

Abstract
Paraneoplastic neurological autoimmunity is often associated with small‐cell lung cancer (SCLC), a highly malignant neuroendocrine tumor. Paraneoplastic autoimmunity often correlates with longer survival. We describe the paraneoplastic neurological manifestations of patients with SCLC with and without SCLC‐predictive autoantibodies and the correlation between autoimmunity and survival. We reviewed the records of 116 patients (51% male) from the Mayo Clinic with histopathologically confirmed SCLC for whom stored serum was available for neural autoantibody testing. Cancer was limited stage in 41%; the median age at diagnosis was 64 years. Paraneoplastic neurological manifestations were recorded in 61% (decreasing frequency: peripheral neuropathy, dysautonomia, cognitive decline, cerebellar ataxia, neuromuscular junction disorder, seizures, cranial neuropathy, movement disorder, brainstem disorder, or myelopathy). Neural autoantibodies, some with pathogenic potential, were detected in the sera of SCLC patients with and without neurological autoimmunity. The most frequent among patients with neurological manifestations were: anti‐neuronal nuclear antibody‐type 1, voltage‐gated calcium channel (VGCC)‐N‐type, VGCC‐P/Q‐type, glutamic acid decarboxylase 65 (GAD65), SOX1, and muscle acetylcholine receptor (AChR); while the most common in patients without neurological manifestations were: GAD65, muscle‐AChR, and VGCC‐P/Q‐type. Neither cancer stage at diagnosis nor survival correlated with neurological manifestations or autoantibody‐positivity, except for shorter survival in patients with myelopathy. The only predictor of longer survival was limited‐stage disease at diagnosis.
Keywords: Anti‐Hu antibody, cancer biomarker, CRMP5 antibody, GABAB receptor antibody, paraneoplastic syndrome
Introduction
Paraneoplastic neurological autoimmunity is associated with small‐cell lung cancer (SCLC), a highly malignant neuroendocrine tumor. The neurological presentations are more diverse than traditionally recognized and can be misdiagnosed as neurodegenerative disorders or cancer progression. The profile of neural autoantibodies in a patient's serum reflects the onconeural antigens expressed by the tumor.1 It is not unusual for multiple autoantibodies and paraneoplastic disorders to co‐exist in one patient.2
More than 90% of patients with newly diagnosed SCLC and neurological impairment are seropositive for one or more SCLC‐predictive autoantibodies; seropositivity for any neural autoantibody has been reported in 40% of SCLC patients without a clinically evident paraneoplastic neurological disorder.2 Paraneoplastic autoimmunity indicates an efficient antitumor immune response. Patients with SCLC and Lambert–Eaton myasthenic syndrome (LEMS) have longer survival than patients without.3 Certain autoantibodies are associated with longer survival in SCLC patients without neurological symptoms.4
The introduction of immune checkpoint inhibitor therapy to oncology practice has been accompanied by an increasing incidence of paraneoplastic autoimmune disorders, both organ and non‐organ specific.5 Ascertainment of a patient's baseline serological profile before initiating this therapy may aid in predicting the risk of autoimmune neurological complications.6
The aim of this study was to describe the neurological manifestations and onconeural autoantibody profiles of patients with SCLC with or without neurological autoimmunity and assess their impact on survival.
Methods
The institutional review board at Mayo Clinic approved this study. Patients with histopathologically confirmed SCLC were identified in the Neuroimmunology Laboratory database. Sera from 71 patients with neurological symptoms were submitted for clinical service evaluation of neural autoantibodies, and sera were collected from 45 patients without neurological symptoms. Sera were collected from patients evaluated clinically at Mayo Clinic from 1980 to 2004 and medical records were reviewed. For patients with multiple specimens, we tested the earliest available specimen. Patients surviving at least seven years after diagnosis of limited disease or three years after diagnosis of extensive disease were considered exceptional survivors, while typical survivors were identified as surviving less than three years with limited and no more than one year with extensive SCLC.
Autoantibody assays
Sera were stored at −30°C and tested in the Mayo Clinic Neuroimmunology Laboratory using clinically validated methods. Radioimmunoprecipitation assays for muscle and ganglionic acetylcholine receptor (AChR), voltage‐gated (Kv1) potassium channel (VGKC)‐complex, glutamic acid decarboxylase 65 (GAD65), and P/Q‐type and N‐type voltage‐gated calcium channel (VGCC) autoantibodies were conducted as previously described.7, 8 Striational antibodies were detected by enzyme‐linked immunosorbent assay. Neuronal nuclear and cytoplasmic immunoglobulin Gs (IgGs, screened by both mouse tissue‐based indirect immunofluorescence and Euroline protein blots, Euroimmun AG, Lübeck, Germany) included anti‐neuronal nuclear antibody (ANNA)‐1, ANNA‐2, ANNA‐3, Purkinje cell antibody (PCA)‐1, PCA‐2, PCA‐Tr, amphiphysin, CRMP5, and SOX1. Autoantibodies specific for N‐methyl‐D‐aspartate (NMDAR), α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPAR), γ‐aminobutyric acid‐B (GABABR), glycine (GlyR) receptors, and AQP4 were tested by cell‐binding immunofluorescence assays on human embryonic kidney (HEK293) cells transfected with plasmids encoding the respective antigens (Euroimmun AG or in‐house).
Statistical analysis
Data were summarized as frequencies and percentages or medians and ranges, as appropriate. Continuous measures were compared between patients using Wilcoxon rank‐sum tests. Dichotomous variables were compared with Fisher's exact tests. Survival time was analyzed using Kaplan–Meier curves. P values < 0.05 were considered statistically significant. Analyses were performed using SPSS 2.0 (IBM Corp., Armonk, NY, USA).
Results
Demographics of the 116 patients are summarized in Table 1. SCLC at cancer diagnosis was limited stage in 41% of patients and extensive in 46%, and information was lacking for 13%. The frequency of neural autoantibodies and autoimmune neurological manifestations did not differ significantly with limited or extensive stage SCLC. Seventy‐five percent of patients received cancer treatment (chemotherapy, radiation, and/or resection), 3% did not, and information was lacking for 22%.
Table 1.
Demographics of the 116 SCLC patients
| Male gender | 59 (51%) |
|---|---|
| Median age (range) | 64 years (37–93) |
| Smoking history | 111 (96%; unknown 3.5%) |
| Disease stage | |
| Limited | 41% |
| Extensive | 46% |
| Unknown | 13% |
| Autoimmune neurological disorder | 71 (61%) |
| Onset before cancer diagnosis | 61 (86%) |
| Cancer treatment | |
| Yes | 87 (75%) |
| No | 4 (3%) |
| Unknown | 25 (22%) |
SCLC, small cell lung cancer.
Serological findings and neurological manifestations
The detection frequencies of neural autoantibodies are summarized in Table 2; serum was available before cytotoxic therapy in 73 patients. ANNA‐1 was the most frequently detected autoantibody among patients with neurological manifestations, followed by VGCC (N‐type and P/Q‐type), GAD65, SOX1, muscle‐AChR, CRMP5, VGKC, ganglionic‐AChR, GABABR, and striational autoantibodies.
Table 2.
Autoantibody frequencies in the 116 SCLC patients
| Autoantibody specificity | With neurological manifestations (71 patients) | Without neurological manifestations (45 patients) | Total | P |
|---|---|---|---|---|
| ANNA‐1 | 34 | 2 | 36 | < 0.001 |
| VGCC, N | 17 | 3 | 20 | 0.01 |
| VGCC, P/Q | 16 | 4 | 20 | NS |
| GAD65 | 16 | 14 | 30 | NS |
| AGNA/SOX1 | 12 | 3 | 15 | NS |
| Muscle AChR | 8 | 4 | 12 | NS |
| CRMP5 | 6 | 0 | 6 | NS |
| VGKC | 3 | 3 | 6 | NS |
| Ganglionic AChR | 2 | 0 | 2 | NS |
| GABAB‐R | 2 | 1 | 3 | NS |
| Striational | 1 | 0 | 1 | NS |
| ANNA‐3 | 0 | 1 | 1 | NS |
AChR, acetylcholine receptor; ANNA, anti‐neuronal nuclear antibody‐type; GAD65, glutamic acid decarboxylase 65; NS, not significant; SCLC, small cell lung cancer; VGCC, voltage‐gated calcium channel; VGKC, potassium voltage‐gated channel. Statistical significance is highlighted in bold.
ANNA‐1 and VGCC‐N‐type autoantibodies were detected more frequently in patients with neurological manifestations than in those without. The VGCC‐P/Q‐type autoantibody was more frequent in patients with LEMS (P = 0.002), muscle‐AChR‐IgG in patients with myasthenia gravis (P = 0.01), and ANNA‐1 in patients with peripheral somatic or autonomic neuropathy (P < 0.001 for both).
Seventy‐one patients had autoimmune neurological manifestations attributable to SCLC (Fig 1). In 86% of cases, neurological symptoms and signs preceded cancer diagnosis. Neurological manifestations often involved multiple levels of the neuraxis and the detected autoantibodies tended to be consistent with the spectrum of manifestations recognized to associate with the neurological phenotype. Peripheral neuropathy was most common (31%, excluding patients whose neuropathy developed after chemotherapy). Dysautonomia was documented in 20 patients, and gastrointestinal dysmotility was a frequent manifestation of ANNA‐1 autoimmunity. Encephalopathy was also common (24%). Autoantibody specificities detected with cerebellar ataxia included ANNA‐1 (5 patients), VGCC‐P/Q‐type (5 patients), and CRMP5 (1 patient); none had amphiphysin‐IgG. Ten of the 13 patients with a neuromuscular junction disorder had Lambert–Eaton myasthenic syndrome, two had myasthenia gravis, and one was not specified. All patients diagnosed with Lambert–Eaton myasthenic syndrome were VGCC‐P/Q‐IgG positive (only 2 had co‐existing SOX1‐IgG) and both patients with myasthenia gravis were muscle‐AChR‐IgG‐positive. Two of the three patients with cranial neuropathy were CRMP5‐IgG‐positive and one was ANNA1‐positive. CRMP5‐IgG and ANNA1‐IgG also were detected in two patients with myelopathy.
Figure 1.
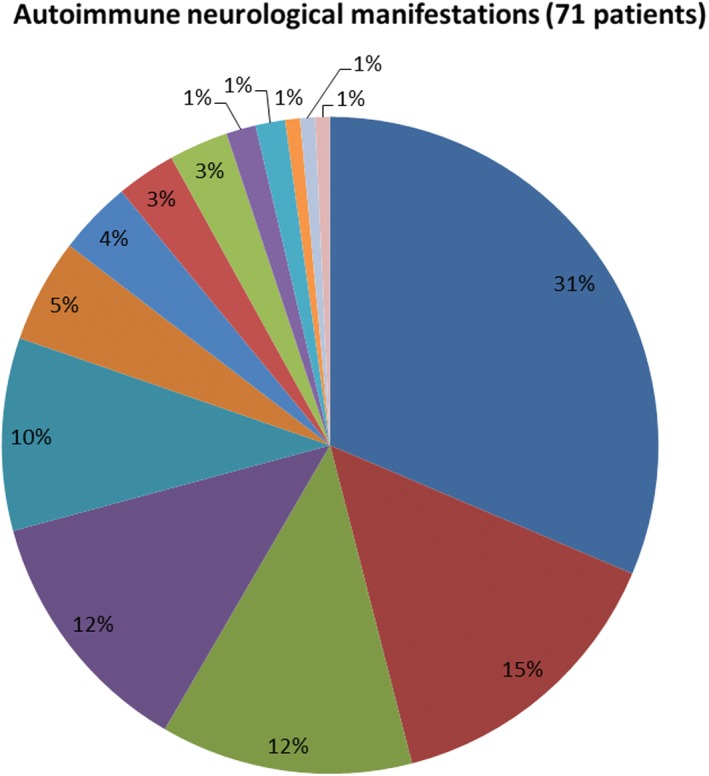
Autoimmune neurological manifestations in 71 patients with small‐ cell lung cancer. ( ) peripheral neuropathy; (
) peripheral neuropathy; ( ) dysautonomia; (
) dysautonomia; ( ) cognitive decline; (
) cognitive decline; ( ) cerebellar ataxia; (
) cerebellar ataxia; ( ) neuromuscular junction disorders; (
) neuromuscular junction disorders; ( ) seizures; (
) seizures; ( ) cranial neuropathy; (
) cranial neuropathy; ( ) movement disorder; (
) movement disorder; ( ) brainstem manifestations; (
) brainstem manifestations; ( ) myelopathy; (
) myelopathy; ( ) psychiatric manifestations; (
) psychiatric manifestations; ( ) opsodonus‐myodonus; (
) opsodonus‐myodonus; ( ) peripheral nerve hyperexcitability; (
) peripheral nerve hyperexcitability; ( ) myopathy.
) myopathy.
Multiple neural autoantibodies were detected in patients without neurological manifestations. GAD65 IgG was the most common specificity, followed by VGCC‐P/Q, muscle‐AChR, SOX 1, Kv1 VGKC‐complex, ANNA‐1, GABABR, and ANNA‐3.
Survival
The overall average survival or follow‐up period was 39 (range: 0–368) months. Twenty‐two patients were exceptional survivors, 66 were typical survivors, and the remainder had an intermediate survival rate. The only independent predictor for longer survival was limited stage disease (tested both as a continuous variable or dichotomous in exceptional versus typical survivors). The presence of neurological signs or detection of a neural autoantibody did not correlate significantly with survival, except for shorter survival in the two patients who had myelopathy.
Discussion
The results of our study confirm that neural autoantibodies are frequently found in patients with SCLC,2 even in neurologically asymptomatic patients, as we previously reported for patients with thymoma.9 Interestingly, IgGs targeting extracellular domains of plasma membrane antigens (e.g. GABAB, muscle‐AChR) and thus having pathogenic potential, were also found in neurologically unaffected patients. The individual patient's autoantibody profile reflects antigens expressed by the tumor and is consistent with the neuroendocrine nature of SCLC.1
Among patients with neurological autoimmunity, the most common clinical manifestations were neuropathy and dysautonomia, the latter frequently manifesting as gastrointestinal dysmotility (an often under‐recognized paraneoplastic disorder). Classic neurological paraneoplastic syndromes included LEMS, encephalitis with seizures, and cerebellar ataxia. The frequency of the neurological presentations we have reported are not representative of their incidence in the SCLC population, but rather reflect the referral bias of patients whose specimens are submitted to our laboratory.
Prospective studies of SCLC patients have reported longer survival in patients with ANNA1‐IgG or other neuronal nuclear autoantibodies or clinical manifestations of Lambert–Eaton myasthenic syndrome.3, 4 Although 22 of the 116 patients in our study had exceptional survival time, we found no correlation between extended survival and any neural autoantibody (more extensive screening than prior studies) or autoimmune neurological disorder. This finding can perhaps be explained by the dampening of the anti‐cancer immune response and its presumed host survival advantage as a result of the cytotoxic chemotherapy ultimately administered to most of the study patients. Other factors potentially limiting interpretation of the study's findings include its retrospective design and the relatively small patient sample. In addition, 37% of the patient samples were collected after chemotherapy, which might have affected IgG‐seropositivity.
We anticipate the frequency of paraneoplastic neurological autoimmunity to increase with the use of checkpoint inhibitor therapy for SCLC. This behooves oncologists to be aware of the spectrum of potential autoimmune neurological sequelae as early recognition and judicious management will optimize oncological and neurological outcomes. Perhaps prospective monitoring of patients’ serum autoantibody profiles before, during, and after such therapy will reveal whether and which neural autoantibodies correlate with long term cancer survival and also might predict a high risk for autoimmune neurological complications.6
Disclosure
No authors report any conflict of interest.
Acknowledgment
Matthew M. Roforth and Jacquelyn A. Grell for technical assistance in performing part of the autoantibody‐detection assays and Mary J. Curtis for secreterial support.
References
- 1. Zekeridou A, Griesmann GE, Lennon VA. Mutated cancer autoantigen implicated cause of paraneoplastic myasthenia gravis. Muscle Nerve 2018; 58: 600–4. [DOI] [PubMed] [Google Scholar]
- 2. Gozzard P, Woodhall M, Chapman C et al. Paraneoplastic neurologic disorders in small cell lung carcinoma: A prospective study. Neurology 2015; 85: 235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maddison P, Gozzard P, Grainge MJ, Lang B. Long‐term survival in paraneoplastic Lambert‐Eaton myasthenic syndrome. Neurology 2017; 88: 1334–9. [DOI] [PubMed] [Google Scholar]
- 4. Gozzard P, Chapman C, Vincent A, Lang B, Maddison P. Novel humoral prognostic markers in small‐cell lung carcinoma: A prospective study. PLoS One 2015; 10: e0143558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen DS, Mellman I. Oncology meets immunology: The cancer‐immunity cycle. Immunity 2013; 39: 1–10. [DOI] [PubMed] [Google Scholar]
- 6. Mammen AL, Rajan A, Pak K et al. Pre‐existing antiacetylcholine receptor autoantibodies and B cell lymphopaenia are associated with the development of myositis in patients with thymoma treated with avelumab, an immune checkpoint inhibitor targeting programmed death‐ligand 1. Ann Rheum Dis 2019; 78: 150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meeusen JW, Klein CJ, Pirko I et al. Potassium channel complex autoimmunity induced by inhaled brain tissue aerosol. Ann Neurol 2012; 71: 417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lennon VA, Kryzer TJ, Griesmann GE et al. Calcium‐channel antibodies in the Lambert‐Eaton syndrome and other paraneoplastic syndromes. N Engl J Med 1995; 332: 1467–74. [DOI] [PubMed] [Google Scholar]
- 9. Zekeridou A, McKeon A, Lennon VA. Frequency of synaptic autoantibody accompaniments and neurological manifestations of thymoma. JAMA Neurol 2016; 73: 853–9. [DOI] [PubMed] [Google Scholar]