for Shannon index). Results were consistent when restricting to adenocarcinoma histology, restricting to patients in batch 2, excluding stage 3–4 cases, or excluding current smokers (p<0.05).
Figure 4. α-diversity in relation to lung tissue type (tumor vs. normal).
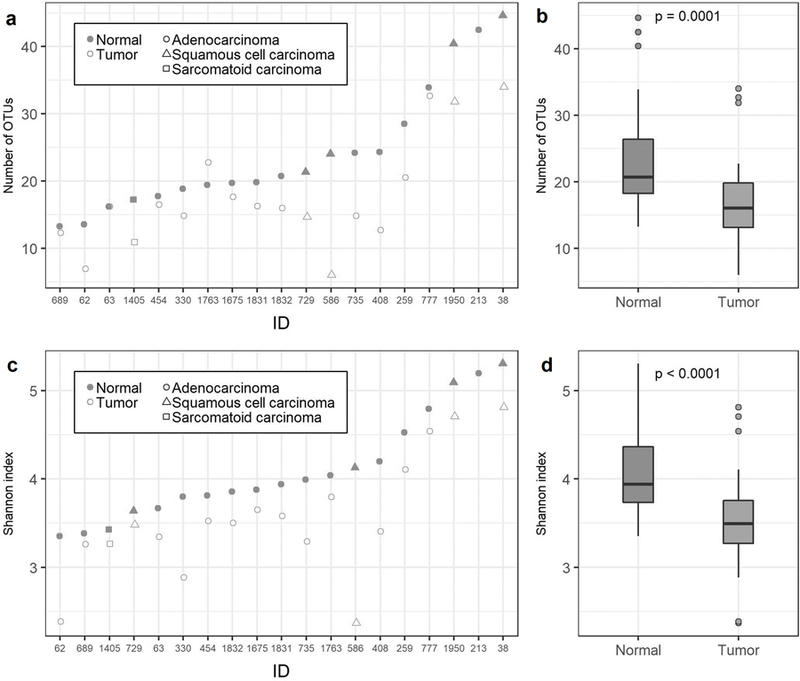
(a) Number of OTUs for tumor/normal pairs by patient ID at an even depth of 63 sequence reads per sample. (b) Distribution of number of OTUs at 63 sequence reads per sample for normal and tumor samples (P-values are from Wilcoxon signed-rank test). (c) Shannon index for tumor/normal pairs by patient ID at an even depth of 63 sequence reads per sample. (d) Distribution of the Shannon index at 63 sequence reads per sample for normal and tumor samples (P-values are from Wilcoxon signed-rank test).
Overall microbiome composition did not differ significantly between paired lung tumor and normal samples according to unweighted and weighted UniFrac distance, Bray-Curtis dissimilarity, or the Jaccard index (Supplementary Figure 6; Supplementary Table 6). Results were consistent when rarefying to an even depth and when restricting to adenocarcinoma histology, restricting to patients in batch 2, excluding stage 3–4 cancers, excluding current smokers, or excluding samples with low sequencing depth. Moreover, paired tumor and normal samples were significantly more alike than random pairings of tumor and normal samples from different patients, according to the unweighted and weighted UniFrac distance, Bray-Curtis dissimilarity, and Jaccard index (all p≤0.02). Lung tumor samples had higher abundance of family Veillonellaceae, lower abundance of genus Cloacibacterium, and lower carriage of family Erysipelotrichaceae, than paired normal samples (p<0.05; Figure 5; Supplementary Table 7); these taxa were not significant after FDR adjustment.
Figure 5. Taxa associated with lung tissue type (tumor vs. normal).
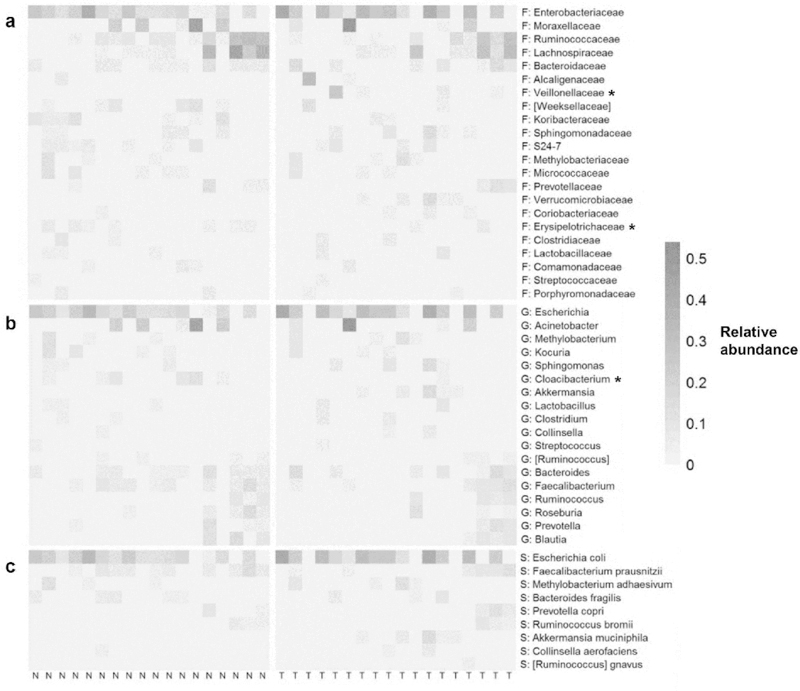
Heatmap shows relative abundance of (a) families, (b) genera, and (c) species in paired normal (N) and tumor (T) samples (only taxa present in >25% of samples are shown). Normal and tumor samples are sorted left to right by patient ID. Taxa with * indicate p<0.05 from Wilcoxon signed-rank test for pair difference in relative abundance, or McNemar’s test for pair difference in carriage (Supplementary Table 6).
DISCUSSION
In this pilot study of the lung microbiome and lung cancer prognosis, we showed, for the first time, that increased diversity and altered composition of the normal lung tissue was associated with reduced disease-free and recurrence-free survival. This important novel observation suggests that the microbiome of normal lung tissue may be used as a biomarker of lung cancer prognosis, which could guide clinical practice to improve survival outcomes for lung cancer patients. We also observed a clear reduction in bacterial richness and diversity in lung tumor samples compared to paired normal tissue samples, indicating dysbiosis of the lung tumor microbiome.
Few studies have reported on the microbiome in lung cancer, and even fewer characterized the microbiome in actual lung tumor tissue. We have reported that lower airway brushes of lung cancer patients (n=39) were enriched in Veillonella and Streptococcus compared to benign lung disease patients (n=36) and healthy controls (n=10) (9). A study of lung cancer attributed to household coal burning in China found that sputum samples of lung cancer cases (n=8) had lower diversity and enrichment of Granulicatella, Abiotrophia, and Streptococcus compared with healthy controls (n=8) (10). Similarly, another study from China reported decreased diversity and increased Streptococcus abundance in bronchial brush specimens from cancerous lung sites compared to paired non-cancerous lung sites (n=24) and healthy controls (n=18) (8). A third report from China found family Veillonellaceae and genera Veillonella, Capnocytophaga, and Selenomonas were more abundant in saliva of lung cancer patients (n=20) compared to controls (n=10) (12). A study in Korea observed that Veillonella and Megasphaera were more abundant in bronchoalveolar lavage fluid from lung cancer patients (n=20) compared to patients with benign lung mass-like lesions (n=8) (11). A study of Italian lung cancer patients found lower bacterial diversity in lung tumor tissue samples (n=31) compared to non-malignant lung tissue (n=165), and no differences in overall composition (β-diversity) between the tumor and non-malignant samples (13). Finally, a recent study of lung tissue samples from lung cancer patients (tumor and adjacent normal) and hospital controls observed increased bacterial diversity in tumor and adjacent normal tissue from lung cancer patients compared with the controls (14).
From this previous literature, it is apparent that the airway and lung microbiome is perturbed in lung cancer patients, which may have implications for prognosis. We observed that greater bacterial diversity and greater abundance of families Bacteroidaceae, Lachnospiraceae, and Ruminococcaceae, and genera Bacteroides, Faecalibacterium, Roseburia, [Ruminococcus], and Ruminococcus in normal lung tissue were associated with reduced survival, while greater abundance of Koribacteraceae and Sphingomonadaceae were associated with increased survival. Interestingly, the majority of our findings were similar for the recurrence-free and disease-free survival outcomes; this may suggest that the normal lung microbiome is related to both recurrences and new primary cancers. Members of Lachnospiraceae and Ruminococcaceae, particularly Roseburia and Faecalibacterium, are known to produce anti-inflammatory short-chain fatty acids (e.g. butyrate) (37), making the association of these bacteria with reduced survival unexpected. Bacteroides abundance in the gut has been associated with impaired antitumor immune responses in melanoma patients (38), and may play a similar cancer-promoting role in the lungs. Though our conclusions are limited by small sample size, these valuable preliminary results suggest that bacteria in resected normal lung tissue may serve as biomarkers of recurrence risk in early stage NSCLC. Moreover, if these identified microbiota are determined to be causally related to cancer recurrence in future investigations, they may serve as novel targets for therapeutic intervention (7) to improve recurrence-free survival in lung cancer patients.
The results of our analysis comparing paired tumor and normal samples are similar to the previous literature in that we observed significant reductions in bacterial diversity and enrichment of Veillonellaceae in lung tumor compared to normal lung tissue, which has been observed by many (8–13), but not all (14), studies comparing lung cancer cases to controls. It is not clear from our observational study whether the identified bacterial differences are causally related to lung carcinogenesis, or are merely reflective of disease processes in the lung. However, there are several mechanisms by which the lung microbiota could contribute to lung carcinogenesis, including genotoxic pathways, bacterial metabolite effects, and induction of host inflammatory pathways (15). For example, intranasal administration of lipopolysaccharide (a membrane component of Gram-negative bacteria) in a mouse model of lung cancer significantly enhanced pulmonary inflammation and lung tumorigenesis (39). We previously showed in a human study that airway Veillonella and Streptococcus were associated with upregulation of ERK and PI3K signaling pathways in the airway, pathways regulating cell proliferation, survival, and differentiation which are upregulated in lung cancer patients (9). Interestingly, we have previously reported that these two genera are enriched in the mouths of current smokers compared to never smokers (40), suggesting a further mechanism by which smoking causes lung cancer. Taken together, there is accumulating support for specific bacteria as biomarkers of lung cancer presence; further study of the causal role of these bacteria in lung carcinogenesis may provide therapeutic targets for lung cancer prevention.
In summary, we showed in a small pilot study that diversity and composition of the normal lung tissue microbiome may be associated with recurrence-free and disease-free survival, and observed differential microbiome signatures between lung tumor and normal tissue that were consistent with previous research. The strengths of our study include the availability of fresh-frozen tumor and normal lung tissue for paired analysis, and prospective long-term follow-up for survival analysis. However, our study conclusions were limited by small sample size and lack of a replication dataset, and therefore findings will require confirmation in a larger study. Additionally, though the 16S rRNA gene sequencing assay provides a snapshot of what bacteria are present in the normal and lung tumor samples, localization of bacteria in these tissue samples (e.g. using fluorescence in situ hybridization (41)) could provide additional insight into bacterial mechanisms of action in lung cancer. Continued study of the role of the lung microbiome in lung cancer may yield several promising future applications, including biomarkers of lung cancer risk, recurrence, and prognosis, and therapeutic targets for lung cancer primary and tertiary prevention.
Supplementary Material
ACKNOWLEDGEMENTS
Lung tissue samples underwent 16S rRNA gene sequencing at the Environmental Sample Preparation and Sequencing Facility at Argonne National Laboratory.
Funding sources:
This work was supported by the National Institutes of Health (NIH) National Cancer Institute (NCI) Early Detection Research Network grant 5U01CA111295–07 to HIP, and NCI grant R01CA164964 to JA. The funders had no involvement in the study design, the collection, analysis, and interpretation of data, the writing of this report, and the decision to submit for publication.
Abbreviations:
- CT
computed tomography
- DFS
disease-free survival
- FDR
false discovery rate
- NSCLC
non-small cell lung cancer
- NTSA
NYU Thoracic Surgery Archives
- OTU
operational taxonomic unit
- PCoA
principal coordinate analysis
- PERMANOVA
permutation multivariate analysis of variance
- OMiSA
optimal microbiome-based survival analysis test
- RFS
recurrence-free survival
- s-OTU
sub-operational taxonomic unit
Footnotes
Conflicts of interest:
All authors declare no potential conflicts of interest.
REFERENCES
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer Journal international du cancer 2015;136(5):E359–86 doi 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA: a cancer journal for clinicians 2017;67(1):7–30 doi 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 3.American Cancer Society. Cancer Facts & Figures 2017 Atlanta, GA: American Cancer Society; 2017. [Google Scholar]
- 4.Detterbeck FC, Chansky K, Groome P, Bolejack V, Crowley J, Shemanski L, et al. The IASLC Lung Cancer Staging Project: Methodology and Validation Used in the Development of Proposals for Revision of the Stage Classification of NSCLC in the Forthcoming (Eighth) Edition of the TNM Classification of Lung Cancer. Journal of Thoracic Oncology 2016;11(9):1433–46 doi 10.1016/j.jtho.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 5.Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, Fagerstrom RM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. The New England journal of medicine 2011;365(5):395–409 doi 10.1056/NEJMoa1102873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dickson RP, Huffnagle GB. The Lung Microbiome: New Principles for Respiratory Bacteriology in Health and Disease. PLoS pathogens 2015;11(7):e1004923 doi 10.1371/journal.ppat.1004923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickson RP, Erb-Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert review of respiratory medicine 2013;7(3):245–57 doi 10.1586/ers.13.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu HX, Tao LL, Zhang J, Zhu YG, Zheng Y, Liu D, et al. Difference of lower airway microbiome in bilateral protected specimen brush between lung cancer patients with unilateral lobar masses and control subjects. International journal of cancer Journal international du cancer 2018;142(4):769–78 doi 10.1002/ijc.31098. [DOI] [PubMed] [Google Scholar]
- 9.Tsay JJ, Wu BG, Badri MH, Clemente JC, Shen N, Meyn P, et al. Airway Microbiota Is Associated with Up-Regulation of the PI3K Pathway in Lung Cancer. American journal of respiratory and critical care medicine 2018. doi 10.1164/rccm.201710-2118OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hosgood HD 3rd, Sapkota AR, Rothman N, Rohan T, Hu W, Xu J, et al. The potential role of lung microbiota in lung cancer attributed to household coal burning exposures. Environmental and molecular mutagenesis 2014;55(8):643–51 doi 10.1002/em.21878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee SH, Sung JY, Yong D, Chun J, Kim SY, Song JH, et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung cancer (Amsterdam, Netherlands) 2016;102:89–95 doi 10.1016/j.lungcan.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 12.Yan X, Yang M, Liu J, Gao R, Hu J, Li J, et al. Discovery and validation of potential bacterial biomarkers for lung cancer. American journal of cancer research 2015;5(10):3111–22. [PMC free article] [PubMed] [Google Scholar]
- 13.Yu G, Gail MH, Consonni D, Carugno M, Humphrys M, Pesatori AC, et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome biology 2016;17(1):163 doi 10.1186/s13059-016-1021-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greathouse KL, White JR, Vargas AJ, Bliskovsky VV, Beck JA, von Muhlinen N, et al. Interaction between the microbiome and TP53 in human lung cancer. Genome biology 2018;19:123 doi 10.1186/s13059-018-1501-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao Q, Jiang F, Yin R, Wang J, Xia W, Dong G, et al. Interplay between the lung microbiome and lung cancer. Cancer letters 2018;415:40–8 doi 10.1016/j.canlet.2017.11.036. [DOI] [PubMed] [Google Scholar]
- 16.Fouhy F, Deane J, Rea MC, O’Sullivan Ó, Ross RP, O’Callaghan G, et al. The Effects of Freezing on Faecal Microbiota as Determined Using MiSeq Sequencing and Culture-Based Investigations. PloS one 2015;10(3):e0119355 doi 10.1371/journal.pone.0119355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw AG, Sim K, Powell E, Cornwell E, Cramer T, McClure ZE, et al. Latitude in sample handling and storage for infant faecal microbiota studies: the elephant in the room? Microbiome 2016;4:40 doi 10.1186/s40168-016-0186-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kia E, Wagner Mackenzie B, Middleton D, Lau A, Waite DW, Lewis G, et al. Integrity of the Human Faecal Microbiota following Long-Term Sample Storage. PloS one 2016;11(10):e0163666 doi 10.1371/journal.pone.0163666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Punt CJ, Buyse M, Kohne CH, Hohenberger P, Labianca R, Schmoll HJ, et al. Endpoints in adjuvant treatment trials: a systematic review of the literature in colon cancer and proposed definitions for future trials. Journal of the National Cancer Institute 2007;99(13):998–1003 doi 10.1093/jnci/djm024. [DOI] [PubMed] [Google Scholar]
- 20.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America 2011;108 Suppl 1:4516–22 doi 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. Isme j 2012;6(8):1621–4 doi 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 2010;7 doi 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nature methods 2013;10(1):57–9 doi 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017;2(2) doi 10.1128/mSystems.00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology 2006;72(7):5069–72 doi 72/7/5069 [pii] 10.1128/AEM.03006-05 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic acids research 2002;30 doi 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Molecular biology and evolution 2009;26(7):1641–50 doi 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. The ISME journal 2011;5(2):169–72 doi 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.GOWER JC. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 1966;53(3–4):325–38 doi 10.1093/biomet/53.3-4.325. [DOI] [Google Scholar]
- 30.Plantinga A, Zhan X, Zhao N, Chen J, Jenq RR, Wu MC. MiRKAT-S: a community-level test of association between the microbiota and survival times. Microbiome 2017;5(1):17 doi 10.1186/s40168-017-0239-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koh H, Livanos AE, Blaser MJ, Li H. A highly adaptive microbiome-based association test for survival traits. BMC genomics 2018;19(1):210 doi 10.1186/s12864-018-4599-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murtagh F, Legendre P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? Journal of Classification 2014;31(3):274–95 doi 10.1007/s00357-014-9161-z. [DOI] [Google Scholar]
- 33.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol 2001;26(1):32–46 doi DOI 10.1046/j.1442-9993.2001.01070.x. [DOI] [Google Scholar]
- 34.Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017;5(1):27 doi 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurtz ZD, Muller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput Biol 2015;11(5):e1004226 doi 10.1371/journal.pcbi.1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandes AD, Reid JNS, Macklaim JM, McMurrough TA, Edgell DR, Gloor GB. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014;2:15- doi 10.1186/2049-2618-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiology Letters 2009;294(1):1–8 doi 10.1111/j.1574-6968.2009.01514.x. [DOI] [PubMed] [Google Scholar]
- 38.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (New York, NY) 2018;359(6371):97–103 doi 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melkamu T, Qian X, Upadhyaya P, O’Sullivan MG, Kassie F. Lipopolysaccharide enhances mouse lung tumorigenesis: a model for inflammation-driven lung cancer. Veterinary pathology 2013;50(5):895–902 doi 10.1177/0300985813476061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu J, Peters BA, Dominianni C, Zhang Y, Pei Z, Yang L, et al. Cigarette smoking and the oral microbiome in a large study of American adults. Isme j 2016. doi 10.1038/ismej.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proceedings of the National Academy of Sciences of the United States of America 2014;111(51):18321–6 doi 10.1073/pnas.1406199111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.