

ABSTRACT
Enteral feeding is a key component of care in neonatal intensive care units (NICUs); however, feeding tubes harbor microbes. These microbes have the potential to cause disease, yet their source remains controversial and clinical recommendations to reduce feeding tube colonization are lacking. This study aims to improve our understanding of the bacteria in neonatal feeding tubes and to evaluate factors that may affect these bacteria. 16S rRNA gene sequencing was used to characterize the bacteria present in pharyngeal, esophageal, and gastric portions of feeding tubes, residual fluid of the tubes, and infant stool using samples from 47 infants. Similar distributions of taxa were observed in all samples, although beta diversity differed by sample type. Feeding tube samples had lower alpha diversity than stool samples, and alpha diversity increased with gestational age, day of life, and tube dwell time. In a subset of samples from 6 infants analyzed by whole metagenome sequencing, there was greater overlap in transferable antimicrobial resistance genes between tube and fecal samples in breast milk fed infants than in formula fed infants. These findings develop our understanding of neonatal feeding tube colonization, laying a foundation for research into methods for minimizing NICU patients’ exposure to antimicrobial resistant microbes.
Keywords: neonatal feeding tube, premature infants, antimicrobial resistance genes, bacteria, 16S rRNA gene sequencing, whole metagenome sequencing
The bacteria and antimicrobial resistance genes found in neonatal feeding tubes overlap with those found in infant stool.
INTRODUCTION
One of every 13 infants born in the United States is admitted to a neonatal intensive care unit (NICU) (Harrison and Goodman 2015). These patients commonly require supportive enteral feedings to maintain adequate caloric intake; however, enteral feeding systems are known to develop microbial biofilms when used in adult, pediatric, and neonatal patients (Mathus-Vliegen et al. 2006, Matlow et al. 2003; Liu et al. 2015). Neonates’ immature immune systems and vulnerability to intestinal dysbiosis put them at a high risk for infections, sepsis, meningitis, and necrotizing enterocolitis (NEC), making the bacterial contamination of their feeding tubes a serious concern (Nanthakumar et al.2000; Mehall et al. 2002a).
Enteral feeding with nasogastric or orogastric feeding tubes is common practice in most NICUs, with as many as 96% of critically-ill neonates receiving enteral feeding (Liu et al. 2015). The average length of time between feeding tube changes varies greatly by facility, ranging from less than one day in some NICUs to several weeks in others (Mehall et al. 2002a; Hurrell et al. 2009; Petersen, Greisen and Krogfelt 2016). Currently, manufacturer guidelines for maximum feeding tube dwell time are based on the rate at which the tube material breaks down in the body, not the rate at which tubes become colonized by microbes (Patel et al. 2014). However, recent interest in the human microbiome has brought greater attention to the issue of microbial feeding tube contamination.
Studies employing both culture-based and molecular microbial identification methods describe diverse biofilms growing on the interior surface of most feeding tubes used in the NICU (Mehall et al. 2002a; Hurrell et al. 2009). Scanning electron microscopy has also shown milk residue and thick biofilms present inside many components of neonatal enteral feeding systems (Hurrell et al. 2009; Gomez et al. 2016; Petersen, Greisen and Krogfelt 2016). In addition to adhering to the feeding tube wall, microbes are also flushed out of the tube and into the stomach during feedings (Hurrell et al. 2009; Gomez et al. 2016; Petersen, Greisen and Krogfelt 2016). Studies suggest that 89% of feeding tubes contaminate the liquid that passes through them and that the remnants of prior meals acquire an average of 107 colony forming units (cfu)/mL before being propelled into the stomach by the next feeding (Hurrell et al. 2009; Petersen, Greisen and Krogfelt 2016).
The bacteria delivered to the gut by contaminated feeding tubes may lead to dysbiosis and poses significant health risks to neonates. Contaminated feeding tubes are associated with feeding intolerance and reduced weight gain as well as acute conditions such as meningitis and NEC (Mehall et al. 2002a; Alkeskas et al. 2015).
Microbial contamination of feeding tubes may also have implications for NICU patients that do not require enteral feeding. Bacterial strains growing in feeding tubes have been documented as the cause of infections in other infants in the NICU, even those without feeding tubes (Mehall et al. 2002b). The potential for bacteria to spread from one patient's feeding tube to other patients is of particular concern as many of the strains isolated from feeding tubes are resistant to a wide array of antimicrobial agents (Hurrell et al. 2009). Antimicrobial resistance is a particular problem in the NICU environment because of the high rates of antimicrobial use in premature infants and the enrichment of antimicrobial resistance in exposed individuals (Gasparrini et al. 2016). In this way, feeding tubes may serve as reservoirs of pathogens that can be transmitted between patients by NICU healthcare providers (Mehall et al. 2002b; Alkeskas et al. 2015).
Several hypotheses of the origins of feeding tube microbes have been proposed. Microbes from the stomach may be introduced to feeding tubes during gastroesophageal reflux or when caregivers aspirate gastric contents into the tube prior to feeding (Hurrell et al. 2009; Petersen, Greisen and Krogfelt 2016). However, the sharing of bacterial strains between infants in the NICU suggests that these bacteria are dispersed by healthcare workers and environmental fomites (Alkeskas et al. 2015; Gomez et al. 2016). The species of bacteria found in feeding tubes are also more commonly associated with nosocomial pathogens than gastric flora (Mehall et al. 2002a). It is also possible that some contaminants originate in the breast milk and formula delivered through feeding tubes, as these sources of nutrition are generally not sterile (Gomez et al. 2016).
The goal of this study was to refine our understanding of biofilm formation on the intracorporeal sections of feeding tubes used in hospitalized neonates. The biofilms found on neonatal enteral feeding tubes are known to be primarily Staphylococcus species and Enterobacteriaceae (Hurrell et al. 2009). However, prior studies did not consider the biogeography of feeding tubes. The microbiome of the esophagus is known to differ from that of the stomach (Hunt and Yaghoobi 2017), creating the potential for the microbial composition of the biofilm found on feeding tubes to differ in the region that reaches the stomach from other sections of the feeding tube as a result of exposure to different microbiomes. The gastric portion of the feeding tube is known to carry the same strains of Enterococcus faecalis and Enterobacter hormaechei as is found in infant stool, and these strains are known to carry antimicrobial resistance genes (ARGs) (Ogrodzki et al. 2017). Differences in biofilm composition between regions of neonatal feeding tubes have not been investigated and our understanding of the relationship between feeding tube biofilm composition and gut microbiota is limited. In this study, 16S rRNA gene sequencing was used to characterize the bacterial composition of biofilms in the gastric, esophageal, and pharyngeal regions of feeding tubes removed from NICU patients, as well as to characterize the bacteria transferred to liquid passing through these same tubes. Whole metagenomic sequencing (WGS) was used to characterize the ARGs present in a subset of gastric and esophageal regions of feeding tubes, and a subset of infant feces.
MATERIALS AND METHODS
Sample collection
Sample collection was conducted at the University of California Davis Medical Center NICU during a 4-month period (August to December 2015). All protocols were approved by the University of California Davis Institutional Review Board and informed consent was obtained from the legal guardians of all study enrollees. Participation in this study did not influence patient care. All infants in the NICU with a feeding tube in place during the sample collection period were eligible for enrollment in the study.
Following their scheduled removal by NICU personnel, feeding tubes were placed in sterile sleeves and frozen at −40°C. Tubes knocked out by infants prior to scheduled removal were not included in the study. Samples of the next stool passed after tube collection were placed in sterile 1.5 mL Eppendorf tubes and frozen at −40°C. The type of nutrition (e.g. breast milk, donor breast milk, formula) given to each subject was recorded on a weekly basis during their participation in the study. Additionally, enrollee's electronic medical records were used to document feeding tube dwell time, route of feeding tube placement, gestational age of the patient, use of acid suppressors or antibiotics, and diagnoses of NEC, sepsis and spontaneous intestinal perforation. All antibiotics were administered intravenously. An additional two unused feeding tubes were collected and processed in the same manner as the used feeding tubes to act as controls for kit contamination.
DNA extraction
Samples were prepared from the residual contents of each tube and from the pharyngeal, esophageal and gastric regions of each tube (Fig. 1). To sample the tube contents, each tube was flushed with 1 mL sterile PBS followed by 0.5 mL of air, and the effluent was collected in a sterile container. The effluent was then centrifuged for 10 minutes at 10 000 rcf and the supernatant was removed until ∼0.2 mL of solution remained, which was used for the DNA extraction. To prepare each tube for DNA extraction, 3 cm segments were removed with sterile scalpels (a new scalpel for each section), split lengthwise and cut into approximately 2 mm pieces. The pharyngeal sample was the 3 cm section beginning 5 cm below the point where the tube exited the body (tube insertion depth). The esophageal sample was the 3 cm section beginning 8 cm above the tube end and ending 5 cm above the tube end. The gastric sample was the final 3 cm of the tube (Fig. 1). The same tube was used to collect effluent and tube sections. There is a possibility that flushing the tube with PBS will disseminate bacteria along the tube; however, should this dissemination occur with PBS it would likely also occur with the breast milk or formula fed to infants. Therefore, flushing the tube with PBS is not expected to change the bacterial communities detected in a manner different from what occurs with routine infant feeding. In addition, the exterior of a subset of tubes was wiped thoroughly with ethanol before collecting the tube effluent and sectioning the tube. The tubes selected for wiping were collected from infants who provided the most total feeding tubes to allow comparison between wiped and unwiped tubes.
Figure 1.
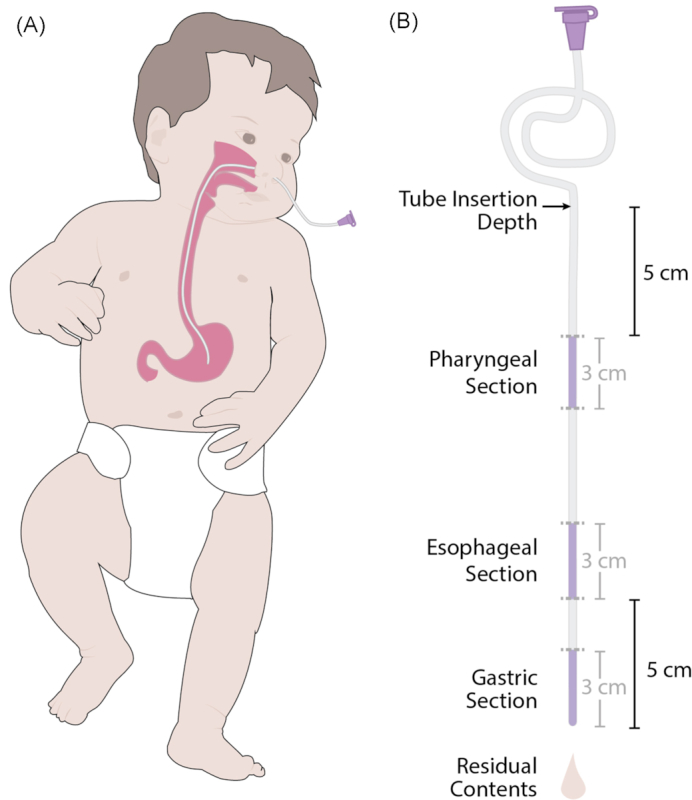
(A) Nasogastric placement of an enteral feeding tube. (B) Diagram showing the four feeding-tube related samples used in this study. The three dark purple segments indicate the three portions of the tube used for DNA sequencing, the fourth sample was the residual contents of the tube.
One hundred milligrams of each fecal sample was used for DNA extraction. If insufficient stool was collected, a sterile swab was used to gather the entire contents of the collection container, and DNA extraction was performed on the swab head.
DNA was extracted from the prepared tube and fecal samples using the Zymo Research Quick-DNA™ Fecal/Soil Microbe Miniprep Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer's instructions.
16S rRNA gene sequencing and bioinformatic processing
The V4 region of the 16S ribosomal RNA gene was amplified and sequenced as previously described (Huda et al. 2014) using barcoded PCR primers F515 (5′-NNNNNNNNGTGTGCCAGC MGCCGCGGTAA-3′) and R806 (5′-GGACTACHVGGGTWTCTAAT-3′). A water negative control was run with every set of 95 samples.
Successful amplification of samples was confirmed with gel electrophoresis. Individually barcoded samples were then pooled together and purified using the Qiagen QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). The amplicon pool was then submitted to the UC Davis Genome Center DNA Technologies Sequencing Core for paired-end library preparation and sequencing using the Illumina MiSeq DNA sequencing system (Illumina, San Diego, CA, USA) using the 250 PE settings.
Reads were demultiplexed using Sabre (Joshi 2011), then imported into QIIME2-2018.4 (Caporaso et al. 2010). Bases before base pair 22 and after base pair 210 were trimmed from the forward read. Bases before base pair 24 and after base pair 210 were trimmed from the reverse read. The trimmed reads were then processed using DADA2 (Callahan et al. 2016). Taxonomy was assigned to representative sequences using the 99% Greengenes (McDonald et al. 2011) pre-generated naïve bayes classifier in QIIME2-2018.4 (Caporaso et al. 2010). A phylogenetic tree was inferred using FastTree (Price, Dehal and Arkin 2009) and midpoint rooted in QIIME2-2018.4 (Caporaso et al. 2010). The resulting replicon sequence variant (RSV) table, phylogenetic tree, representative sequences and taxonomic assignments were exported from QIIME for use in downstream analysis in R.
Whole metagenomic sequencing and bioinformatic processing
Gastric tube, esophageal tube, and fecal samples were selected from six infants for WGS. Infants were selected for inclusion in the WGS substudy using the following criteria: (i) sufficient remaining DNA from the gastric tube section and the esophageal tube section and from the fecal sample collected immediately after tube removal for WGS, (ii) three infants were chosen based on a history of exclusive breast milk feeding and three infants were chosen based on a history of mixed breast milk and formula feeding (two infants) or exclusive formula feeding (one infant) and (iii)i nfants in the two feeding groups were frequency matched on history of sepsis (one infant in each group had a history of sepsis prior to tube removal). Only a single feeding tube and stool sample per infant was included. The 18 selected samples from 6 infants were then sent to the UC Berkeley Functional Genomics Laboratory for library preparation and Illumina HiSeq 4000 sequencing using 150 PE settings, as previously described (Taft et al. 2018).
Host DNA subtraction of resulting reads was completed using BMTagger (Rotmistrovsky and Agarwala 2011) and the GRCh38 build of the human genome. To ensure read quality, trimming was completed using Trimmomatic v. 0.36 (Bolger, Lohse and Usadel 2014) with a sliding window of 4 base pairs, a minimum average quality of 15 and a minimum length of 99 base pairs. Reads where both pairs were retained after trimming were then merged using FLASH v 1.2.11 (Magoc and Salzberg 2011). Successfully merged reads were then analyzed using AMR++ (Resistome Analyzer v. 1) and the MEGARes v 1.01 database with default settings (Lakin et al. 2017) to determine the total number of reads mapping to ARGs. ARG reads were then normalized using the method described by Li et al. (Li et al. 2015) as implemented by (Taft et al. 2018). To identify transferable ARGs, merged reads were assembled using MEGAHIT v. 1.0.6 (Li et al. 2015b) and contigs were then analyzed using ResFinder 3.0 (Zankari et al. 2012).
Statistical analysis
All statistical analysis was completed in R 3.4.3 statistical software (R Core Team 2016) and QIIME2-2018.4 (Caporaso et al. 2010). For the 16S rRNA gene sequencing analysis, the sequencing depth of samples in the RSV table was checked, and all samples within five times the sequencing depth of the highest negative control were excluded. Sequences were then rarefied to the read depth of the sample with the fewest reads using the vegan package (Dixon 2003; Oksanen et al. 2017). Correlation between feeding tube sections and fecal samples in all phyla and all families that had a median relative abundance >0% were tested using Kendall's tau. This analysis excluded samples where a matching sample was not available, e.g. a correlation between stool samples and the pharyngeal tube section will exclude the pharyngeal tube section from an infant who did not have a stool sample collected on the day or just after the feeding tube was removed. Alpha diversity (the within sample diversity, e.g. the total number of different species detected in a sample and their evenness) was analyzed using the Shannon index by Generalized Estimating Equation models using the vegan (Oksanen et al. 2017) and gee (https://CRAN.R-project.org/package=gee) R packages to test for associations between alpha diversity and clinical parameters using a backwards elimination approach. Included parameters were tube section, day of life at collection, milk type, tube in-dwelling duration, gestational age, infant given acid suppressors, infant history of sepsis, infant history of NEC, infant history of intestinal perforation and whether the infant received antibiotics at birth. Tube section was considered the key variable, and was included in all models.
Beta diversity (the between sample diversity, e.g. the number of species found in both of two different samples) was analyzed using unweighted (Lozupone and Knight 2005) UniFrac distance on the rarefied RSV table as implemented in the GUniFracs package (Chen 2012). Results were visualized using non-metric multidimensional scaling (NMDS) as implemented in vegan. Differences in beta diversity by tube section were tested using PERMANOVA (Anderson 2001) as implemented by the adonis command in vegan, with strata specified to subject ID to control for repeated measures after testing for differences in beta-dispersion by tube section.
To determine if wiping the exterior of the tubes would influence the bacteria detected, wiped tubes were matched to unwiped tubes collected from the same infant as close as possible in time. Fecal samples were excluded from the wiped/unwiped tube comparison. Only tube section and content samples that were successfully sequenced in both the wiped and the matched unwiped tube were included in the analysis. Alpha diversity as calculated with the Shannon index was compared between the wiped and unwiped tubes using a paired t-test. Beta diversity was compared between wiped and unwiped tube sections using the unweighted UniFrac distance measure and PERMANOVA, as implemented by the adonis command with strata specified to subject ID.
The resistome as measured by the normalized abundance of reads in the AMR++ results was compared between feeding tube samples and stool samples using Bray–Curtis dissimilarity and PERMANOVA, and visualized using NMDS in vegan. As a result of the small sample size for the WGS subset, only feeding tube section and feeding type were included in the PERMANOVA model. Subject was used as the strata in the model to account for repeated measure sampling. To test for differences in total normalized ARGs by tube section, Friedman's test was run using subject ID as the blocking variable. Jaccard dissimilarity was calculated between tube sections and fecal samples from the same infant to describe the degree of difference in the resistome from the different sample types.
The median number of transferable ARGs and the number of transferable ARG classes detected by ResFinder was calculated for each of the three sample types. Jaccard dissimilarity was used to calculate the degree of overlap between the different sample types for both ARGS and classes, and a Wilcoxon paired test was used to determine if the degree of overlap between the stool samples and the tube section samples was different than the degree of overlap between the tube sections. The total number of transferable ARGs and total number of transferable ARG classes detected in each tube section and the stool samples were compared using a Kruskal–Wallis test. Kruskal–Wallis tests were also used to compare the total number of ARGs in infants fed exclusively breast milk and infants fed at least some formula for each of the three sample types.
RESULTS
Study subjects and samples
A total of 51 infants were enrolled during the study period, 47 of whom had clinical information collected and feeding tubes and stool samples successfully sequenced. This resulted in a sample size of 97 feeding tubes and 92 stool samples. Clinical data for study participants is presented in Table 1. None of the infants received probiotics during the study period. A total of 35 infants received antibiotics during the study as summarized in Table S1 (Supporting Information).
Table 1.
Demographic characteristics of the 47 study infants with collected feeding tubes.
| Demographic characteristics | Mean (standard deviation) | Percentage of infants |
|---|---|---|
| Gestational age | 32.3 weeks (5.2) | |
| Birthweight | 1965 grams (1133) | |
| Feeding tube dwell time | 9.6 days (8.7) | |
| Nutrition type | Breast milk | 72% |
| Formula | 11% | |
| Mixed | 17% | |
| Disease history | Sepsis | 19% |
| NEC | 6% | |
| Spontaneous intestinal perforation | 6% | |
| Medications | Antibiotics at birth | 74% |
| Acid suppressors | 4% |
Marker gene sequencing
A total of 494 samples were sequenced (3 tube sections per tube, the residual contents of each tube and the 92 stool samples, plus 8 kit controls from the unused tubes and 6 negative controls). Post-processing with QIIME2, sample read depth ranged from 0 reads to 76 998 reads, with a gap between samples with read depth under 1200 reads and read depth over 4000 reads. Read depth in the negative controls ranged from 0 reads to 284 reads. Read depth of kit controls ranged from 3111 reads to 6414 reads. Kit controls contained reads from 17 replicon sequence variants (RSVs). Of these RSVs in the kit control samples, 6 RSVs were detected in all 8 kit control samples, 1 was detected in two kit control samples with fewer than 100 total detected reads and the remaining 10 were present in only 1 sample with fewer than 10 detected reads. All RSVs detected in the kit controls were Proteobacteria, and all but one of the RSVs were Gammaproteobacteria. The remaining RSV was Betaproteobacteria, and found in only one of the 8 kit control samples. RSVs detected in all 8 of the kit controls were excluded from additional analysis. After excluding these 6 kit contamination associated RSVs, read depth of samples ranged from 0 reads to 75 522 reads. Samples with a sequencing depth of less than 1500 reads after excluding RSVs detected in the kit control samples were excluded from additional analysis, leaving 378 samples included in the study (73 pharyngeal tube sections, 66 esophageal tube sections, 69 gastric tube sections, 81 tube contents and 89 stool samples). Samples were rarefied to 1528 reads, which was the read depth of the lowest included sample after excluding kit contamination associated RSVs. There were 34 tube samples from 4 infants included in the wiped and unwiped tube comparison.
A total of eight phyla were detected in the samples: Actinobacteria, Bacteroidetes, Cyanobacteria, Firmicutes, Fusobacteria, Proteobacteria, Tenericutes and Thermi. In addition, some sequences were identified in tube sections and stool samples did not classify as any bacterial phylum. The phyla present were generally similar between stool samples and feeding tube sections (Fig. 2). Only three phyla had a median relative abundance greater than zero: Actinobacteria (0.13% median relative abundance), Firmicutes (51% median relative abundance) and Proteobacteria (35% median relative abundance). After Bonferroni correction for multiple comparison, only the correlation of phylum Actinobacteria between stool samples and pharyngeal tube sections was non-significant. All other phyla level correlations between stool samples and tube sections and between the different tube sections were significant (Table 2). The correlation between different tube sections was generally stronger than the correlation between tube sections and stool samples. In the wiped and unwiped tube comparison subset, there were only four phyla detected: Actinobacteria, Firmicutes, Proteobacteria and Thermi. Phylum Thermi was detected at low levels (∼1% relative abundance) in only a single wiped sample. The other three phyla were detected in all samples, suggesting that the phyla present in the wiped and unwiped tubes are generally similar.
Figure 2.

Relative abundance of phyla in feeding tube sections and stool samples. Plots are aligned such that the samples from the same feeding tube and the next passed stool sample are aligned with each other. If the next passed stool sample after removal of a feeding tube occurred after another collected feeding tube was in place, that stool sample was given its own space on the plots.
Table 2.
Correlation of taxa between tube sections using Kendall's tau. Value in parentheses is the P-value for the correlation. A P-value less than 0.0025 is significant after Bonferroni correction for multiple comparison for family level comparisons, and a P-value less than 0.0042 is significant after Bonferroni correction for multiple comparisons for phyla level comparisons.
| Sample pairs | Phylum: Actinobacteria | Phylum: Firmicutes | Phylum: Proteobacteria | Family: Enterobacteriaceae | Family: Enterococcaceae | Family: Staphylococcaceae | Family: Streptococcocaceae |
|---|---|---|---|---|---|---|---|
| Stool-pharyngeal | 0.18 (P = 0.044) | 0.28 (P = 0.00087) | 0.28 (P = 0.0012) | 0.39 (P < 0.0001) | 0.46 (P < 0.0001) | 0.33 (P = 0.00014) | 0.41 (P < 0.0001) |
| Stool-esophageal | 0.37 (P = 0.00025) | 0.39 (P < 0.0001) | 0.43 (P < 0.0001) | 0.53 (P < 0.0001) | 0.42 (P < 0.0001) | 0.19 (P = 0.045) | 0.35 (P = 0.00024) |
| Stool-gastric | 0.31 (P = 0.0015) | 0.55 (P < 0.0001) | 0.44 (P < 0.0001) | 0.48 (P < 0.0001) | 0.43 (P < 0.0001) | 0.45 (P < 0.0001) | 0.42 (P < 0.0001) |
| Stool-contents | 0.24 (P = 0.0097) | 0.36 (P < 0.0001) | 0.35 (P < 0.0001) | 0.42 (P < 0.0001) | 0.48 (P < 0.0001) | 0.42 (P < 0.0001) | 0.33 (P < 0.0001) |
| Pharyngeal-esophageal | 0.53 (P < 0.0001) | 0.47 (P < 0.0001) | 0.52 (P < 0.0001) | 0.59 (P < 0.0001) | 0.66 (P < 0.0001) | 0.37 (P < 0.0001) | 0.57 (P < 0.0001) |
| Pharyngeal -gastric | 0.51 (P < 0.0001) | 0.50 (P < 0.0001) | 0.60 (P < 0.0001) | 0.70 (P < 0.0001) | 0.66 (P < 0.0001) | 0.41 (P < 0.0001) | 0.54 (P < 0.0001) |
| Pharyngeal-contents | 0.39 (P < 0.0001) | 0.50 (P < 0.0001) | 0.56 (P < 0.0001) | 0.63 (P < 0.0001) | 0.67 (P < 0.0001) | 0.36 (P < 0.0001) | 0.50 (P < 0.0001) |
| Esophageal-gastric | 0.70 (P < 0.0001) | 0.69 (P < 0.0001) | 0.68 (P < 0.0001) | 0.68 (P < 0.0001) | 0.74 (P < 0.0001) | 0.48 (P < 0.0001) | 0.69 (P < 0.0001) |
| Esophageal-contents | 0.52 (P < 0.0001) | 0.48 (P < 0.0001) | 0.50 (P < 0.0001) | 0.52 (P < 0.0001) | 0.66 (P < 0.0001) | 0.42 (P < 0.0001) | 0.45 (P < 0.0001) |
| Gastric-contents | 0.55 (P < 0.0001) | 0.55 (P < 0.0001) | 0.63 (P < 0.0001) | 0.66 (P < 0.0001) | 0.70 (P < 0.001) | 0.47 (P < 0.0001) | 0.57 (P < 0.0001) |
A total of 54 different bacterial families were detected in feeding tube and stool samples, in addition to some sequences that did not classify as any bacterial family. Of these, only four families had median relative abundance greater than zero: Enterococcaceae (median relative abundance of 0.26%), Streptococcaceae (2.8%), Staphylococcaceae (3.1%) and Enterobacteriaceae (18%). At the family level, only the correlation of family Staphylococcaceae between stool samples and esophageal tube sections was non-significant after Bonferroni correction. All other tested correlations at the family level were significant (Table 2). In the wiped and unwiped tube comparison subset, there were 27 different families detected. Of these, 15 families were found in both the wiped and unwiped tubes, 8 families were found in only the wiped tubes and 4 families were found only in the unwiped tubes. Of these, 4 of the families in only the wiped tubes and 3 of the families in only the unwiped tubes were present in only a single sample at a relative abundance of less than 1%. Family Chromatiaceae was present in two wiped tube samples from two different infants at a relative abundance of less than 1%. Family Clostridiaceae was present in three samples from three different wiped feeding tubes collected from two infants at a relative abundance of less than 1%. Family Thermaceae was present at a relative abundance of 1.2% from a single wiped tube sample. Family Planococcaceae was present at a relative abundance of 1.9% from a single wiped tube sample. Finally, family Xanthomonadaceae was present at a relative abundance of 6.9% in a single unwiped tube sample. Overall, the bacteria detected in the wiped and unwiped tubes were generally similar at the family level.
Alpha diversity
Using the Shannon index, all tube sections and the tube contents, namely, the pharyngeal sections ( P < 0.0001), the esophageal sections ( P < 0.0001), the gastric sections ( P = 0.034) and the residual tube contents (P < 0.0001) had lower alpha diversity than the fecal samples (Fig. 3). Additional variables significantly associated with alpha diversity were day of life at sample collection (Shannon index increased with age, P < 0.0001), tube duration (Shannon index increased with longer duration, P < 0.0001), gestational age (Shannon index increased with greater gestational age, P = 0.0073), breast milk feeding compared to formula feeding (Shannon index increased with breast milk feeding, P = 0.0082), feeding mix of breast milk and formula compared to formula only (Shannon index increased with mixed feeding, P = 0.0025), history of NEC (higher Shannon index in infants with a history of NEC, P = 0.00013) and history of sepsis (lower Shannon index in infants with a history sepsis, P = 0.00013) . All other clinical parameters were dropped from the model. There were no significant differences in alpha diversity between the wiped and unwiped tubes in the subset comparison using a paired t-test ( P = 0.58).
Figure 3.

Alpha diversity as measured by Shannon index. The tube contents and sections had significantly lower alpha diversity than the stool samples.
Beta diversity
The three dimensional NMDS ordination generated using the unweighted UniFrac distance matrix showed separation on the third axis between the fecal samples and the feeding tubes, but no obvious separation by tube section or between tube contents and the tube sections (Fig. 4). There was a significant difference in beta-dispersion of the unweighted UniFrac results between the different tube sections (P = 0.00875); however, because the larger groups tended to have larger dispersion this will result in a test that is too conservative (Anderson and Walsh 2013) (89 fecal samples with average distance to median of 0.447, 73 pharyngeal tube sections with average distance to median of 0.440, 66 esophageal tube sections with average distance to median of 0.381, 69 gastric tube sections with average distance to median of 0.422, and 81 tube content samples with average distance to median of 0.443). The PERMANOVA results found a significant difference by tube section (P = 0.001). There was no significant difference in beta diversity between the wiped and unwiped tubes in the subset comparison (P = 0.75).
Figure 4.

Non-metric multidimensional scaling ordination based on unweighted UniFrac distance. The stool samples appear to separate from the feeding tube samples on the third axis, but there is no obvious clustering by feeding tube section on any axis.
WGS
Table S2 (Supporting Information) provides infant demographic information by feeding type. Briefly, no infants in the WGS substudy had NEC, all infants were exposed to antibiotics at birth (ampicillin and gentamicin for 2 days), one infant was exposed to a second course of antibiotics prior to tube collection (cefepime for 10 days starting at day of life 15) and gestational age was similar between the three infants fed exclusively breast milk and the three infants fed at least some formula.
Normalized abundance of ARGs
There was visible separation of the stool samples and feeding tube sections in the NMDS of the resistome (Fig. 5). The PERMANOVA confirmed this result, demonstrating significant differences by sample type (P = 0.012) and by feeding type (P = 0.012). This indicated that the abundance of ARGs as estimated by number of reads per 16S read copy was different between stool samples and feeding tube sections, and that the abundance of ARGs was associated with whether infants are fed exclusive breast milk or at least some formula. There was no significant difference in total ARG abundance by tube section (P = 0.846). However, the median Jaccard dissimilarity between stool samples and the gastric tube samples was 0.92, the median Jaccard dissimilarity between stool samples and the esophageal tube section was 0.97, and the median Jaccard dissimilarity between the gastric and esophageal tube sections was 0.92. As Jaccard dissimilarity ranges from 0 to 1, with completely identical samples having a value of 0 and samples with no overlap having a value of 1, this suggested that the resistome present in stool samples and in different tube sections varies widely.
Figure 5.
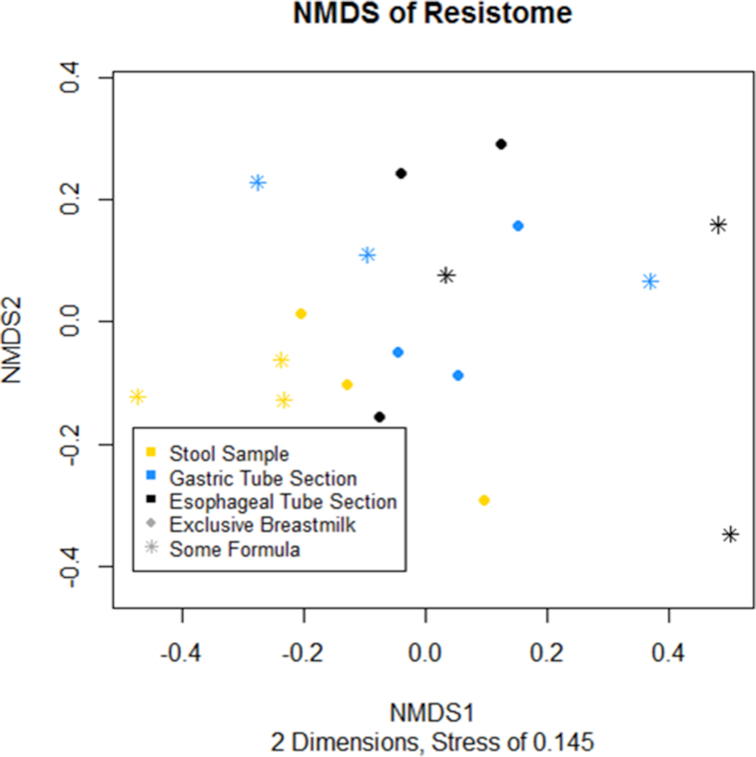
Non-metric multidimensional scaling ordination of the resistome, calculated using normalized AMR++ results and Bray–Curtis Dissimilarity.
Transferable ARGs
There were a median of 8 transferable ARGs in the stool metagenomes, including one sample which had no detected transferable ARGs. The gastric section of the feeding tubes had a median of 9.5 transferable ARGs detected per sample and the esophageal section of the feeding tube had a median of 11 transferable ARGs per sample. The esophageal and gastric feeding tube sections had a median of 5 transferable ARG classes present per sample, while the stool samples had a median of 5.5. Table S3 (Supporting Information) lists the identity of ARGs found in each sample, subject 5 is the individual who received additional antibiotic treatment prior to tube collection. The median Jaccard dissimilarity in transferable ARGs between stool samples and gastric tube section samples was 0.84, between stool samples and esophageal tube section samples was 0.98, and between gastric and esophageal tube sections was 0.81. The median Jaccard dissimilarity in transferable ARG classes between stool samples and gastric tube section samples was 0.54, between stool samples and esophageal tube samples was 0.38, and between gastric and esophageal tube sections was 0.38. There were no significant differences in Jaccard dissimilarity when comparing ARGs or ARG classes between the two tube sections to the Jaccard dissimilarity between the stool samples and the gastric tube sections or the stool samples and the esophageal tube sections. There was also no significant difference in the number of ARG classes detected in each of the tube sections and the stool samples (P = 0.79) or in the number of ARGs detected in each of the tube sections and the stool samples (P = 0.91). Figure 6 shows the overlap in ARGs between the three samples for each infant. Despite the lower Jaccard dissimilarity than that detected in the normalized abundance analysis, there are still relatively few ARGs that overlap between the three sample types. There were also no significant differences in the number of ARGs by feeding type in stool samples (P = 0.29), gastric tube sections (P = 0.64) or esophageal tube sections (P = 0.39). There were no significant differences by feeding type in the number of ARG classes detected in stool samples (P = 0.30), gastric samples (P = 0.95) or esophageal samples (P = 0.62).
Figure 6.

Venn diagrams showing the overlap of ARGs by tube section in each infants. The top row contains diagrams for infants fed exclusively breast milk, while the bottom row infants were fed at least some formula.
DISCUSSION
The identification of Proteobacteria, Firmicutes and Actinobacteria as the main components of feeding tube biofilms supports the findings of prior studies. These phyla have been previously reported as the most abundant components of bacterial populations in NICU patient feeding tubes, enteral feeding system extension tubing and neonates’ gastroesophageal microbiota (Mehall et al. 2002a; Matlow et al. 2003; Hurrell et al. 2009; Milisavljevic et al. 2013; Gomez et al. 2016; Petersen Greisen and Krogfelt 2016). Similar to previous studies, staphylococci, enterococci and Enterobacteriaceae were identified as the most abundant types of bacteria in NICU enteral feeding systems and the neonatal gastroesophageal microbiome (Mehall et al. 2002a; Milisavljevic et al. 2013; Gomez et al. 2016; Petersen, Greisen and Krogfelt 2016). In addition, this study found frequent colonization with streptococci. As NICUs may have very different background colonization of the preterm infant gut microbiome (Taft et al. 2014), the streptococci found in feeding tubes in our study may reflect natural variation in colonization patterns between NICUs. In a previous bacterial surface colonization study in this NICU we found sporadically elevated relative abundances of streptococci on neonate-associated surfaces (Bokulich, Mills and Underwood 2013).
The most abundant phyla and families detected in feeding tubes correlated with the levels of those same phyla and families in stool samples obtained from patients. This result suggests that the patient gut, rather than the hospital environment, is the main factor shaping feeding tube biofilm composition as skin is the dominant source of microbes in the NICU hospital environment (Brooks et al. 2018), and the dominant taxa in the feeding tubes correlated with gut microbes. This supports the hypothesis that the bacteria in feeding tubes originate from the gut microbiome, possibly colonizing the tube when gastric content is aspirated by healthcare providers prior to feeding (Hurrell et al. 2009; Petersen, Greisen and Krogfelt 2016). Previous studies have identified the same strains of bacteria in neonates’ feeding tubes as in their stool and shown that these strains can carry the same resistance phenotype, further evidence of a close relationship between feeding tube and gut microbiota (Gomez et al. 2016; Petersen, Greisen and Krogfelt 2016; Ogrodzki et al. 2017). Furthermore, the microbes comprising the biofilm found on different tube sections and the microbes found in the tube contents were correlated with each other, suggesting that at least the major taxa found in feeding tubes remain at similar levels across the entire tube.
The practice of aspirating gastric contents prior to feedings may also explain the similarity of bacterial populations in the gastric, esophageal and pharyngeal regions of the feeding tubes. Checking the gastric aspirate involves drawing fluid from the stomach all the way up the feeding tube, thereby exposing the entire length of the feeding tube to the gastric microbiota. This mechanism of tube inoculation could also explain why no differences in bacterial populations were observed between the gastric section and other sections of the tube, despite the reduced pH in the stomach. Alternatively, this apparent lack of effect due to stomach acid could be caused by neonates’ often elevated gastric pH (Hurrell et al. 2009). Further support for the conclusion that tube communities originate from infant microbes is that the alpha diversity of tube sections increased with infant age, even with tube dwell time included in the model. The alpha diversity of infant microbiomes increases with age as more species join the gut community (Hill et al. 2017); however, the number of environmental species of bacteria to which a feeding tube is exposed would not necessarily be expected to change as infants grow older.
In contrast with these results, PERMANOVA analysis based on unweighted UniFrac found significant differences in the communities by feeding tube section, and the results of the non-metric multidimensional ordination suggest that the detected differences may be driven primarily by the difference between stool samples and the feeding tube sections. As unweighted UniFrac distance is heavily influenced by rare taxa, this suggests that while the major taxa are similar between stool samples and feeding tube sections, the rarer taxa diverge between these sample sets. Whether this difference represents species present in the feeding tubes not present in the gut, species present in the gut not present in feeding tubes, differences in minor species in different sections of the feeding tubes or all of these factors remains unclear. Despite the correlation in taxa and the association of tube alpha diversity with infant age, the causal nature of the relationship between taxa in feeding tubes and taxa present in stool also remains unsettled. Even though the differences between the infant stool samples and the feeding tube samples may be driven by the presence of rare species, these differences may have important implications for infant health because the overlap in the resistome between infant stool samples and feeding tube sections is low. This lack of overlap suggests that feeding tubes are a source of exposure to ARGs that are not already a part of the infant gut microbiome. As biofilms are sites of increased horizontal gene transfer (Madsen et al. 2012; Juma and Forsythe 2015), the presence of ARGs in feeding tubes not found in infant stool samples potentially increases the risk of additional infant gut carriage of resistant organisms. The carriage of antimicrobial resistance organisms is associated with increased risk of severe infection and death (Kluytmans, van Belkum and Verbrugh 1997; Lautenbach et al. 2001; Molbak 2004), potentially exacerbating the already increased risk of infection experienced by preterm infants.
The alpha diversity of the feeding tube sections increased with increasing dwell time, suggesting that as tubes remain in place the number of different species inhabiting the feeding tubes continues to increase. As such, shorter dwell times for feeding tubes may reduce the number of taxa infants who are exposed through feeding tubes; and as tubes carried distinct resistomes from infant stool samples, this could reduce infant exposure to ARGs. However, feeding tube biofilms form in a matter of hours, replacing feeding tubes causes measurable pain and distress, and the tube insertion procedure comes with its own risks (Kristoffersen et al. 2011). Accordingly, the problem of feeding tube contamination likely cannot be entirely solved by more frequent feeding tube changes, and more research is needed to understand where the ARGs present in feeding tubes originate and under which circumstances those genes may incorporate into the infant resistome.
DATA AVAILABILITY
Demultiplexed 16S rRNA gene sequencing reads and host-subtracted whole metagenome sequencing data is available on SRA (SUB4491964).
Supplementary Material
ACKNOWLEDGEMENTS
This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by National Institutes of Health S10 Instrumentation Grants S10RR029668 and S10RR027303.
FUNDING
This study was supported by National Institutes of Health awards [F32HD093185] (DHT) and [AT008759] (DAM).
Conflict of interest
DAM is a cofounder of Evolve Biosystems, a company focused on diet-based manipulation of the gut microbiota. Evolve Biosystems played no role in the origination, design, execution, interpretation or publication of this work.
REFERENCES
- Alkeskas A, Ogrodzki P, Saad M et al.. The molecular characterisation of Escherichia coli K1 isolated from neonatal nasogastric feeding tubes. BMC Infect Dis. 2015;15:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MJ. A new method for non‐parametric multivariate analysis of variance. Austral ecology. 2001;26:32–46. [Google Scholar]
- Anderson MJ, Walsh DC. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing? Ecological Monographs. 2013;83:557–74. [Google Scholar]
- Bokulich NA, Mills DA, Underwood MA. Surface microbes in the neonatal intensive care unit: changes with routine cleaning and over time. J Clin Microbiol. 2013;51:2617–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B, Olm MR, Firek BA et al.. The developing premature infant gut microbiome is a major factor shaping the microbiome of neonatal intensive care unit rooms. Microbiome. 2018;6:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ et al.. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. GUniFrac: Generalized UniFrac Distances. 2012. [Google Scholar]
- Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–30. [Google Scholar]
- Gasparrini AJ, Crofts TS, Gibson MK et al.. Antibiotic perturbation of the preterm infant gut microbiome and resistome. Gut microbes. 2016;7:443–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez M, Moles L, Melgar A et al.. Early gut colonization of preterm infants: effect of enteral feeding tubes. J Pediatr Gastroenterol Nutr. 2016;62:893–900. [DOI] [PubMed] [Google Scholar]
- Harrison W, Goodman D. Epidemiologic trends in neonatal intensive care, 2007–2012. JAMA Pediatr. 2015;169:855–62. [DOI] [PubMed] [Google Scholar]
- Hill CJ, Lynch DB, Murphy K et al.. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET cohort. Microbiome. 2017;5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huda MN, Lewis Z, Kalanetra KM et al.. Stool microbiota and vaccine responses of infants. Pediatrics. 2014;134:e362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RH, Yaghoobi M. The esophageal and gastric microbiome in health and disease. Gastroenterol Clin North Am. 2017;46:121–41. [DOI] [PubMed] [Google Scholar]
- Hurrell E, Kucerova E, Loughlin M et al.. Neonatal enteral feeding tubes as loci for colonisation by members of the Enterobacteriaceae. BMC Infect Dis. 2009;9:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N. sabre - A Barcode Demultiplexing and Trimming Tool for FastQ Files. GitHub, 2011. [Google Scholar]
- Juma NA, Forsythe SJ. Microbial biofilm development on neonatal enteral feeding tubes. In: Donelli G.(ed). Biofilm-Based Healthcare-Associated Infections: Volume I. US: Springer International Publishing, Cham, 2015,113–21. [DOI] [PubMed] [Google Scholar]
- Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev. 1997;10:505–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristoffersen L, Skogvoll E, Hafstrom M. Pain reduction on insertion of a feeding tube in preterm infants: a randomized controlled trial. Pediatrics. 2011;127:e1449–54. [DOI] [PubMed] [Google Scholar]
- Lakin SM, Dean C, Noyes NR et al.. MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 2017;45:D574–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenbach E, Patel JB, Bilker WB et al.. Extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae: risk factors for infection and impact of resistance on outcomes. Clin Infect Dis. 2001;32:1162–71. [DOI] [PubMed] [Google Scholar]
- Li B, Yang Y, Ma L et al.. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 2015a;9:2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu C-M, Luo R et al.. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015b;31:1674–6. [DOI] [PubMed] [Google Scholar]
- Liu J, Kong K, Tao Y et al.. Optimal timing for introducing enteral nutrition in the neonatal intensive care unit. Asia Pac J Clin Nutr. 2015;24:219–26. [DOI] [PubMed] [Google Scholar]
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen JS, Burmølle M, Hansen LH et al.. The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol Med Microbiol. 2012;65:183–95. [DOI] [PubMed] [Google Scholar]
- Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathus-Vliegen EM, Bredius MW, Binnekade JM. Analysis of sites of bacterial contamination in an enteral feeding system. JPEN J Parenter Enteral Nutr. 2006;30:519–25. [DOI] [PubMed] [Google Scholar]
- Matlow A, Wray R, Goldman C et al.. Microbial contamination of enteral feed administration sets in a pediatric institution. Am J Infect Control. 2003;31:49–53. [DOI] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J et al.. An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2011;6:610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehall JR, Kite CA, Gilliam CH et al.. Enteral feeding tubes are a reservoir for nosocomial antibiotic-resistant pathogens. J Pediatr Surg. 2002a;37:1011–2. [DOI] [PubMed] [Google Scholar]
- Mehall JR, Kite CA, Saltzman DA et al.. Prospective study of the incidence and complications of bacterial contamination of enteral feeding in neonates. J Pediatr Surg. 2002b;37:1177–82. [DOI] [PubMed] [Google Scholar]
- Milisavljevic V, Garg M, Vuletic I et al.. Prospective assessment of the gastroesophageal microbiome in VLBW neonates. BMC Pediatr. 2013;13:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molbak K. Spread of resistant bacteria and resistance genes from animals to humans–the public health consequences. J Vet Med B. 2004;51:364–9. [DOI] [PubMed] [Google Scholar]
- Nanthakumar NN, Fusunyan RD, Sanderson I et al.. Inflammation in the developing human intestine: a possible pathophysiologic contribution to necrotizing enterocolitis. Proc Natl Acad Sci USA. 2000;97:6043–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodzki P, Cheung CS, Saad M et al.. Rapid in situ imaging and whole genome sequencing of biofilm in neonatal feeding tubes: a clinical proof of concept. Sci Rep. 2017;7:15948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Friendly M et al.. vegan: Community Ecology Package. 2017. [Google Scholar]
- Patel AL, Trivedi S, Bhandari NP et al.. Reducing necrotizing enterocolitis in very low birth weight infants using quality-improvement methods. J Perinatol. 2014;34:850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SM, Greisen G, Krogfelt KA. Nasogastric feeding tubes from a neonatal department yield high concentrations of potentially pathogenic bacteria- even 1 d after insertion. Pediatr Res. 2016;80:395–400. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2016. [Google Scholar]
- Rotmistrovsky K, Agarwala R. BMTagger:best match tagger for removing human reads from metagenomics datasets. 2011. [Google Scholar]
- Taft DH, Ambalavanan N, Schibler KR et al.. Intestinal microbiota of preterm infants differ over time and between hospitals. Microbiome. 2014;2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft DH, Liu J, Maldonado-Gomez MX et al.. Bifidobacterial dominance of the gut in early life and acquisition of antimicrobial resistance. mSphere. 2018;3:e00441–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zankari E, Hasman H, Cosentino S et al.. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Demultiplexed 16S rRNA gene sequencing reads and host-subtracted whole metagenome sequencing data is available on SRA (SUB4491964).