

SUMMARY
CDK4/6 inhibition is now part of the standard armamentarium for patients with estrogen receptorpositive (ER+) breast cancer, so that defining mechanisms of resistance is a pressing issue. Here, we identify increased CDK6 expression as a key determinant of acquired resistance after palbociclib treatment in ER+ breast cancer cells. CDK6 expression is critical for cellular survival during palbociclib exposure. The increased CDK6 expression observed in resistant cells is dependent on TGF-b pathway suppression via miR-432-5p expression. Exosomal miR-432-5p expression mediates the transfer of the resistance phenotype between neighboring cell populations. Levels of miR-432-5p are higher in primary breast cancers demonstrating CDK4/6 resistance compared to those that are sensitive. These data are Furthermore confirmed in pre-treatment and post-progression biopsies from a parotid cancer patient who had responded to ribociclib, demonstrating the clinical relevance of this mechanism. Finally, the CDK4/6 inhibitor resistance phenotype is reversible in vitro and in vivo by a prolonged drug holiday.
Graphical Abstract
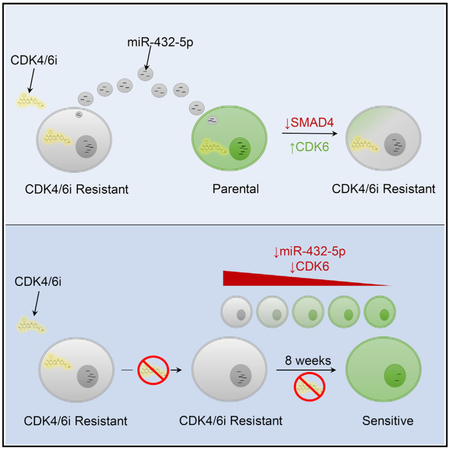
In Brief
Cornell et al. demonstrate a mechanism of acquired CDK4/6 inhibitor resistance that is independent of inherent genetic mutations, is conferred through extracellular signaling, and is reversible in vitro and in vivo. Resistance was mediated by exosomal miRNA, causing increased expression of CDK6 to overcome G1 arrest and promote cell survival.
INTRODUCTION
Cyclin D-dependent kinase activity is thought to be a driving factor for carcinogenesis in >80% of hormone receptor-positive breast cancers (Massagué, 2004), providing rationale for the inhibition of the cell-cycle kinases, cyclin-dependent kinase 4 (CDK4) and CDK6, in this breast cancer subset (Arnold and Papanikolaou, 2005; Elsheikh et al., 2008; Perou et al., 2000; The Cancer Genome Atlas Network, 2012; Velasco-Velázquez et al., 2011). The use of potent and highly selective CDK4/6 inhibitors, including palbociclib, ribociclib, and abemaciclib, has transformed the treatment of metastatic estrogen receptor-positive (ER+), human epidermal growth factor receptor 2-negative (HER2−) breast cancer based on prolonged progression-free survival when these agents are combined with hormone treatment compared to hormone therapy alone (Cristofanilli et al., 2016; Finn et al., 2016; Goetz et al., 2017; Hortobagyi et al., 2016; Sledge et al., 2017). In addition, abemaciclib has been approved as a monotherapy for patients with advanced ER+ breast cancer who have progressed on prior endocrine therapy and chemotherapy (Dickler et al., 2017). CDK4/6 inhibition may also have activity in HER2-driven breast cancer and in triplenegative breast cancers that retain expression of the retinoblastoma (RB) protein (Roberts et al., 2012; Yu et al., 2006).
CDK4/6 inhibitor-based treatment is complicated by the development of acquired resistance. To date, resistance mechanisms have not been extensively investigated. In leukemia models, reduced p27Kip1 expression and elevated CDK2 activity can overcome palbociclib-mediated G1 arrest (Wang et al., 2007). In breast cancer models, RB loss, amplification of CCNE1 (Herrera-Abreu et al., 2016), CDK6 (Yang et al., 2017), or FGFR1 (Formisano et al., 2017) and increased pyruvate dehydrogenase kinase 1 (PDK1) activity (Jansen et al., 2017) are also mechanisms by which the cancer cell can bypass CDK4/6 inhibitor-mediated G1 arrest. In analyses of tumor or liquid biopsies from breast cancer patients treated with CDK4/6 inhibitors, high cyclin E expression may define populations with intrinsic resistance (Turner et al., 2018), while acquired RB1 or PIK3CA mutation and fibroblast growth factor receptor (FGFR) pathway activation have been identified in post-progression samples (Condorelli et al., 2018; Formisano et al., 2017; Mao et al., 2018; O’Leary et al., 2018).
Here, we present a previously unreported mechanism by which resistance to CDK4/6 inhibitor treatment arises. Acquired resistance is centered on increased CDK6 protein concentration as the key determinant, achieved via the suppression of the transforming growth factor β (TGF-β) pathway mediated by microRNA (miRNA) expression. Consequently, resistance is transmissible by extracellular signaling and is reversible both in vitro and in vivo.
RESULTS
CDK4/6 Inhibitor-Resistant Cells Have Increased CDK6 and Cyclin D1 Expression
We generated palbociclib-resistant T47D ER+ breast cancer cells by continuous exposure to 100 nM drug and weekly monitoring of cell-cycle analysis by the quantification of DNA content. Initial exposure (week 0) led to a profound G1 arrest with >90% of cells in the G1 phase of the cell cycle. G1 arrest persisted to this degree for 5 weeks in the presence of palbociclib, and thereafter began to gradually decrease as cells took on a normal cell-cycle profile (Figure 1A). After 12 weeks of continuous palbociclib exposure, the cell-cycle distribution was indistinguishable from that of parental cells, as was the rate of proliferation, and they were deemed resistant (Figures 1A and 1B). The palbociclib concentration was then gradually escalated; each gradation (100 → 200 → 300 → 500 nM) was applied once a normal cell-cycle profile was achieved, until cells were resistant to 500 nM palbociclib (Figure 1A). While cells resistant to each concentration of palbociclib had indistinguishable cell-cycle profiles, those resistant to 300 and 500 nM drug (R300 and R500) had a slower doubling time (parental = 23.6 h, R100 = 23.6 h, R200 = 23.6 h, R300 = 28.8 h, R500 = 31.8 h; Figure 1B). Similar palbociclib-resistant derivatives were generated from MCF7 and ZR-75-1 ER+ breast cancer cells, as well as SKBR3 and BT-20 cells, representative of RB-expressing HER2-amplified and triple-negative breast cancer subsets, respectively (Figures S1A–S1F).
Figure 1. Generated CDK4/6 Inhibitor-Resistant Cell Lines Have Dramatically Increased CDK6 Protein Expression.

(A) Flow cytometry analysis of the cell-cycle profile in T47D cells after prolonged treatment (up to 12 weeks) with 100 nM palbociclib to establish resistance. Once resistant, the palbociclib dose was gradually increased from 100 nM (R100) to 500 nM (R500) and cells were monitored until resistance was confirmed by the cell-cycle profile.
(B) Growth rate of resistant T47D cells compared to parental cells.
(C) Real-time qPCR analyzing the fold change in mRNA expression between parental and resistant T47D cells (resistant to 100 nM palbociclib). Data are reported as the means ± SEMs of 3 independent experiments. *p < 0.05, ***p < 0.001.
(D) Whole-genome expression array analysis of the fold change in mRNA expression between parental and resistant T47D cells (resistant to 100 nM palbociclib). Data are reported as the means ± SEMs of 3 independent experiments. *p < 0.05, ***p < 0.001.
(E) Western blot analysis of both T47D and MCF7 resistant cells.
Analysis of cell-cycle genes via qPCR revealed a significant increase in CDK6 and CCND1 expression in resistant (R100) versus parental cells. These increases in mRNA expression were not accompanied by gene amplification as there was no variation in the copy number of these genes (Figure S2). No significant changes were observed in the remaining cyclin and CDK genes (Figure 1C). We also analyzed multiple genes related to cell cycle, growth, and/or CDK4/6 inhibitor resistance (Figure 1D). There were significant, albeit small (<2-fold), changes in the expression of HRAS, KRAS, MEK1, AKT1, PIK3R3, and PTEN in resistant cells. In correlation with gene expression, the greatest changes in protein expression were increased CDK6 and cyclin D1, observed in both T47D and MCF7 cells, with the expression increasing stepwise in cells that were resistant to higher concentrations of palbociclib (Figure 1E). A small stepwise increase in cyclin E levels was also observed, along with a progressive decrease in CDK1 expression. Phosphorylation of RB at the CDK4/6 site Ser807/Ser811, as well as at Thr356, was maintained in all resistant cells (Figure 1E).
CDK6 Knockdown Re-sensitizes Resistant Cells, and Overexpression of CDK6 Confers Resistance in Parental Cells
To determine the contribution of CDK6 to palbociclib resistance, we manipulated CDK6 expression in both parental and resistant T47D cells. Neither overexpression of CDK4 or CDK6 nor depletion of CDK6 significantly influenced the cell-cycle profile of parental T47D cells (Figure 2A). Substantial overexpression of CDK4 (↑CDK4) and CDK6 (↑CDK6) was achieved in parental cells and confirmed by western blot (Figure 2B). In addition, robust knockdown of CDK6 was confirmed in resistant cell lines (Figure 2C). Of note, depletion of CDK6 in resistant cells lines reduced CDK6 protein expression to a level approximating that seen in parental cells (Figure 2C).
Figure 2. Resistance to CDK4/6 Inhibition Is Mediated by High CDK6 Expression.

(A) Flow cytometry analyses of the cell-cycle profile of knockdown and overexpression T47D cells (↑CDK4 and ↑CDK6). Data are reported as the means ± SEMs of 3 independent experiments.
(B) Western blot analysis of CDK4/6 overexpression in parental cells.
(C) Western blot analysis of CDK6 knockdown in resistant T47D cells.
(D) Cell-cycle analysis of resistant T47D cells ± CDK6 depletion. Asterisks represent a significant difference between shCDK6 and shNT G1 populations: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are reported as the means ± SEMs of 3 independent experiments.
(E) Cell-cycle analysis of parental, ↑CDK4 and ↑CDK6 T47D cells during a 14-day period in the presence or absence of palbociclib treatment. Data are presented as the combined percentage of cells in S + G2 phase of the cell cycle.
See also Figures S3 and S4.
Non-target (NT) short hairpin RNA (shRNA) had no effect on the cell-cycle profile of resistant cells (R100–R500). shRNA-mediated depletion of CDK6 had no effect on the cell-cycle profile in parental cells (Figure 2A) but re-sensitized all of the resistant cells (maintained in palbociclib), which was evident in the significant increase in the G1 population in these cells (Figure 2D). In contrast, depletion of CDK4 did not re-sensitize resistant cells (Figure S3). Treatment of parental T47D cells with 100 or 300 nM palbociclib caused sustained G1 arrest, as demonstrated by the low percentage of cells in S and G2 phases of the cell cycle, which persisted for 14 days. CDK4-overexpressing cells treated with 100 nM palbociclib returned to a normal cell-cycle profile after 14 days of continuous treatment, although they remained stalled in the presence of 300 nM palbociclib. In contrast, CDK6-overexpressing cells became resistant to both 100 and 300 nM palbociclib within 10 days (Figure 2E). Furthermore, CDK6 overexpression in parental T47D or MCF7 cells significantly increased the growth inhibition 50 (GI50) not only of palbociclib but also ribociclib and abemaciclib in growth inhibition assays (Figures S4A and S4B).
CDK6 Contributes to the Survival of ER+ Breast Cancer Cells after Palbociclib Exposure
To broadly assess the effects of cell-cycle proteins on the response to palbociclib, we performed clonogenic survival assays after knockdown of a variety of cyclins and CDKs in parental T47D cells. Robust knockdown was achieved with each of the shRNAs (Figure 3A). shRNA-mediated depletion of CDK6 resulted in significantly decreased survival after palbociclib treatment compared to cells expressing a non-target shRNA control, or cells expressing shRNAs targeting other cell-cycle proteins (Figures 3B and 3C).
Figure 3. CDK6 Expression and Activity Contributes to Cell Survival after Palbociclib Exposure.

(A) Western blot analysis of shRNA-mediated knockdown of cell-cycle proteins in T47D cells.
(B) Clonogenic survival assay after 24 h of palbociclib exposure on shRNA-expressing T47D cells. Data are reported as the means ± SEMs of 3 independent experiments.
(C) Confirmatory CDK6 knockdown and clonogenic survival assay with an additional CDK6 shRNA. Survival was significantly decreased in both shCDK6-1 and -2 compared to shNT (p < 0.0001). Data are reported as the means ± SEMs of 3 independent experiments.
(D) Western blot of T47D cells with CRISPR/Cas9 knockout of CDK6 using an sgRNA targeting the 5′ UTR, followed by ectopic expression of CDK6 mutants. Clonogenic survival assay of CRISPR/Cas9 knockout CDK6, and mutant add-back lines treated with escalating dose of palbociclib.
(E) Cell-cycle analysis of shCDK6 T47D cells ± 100 nM palbociclib treatment for 24 h. Data are reported as the means ± SEMs of 3 independent experiments. ****p < 0.0001.
(F) Annexin V apoptosis assay using shSCR, shCDK6, and sgCDK6 T47D cells treated with escalating doses of palbociclib for 48 h. Data are reported as the means ± SDs of 2 independent experiments.
To determine whether the kinase activity of CDK6 was required for the survival of T47D cells after palbociclib exposure, we used CRISPR/Cas9-mediated knockout using a single guide RNA (sgRNA) targeting the 5′ UTR, and then overexpressed wild-type CDK6 or one of several mutant kinases (Figure 3D). A similar decrease in survival after palbociclib treatment was evident for both CDK6 knockout cells (sgK6–7, lethal concentration 50 [LC50] = 34.2 nM) and kinase dead CDK6-expressing cells (K43M, LC50 = 26.6 nM), compared to control (sgNT, LC50 = 66.2) and wild-type (WT, LC50 = 229.0 nM) cells. In addition, the overexpression of CDK6 proteins that are INK4 insensitive (R31C, LC50 = 632.0 nM) or constitutively active (S178P LC50 = 269.0 nM) substantially increased survival after palbociclib treatment (Figure 3D). CDK6 knockdown alone had no effect on the cell-cycle profile; however, CDK6-depleted cells treated with palbociclib had significantly more sub-G1 DNA content than control cells (Figure 3E), likely due to a marked increase in apoptosis in both CDK6 knockdown and knockout cells. In contrast, palbociclib had no effect on the apoptotic fraction of shNT-expressing cells (Figure 3F).
These results suggest that the low-level CDK6 expression in parental T47D cells is critical for survival in response to palbociclib, perhaps explaining the propensity of cells to overexpress CDK6 under the selective pressure of the drug as acquired resistance emerges.
CDK4/6 Inhibitor Resistance Is Mediated by Extracellular Signaling
While generating resistant lines, we observed that the population of cells appeared to overcome CDK4/6 inhibition as a whole, rather than forming distinct colonies of resistant cells. This phenomenon led us to believe that resistance was being mediated by extracellular factors. To test this hypothesis, we combined GFP-expressing parental cells with non-fluorescent T47D cells, either parental or resistant, in the presence or absence of palbociclib. When parental cells were mixed together, they behaved as expected, displaying a normal cell-cycle distribution when untreated and G1 arrest with CDK4/6 inhibition. In contrast, the co-culture of GFP-expressing parental cells and non-fluorescent resistant cells for 48 h led to a resistant phenotype in the GFP-expressing parental cells, demonstrated by the lack of cell-cycle arrest after 100 nM palbociclib treatment (Figure 4A). Quantification of DNA content demonstrated that co-culturing resistant cells with GFP-expressing parental cells resulted in parental cells becoming resistant to all concentrations of palbociclib within 96 h of coculture (Figure 4B).
Figure 4. CDK4/6 Inhibitor Resistance Is Transmitted via Exosomal Signaling.

(A) A schematic representation of the assay to test resistance transmission from a resistant cell to a non-resistant population. Parental cells were engineered to express GFP and mixed with non-fluorescent parental or resistant cells and incubated for 48 h. Cells were then sorted by fluorescence-activated cell sorting (FACS) based on GFP status, and the cell-cycle profiles of both GFP+ and GFP− populations were analyzed.
(B) Cell-cycle analysis of parental, GFP+ T47D, and MCF7 cells co-cultured for 96 h with palbociclib-resistant (100–500 nM) cells. Adjacent bars represent cells that were co-cultured. The palbociclib concentration in the medium was maintained at the level of the resistant cells. Data are reported as the means ± SEMs of 3 independent experiments.
(C) Western blot analysis of CDK6 protein expression. Panel 1: parental GFP cells were co-cultured with either parental or resistant cells for 48 h, then FACS sorted by GFP expression. Panel 2: T47D cells were treated with palbociclib for up to 6 days. Panel 3: parental T47D cells were incubated with conditioned medium from resistant cells. The medium contained 100 nM palbociclib and was replaced daily. Panel 4: exosomes from resistant T47D cell medium were harvested and subsequently added to parental cells daily for 6 days.
(D) Excreted cytokine assay performed on conditioned medium from resistant versus parental cell T47D cells. Data are reported as the means ± SDs cytokine expression of 2 independent experiments.
After co-culturing the cells for 48 h, we sorted the cells based on GFP expression and performed western blot analysis. Parental T47D or MCF7 cells that had been co-cultured with resistant cells gained substantial CDK6 expression, comparable to that of resistant cells (Figure 4C, first panel). This increase was not simply due to drug treatment, since prolonged palbociclib exposure up to 6 days had no observable effect on CDK6 expression (Figure 4C, second panel). Growing parental T47D cells in resistant cell-conditioned media also caused a marked increase in CDK6 expression during a 6-day time period (Figure 4C, third panel). Finally, treating parental T47D cells with purified exosomes from the media of resistant cells elicited a similar increase in CDK6 protein expression (Figure 4C, fourth panel).
To further confirm the role of exosomes in resistance transmission, we repeated the GFP-co-culture assay while inhibiting exosome production using manumycin A or GW4869 (Figure S5A). Both manumycin A and GW4869 treatments resulted in a significant increase in the G1 population of GFP-positive parental T47D cells, indicating a perturbation of resistance transmission by inhibiting exosome biogenesis (Figure S5B).
We also compared the excretion of numerous extracellular signaling cytokines; there were no significant or >2-fold changes in resistant compared with parental T47D cells (Figure 4D).
Exosomal miR-432-5p Mediates CDK4/6 Inhibitor Resistance
We next examined the miRNA profile of parental and resistant T47D cells, as well as parental cells grown in media conditioned by resistant cells. Palbociclib-resistant cells and parental cells grown in resistant cell-conditioned media shared a more similar expression profile compared to that of parental cells (Figure 5A). Numerous miRNAs were differentially expressed to a significant degree when comparing parental and resistant cells. miRNA target prediction algorithms revealed that 8 miRNAs that were significantly downregulated and 2 that were up-regulated in resistant cells were predicted to bind CDK6 mRNA (Figure 5B).
Figure 5. Resistance Is Mediated by Exosomal miR-432-5p.

(A) Hierarchal clustering of 30 miRNAs from miRNA expression profiling that display large-magnitude changes between parental, resistant, and parental T47D cells treated for 48 h with resistant cell medium.
(B) Significantly changed miRNAs grouped by expression in resistant relative to parental cells, sorted by significance. Highlighted miRNAs are significantly predicted to target CDK6 mRNA.
(C) Exosomes were harvested from the media of parental and resistant T47D cells. Real-time qPCR was performed to detect each of the miRNAs listed previously. The expression of detectable miRNAs is presented as fold change in resistant versus parental exosomes.
(D) Western blot analysis of CDK6 protein in resistant and parental T47D and MCF7 cells overexpressing selected miRNAs.
(E) Cell-cycle analysis of parental and miR-432-5p overexpressing T47D cells ± 100 nM palbociclib. Data are reported as the means ± SEMs of 3 independent experiments.
(F and G) GFP+ parental T47D cells were co-cultured for 48 h with either parental or miR-432-5p overexpressing cells (↑miR-432-5p) ± 100 nM palbociclib. Cells were then flow sorted by GFP status and analyzed for cell-cycle distribution (F). Data are reported as the means ± SEMs of 3 independent experiments (G).
(H and I) Resistant T47D and MCF7 (R100) cells were transfected with an miR-432-5p inhibitor and then incubated for 96 h before being analyzed by western blot (H) to determine CDK6 protein levels or by flow cytometry (I) to determine the cell-cycle distribution. Data are reported as the means ± SEMs of 3 independent experiments.
Analysis of differentially expressed miRNAs in the purified exosomes of parental and resistant T47D cells revealed a >100-fold difference in 5 miRNAs: miR-1973, miR-432-5p, miR-874-3p, miR-4695-3p, and miR-186-5p, 3 of which were decreased and predicted to target CDK6 mRNA (Figure 5C). To determine whether these miRNAs were involved in increased CDK6 expression and palbociclib resistance, we aimed to stably express each miRNA in both parental and resistant T47D and MCF7 cells. Plasmid-driven expression of miR-1973, 4695-3p, and 186-5p was unsuccessful, however, so we opted to overexpress miR-181a-5p, as it was the most significantly increased miRNA in resistant cells (Figure 5B), and miR-432-5p and miR-874-3p. miR-432-5p-overexpressing T47D and MCF7 cells both had markedly increased CDK6 protein expression. The overexpression of miR-874-3p produced inconsistent results, with a marked decrease in CDK6 protein in resistant T47D and parental MCF7 cells and no effect in resistant MCF7 cells (Figure 5D).
As it seemed most likely that transmissible resistance was driven by the increased expression of an miRNA, rather than by the decreased expression, we focused primarily on miR-432-5p. Parental cells overexpressing miR-432-5p behaved much like resistant cells–in other words, they did not arrest to 100 nM palbociclib (Figure 5E), had significantly increased palbociclib and ribociclib GI50s (Figures S4A and S4B), and could confer resistance in parental cells by co-culture (Figures 5F and 5G). Furthermore, when resistant T47D and MCF7 cells were transfected with an miR-432-5p inhibitor (an antisense RNA), CDK6 levels decreased and G1 arrest increased (Figures 5H and 5I).
miR-432-5p Increases CDK6 Protein Expression by Targeting the TGF-β Pathway
To determine the target of miR-432-5p, we performed miRNA: mRNA pull-down using a synthetic, biotin-labeled miR-432-5p and RNA-seq to identify all of the interacting mRNAs. Analysis of the pull-down mRNAs revealed numerous genes of the TGF-β pathway to be significantly enriched. Comparison between miRNA:mRNA pull-down and whole-genome mRNA expression revealed 2 genes, TGFBR3 and SMAD4, both of which were enriched by pull-down and downregulated in T47D resistant cells (Figure 6A). Using target prediction algorithms, miR-432-5p was significantly predicted to bind the 3′ UTR of both TGFBR3 and SMAD4 (Figure 6B). To further explore the role of the TGF-β pathway, we performed pathway enrichment analysis on multiple datasets, including mRNA and miRNA cell line arrays and miRNA-seq in patient biopsies. In each analysis we found a highly significant enrichment of genes and/or miRNAs involved in TGF-β pathway signaling (Figure 6C).
Figure 6. miR-432-5p Expression Is Significantly Higher in a Post-CDK4/6 Inhibitor-Treated Biopsy and Reduces TGF-β Pathway Signaling via Downregulation of SMAD4.

(A) Biotin-labeled miRNA-432-5p was used for mRNA pull-down in parental T47D cells. Fold enrichment is expressed relative to control RNA and correlated with mRNA expression data. Each experiment was performed in triplicate.
(B) Predicted miRNA binding of SMAD4 and TGFBR3 3′ UTR.
(C) GeneGo pathway enrichment analysis was performed on multiple datasets to compare enrichment in resistant versus sensitive patient biopsies (miRNA-seq), resistant versus sensitive T47D cells (whole-genome mRNA and miRNA arrays), and miRNA:mRNA capture versus total RNA (biotin-labeled miR-432-5p mRNA capture followed by RNA-seq).
(D) miRNA expression analysis was carried out by real-time qPCR from patient tumor biopsies pre-and post-CDK4/6 inhibitor (ribociclib) treatment. miRNAs were ranked based on the fold change in the post-treatment biopsy. Data are reported as the means ± SEMs of 3 independent experiments. ****p < 0.0001.
(E) Fold change in the mRNA expression of select genes in resistant cells and the post-CDK4/6 inhibitor treatment biopsy. Data are reported as the means ± SEMs of 3 independent experiments.
(F) Western blot showing the protein levels of TGF-β pathway components in parental, resistant, miR-scramble, and miR-432-5p expressing T47D and MFC7 cells.
(G) Confirmation of SMAD4 overexpression in resistant T47D cells by western blot.
(H) Cell-cycle analysis of SMAD4 overexpressing (↑SMAD4) palbociclib-resistant T47D cells. Data are reported as the means ± SEMs of 3 independent experiments. *p < 0.05, **p < 0.01. Indicated significance is between G1 populations of R100 versus R100 + ↑SMAD4.
miR-432-5p Expression Is Higher in Biopsies from ER+ Breast Cancer Patients with Intrinsic or Acquired CDK4/6 Inhibitor Resistance Compared to Those from Patients with Sensitive Disease
Using miRNA-seq, we analyzed 44 tumor biopsies from patients who received CDK4/6 inhibitor treatment either with hormone therapy or as a monotherapy for metastatic ER+, FIER2− breast cancer. Biopsies obtained pre-treatment were phenotypically stratified based on the response to CDK4/6 inhibitor treatment as either sensitive or intrinsically resistant, while those obtained post-progression were defined as having acquired resistance (Wander et al., 2018). When comparing all of the resistant (intrinsic and acquired) versus sensitive tumors, pathway enrichment analysis highlighted a highly significant enrichment of the “TGF-β signaling via miRNA in breast cancer” pathway (p = 5.05E–19, false discovery rate [FDR] = 3.53E–18; Figure 6C). In addition, there was a >1.8-fold increase in miR-432-5p, a >1.5-fold increase in CDK6, and a significant >2.8-fold decrease in SMAD4 expression among resistant tumors (Figure S6), suggesting that the resistance mechanism identified in vitro may be operating in primary tumors.
miR-432-5p Is Highly Expressed in a Post-progression Biopsy from a Patient Treated with CDK4/6 Inhibition
To obtain more definitive evidence of the clinical relevance of miR-432-5p-mediated resistance, we next analyzed pre-treatment and post-progression biopsies from a patient with parotid cancer harboring CDKN2A/B loss, who had achieved a partial response to the CDK4/6 inhibitor ribociclib (Infante et al., 2016). The post-progression biopsy had a significant increase in several of the previously investigated miRNAs. Most notably, miR-432-5p expression was significantly higher (p < 0.0001), with an 88-fold increase relative to the pre-treatment biopsy (Figure 6D).
To correlate the data with resistant cells and the patient biopsies, we analyzed the mRNA expression of genes involved in the cell cycle and TGF-β pathway by real-time qPCR and calculated the fold change in resistant versus parental cells and post- versus pre-treatment biopsies. Both resistant cell lines, as well as the post-treatment patient biopsy, demonstrated a significant decrease in SMAD4 mRNA expression that was accompanied by increased CDK6 expression (Figure 6E). These data were confirmed by western blot. Both T47D and MCF7 palbociclib-resistant and miR-432-5p-overexpressing cells had markedly lower SMAD4 protein expression than did parental cells (Figure 6F). To further confirm that lowered SMAD4 level was a critical component of palbociclib resistance, we overexpressed SMAD4 in resistant cells; this resulted in decreased CDK6 expression (Figure 6G) and restored susceptibility to CDK4/6 inhibitor-mediated cell-cycle arrest (Figure 6H).
These data suggest antagonism between inhibition of the TGF-β pathway and CDK4/6 inhibition. To confirm this expectation, we performed synergy studies with the TGF-β inhibitor galunisertib and palbociclib. Increasing doses of galunisertib reduced the growth-inhibitory effect of palbociclib and were significantly antagonistic in both parental and resistant T47D cells (Figures S7A and S7B). Galunisertib treatment had no effect on the growth of parental or resistant cells when used alone (Figure S7C); however, it did prevent G1 arrest when used in combination with palbociclib (Figure S7D).
Acquired Resistance Is Reversed by Drug Removal
We next sought to determine whether the resistance acquired by this mechanism was reversible. Resistant T47D and MCF7 cells were incubated in drug-free media for up to 7 weeks and rechallenged weekly with 100 nM palbociclib for 24 h, followed by analysis of DNA content for cell-cycle position. After 6 weeks in drug-free media, rechallenge with palbociclib resulted in a cell-cycle arrest that was indistinguishable from parental cells treated with the same concentration (Figure 7A). Analysis of resistant cell lysates via western blot revealed that the removal of palbociclib caused CDK6 protein to decrease over time. In correlation with the cell-cycle effects, the CDK6 protein level was reduced to a level comparable to that in parental T47D or MCF7 cells after 7 weeks in drug-free media (Figure 7B). Palbociclib-resistant cells that had been cultured in drug-free media for ≥7 weeks were labeled “ex-resistant.” The ex-resistant cells also had a significantly lower palbociclib GI50 compared to resistant cells, which was not significantly different from that of parental cells (Figures S4A and S4B).
Figure 7. Resistance Is Reversible by Prolonged Drug Absence.

(A) Cell-cycle analysis of T47D control-treated cells, palbociclib-treated cells, and cells grown in the absence of drug for up to 7 weeks before being re-challenged with 100 nM palbociclib for 24 h.
(B) Western blot analysis of CDK6 protein expression in T47D and MCF7 cells that have been removed from palbociclib-containing media for up to 7 weeks.
(C) Gene expression analysis by a whole-genome array of parental, resistant, and ex-resistant T47D cells. Ex-resistant cells are resistant cells that have had palbociclib exposure removed for a minimum of 7 weeks. Data are presented as means ± SEMs normalized to parental gene expression for 3 independent experiments. ****p < 0.0001.
(D) CDK6, CCND1, and miR-432 expression analysis by qPCR in parental, resistant, and ex-resistant cells. Data are presented as means ± SEMs 2−ΔΔCT normalized to parental gene expression, performed in triplicate. ***p < 0.001, ****p < 0.0001.
(E) Xenografts were established in the presence of palbociclib treatment using palbociclib-resistant (R100) MCF7 cells. Palbociclib treatment of 100 mg/kg/day was maintained until day 36, then removed for 28 days before being re-introduced. Data are relative tumor volume (RTV) of each individual animal as well as the overall average RTV during the course of 90 days (n = 13 at day 0; n = 8 at day 90).
(F) Gene expression analysis by qPCR in tumors taken from mice at days 36 and 64. Data are presented as means ± SEMs2−ΔΔCT normalized to day 36 tumor gene expression (n ≥ 3). *p < 0.05, **p < 0.01.
Analysis of gene expression in the ex-resistant compared to resistant and parental T47D cells revealed several significant changes. Most notably, there was a highly significant decrease in the expression of CDK6, CCND1, and CCNE1, as well as a significant increase in RB1 (p < 0.0001) expression in ex-resistant compared to resistant cells (Figures 7C and 7D). While many cell-cycle-related genes in ex-resistant cells returned to a similar level as that present in parental cells, hierarchal clustering of 100 significantly changed genes revealed that ex-resistant cells were more closely related to resistant cells than to parental cells (Figure S8). In addition, we analyzed the expression of miR-432-5p in ex-resistant T47D cells. Relative to resistant cells, expression of the miRNA was markedly decreased, correlating with reduced CDK6 and CCND1 expression, and the re-sensitization of these cells to palbociclib (Figure 7D).
To determine whether reversible CDK4/6 inhibitor resistance could be modeled in vivo, palbociclib-resistant xenografts were established by implanting resistant MCF7 cells into mice, followed by immediate palbociclib treatment. Once resistant tumors were established and growing (day 36), treatment was discontinued for the next 28 days. After this prolonged treatment holiday, treatment was re-introduced and caused a marked decrease in tumor burden in the previously palbociclib-resistant tumors (Figure 7E). Tumor samples were collected on day 36, representative of resistant tumors on the final day of palbociclib treatment, and on day 64, representative of ex-resistant tumors before palbociclib reintroduction. Gene expression analysis of day 36 and day 64 tumors revealed a marked decrease in both CDK6 and CCND1 expression in day 64 tumors (ex-resistant) relative to day 36. There was also a significant decrease in miR-432-5p expression on day 64 compared to day 36 (Figure 7F).
DISCUSSION
Inhibition of CDK4/6 is now a mainstay of ER+ breast cancer treatment, with concerted efforts under way to understand resistance. Here, through the generation of cell lines with acquired palbociclib resistance, we have highlighted CDK6 as a key protein mediating cell-cycle progression in the presence of the inhibitor.
CDK6 governs not only resistance but also the initial response to CDK4/6 inhibition. shRNA-mediated depletion of the small amount of CDK6 in parental cells converted the response to palbociclib from cytostatic to cytotoxic. Despite the relative equipotency of palbociclib against CDK4 and CDK6 (Fry et al., 2004), there was no effect on survival after treatment when CDK4 or D-cyclins were depleted from parental cells. Hence, the low level of CDK6 activity in parental cells is required for survival in response to palbociclib, and when enhanced in expression, caused resistance to cell-cycle arrest and reduced growth inhibition. The resistance phenotype required the presence of an active kinase domain. It is possible that CDK6 is a general survival factor in ER+ breast cancer cells, protecting from stresses that include not only direct cell-cycle blockade but also disruption of the hormonal axis (Alves et al., 2016). Further work will be required to determine whether CDK6 also protects against DNA damage-induced cell death, as has been described in ovarian cancer cells (Dall’Acqua et al., 2017).
During the generation of resistant cells, we did not observe clonally expanding populations, in direct contrast to present models of kinase inhibitor resistance in which a subpopulation of cells harboring an inherent mutation emerges under selective pressure. With continuous exposure to palbociclib, the entire population of cells remained in G1 phase of the cell cycle without an increase in cell death, followed by cells gradually cycling in unison. This suggested that resistance was not reliant on an inherent genetic alteration present in low allelic burden, but rather a feedback loop involving some degree of extracellular signaling that drives the necessary CDK6 expression.
This hypothesis was further supported by the reversibility of resistance, which could not occur had resistance arisen due to a permanent genetic event. While the expression of many cell-cycle genes returns to levels similar to those in parental cells, hierarchical clustering of gene expression analysis data showed ex-resistant cells to be more closely related to resistant cells than parental cells, suggesting that the cells do not fully revert after treatment is removed. However, the ex-resistant cells do reestablish a similar expression of cell-cycle-related genes, and specifically CDK6, accounting for their re-sensitization to palbociclib. We further confirmed this phenomenon in vivo. Following a drug holiday, re-exposure to palbociclib caused the substantial regression of all but 1 tumor, indicating that a phenotypic change can occur by removing CDK4/6 inhibition and that treatment holidays could be a useful clinical strategy. Further investigation of this phenomenon is required to determine whether cells will ever return to a state that is indistinguishable from parental cells on a genetic level and to determine whether these changes can be exploited to gain a therapeutic advantage. In the case of mantle cell lymphoma cells, the altered gene expression pattern of cells released from an acute palbociclib-mediated G1 arrest created acquired vulnerabilities, including susceptibility to signal transduction inhibitors (Chiron et al., 2013; Di Liberto et al., 2016).
During co-culture experiments, the acquisition of resistance occurred within 96 h, in stark contrast to the 12-week period of continuous drug exposure required to initially derive the resistant lines, which supports a role for extracellular signaling. Resistance was unrelated to cytokine expression and dependent on exosomes, leading us to investigate miRNAs, as numerous recent publications have identified exosomal miRNA signaling as a biomarker and key pathway regulator (Choi et al., 2014; Hannafon et al., 2016; Kosaka et al., 2010; Mittelbrunn et al., 2011; Montecalvo et al., 2012; Rabinowits et al., 2009). The miRNA profile of resistant cells was more closely related to that of parental cells cultured in resistant cell medium for 48 h than it was to parental cells in standard medium, highlighting the importance of extracellular miRNA signaling in regulating both mRNA and miRNA expression.
Of the 20 most significantly increased miRNAs in resistant cells, only 2 were predicted to target CDK6, as opposed to 8 of the decreased miRNAs. Although investigating the downregulated miRNAs predicted to target CDK6 will be of interest, it is unlikely that these miRNAs are related to the mechanism of transmitted resistance. Instead, we found that overexpression of miR-432-5p, one of the significantly upregulated miRNAs in resistant cells targeting SMAD4 and the TGF-β pathway, phenocopied resistant lines generated after several weeks of continuous drug exposure. To this end, overexpression of SMAD4 reduced CDK6 expression, as previously reported (Tsubari et al., 1999; Zhang et al., 2001), and reversed CDK4/6 inhibitor resistance.
To validate the importance of miR-432-5p expression clinically, we examined primary breast cancer samples. Although differences in miR-432-5p and CDK6 expression between CDK4/6 inhibitor-resistant and sensitive populations only trended toward significance, the small sample size and variable timing of post-progression biopsies could have affected results from patients with acquired resistance. Ideally, paired pre- and post- progression biopsies will be required to rigorously show increased miRNA expression as a determinant of acquired resistance. In spite of these limitations, we observed a highly significant enrichment of miRNAs targeting the TGF-β pathway in the resistant patient group, which is indicative of TGF-β pathway suppression in these patients. Additional work will be required to determine whether high miR-432-5p expression may occur along with other alterations conferring resistance or whether this mechanism is mutually exclusive with other such alterations. We were able to assay paired samples from a patient whose tumor had responded to ribociclib, in which we identified a highly significant increase in miR-432-5p and CDK6 in the post-progression biopsy, lending further support for the occurrence of this mechanism of resistance in primary patient samples.
Our results suggested that TGF-β pathway inhibition would be antagonistic with CDK4/6 inhibition, which we confirmed in combinatorial experiments. Previous work in pancreatic cancer cell lines indicated that palbociclib can induce TGF-β pathway-mediated epithelial-mesenchymal transition (EMT), which is prevented by TGF-βR1 inhibition, so that combined CDK4/6 and TGF-βR1 inhibition may be beneficial (Liu and Korc, 2012). Notably, we did not observe evidence of palbociclib-induced EMT in the breast cancer cell lines. These results demonstrate the complexity of the TGF-β pathway and its interface with CDK4/6 inhibition, which may be context dependent.
In summary, the data presented here demonstrate a clinically relevant mechanism of CDK4/6 inhibitor resistance whereby increased exosomal expression of miR-432-5p causes a down-regulation of TGF-β pathway signaling via SMAD4 knockdown, which in turn results in an increase in CDK6 expression, allowing cells to overcome G1 arrest. While multiple studies have focused on the regulatory properties of exosomal miRNA, the data presented here represent a mechanism of acquired drug resistance that is dependent on excreted miRNA that can be reversed by miRNA inhibition.
It will be important to determine whether this mechanism of resistance is found in other patients with acquired CDK4/6 inhibitor resistance. As such resistance is reversible, re-challenge with a CDK4/6 inhibitor may be prove beneficial after an adequate drug holiday. In addition, as resistance is mediated by exosomal signaling, analysis of patient plasma exosomes may identify emerging resistance before radiological progression and favorably affect patient management.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Geoffrey I. Shapiro (geoffrey_shapiro@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Metastatic breast and parotid cancer patient biopsies
Tumor biopsy specimens were obtained from 44 patients receiving care for metastatic hormone-receptor positive (HR+) HER2 negative breast cancer at the Dana-Farber Cancer Institute as part of voluntary research protocol 05-246. Informed consent was obtained in accordance with institutional review board (IRB) approval and the Declaration of Helsinki. Extra tissue was donated when available for miRNA extraction and quantification. All patients received CDK4/6 inhibitors in the metastatic setting. Clinical records were reviewed to determine the duration on therapy, radiographic response, and reason for discontinuation. Collated data were de-identified and biopsy samples were phenotypically described as follows: sensitive – any biopsy obtained within 32 days of starting therapy or any time prior to therapy initiation in patients who had a response (via radiographic assessment on any intervening staging CT study) or achieved stable disease for at least six months; acquired-resistant – any biopsy obtained within 32 days of drug discontinuation or anytime thereafter in a patient who achieved a response via radiographic imaging or stability for at least six months; and intrinsic-resistant – any biopsy obtained within 120 days of initiating therapy or anytime thereafter in a patient who had progression on their first interval re-staging study or achieved less than six months of stable disease. More information on this cohort is found in: (Mao et al., 2018; Wander et al., 2018). Informed consent was also obtained for the pre-and post-progression biopsies obtained from the patient with parotid cancer.
Mice
Female nude mice (NCr-Foxn1NU, Tactonic Laboratories, RRID:IMSR_TAC:ncrnu), approximately 8 weeks old were maintained and handled in isolators under specific pathogen-free conditions for tissue distribution and efficacy studies in accordance with local guidelines and therapeutic interventions approved by the Animal Care and Use Committee of Dana-Farber Cancer Institute. Mice were implanted with 17β-estradiol pellets (0.36 mg, 90-day release, Innovative Research of America) using a precision trochar 1 day prior to cell implantation. 7 × 106 palbociclib-resistant MCF7 cells in 100 μL culture medium + 50% matrigel (BD Biosciences) were subcutaneously implanted into the flanks of the mice (n = 20). Twenty-four hours after cell implantation, palbociclib treatment was initiated. Mice were weighed every other day and sacrificed if > 20% of day 0 body weight was lost. Mice were sacrificed if tumors reached 20 mm in any direction. Palbociclib was administered daily via oral gavage at 100 mg/kg, dissolved in 10 mM sodium lactate buffer, pH 4. Once tumors were established to be growing in the presence of drug (day 36, average tumor size roughly 3 mm × 3 mm, n = 16), palbociclib treatment was ceased for 28 days. After 28 days with no treatment, palbociclib treatment was resumed (n = 12). Tumors were measured throughout the experiment and tumor volume calculated using the formula (width2 × length)/2. Relative tumor volume was calculated by normalizing to day 21 tumor volume. Mice without palpable tumors were taken off the study at this point.
Cell lines
MCF7, T47D, BT-20, SK-BR-3, ZR-75-1 and 293T cells were purchased from American Type Culture Collection (ATCC). Cells were maintained in a humidified incubator at 37°C and 5% CO2. Cells were routinely tested and deemed free of Mycoplasma contamination (MycoAlert Assay, Lonza). T47D and MCF7 cells were maintained as monolayers in Dulbecco’s modified Eagles medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 2 mM penicillium:streptomycin solution and 0.2 Units/ml bovine insulin. 293T cells were grown in Dulbecco’s modified Eagles medium supplemented with 10% fetal bovine serum. SK-BR-3 cells were grown in McCoy’s 5a Medium Modified with 10% FBS. ZR-75-1 cells were grown in RPMI-1640 with 10% FBS. BT-20 cells were gown in Eagle’s Minimum Essential Medium with 10% FBS. All cell media was supplemented with 100 IU/ml penicillin-streptomycin.
METHOD DETAILS
Western Blot
Cells were trypsinized, centrifuged and washed with PBS before lysing with RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS, Boston BioProducts) containing protease and phosphatase inhibitor cocktails (Calbiochem). Lysates were sonicated twice and incubated on ice for 30 mins, then centrifuged at 16,000 g for 10 min. Protein concentration was quantified using a Pierce-BCA assay with a standard curve. Protein separation by gel electrophoresis using 4%–20% agarose gels and transferred to PVDF. Membranes were immunoblotted for 1 hour at room temp or overnight at 4°C with primary antibodies, then washed and probed with anti-mouse or anti-rabbit IgG secondary antibodies (GE Healthcare) for 1 hour at room temperature. Chemiluminescence was detected using X-ray films. Extracellular Cytokines were analyzed using the Human Cytokine Array Kit performed per manufactures instructions (R&D Systems). Parental and palbociclib-resistant cells were seeded in parallel and medium harvested after 24 hours for processing.
FLOW CYTOMETRY
For cell cycle analysis, cells were harvested, washed with PBS and fixed with 80%, −20°C ethanol for 30 minutes on ice. Cells were then washed with PBS and stained with propidium iodide/RNase solution (BD Biosciences) for 15-30 minutes at 37°C in subdued light. Cells were subsequently filtered to ensure a single cell population and analyzed using a BD Fortessa cytometer with FACS Diva software (BD Biosciences).
Annexin V apoptosis assays were performed and analyzed in accordance with the manufacturer’s instructions (BD Bioscience). For transferable resistance assays, parental cells expressing the GFP protein in the pLEX-307 backbone were mixed 1:1 with non-fluorescent cells. Cells were sorted based on GFP status for subsequent cell cycle analysis or western blotting.
CLONOGENIC SURVIVAL ASSAY
Cells (1 × 105) were seeded in 6-well plates and incubated for 24 hours and exposed to DMSO or palbociclib for 24 hours and then reseeded in 90 cm dishes in drug-free medium. Cells were incubated for 14 to 21 days to allow colony formation. Colonies were fixed and permeabilized using fixation solution (75% methanol, 25% Acetic acid), stained with 1% crystal violet and counted. Survival was determined by first calculating the plating efficiency (number of colonies/cells seeded) and then normalizing to the DMSO-treated control.
GROWTH INHIBITION ASSAY
Cells were seeded into 96-well plates (250 – 1000 cells/well) and incubated for 24 hours. Growth medium was then replaced with medium containing either DMSO, palbociclib, ribociclb, abemaciclib, or galunisertib and plates incubated for 5-7 days to allow a minimum of 3 cell doublings in DMSO-treated control cells. Growth was quantified using CellTiter-Glo as per the manufacturer’s instructions. Relative growth percentage was obtained by first subtracting background (CellTiter-Glo reagent in medium) and day 0 values, then normalizing to the DMSO-treated control. BLISS Synergy/antagonism score was modeled using Combenefit (version 2.021) software (Di Veroli et al., 2016).
Exosome purification and quantification
Exosomes were purified from tissue culture medium using either differential ultracentrifugation or Total Exosome Isolation Reagent as per the manufacturer’s instructions (Thermo Fisher Scientific). Exosomes were quantified using EXOCET quantification assay as per manufacturer’s instructions (SystemBiosciences).
RNA extraction
Cultured Cells
RNA was extracted from cultured cells using a combined TRIZOL/column purification. Briefly, cells were lysed by addition of TRIZOL to the cell pellet, mixed thoroughly and incubated at room temperature for 10 minutes. Chloroform was then added and mixed thoroughly, then centrifuged for 15 minutes at 12,000 g, 4°C. The aqueous layer was then removed, mixed with an equal volume 70% ethanol, and transferred to an miRNA or mRNA PureLink column. RNA purification was then performed as per PureLink kit instructions (Thermo Fisher Scientific).
Exosomes
RNA extraction from isolated exosomes was performed using Total Exosome RNA & Protein Isolation Kit (Thermo Fisher Scientific) as per the manufacturer’s instructions.
Xenografts
Xenografts were harvested and snap frozen. Approximately 10 mg of tissue was homogenized using a handheld electric tissue homogenizer in TRIZOL reagent. Once homogenized, RNA was extracted using the combined TRIZOL/column purification protocol described earlier.
Patient samples
Patient biopsies were snap frozen and RNA extraction was performed using AllPrep DNA/RNA Mini Kit as per the manufacturer’s instructions (QIAGEN). RNA from formalin-fixed paraffin-embedded tissue was extracted using RecoverAll Total Nucleic Acid Isolation Kit for FFPE tissues (Life Technologies). RNA quality was determined using a Bioanalyzer.
mRNA and miRNA quantification
For mRNA-based qPCR, reverse transcription was performed using High Capacity cDNA Reverse Transcription Kit (Life Technologies). For miRNA-based qPCR, reverse transcription was performed using the miRCURY LNA universal RT kit (Exiqon). qPCR was performed using a 2X Power SYBR green master mix (Applied Biosystems) and the following primers: CCND1, CCND3, CDK4, CDK6, TGFBR1, TGFBR2, TGFBR3, SMAD3, SMAD4, GAPDH, and miR-432-5p. SYBR green signal was detected using a Lighcycler 480 system (Roche). Relative gene expression was calculated using the 2–ΔΔCT method (Livak and Schmittgen, 2001).
mRNA Array
Gene expression analysis was performed with Affymetrix Human Genome U133A 2.0 library as per manufacturer’s instructions. Differential expression analysis was performed with RMA normalization using the Transcriptome Analysis Console software (Applied Biosystems, version 4.0.1.36).
Copy Number Variation
Copy number variation was analyzed by nanoString using the nCounter v2 Cancer CNV Assay. Copy number estimations are expressed as resistant cell/parental cell. Copy number gains were taken as a ratio of > 2.5. Copy number loss was taken as a ratio of < 1.5, as per the manufacturer’s instructions.
Biotin Labeled miRNA-mRNA pulldown
miRNA:mRNA pulldown and was performed as previously described (Wani and Cloonan, 2014). Briefly, biotin labeled miR-432-5p (Exiqon) was transfected into T47D cells and incubated for 24 hours at 37°C. Cells were then lysed and biotin- labeled miRNA:mRNA duplexes were captured using Dynabeads MyOne Streptavidin C1, which were washed and mRNA purified. mRNA was amplified using Illumina Total prep RNA Amplification kit and cRNA hybridized onto an Illumina Human HT-12 array. Data was expressed as reads per kilobase of transcript per million mapped reads (FKPM) and pulldown compared to total RNA. Significantly enriched mRNAs were then subjected to pathway analysis and correlated with gene expression data.
RNAseq
cDNA was synthesized using SmartSeq v4 reagents (Clontech) from 5ng of RNA. Full length cDNA was fragmented to a mean size of 150bp with a M220 ultra-sonicator (Covaris) and Illumina libraries were prepared from 2ng of sheared cDNA using Thruplex DNAseq reagents (Rubicon Genomics) according to the manufacturer’s protocol. The finished dsDNA libraries were quantified by Qubit fluorometer (Thermo Fisher Scientific), TapeStation 2200 (Agilent), and RT-qPCR using the library quantification kit (Kapa Biosystems). Uniquely indexed libraries were pooled in equimolar ratios and sequenced on an Illumina MiSeq run with single-end 50bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities. Sequenced reads were aligned to the hg19 reference genome assembly and gene counts were quantified using STAR (v2.5.1b) aligner (Dobin et al., 2013). Normalized read counts (RPKM) were calculated using Cufflinks (v2.2.1) software (Trapnell et al., 2010).
miRNA Profiling
For miRNA arrays, 750 ng total RNA from both sample and reference was labeled with Hy3 and Hy5 fluorescent label, respectively, using the miRCURY LNA microRNA Hi-Power Labeling Kit, Hy3/Hy5 (Exiqon) following the procedure described by the manufacturer. The Hy3-labeled samples and a Hy5-labeled reference RNA sample were mixed pair-wise and hybridized to the miRCURY LNA microRNA Array 7th Gen (Exiqon), which contains capture probes targeting all microRNAs for human, mouse or rat registered in the miRBASE 18.0. The hybridization was performed according to the miRCURY LNA microRNA Array Instruction manual using a Tecan HS4800 hybridization station (Tecan). After hybridization, the microarray slides were scanned and stored in an ozone-free environment (ozone level below 2.0 ppb) in order to prevent potential bleaching of the fluorescent dyes. The miRCURY LNA microRNA Array slides were scanned using the Agilent G2565BA Microarray Scanner System (Agilent Technologies) and image analysis was carried out using the ImaGene® 9 (miRCURY LNA microRNA Array Analysis Software, Exiqon). The quantified signals were background corrected (Normexp with offset value 10) and normalized using the global Lowess (LOcally WEighted Scatterplot Smoothing) regression algorithm.
miRNaseq
Libraries were prepared using NEBNext Small RNA library preparation kit (New England Biolabs) from 80ng of purified total RNA according to the manufacturer’s protocol. Completed libraries were size selected from 105-155bp on a Pippin Prep (Sage Biosciences) using a 3% agarose gel according to the manufacturer’s protocol. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on a NextSeq500 (Illumina) with single-end 75bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities. Fastq files were generated and sample data demultiplexed using Illumina bcl2fastq (v2.18). Raw fastq files were first trimmed to remove Illumina adaptor sequences using cutadapt software with the following flags: Cutadapt – adaptor = AGATCGGA AGAGCACACGTCTGAACTCCAGTCAC-minimum-length = 8-m 17-overlap = 8. Trimmed reads were collapsed using the seqcluster collapse command from the seqbuster package (Pantano et al., 2010, 2011) and mapped to human miRNA sequences from miRBase (version 21) using miraligner with following flags: Miraligner–sub 1–trim 3–add 3–s hsa–freq. Count frequencies were extracted from miralign output and normalized to counts per million (CPM).
RNA Interference
Cells were reverse-transfected with either Non-Target, CDK4, or CDK6 siRNA (Dharmacon) using lipofectamine 3000 according to the manufacturer’s instructions. Protein expression was analyzed by western blot after 72 hours.
Lentivirus production and Stable Cell Line generation
Cells stably expressing shRNA, miRNA or cDNA were generated using lentiviral transfection, as previously described using the pLKO.1 (Moffat et al., 2006) or pLEX-307 backbone. Briefly, 293T cells were transfected with Ps-PAX2, VSV-G, and an expression vector using lipofectamine 3000 (Thermo Fisher Scientific). Transfected 293T cells were incubated for 16 hours and then transferred into 30% FBS containing medium to enhance viral production. After 24 hours, medium containing virus was harvested, centrifuged at 500 g for 10 mins, filtered through a 0.45 μM filter and stored at −80°C. Target cells were incubated in standard medium containing 8 μg/ml polybrene and viral medium added to make a final ratio of 1 in 5. Multiple stable cell lines were generated for each protein knockdown; shRNA sequences were obtained from the RNAi Consortium database (Moffat et al., 2006). GFP, SMAD4, and CDK6 GeneArt cDNA sequences (WT, K43M (Hu et al., 2011), S178P (Bockstaele et al., 2009) and R31C (Hu et al., 2011) were purchased with attB1/2 sequences added and cloned into the pLEX-307 backbone using the gateway cloning system (Thermo Fisher Scientific). For miRNA overexpression, approximately 500 bp of DNA encompassing each miRNA sequence was PCR purified and cloned into the pLEX-307 backbone. miRNA mimics (Thermo Fisher Scientific) were transfected into cells as a positive control for miRNA detection. Protein expression/miRNA expression/knockdown was confirmed after 5 days of selection in puromycin (1 μg/ml for both T47D and MCF7). CRISPR/Cas9 knockouts were performed as previously described (Sanjana et al., 2014), and the following sgRNA sequences cloned into the LentiCRIPSR v2 vector - sgCDK6- 7 (targeting the 5′UTR): CCGCTCCACCCGCTCATCGT, Non-Target sgRNA: CGTGATGGTCTCGATTGAGT. Virus was generated as described and cells were selected using 1 μg/ml puromycin and protein knockout confirmed after approximately 12 days.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical comparisons were performed using un-paired t tests or ANOVA unless otherwise stated. P value < 0.05 was considered significant (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). Statistics and number of biological repeats can be found in the relevant figure legends. Statistical analyses were completed using Graphpad Prism 7. Pathway analysis was performed using MetaCore platform.
DATA AND SOFTWARE AVAILABILITY
Sequencing data have been deposited in the Gene Expression Omnibus (GEO) under the accession number GEO: GSE117748.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CDK1 | Santa Cruz Biotechnology | Cat# sc-954; RRID:AB_631207 |
| anti-CDK2 | Santa Cruz Biotechnology | Cat# sc-163; RRID:AB_631215 |
| anti-CDK4 | Cell Signaling | Cat# 12790; RRID:AB_2631166 |
| anti-CDK4 | Santa Cruz Biotechnology | Cat# sc-23896; RRID:AB_627239 |
| anti-CDK6 | Cell Signaling | Cat# 13331; RRID:AB_2721897 |
| anti-CDK6 | Santa Cruz Biotechnology | Cat# sc-7961; RRID:AB_627242 |
| anti-cyclin D1 | Cell Signaling | Cat# 2922; RRID:AB_2228523 |
| anti-cyclin D2 | Cell Signaling | Cat# 3741; RRID:AB_2070685 |
| anti-cyclin D3 | Cell Signaling | Cat# 2936; RRID:AB_2070801 |
| anti-cyclin E | Santa Cruz Biotechnology | Cat# sc-247; RRID:AB_627357 |
| anti-p21 | Cell Signaling | Cat# 2947; RRID:AB_823586 |
| anti-p27 | Cell Signaling | Cat# 2552; RRID:AB_10693314 |
| anti-pRb-Ser807/811 | Cell Signaling | Cat# 9308; RRID:AB_331472 |
| anti-pRb-thr356 | Abcam | Cat# ab4780; RRID:AB_304617 |
| anti-Rb | Cell Signaling | Cat# 9309; RRID:AB_823629 |
| anti-SMAD4 | Cell Signaling | Cat# 38454; RRID:AB_2728776 |
| anti-TGFβ | Santa Cruz Biotechnology | Cat# sc-146; RRID:AB_632486 |
| anti-TGFβ-R1 | Santa Cruz Biotechnology | Cat# sc-398; RRID:AB_632493 |
| anti-TGFβ-R2 | Santa Cruz Biotechnology | Cat# sc-400; RRID:AB_632497 |
| anti-TGFβ-R3 | Abcam | Cat# ab78421; RRID:AB_2202598 |
| anti-V5 | GenScript | Cat# A01733; RRID:AB_2622219 |
| anti-β-Actin | GenScript | Cat# A00702; RRID:AB_914102 |
| anti-mouse IgG HRP secondary | GE Healthcare | Cat# 45000679; RRID:AB_772210 |
| anti-rabbit IgG HRP secondary | GE Healthcare | Cat# 45000682; RRID:AB_2722659 |
| Biological Samples | ||
| 44 breast cancer biopsies | Dana-Farber Cancer Institute | part of voluntary research protocol 05-246 |
| Paired parotid cancer biopsies | See Infante et al., 2016 | NCT01237236 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Palbociclib | Selleckchem | Cat# S1116 |
| Palbociclib (bulk) | MedChem Express | Cat# HY-A0065 |
| Ribociclib | Selleckchem | Cat# S7440 |
| Abemaciclib | Selleckchem | Cat# S5716 |
| Puromycin Dihydrochloride | Sigma Aldrich | Cat# P8833 |
| DMEM | Thermo Fisher Scientific | Cat# 11965118 |
| RPMI 1640 | Thermo Fisher Scientific | Cat# 11875119 |
| McCoy’s 5A | Thermo Fisher Scientific | Cat# 16600082 |
| EMEM | Thermo Fisher Scientific | Catt# 11095080 |
| DMSO | Sigma Aldrich | Cat# D8418 |
| Lipofectime 3000 | Thermo Fisher Scientific | Cat# L3000008 |
| Trizol | Thermo Fisher Scientific | Cat# 15596026 |
| Galunisertib | Selleckchem | Cat# S2230 |
| Insulin | Sigma Aldrich | Cat# I9278 |
| FBS | Gemini Bio-Products | Cat# 900-108 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15140163 |
| Ampicillin | Sigma Aldrich | Cat# A5354 |
| Kanamycin | Thermo Fisher Scientific | Cat# 15160054 |
| LB Agar | Thermo Fisher Scientific | Cat# DF040117 |
| Protease Inhibitor Cocktail Set I | Calbiochem | Cat# 539131 |
| Phosphatase Inhibitor Cocktail Set I | Calbiochem | Cat# 524624 |
| Phosphatase Inhibitor Cocktail Set II | Calbiochem | Cat# 524625 |
| RIPA Buffer | Boston BioProducts | Cat# BP-115 |
| Total Exosome Isolation Reagent | Thermo Fisher Scientific | Cat# 4478359 |
| Crystal Violet | Sigma Aldrich | Cat# C6158 |
| Sodium L-lactate | Sigma Aldrich | Cat# L7022 |
| Propidium Iodide/Rnase buffer | Thermo Fisher Scientific | Cat# BDB550825 |
| GW4869 | Sigma Aldrich | Cat# D1692 |
| Manumycin A | Santa Cruz Biotechnology | Cat# 200857 |
| Chloroform | Sigma Aldrich | Cat# 288306 |
| Critical Commercial Assays | ||
| Human Cytokine Array Kit | R&D Systems | Cat# ARY005 |
| miRNA Profiling Array (7th Gen) | Exiqon | Cat# 208500 |
| Affymetrix Human Genome U133A 2.0 array | Affymetrix | Cat# 900468 |
| SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing | CloneTech | Cat# 634888 |
| NEBNext Small RNA Library Prep Set for Illumina | New Englend BioLabs | Cat# E7330S |
| Annexin V: FITC Apoptosis Detection Kit | Thermo Fisher Scientific | Cat# BDB556547 |
| AllPrep DNA/RNA Mini Kit | QIAGEN | Cat# 80204 |
| PureLink RNA Mini Kit | Thermo Fisher Scientific | Cat# 12183025 |
| Total Exosome RNA & Protein Isolation Kit | Thermo Fisher Scientific | Cat# 4478545 |
| miRCURY LNA RT Kit | Exiqon/QIAGEN | Cat# 339340 |
| nanoString nCounter v2 Cancer CNV Assay | nanoString | Cat# XT-CSO-CAN2-12 |
| EXOCET Exosome Quantitation Kit | System Biosciences | Cat# EXOCET96A-1 |
| CellTiter-Glo | Promega | Cat# G7572 |
| ThruPLEX DNA-Seq Kit | Rubicon Genomics | Cat# R400675 |
| Library Quantification Kit | Kapa Biosystems | Cat# 07960140001 |
| Deposited Data | ||
| Gene expression array 1 | Gene Expression Omnibus | GEO: GSE117748, GSE117742 |
| Gene expression array 2 | Gene Expression Omnibus | GEO: GSE117748, GSE117743 |
| miRNA array | Gene Expression Omnibus | GEO: GSE117748, GSE117745 |
| RNaseq | Gene Expression Omnibus | GEO: GSE117748, GSE117746 |
| miRNaseq | Gene Expression Omnibus | GEO: GSE117748, GSE117747 |
| Experimental Models: Cell Lines | ||
| MCF7 | ATCC | Cat# HTB-22 |
| T47D | ATCC | Cat# HTB-133 |
| BT-20 | ATCC | Cat# HTB-19 |
| SK-BR-3 | ATCC | Cat# HTB-30 |
| ZR-75-1 | ATCC | Cat# CRL-1500 |
| 293T | ATCC | Cat# CR-3216 |
| Experimental Models: Organisms/Strains | ||
| NCr-Foxn1NU nude mice | Taconic | Cat# NCRNU-F; RRID:IMSR_TAC:ncrnu |
| Oligonucleotides | ||
| hsa-miR-432-5p miRVana miRNA mimic | Thermo Fisher Scientific | Cat# 4464066 |
| miR-432-5p LNA PCR primer set | Exiqon/QIAGEN | Cat# 204776 |
| Synthetic microRNA Inhibitor, Human hsa-miR-432-5p | Sigma Aldrich | Cat# HSTUD0572 |
| Primers for qPCR, see Table S1 | Thermo Fisher Scientific | Cat# A15612 |
| CDK6 sgRNA-7: CCGCTCCACCCGCTCATCGT | Thermo Fisher Scientific | Cat# A15612 |
| Non-Target sgRNA: CGTGATGGTCTCGATTGAGT | Thermo Fisher Scientific | Cat# A15612 |
| ON-TARGETplus CDK6 siRNA set of 4 | Dharmacon | Cat# LU-003240-00-0002 |
| ON-TARGETplus CDK4 siRNA set of 4 | Dharmacon | Cat# LU-003238-00-0002 |
| ON-TARGETplus NT siRNA | Dharmacon | Cat# D0018100105 |
| Recombinant DNA | ||
| psPAX2 | Addgene | Cat# 12260; RRID:Addgene_12260 |
| pCMV-VSV-G | Addgene | Cat# 8454; RRID:Addgene_8454 |
| lentiCRISPR v2 | Addgene (Sanjana et al., 2014) | Cat# 52961; RRID:Addgene_52961 |
| pLEX_307 | Addgene | Cat# 41392; RRID:Addgene_41392 |
| pLKO.1 | Addgene (Moffat et al., 2006) | Cat# 10878; RRID:Addgene_10878 |
| Gene art sythetic constructs - CDK6 cDNA | Thermo Fisher Scientific | NA |
| Gene art sythetic constructs - GFP cDNA | Thermo Fisher Scientific | NA |
| Gene art sythetic constructs - SMAD4 cDNA | Thermo Fisher Scientific | NA |
| shCDK1 pLKO.1 | Harvard Plasmid Core | Cat# TRCN0000000583 |
| shCDK2 pLKO.1 | Harvard Plasmid Core | Cat# TRCN0000039958 |
| shCDK4 pLKO.1 | Harvard Plasmid Core | Cat# TRCN0000010472 |
| shCDK6-1 pLKO.1 | Harvard Plasmid Core | Cat# TRCN0000039744 |
| shCDK6-2 pLKO.1 | Harvard Plasmid Core | Cat# TRCN0000039747 |
| shCCND1 pLKO.1 | Harvard Plasmid Core | Cat# TRCN0000040039 |
| shCCND3 pLKO.1 | Harvard Plasmid Core | Cat# TRCN0000003827 |
| shCCNE1 pLKO.1 | Harvard Plasmid Core | Cat# TRCN0000045302 |
| Non-Target control - shNT pLKO.1 | Dharmacon | Cat# RHS6848 |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Transcriptome Analysis Console (version 4.0.1.36) | Applied Biosystems | https://www.thermofisher.com/us/en/home/global/forms/life-science/download-tac-software.html |
| Combenefit (version 2.021) | Di Veroli et al., 2016 | https://sourceforge.net/projects/combenefit/ |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| bcl2fastq | Illumina | https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html |
| Seqbuster | Pantano et al., 2010, 2011 | https://github.com/lpantano/seqbuster |
| Cufflinks | Trapnell et al., 2010 | http://cole-trapnell-lab.github.io/cufflinks/ |
Highlights.
CDK4/6 inhibitor resistance is mediated by increased CDK6 expression
Resistance can be conferred between cell populations via extracellular signaling
Increased exosomal miR-432-5p suppresses the TGF-β pathway, increasing CDK6 levels
Resistance is reversible by prolonged drug removal in vivo
ACKNOWLEDGMENTS
This work was supported by a Developmental Research Project grant as part of the Dana-Farber/Harvard Cancer Center Specialized Program of Research Excellence (SPORE) in Breast Cancer, NIH grant P50 CA168504 (G.I.S.), a Dana-Farber Cancer Institute (DFCI) Department of Medical Oncology Discovery Grant (L.C. and G.I.S.), Susan G. Komen Career Catalyst Research Grant CCR15333343 (N.W.), American Association for Cancer Research (AACR) NextGen Grant for Transformative Cancer Research 1620-38-WAGL (N.W.), as well as grants from the V Foundation (N.W.) and The Cancer Couch Foundation (N.W.). S.A.W. was supported by Dana-Farber Cancer Institute T32CA009172 Graduate Training in Cancer Research grant, a Dana-Farber Cancer Institute Wong Family Award, and an American Society of Clinical Oncology (ASCO) Young Investigator Award (YIA).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online athttps://doi.org/10.1016/j.celrep.2019.02.023.
DECLARATION OF INTERESTS
G.I.S. has received research funding from Eli Lilly, Merck KGaA/EMD-Serono, Merck, and Sierra Oncology. He has served on advisory boards for Pfizer, Eli Lilly, G1 Therapeutics, Roche, Merck KGaA/EMD-Serono, Sierra Oncology, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Astex, Almac, Ipsen, Bayer, Angiex, and Daiichi Sankyo. The Dana-Farber Cancer Institute has received funding from Pfizer and Array BioPharma for the conduct of investigator-initiated clinical trials of palbociclib led by G.I.S. N.W. is a consultant for Novartis, Eli Lilly, and Foundation Medicine, an equity holder in Foundation Medicine, and receives sponsored research support from Novartis, Merck, and Puma Biotechnology. S.A.W. is a consultant for Foundation Medicine, Eli Lilly, and InfiniteMD, and holds equity in InfiniteMD. L.C. and G.I.S. have a pending patent related to this work entitled “Compositions and Methods for Predicting Response and Resistance to CDK4/6 Inhibition.” This application claims the benefit of priority under 35 U.S.C. § 119(e) to U. S. Provisional Application No. 62/538,319, filed July 28, 2017.
REFERENCES
- Alves CL, Elias D, Lyng M, Bak M, Kirkegaard T, Lykkesfeldt AE, and Ditzel HJ (2016). High CDK6 Protects Cells from Fulvestrant-Mediated Apoptosis and is a Predictor of Resistance to Fulvestrant in Estrogen Receptor-Positive Metastatic Breast Cancer. Clin. Cancer Res 22, 5514–5526. [DOI] [PubMed] [Google Scholar]
- Arnold A, and Papanikolaou A (2005). Cyclin D1 in breast cancer pathogenesis. J. Clin. Oncol 23, 4215–4224. [DOI] [PubMed] [Google Scholar]
- Bockstaele L, Bisteau X, Paternot S, and Roger PP (2009). Differential regulation of cyclin-dependent kinase 4 (CDK4) and CDK6, evidence that CDK4 might not be activated by CDK7, and design of a CDK6 activating mutation. Mol. Cell. Biol 29, 4188–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiron D, Martin P, Di Liberto M, Huang X, Ely S, Lannutti BJ, Leonard JP, Mason CE, and Chen-Kiang S (2013). Induction of prolonged early G1 arrest by CDK4/CDK6 inhibition reprograms lymphoma cells for durable PI3Kδ inhibition through PIK3IP1. Cell Cycle 12, 1892–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YE, Pan Y, Park E, Konstantinopoulos P, De S, D’Andrea A, and Chowdhury D (2014). MicroRNAs down-regulate homologous recombination in the G1 phase of cycling cells to maintain genomic stability. eLife 3, e02445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, Dubois J, Nagy RJ, Lanman RB, Iafrate AJ, et al. (2018). Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol 29, 640–645. [DOI] [PubMed] [Google Scholar]
- Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, et al. (2016). Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol 17, 425–439. [DOI] [PubMed] [Google Scholar]
- Dall’Acqua A, Sonego M, Pellizzari I, Pellarin I, Canzonieri V, D’Andrea S, Benevol S, Sorio R, Giorda G, Califano D, et al. (2017). CDK6 protects epithelial ovarian cancer from platinum-induced death via FOXO3 regulation. EMBO Mol. Med 9, 1415–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Liberto M, Huang X, Elemento O, Eng K, Blum KA, Bartlett NL, Park SI, Ruan J, Maddocks KJ, Inghirami G, et al. (2016). PIK3IP1 Inhibition of PI3K in G1 Arrest Induced By CDK4 Inhibition Reprograms MCL for Ibrutinib Therapy. Blood 128, 610. [Google Scholar]
- Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, and Jodrell DI (2016). Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics 32, 2866–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickler MN, Tolaney SM, Rugo HS, Cortés J, Diéras V, Patt D, Wildiers H, Hudis CA, O’Shaughnessy J, Zamora E, et al. (2017). MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR+/HER2− Metastatic Breast Cancer. Clin. Cancer Res 23, 5218–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsheikh S, Green AR, Aleskandarany MA, Grainge M, Paish CE, Lambros MB, Reis-Filho JS, and Ellis IO (2008). CCND1 amplification and cyclin D1 expression in breast cancer and their relation with proteomic subgroups and patient outcome. Breast Cancer Res. Treat 109, 325–335. [DOI] [PubMed] [Google Scholar]
- Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, et al. (2016). Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med 375, 1925–1936. [DOI] [PubMed] [Google Scholar]
- Formisano L, Lu Y, Jansen VM, Bauer JA, Hanker AB, Sanders ME, González-Ericsson P, Kim S, Arnedos M, André F, and Arteaga CL (2017). Abstract 1008: gain-of-function kinase library screen identifies FGFR1 amplification as a mechanism of resistance to antiestrogens and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. 77, 1008.28011623 [Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, and Toogood PL (2004). Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther 3, 1427–1438. [PubMed] [Google Scholar]
- Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, Park IH, Trédan O, Chen SC, Manso L, et al. (2017). MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol 35, 3638–3646. [DOI] [PubMed] [Google Scholar]
- Hannafon BN, Trigoso YD, Calloway CL, Zhao YD, Lum DH, Welm AL, Zhao ZJ, Blick KE, Dooley WC, and Ding WQ (2016). Plasma exosome microRNAs are indicative of breast cancer. Breast Cancer Res. 18, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M, Rodriguez O, Grueso J, et al. (2016). Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 76, 2301–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, Campone M, Blackwell KL, André F, Winer EP, et al. (2016). Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med 375, 1738–1748. [DOI] [PubMed] [Google Scholar]
- Hu MG, Deshpande A, Schlichting N, Hinds EA, Mao C, Dose M, Hu GF, Van Etten RA, Gounari F, and Hinds PW (2011). CDK6 kinase activity is required for thymocyte development. Blood 117, 6120–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante JR, Cassier PA, Gerecitano JF, Witteveen PO, Chugh R, Ribrag V, Chakraborty A, Matano A, Dobson JR, Crystal AS, et al. (2016). A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clin. Cancer Res 22, 5696–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen VM, Bhola NE, Bauer JA, Formisano L, Lee KM, Hutchinson KE, Witkiewicz AK, Moore PD, Estrada MV, Sánchez V, et al. (2017). Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Cancer Res. 77, 2488–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, and Ochiya T (2010). Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem 285, 17442–17452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, and Korc M (2012). Cdk4/6 inhibition induces epithelial-mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol. Cancer Ther 11, 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Mao P, Kusiel J, Cohen O, and Wagle N (2018). Abstract PD4-01: The role of FGF/FGFR axis in resistance to SERDs and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. 78, PD4–PD01. [Google Scholar]
- Massagué J (2004). G1 cell-cycle control and cancer. Nature 432, 298–306. [DOI] [PubMed] [Google Scholar]
- Mittelbrunn M, Gutiérrez-Vázquez C, Villarroya-Beltri C, González S, Sánchez-Cabo F, González MA, Bernad A, and Sánchez-Madrid F (2011). Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun 2, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, Baty CJ, Gibson GA, Erdos G, Wang Z, et al. (2012). Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 119, 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, André F, Loibl S, Loi S, Garcia-Murillas I, et al. (2018). The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 8, 1390–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantano L, Estivill X, and Martí E (2010). SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res. 38, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantano L, Estivill X, and Martí E (2011). A non-biased framework for the annotation and classification of the non-miRNA small RNA transcriptome. Bioinformatics 27, 3202–3203. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. (2000). Molecular portraits of human breast tumours. Nature 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Rabinowits G, Gerçel-Taylor C, Day JM, Taylor DD, and Kloecker GH (2009). Exosomal microRNA: a diagnostic marker for lung cancer. Clin. Lung Cancer 10, 42–46. [DOI] [PubMed] [Google Scholar]
- Roberts PJ, Bisi JE, Strum JC, Combest AJ, Darr DB, Usary JE, Zamboni WC, Wong KK, Perou CM, and Sharpless NE (2012). Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J. Natl. Cancer Inst 104, 476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledge GW Jr., Toi M, Neven P, Sohn J, Inoue K, Pivot X, Burdaeva O, Okera M, Masuda N, Kaufman PA, et al. (2017). MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol 35, 2875–2884. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubari M, Taipale J, Tiihonen E, Keski-Oja J, and Laiho M (1999). Hepatocyte growth factor releases mink epithelial cells from transforming growth factor β1-induced growth arrest by restoring CDK6 expression and cyclin E-associated Cdk2 activity. Mol. Cell. Biol 19, 3654–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, DeMichele A, Harbeck N, André F, Zhang Z, et al. (2018). Abstract CT039: Cyclin E1 (CCNE1) expression associates with benefit from palbociclib in metastatic breast cancer (MBC) in the PALOMA3 trial. Cancer Res. 78, CT039. [Google Scholar]
- Velasco-Velázquez MA, Li Z, Casimiro M, Loro E, Homsi N, and Pestell RG (2011). Examining the role of cyclin D1 in breast cancer. Future Oncol. 7, 753–765. [DOI] [PubMed] [Google Scholar]
- Wander SA, Cohen O, Johnson GN, Kim D, Luo F, Mao P, Nayar U, Helvie K, Marini L, Freeman S, et al. (2018). Whole exome sequencing (WES) in hormone-receptor positive (HR+) metastatic breast cancer (MBC) to identify mediators of resistance to cyclin-dependent kinase 4/6 inhibitors (CDK4/6i). J. Clin. Oncol 36 (No. 15 Suppl), 12016. [Google Scholar]
- Wang L, Wang J, Blaser BW, Duchemin A-M, Kusewitt DF, Liu T, Caligiuri MA, and Briesewitz R (2007). Pharmacologic inhibition of CDK4/6: mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood 110, 2075–2083. [DOI] [PubMed] [Google Scholar]
- Wani S, and Cloonan N (2014). Profiling direct mRNA-microRNA interactions using synthetic biotinylated microRNA-duplexes. bioRxiv. 10.1101/005439. [DOI] [Google Scholar]
- Yang C, Li Z, Bhatt T, Dickler M, Giri D, Scaltriti M, Baselga J, Rosen N, and Chandarlapaty S (2017). Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36, 2255–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Sicinska E, Geng Y, Ahnström M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stål O, and Sicinski P (2006). Requirement for CDK4 kinase function in breast cancer. Cancer Cell 9, 23–32. [DOI] [PubMed] [Google Scholar]
- Zhang F, Taipale M, Heiskanen A, and Laiho M (2001). Ectopic expression of CDK6 circumvents transforming growth factor-beta mediated growth inhibition. Oncogene 20, 5888–5896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been deposited in the Gene Expression Omnibus (GEO) under the accession number GEO: GSE117748.