

Abstract
Aromatic compounds are a common carbon and energy source for many microorganisms, some of which can even degrade toxic chloroaromatic xenobiotics. This comparative study of aromatic metabolism in 32 Betaproteobacteria species describes the links between several transcription factors (TFs) that control benzoate (BenR, BenM, BoxR, BzdR), catechol (CatR, CatM, BenM), chlorocatechol (ClcR), methylcatechol (MmlR), 2,4-dichlorophenoxyacetate (TfdR, TfdS), phenol (AphS, AphR, AphT), biphenyl (BphS), and toluene (TbuT) metabolism. We characterize the complexity and variability in the organization of aromatic metabolism operons and the structure of regulatory networks that may differ even between closely related species. Generally, the upper parts of pathways, rare pathway variants, and degradative pathways of exotic and complex, in particular, xenobiotic compounds are often controlled by a single TF, while the regulation of more common and/or central parts of the aromatic metabolism may vary widely and often involves several TFs with shared and/or dual, or cascade regulation. The most frequent and at the same time variable connections exist between AphS, AphR, AphT, and BenR. We have identified a novel LysR-family TF that regulates the metabolism of catechol (or some catechol derivative) and either substitutes CatR(M)/BenM, or shares functions with it. We have also predicted several new members of aromatic metabolism regulons, in particular, some COGs regulated by several different TFs.
Keywords: comparative genomics, transcription factor binding site (TFBS), aromatic metabolism regulation, benzoate, phenol, biphenyl, catechol, chlorocatechol
Introduction
Aromatic compounds such as phenol, toluene, xylene, benzoate are the second most abundant class of organic compounds after carbohydrates and a common carbon and energy source for many microorganisms (Arai et al., 1999; López Barragán et al., 2004; Gescher et al., 2005; Carmona et al., 2009; Li et al., 2010; Marín et al., 2010; Valderrama et al., 2012). In natural habitats, they are accumulated mainly due to the degradation of plant-derived molecules (e.g., lignin) (Li et al., 2010). On the other hand, stability of the benzene ring together with massive production and release of various xenobiotic aromatic compounds into the environment makes them one of the most significant classes of pollutants (López Barragán et al., 2004; Carmona et al., 2009). Notably, some bacteria can grow on toxic chloroaromatic compounds, e.g., (poly)chlorinated phenol or biphenyl derivatives, which are serious environmental pollutants generated by human industrial activities (Ohtsubo et al., 2001; Chávez et al., 2004; Takeda et al., 2004). For example, soil bacteria can degrade 2,4-dichlorophenoxyacetic acid (2,4-D), a widely used herbicide (Vedler et al., 2004; Trefault et al., 2009). Thus, bacterial metabolism of aromatic compounds and its regulation is an important research object.
Structurally diverse aromatic compounds are usually converted to a limited number of simpler common intermediates, such as benzoate or catechol, which are subsequently channeled into the tricarboxylic acid (TCA) cycle (Cowles et al., 2000; Tover et al., 2000, 2001; Carmona et al., 2009; Li et al., 2010; Marín et al., 2010). Hence, aromatic biodegradative routes may be divided into the upper and lower pathways, the latter connected with the few central intermediates, and combinations of various upper pathways with standard lower pathways can sufficiently broaden substrate specificities of bacteria and are an important factor in their adaptation to xenobiotics in the environment (Klemba et al., 2000).
Genes of various aromatic metabolic pathways, especially those involved in the degradation of xenobiotic compounds, are often located on plasmids, thus allowing for their efficient lateral transfer and adaptation of bacteria to novel compounds, expanding their catabolic abilities (Arai et al., 1999; Klemba et al., 2000; Vedler et al., 2004). Moreover, sets of aromatic metabolism genes are often organized in large supraoperonic clusters and associated with mobile DNA elements, in particular ICE (integrative and conjugative elements, characterized by insertion into tRNAGlygenes), that are likely responsible for their transfer and insertion (Laemmli et al., 2000; Carmona et al., 2009; Lechner et al., 2009; Zamarro et al., 2016; Miyazaki et al., 2018).
While various aromatic metabolic pathways, their biochemistry and regulation are rather well-studied in many bacteria, not much is known about connections and interactions between the regulators of these pathways. Here we describe the links between several transcription factors (TFs) that control different steps of aromatic metabolism (including dual and cascade regulation) and demonstrate variability in the organization of the aromatic metabolism operons and the structure of the regulatory networks even between closely related species.
Phenol, Biphenyl, and Toluene Metabolism
The aph and bph genes encode proteins of the phenol and biphenyl (or polychlorinated biphenyl) degradation, respectively (Arai et al., 1999, 2000; Ohtsubo et al., 2001; Takeda et al., 2004).
In the aph-encoded pathway, phenol is converted into catechol via multi-component phenol hydroxylase AphKLMNOP (and ferredoxin AphQ), which further goes through the meta-cleavage by the catechol-2,3-dioxygenase AphB (Arai et al., 1999, 2000). Resulting 2-hydroxymuconate semialdehyde can be converted yielding 2-hydroxypenta-2,4-dienoate and formate either directly by a 2-hydroxymuconic semialdehyde hydrolase (COG596, MhpC/DmpD/XylF) or through 2-hydroxymuconate and 4-oxalocrotonate via the 2-hydroxymuconic semialdehyde dehydrogenase AphC, 4-oxalocrotonate tautomerase AphI, and 4-oxalocrotonate decarboxylase AphH (Figure 1 and Supplementary Table S1) (Arai et al., 2000).
FIGURE 1.

Scheme of benzoate, phenol, biphenyl, toluene, 2,4-dichlorophenoxyacetate, 3-chlorocatechol and 4-methylcatechol utilization and its regulation in Betaproteobacteria. Dashed arrows denote transporters, arrow color denotes regulation by the following TFs: AphS/BphS – red, AphR – orange, AphT – green, BenR – pink, BoxR/BzdR – brown, CatR and/or CatM/BenM – light blue, CatR∗ – dark blue, ClcR and TfdR(S) – magenta.
Biphenyl is converted via the biphenyl 2,3-dioxygenase BphA into biphenyl dihydrodiol (2,3-dihydroxy-4-phenylhexa-4,6-diene), which is then transformed into 2,3-dihydroxybiphenyl by the 2,3-dihydrobiphenyl-2,3-diol dehydrogenase BphB (Ohtsubo et al., 2001; Takeda et al., 2004). The 2,3-dihydroxybiphenyl ring is cleaved by the 2,3-dihydroxybiphenyl-1,2-dioxygenase BphC, and the resulting intermediate (2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate) is split into benzoate and 2-hydroxypenta-2,4-dienoate by the 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate hydrolase BphD (Ohtsubo et al., 2001; Takeda et al., 2004).
Further reactions are common for the phenol and biphenyl utilization pathways: 2-hydroxypenta-2,4-dienoate is converted to acetyl-CoA and pyruvate by the consecutive action of the 2-hydroxypenta-2,4-dienoate hydratase AphE/BphE, 4-hydroxy-2-ketovalerate aldolase AphG/BphF, and acetaldehyde dehydrogenase AphF/BphG (Figure 1 and Supplementary Table S1) (Arai et al., 2000; Ohtsubo et al., 2001; Takeda et al., 2004).
Similarly to reactions with phenol, toluene can be converted via multi-component toluene-2-monoxygenase (tbmABCDEF, e.g., in Burkholderia vietnamiensis G4), toluene-3-monooxygenase (tbuA1UBVA2C, e.g., in Ralstonia pickettii PKO1) or toluene-4-monooxygenase (tmoABCDE, e.g., in Pseudomonas mendocina KR1) to o-cresol, m-cresol, or p-cresol, respectively, which are subsequently metabolized through ortho- or meta-cleavage pathways (Yen et al., 1991; Byrne et al., 1995; Byrne and Olsen, 1996; Leahy et al., 1997; Kahng et al., 2000).
In general, the aph and bph genes are organized into long operons, and their expression is controlled by one or more TFs, usually transcribed divergently from the regulated genes (Arai et al., 1999, 2000; Ohtsubo et al., 2001; Teramoto et al., 2001; Takeda et al., 2004). Regulators of the aromatic catabolism are usually activators, though there are examples of repressor-mediated regulation (Ohtsubo et al., 2001). For example, in Comamonas testosteroni TA441, the aph gene cluster is regulated by the GntR-family repressor AphS, activator AphR from the XylR/DmpR subclass of the NtrC family (TFs from this group often control multi-component phenol hydroxylases), and the LysR-family activator AphT (CatR/ClcR-type) (Arai et al., 1999, 2000; Teramoto et al., 2001). AphS and AphR control expression of their own aphRS operon and divergently transcribed genes of phenol hydroxylase and catechol-2,3-dioxygenase, while other aph genes of the lower pathway are under dual regulation by AphR and AphT (Arai et al., 1999, 2000; Teramoto et al., 2001). Toluene metabolism genes in some bacteria also are regulated by homologous TFs from the XylR/DmpR group (e.g., TbuT in R. pickettii PKO1, TbmR in B. vietnamiensis G4) (Byrne and Olsen, 1996; Leahy et al., 1997; Kahng et al., 2000).
AphS is highly similar to the repressor of the bph genes, BphS, described in, e.g., Burkholderia xenovorans LB400 and Ralstonia eutropha A5 (not to be confused with BphS from the BphST two-component system, e.g., in Rhodococcus sp. RHA1) (Arai et al., 1999; Ohtsubo et al., 2001; Takeda et al., 2004). The binding motif of AphS and BphS is a palindrome with the consensus AwATATCGATATwT; multiple sites have been identified upstream of the regulated genes, which implies cooperative TF binding (Arai et al., 1999; Ohtsubo et al., 2001). The putative AphR binding motif, GCTTGATCATTTGATCATGC, predicted in Comamonas testosteroni TA441, is similar to the recognition sites of DmpR, XylR, and HbpR that are TFs from the same family (Arai et al., 1999; Jaspers et al., 2001; Tropel and van der Meer, 2002).
Benzoate and (Chloro/Methyl)Catechol Metabolism
Bacteria employ three different strategies for the degradation of benzoate – aerobic, anaerobic, and the hybrid one, which has features of both aerobic and anaerobic pathways (Valderrama et al., 2012; Chen et al., 2014). The classical aerobic pathway starts with hydroxylation of benzoate by mono- and dioxygenases into catechol, protocatechuate, or gentisate, e.g., in a two-step process catalyzed by BenABC and BenD (Collier et al., 1998; Cowles et al., 2000; Gescher et al., 2005, 2006; Li et al., 2010; Chen et al., 2014). These compounds are further cleaved by various dioxygenases either between the two hydroxyl groups (ortho-cleavage, β-ketoadipate pathway) or next to one of the hydroxyls (meta-cleavage) (Collier et al., 1998; Cowles et al., 2000; Gescher et al., 2002, 2005; Li et al., 2010; Chen et al., 2014). The ortho-cleavage pathways of catechol, 3-chlorocatechol, and 2,4-D (encoded by the cat, clc, and tfd genes, respectively) are parallel catabolic pathways that convert these compounds into the common intermediate β-ketoadipate, and further channel it into the TCA cycle, namely into acetyl-CoA and succinyl-CoA (Parsek et al., 1994; McFall et al., 1997b; Chugani et al., 1998; Laemmli et al., 2000; Vedler et al., 2004; Ezezika et al., 2006; Li et al., 2010). This conversion involves aromatic ring cleavage into (chloro)muconate and formation of corresponding lactones, carried out by homologous enzymes – dioxygenases CatA, ClcA, and TfdC, and cycloisomerases CatB, ClcB, and TfdD, respectively (Figure 1 and Supplementary Table S1) (McFall et al., 1997a,b; Chugani et al., 1998; Tover et al., 2001).
The pathways mentioned above are regulated by homologous TFs from the LysR family: the cat genes are controlled by CatR or CatM (e.g., in Pseudomonas putida and Acinetobacter baylyi ADP1, respectively); the clc genes, by ClcR (e.g., in P. putida); and the tfd genes (e.g., in Ralstonia eutropha JMP134), by TfdR and an almost identical paralog, TfdS (Parsek et al., 1994; Leveau and van der Meer, 1996; Chugani et al., 1997, 1998; McFall et al., 1997a,b; Collier et al., 1998; Laemmli et al., 2000; Tover et al., 2000, 2001; Ezezika et al., 2006). Moreover, they are not only homologous, but share functions and can partially substitute each other (Parsek et al., 1994; McFall et al., 1997b; Trefault et al., 2009). The genes of all these LysR-family TFs are divergently transcribed from the regulated operons (Parsek et al., 1994; Leveau and van der Meer, 1996; Chugani et al., 1997, 1998; McFall et al., 1997a,b; Laemmli et al., 2000). The binding motifs of these TFs are similar inverted repeats with the consensus AkACC–N5–GGTAT, conforming to the general LysR-family motif T–N11–A (Parsek et al., 1994; Chugani et al., 1997, 1998; McFall et al., 1997a,b; Laemmli et al., 2000; Tover et al., 2000).
Benzoate metabolism is regulated by the AraC/XylS-type activator BenR (e.g., in Pseudomonas putida) and LysR-family activator BenM (e.g., in Acinetobacter baylyi ADP1) (Collier et al., 1998; Cowles et al., 2000; Ezezika et al., 2006; Li et al., 2010). BenR binding sites are repeated sequences with consensus TGCA–N6–GGNTA (Cowles et al., 2000; Li et al., 2010). Highly homologous regulators BenM and CatM bind similar DNA motifs and can partially substitute each other, playing overlapping roles in controlling the expression of the ben and cat genes, which are involved in the benzoate consumption and catabolism in response to different effectors (Collier et al., 1998; Ezezika et al., 2006; Li et al., 2010).
The first step of the anaerobic benzoate degradation is its activation to benzoyl-CoA by benzoate-CoA ligase BzdA (Geissler et al., 1988; Gescher et al., 2002; López Barragán et al., 2004; Barragán et al., 2005; Durante-Rodríguez et al., 2008; Carmona et al., 2009; Valderrama et al., 2012). The next reaction is reductive dearomatization of the aromatic ring by benzoyl-CoA reductase (BcrCBAD/BadDEFG/BzdNOPQ with ferredoxin Fdx/BadB/BzdM as an electron donor), yielding cyclohex-1,5-diene-1-carbonyl-CoA or cyclohex-1-ene-carbonyl-CoA (Koch and Fuchs, 1992; López Barragán et al., 2004; Durante-Rodríguez et al., 2008; Carmona et al., 2009; Valderrama et al., 2012; Chen et al., 2014).
Through a series of reactions including hydrolytic ring cleavage (carried out by the hydratase Dch/BadK/BzdW, dehydrogenase Had/BadH/BzdX, hydrolase Oah/BadI/BzdY), these intermediates are subsequently converted into 3-hydroxypimelyl-CoA or pimelyl-CoA, and finally converted via β-oxidation-like reactions and decarboxylation into acetyl-CoA, which is channeled into the central metabolism (Figure 1 and Supplementary Table S1) (Carmona et al., 2009; López Barragán et al., 2004; Valderrama et al., 2012; Chen et al., 2014).
Another way of the benzoate catabolism is the aerobic hybrid pathway (CoA-dependent epoxide box pathway), starting with the activation of benzoate to benzoyl-CoA by benzoate-CoA ligase BclA, as in the anaerobic metabolism (Gescher et al., 2002; Valderrama et al., 2012; Chen et al., 2014). Some bacteria, for example, Thauera and Magnetospirillum strains, have one benzoate-CoA ligase for both bzd and box pathways and a single regulator, while others, e.g. Azoarcus sp. CIB and Azoarcus aromaticum EbN1 strains, have two different ligases and regulators, for the aerobic (BclA and BoxR) and anaerobic (BzdA and BzdR) pathways, respectively (Carmona et al., 2009; Valderrama et al., 2012). Benzoyl-CoA oxygenase BoxAB and dihydrolase BoxC subsequently oxidatively dearomatize and hydrolytically cleave benzoyl-CoA into 3,4-dehydroadipyl-CoA semialdehyde (Gescher et al., 2002, 2005, 2006; Valderrama et al., 2012; Chen et al., 2014). The latter is oxidized by 3,4-dehydroadipyl-CoA semialdehyde dehydrogenase BoxD to 3,4-dehydroadipyl-CoA (Gescher et al., 2006; Chen et al., 2014), which is further converted through several coenzyme A thioesters to succinyl-CoA and acetyl-CoA via reactions similar to the β-oxidation and β-ketoadipate pathway (Figure 1 and Supplementary Table S1) (Gescher et al., 2002, 2006; Valderrama et al., 2012; Chen et al., 2014). Both aerobic and anaerobic metabolic pathways for the benzoate degradation may occur simultaneously (Valderrama et al., 2012; Chen et al., 2014). The box pathway is more widely distributed among Betaproteobacteria than in the other taxa (Chen et al., 2014).
BoxR and BzdR are closely related repressor proteins that have an N-terminal DNA-binding domain homologous to that of xenobiotic response element (XRE) transcriptional regulators and a C-terminal domain similar to shikimate kinase (PF01202) (Barragán et al., 2005; Durante-Rodríguez et al., 2008, 2010; Valderrama et al., 2012). BzdR controls expression of the adjacent bzd genes involved in the anaerobic degradation of benzoate; the genes are typically organized in a single operon, e.g., bzdNOPQMSTUVWXYZA in Azoarcus sp. CIB (López Barragán et al., 2004; Barragán et al., 2005; Durante-Rodríguez et al., 2008, 2010; Valderrama et al., 2012). BoxR represses the aerobic hybrid pathway of the benzoate utilization encoded by the box genes (Valderrama et al., 2012).
The BoxR and BzdR regulators are highly similar not only in sequence, but also in the DNA recognition mechanism, they act synergistically and cross-regulate the box and bzd genes, possibly as an adaptation to changing oxygen concentrations (Carmona et al., 2009; Valderrama et al., 2012). Both BoxR and BzdR bind promoter regions containing several repeats of the TGCA sequence that usually form longer palindromes, possibly cooperatively binding several BzdR dimmers (Barragán et al., 2005; Durante-Rodríguez et al., 2008, 2010; Valderrama et al., 2012). This conforms to the fact that other TFs from the HTH-XRE family also bind short repeated sequences that, in most cases, are located within palindromic regions (Barragán et al., 2005; Durante-Rodríguez et al., 2008). These TGCA sequences are separated by 1, 6, or 15 nucleotides (Barragán et al., 2005; Valderrama et al., 2012).
Degradation of 4-methylcatechol via ortho-cleavage usually leads to the formation of 4-methylmuconolactone, which bacteria are rarely able to utilize (Marín et al., 2010). The process involves 4-methylmuconolactone isomerization and rearrangement of the double bond by methylmuconolactone isomerases MmlI and MmlJ, while resulting 4-methyl-3-ketoadipate enol-lactone is converted into 4-methyl-3-ketoadipate via 4-methyl-3-ketoadipate enol-lactone hydrolase MmlL (Marín et al., 2010). Further metabolism is analogous to the reactions of the β-ketoadipate pathway: 4-methyl-3-ketoadipyl-CoA-transferase MmlFG and thiolase MmlC consequently transform 4-methyl-3-ketoadipate into methylsuccinyl-CoA and acetyl-CoA (Figure 1 and Supplementary Table S1) (Marín et al., 2010). The mml operon includes mmlR that encodes a LysR-family TF likely regulating the operon transcription (Marín et al., 2010).
Goals
Here, we apply comparative genomics methods to characterize the regulation of the main aromatic metabolic pathways in a representative set of Betaproteobacteria. We reconstruct corresponding regulons and predict new regulon members and their function, and describe the interactions between the studied TFs.
Materials and Methods
For the comparative study we selected 32 genomes available for the analysis with RegPredict (Novichkov et al., 2010), which we consider to be a relatively unbiased, representative set of Betaproteobacteria (Supplementary Table S2). The genomes were downloaded from GenBank (Benson et al., 1999).
Homologs of proteins were identified by PSI-BLAST (Altschul et al., 1997) (E-value cutoff, 10-20), and orthologs were determined by construction of phylogenetic trees and analysis of the genomic context (e.g., co-localization with genes of a certain metabolic pathway in most genomes). Amino acid and nucleotide sequence alignments were performed using the MUSCLE tool (default parameters) (Edgar, 2004). Phylogenetic trees were built using PhyML (default parameters) (Dereeper et al., 2008) and visualized with Dendroscope (Huson et al., 2007).
Candidate binding sites were identified (or confirmed if they had been previously predicted) by phylogenetic footprinting, that is, manual inspection of alignments of orthologous upstream regions with identification of consecutive conserved nucleotide positions, following the assumption that binding sites are more conserved than intergenic regions in general (Rodionov, 2007). Nucleotide position weight matrices (PWMs) for each TF were constructed by the SignalX program as previously described (Gelfand et al., 2000), using training sets of upstream regions of genes presumably belonging to the respective regulons. PWMs were then used to search for additional regulon members.
Computational search for candidate binding sites in upstream gene regions [350 nucleotides (nt) upstream and 50 nt downstream relative to the annotated gene start] was performed using the GenomeExplorer program package (Mironov et al., 2000). Score thresholds for the identification of sites were selected so that candidate sites upstream of functionally relevant genes were accepted, while the fraction of genes preceded by candidate sites did not exceed 5% in each studied genome. Weaker sites (with scores 10% less than the threshold) were also taken into account if their positions were similar to positions of stronger sites upstream of orthologous genes and/or there were no stronger competing sites in the same intergenic region. New candidate members were assigned to a regulon if they were preceded by candidate binding sites in several genomes. The reconstructed regulons were extended to include all genes in putative operons, the latter defined as the strings of genes transcribed in the same direction, with intergenic distances not exceeding 200 nt, when such organization persisted in several genomes. Motif logos were constructed using WebLogo (Crooks et al., 2004).
Results
Metabolic pathways of aromatic compounds are diverse, and their regulation is often complex and evolutionally labile. Here we analyze the utilization pathways of phenol, biphenyl, benzoate, catechol and its chloro- and methyl-derivatives, describe their regulation in Betaproteobacteria, reconstruct the regulons, identify new regulon members, and predict their metabolic functions.
Regulation of the Phenol, Biphenyl, Toluene and Benzoate Metabolism by AphS/BphS, AphR, AphT, and BenR
AphS and BphS are very close homologs and their binding motifs have a similar structure. Hence, here these TFs have been assigned names AphS or BphS according to the branching of the phylogenetic tree (Supplementary Figure S1) and co-localization of the regulatory genes with either phenol or biphenol metabolic genes (genomic context).
Phylogenetic footprinting confirmed, and further site search refined the previously described AphS/BphS binding motif (Ohtsubo et al., 2001), which is an even 14 nt palindrome with the consensus AAATmTCGAkATTT (Figure 2A).
FIGURE 2.

LOGO diagrams of transcription factor binding motifs. (A) AphS/BphS, AphR, BenR, and BoxR/BzdR. (B) LysR-family regulators of the aromatic metabolism. The LOGO diagrams were built using all the predicted binding sites for the respective TF in all studied genomes (see Supplementary Table S3), the exact numbers of sites are given in parentheses.
Among the studied bacteria, BphS was identified only in Burkholderia xenovorans LB400 and Polaromonas naphthalenivorans CJ2, which also lack AphR, AphT, and aph genes of phenol utilization, and in Azoarcus sp. BH72 (see below). The BphS regulon in B. xenovorans LB400 and P. naphthalenivorans CJ2 consists of one operon, bphSA1A2XA3A4BCKEGFD, that is comprised of the regulatory gene and genes encoding the complete pathway of the biphenyl conversion into pyruvate and acetyl-CoA (Figure 3 and Supplementary Tables S1, S2). Close homologs (possibly orthologs) of some bph genes are identified in B. petrii DSM 12804, which has neither AphS/BphS, AphR, nor AphT; they are clustered together with the clc genes, but are not regulated by either ClcR or CatR, and may be under regulation of an adjacent AraC-family TF (Bpet3746).
FIGURE 3.

Operon organization and regulation of aromatic metabolism genes regulated by AphS/BphS and AphR in Burkholderia spp., P. naphthalenivorans CJ2, Ralstonia spp., and V. eiseniae EF01-2. Color denotes regulation by the following TFs: AphS/BphS – red, AphR – orange. Double coloring denotes dual regulation. Genes/operons are shown by arrows, black arrows denote pseudogenes, gray – transposases; “hp” indicates hypothetical proteins. Genome abbreviations as in Supplementary Table S2.
Interactions between regulators AphS, AphR, AphT, and BenR, and the operon organization of their regulated genes vary, and the exposition below is structured according to these interactions, starting with bacteria that have only AphS and AphR TFs as the regulators of the phenol metabolism, and continued with bacteria where the phenol metabolism is regulated by AphS, AphR, and AphT.
Bacteria having AphS always have AphR as well, their genes usually forming the aphRS operon. Burkholderia sp. 383, Ralstonia eutropha JMP134, Ralstonia pickettii 12J, and Verminephrobacter eiseniae EF01-2 have only AphS and AphR as the phenol regulators, and their aph metabolic genes are organized in a long operon under dual regulation (Figure 3 and Supplementary Tables S2, S3).
Moreover, in R. eutropha JMP134 and R. pickettii 12J, the toluene transport and metabolic genes tbuX (Kahng et al., 2000; van den Berg et al., 2015) and tmoABCDEF, localized between aphRS and other aph genes, are regulated not only by their own repressor TbuT, but by both AphS and AphR. In Ralstonia metallidurans CH34 that lacks AphS, AphR, and TbuT, the tmoABCDEF genes are likely co-transcribed with the aph genes as a single operon, possibly under regulation of the AphR/TbuT homolog (Rmet_1305, Supplementary Figure S2), though no binding motif of this TF could be identified. Burkholderia vietnamiensis G4 features the same organization of this operon, except having only tmoA out of all toluene metabolic genes, possibly as a result of disruption of the tmo gene cluster by transposases. This operon is also likely regulated by an AphR/TbuT homolog, though no AphR-type binding site has been found (Figure 3, Supplementary Figure S2, and Supplementary Table S2). In Dechloromonas aromatica RCB, the tmoABCDEF–tbuX operon is regulated by AphS and is localized in a large locus of aromatic metabolism genes, close to an operon controlled by AphT (see below).
Most aph genes in Azoarcus sp. BH72 are predicted to be regulated by AphS (azo2437), except aphKLMN that is under dual control by AphS and AphR; BphS in this species (azo1969) likely regulates its own expression and the divergent operon, comprised of homologs of bphD and bphC, and a pfam13924 hypothetical protein with a lipocalin-like domain (Figure 4 and Supplementary Table S2).
FIGURE 4.

Operon organization and regulation of aromatic metabolism genes regulated by AphS/BphS, AphR, and AphT in other Comamonadaceae and Azoarcus sp. BH72. Color denotes regulation by the following TFs: AphS/BphS – red, AphR – orange, AphT – green. Double coloring denotes dual regulation. Genes/operons are shown by arrows, black arrows denote pseudogenes, gray – transposases. Genome abbreviations as in Supplementary Table S2.
All metabolic aph genes in R. eutropha H16, as well as the second copy of aphKLMNOP genes with catA (whose orthologs are generally regulated by CatR(M)/BenM) in R. eutropha JMP134 and R. metallidurans CH34 are presumably regulated by AphR/TbuT homologs with the binding sites similar to the AphR motif (Figure 3, Supplementary Figure S2, and Supplementary Tables S2, S3).
Moreover, in Burkholderia sp. 383, AphR likely also controls several adjacent operons formed by COG2072 flavin-containing monooxygenase, COG657 esterase/lipase, COG2303 dehydrogenase possibly associated with lipid metabolism; COG115 aminotransferase or putative 4-amino-4-deoxychorismate lyase (yielding 4-aminobenzoate), a DUF1302 protein, PF07044 and COG1033 exporters, and COG4447 glycosyl hydrolase (their possible role in aromatic metabolism discussed further); COG1012 aldehyde dehydrogenase; COG2084 3-hydroxyacid dehydrogenase, COG5285 phytanoyl-CoA dioxygenase, COG2814 efflux permease and hypothetical protein; COG1 (HemL) aminotransferase/aminomutase (Figure 3 and Supplementary Tables S2, S3).
AphT was identified only in several Commamonadaceae and Rhodocyclales (Figure 4, 5 and Supplementary Table S2). The predicted AphT binding motif is an odd palindrome that conforms to the general LysR-family consensus (T–N11–A) and is similar to the CatR and ClcR motifs (Figure 2B).
FIGURE 5.

Operon organization and regulation of aromatic metabolism genes regulated by AphS/BphS, AphR, AphT and BenR in D. aromatica RCB and Thauera sp. MZ1T. Color denotes regulation by the following TFs: AphS/BphS – red, AphR – orange, AphT – green, BenR – pink. Double coloring denotes dual regulation. Genes/operons are shown by arrows, black arrows denote pseudogenes. Operons flanked with double slashes belong to the same locus. Genome abbreviations as in Supplementary Table S2.
In some bacteria, AphT complements AphS and AphR in regulation of the aph genes. AphS, AphR and its homologs always regulate genes of phenol hydroxylase subunits, while other aph metabolic genes can be controlled by AphT (Supplementary Table S2). Notably, in Leptothrix cholodnii SP-6, the aph genes that are controlled by AphT and those under dual AphS and AphR regulation are in different loci, while in Acidovorax sp. JS42, all aph genes, including aphR, aphS, and two copies of aphT, are in the same locus, forming three adjacent pairs of divergent operons (Figure 4 and Supplementary Table S2). In Methylibium petroleiphilum PM1 that has neither AphS, nor AphR, the regulation is similarly divided between AphT and an AphR/TbuT homolog (Supplementary Figure S2), genes forming four operons in a single locus (Mpe_A2286–2265). The second cluster of the aph genes in M. petroleiphilum PM1 is also organized in several operons, some of which are regulated by another AphR/TbuT homolog (Supplementary Figure S2) or by a second copy of AphT. This AphT paralog was possibly earlier involved in the control of aphCEH, and, as in Dechloromonas aromatica RCB, of the close homologs (but not orthologs, data not shown) of aphIFG, and of a COG1028 gene. But the operon has been disrupted by a transposase, thus only the first gene, encoding 5-carboxymethyl-2-hydroxymuconate semialdehyde dehydrogenase (Mpe_A3328), that is preceded by a strong AphT-binding site, is currently under its regulation (Figure 4 and Supplementary Tables S2, S3).
In Delftia acidovorans SPH-1, that has no aph metabolic genes, AphT is predicted to regulate its own gene and a divergent operon comprised of a COG3181 transporter (only weakly similar to COG3181 proteins in other studied aromatic regulons, data not shown), a COG3485 enzyme related to catechol 1,2-dioxygenases, but likely encoding dioxygenase of some catechol derivative, such as 3,4-dihydroxybenzoate or 3,4-dihydroxyphenylacetate, and COG3971 2-oxo-hepta-3-ene-1,7-dioic acid hydratase, an enzyme of the lower 3,4-dihydroxyphenylacetate utilization pathway (Figure 4 and Supplementary Tables S2, S3). Thus, this TF likely regulates the 3,4-dihydroxyphenylacetate utilization.
AphT in D. aromatica RCB controls two operons in a large aromatic metabolism gene cluster (Figure 5 and Supplementary Table S2). One of them consists of COG3836 2,4-dihydroxyhept-2-ene-1,7-dioic acid aldolase that is an enzyme of the lower 3,4-dihydroxyphenylacetate utilization pathway, one of three copies of aphB and aphC, close homologs (but not orthologs according to the phylogenetic tree, data not shown) of aphF, aphG, and aphI, and, finally, COG1028 3-ketoacyl-ACP reductase that may perform some intermediate reaction of the pathway. The second operon, which is also controlled by AphS, is comprised of COG4447 glycosyl hydrolase, COG1033 and PF07044 exporters, and a DUF1302 protein; in Methylibium petroleiphilum PM1 these four genes, together with the co-transcribed aph genes, are also regulated by AphT (Figure 4 and Supplementary Table S2). A similar composition was observed in Azoarcus sp. BH72, where AphT regulates itself and the divergent operon comprised of distant homologs of the four aforementioned genes, as well as genes encoding COG589 universal stress protein, methyl-accepting chemotaxis transducer, and regulator LapR (Figure 4 and Supplementary Table S2). These proteins are possibly involved in sensing some aromatic compounds (as it has been shown that several methyl-accepting chemotaxis proteins are induced while growing on aromatic substrates), coping with their toxic concentrations, and solvent stress response (Wöhlbrand et al., 2008; Carmona et al., 2009; Büsing et al., 2015). LapR, a close homolog of AphR, has been described as an alkylphenol metabolism regulator (Jeong et al., 2003), hence it should control transcription of the adjacent lap genes of the alkylphenol metabolism (close homologs of aph), forming a regulatory cascade AphT–LapR, but no candidate LapR (AphR-type) binding sites have been identified upstream of the lap genes.
AraC-family regulator BenR was found only in two Rhodocyclales, D. aromatica RCB and Thauera sp. MZ1T. Therefore, to build a reliable PWM we also used predicted BenR binding sites from five species of Pseudomonas spp., Gammaproteobacteria (P. aeruginosa, P. entomophila, P. fluorescens, P. putida, P. stutzeri; data not shown). In D. aromatica RCB and Thauera sp. MZ1T, BenR controls not only benzoate metabolic and transport ben genes, but also complements AphS and AphR in the regulation of several aph phenol metabolic genes. Moreover, benR is directly regulated by AphS; in Thauera sp. MZ1T, in an operon with COG251 enamine deaminase gene. In both these bacteria, BenR regulates two divergent operons: benABCD and an operon containing COG4231 2-ketoacid ferredoxin oxidoreductase (distantly homologous to the known phenyl- or other arylpyruvate ferredoxin oxidoreductases), aphQ (only in Thauera sp. MZ1T), aphBCEH, benzoate ABC transporter, and the gene encoding a COG3945 protein (whose possible role is described below in the BoxR section). In D. aromatica RCB, this divergon is also regulated by AphS. Other aph genes (co-localized with BenR-regulated genes in Thauera sp. MZ1T and located in the different locus in D. aromatica RCB) are regulated by AphS either with or without AphR, as well as by AphT in D. aromatica RCB (Figure 5 and Supplementary Tables S2, S3). Among the studied genomes, these two bacteria have the most complex interaction of aromatic regulators, where AphS can share regulation of metabolic genes with AphR, AphT, and BenR, while simultaneously directly controlling expression of these TFs.
Regulation of the Benzoate Metabolism by BoxR and BzdR
Among the Bordetella spp. studied here, boxR and other box genes, as well as most other aromatic metabolism genes, are found only in Bordetella petrii DSM 12804. Among Burkholderia spp., boxR and other box genes are present in Burkholderia phymatum STM815 and Burkholderia xenovorans LB400; the latter has two paralogs of boxR and box genes, apparently resulting from a recent duplication (e.g., see the BoxR phylogenetic tree, Supplementary Figure S3). Cupriavidus taiwanensis and Ralstonia eutropha H16 also have two copies of boxR and metabolic box genes, but in these cases, according to the phylogenetic tree, it is more likely a result of horizontal gene transfer. Only Azoarcus sp. EbN1 among the studied bacteria has both boxR and bzdR, named according to their co-localization with the regulated genes: one of these paralogous TFs is co-localized with box genes, while the other one is localized within the anaerobic benzoate metabolism gene cluster bzdR–hypothetical_protein–bzdNOPQMSTUVWXYZA–benzoate_ABC_transporter–benK. Except for benzoate-CoA ligase, which is a common enzyme for both anaerobic and aerobic epoxide pathways, the bzd genes are absent in other bacteria from the studied set. Tmz1t_2954–2945 genes in Thauera sp. MZ1T have orthologs in Thauera aromatica annotated as had–oah–dch–bcrCBAD–fdx (López Barragán et al., 2004), but these genes are rather distantly homologous to the bzd genes from Azoarcus sp. EbN1, and we have not found any candidate BzdR/BoxR-binding sites upstream of this operon in Thauera sp. MZ1T.
BoxR-regulated genes in all studied bacteria are organized in divergent clusters, where one part almost always contains separately transcribed (or first in the operon) boxR and an adjacent operon including boxCBA and, optionally, genes encoding COG1024/COG447, COG1250, COG2030 proteins and COG183 3-ketoadipyl-CoA thiolase (Figure 6, 7 and Supplementary Tables S2, S3). The other part of the divergon is formed by boxD, bclA, genes encoding COG2267/COG596 lactonase of 3-hydroxyadipyl-CoA lactone (one of additional intermediates of the benzoate utilization via the box pathway; Zaar et al., 2001; Gescher et al., 2002), a pfam16155 protein with the cupin-like fold homologous to 4-hydroxylaminobenzoate lyase/2-hydroxylaminobenzoate mutase (likely involved in the metabolism of some benzoate derivative, e.g., protocatechuate; Gescher et al., 2002), and benzoate transporters; some of these genes may be missing and the gene order in an operon may vary depending on the genome (Figure 6, 7 and Supplementary Tables S2, S3).
FIGURE 6.

Operon organization and regulation of aromatic metabolism genes regulated by BoxR/BzdR in B. petrii DSM 12804, Burkholderia spp., and Ralstonia spp. Brown color denotes regulation by BoxR/BzdR. Genes/operons are shown by arrows, black arrows denote pseudogenes, gray – transposases. Genome abbreviations as in Supplementary Table S2.
FIGURE 7.

Operon organization and regulation of aromatic metabolism genes regulated by BoxR/BzdR in other studied Betaproteobacteria. Brown color denotes regulation by BoxR/BzdR. Genes/operons are shown by arrows, gray arrows denote transposases; “hp” indicates hypothetical proteins. Genome abbreviations as in Supplementary Table S2.
BoxR regulons in most studied bacteria include putative benzoate ABC transporter genes, whose role in the benzoate uptake has been predicted, but not experimentally demonstrated (Gescher et al., 2002; López Barragán et al., 2004; Carmona et al., 2009; Chen et al., 2014). Notably, in D. aromatica RCB, which lacks BoxR, this ABC transporter is located within another benzoate metabolic gene cluster and is under dual regulation by BenR and AphS. Resembling regulation is observed in Thauera sp. MZ1T, which has two paralogous copies of this transporter: one is under control of BenR, the other is regulated by BoxR (Figure 5, 7 and Supplementary Table S2). Polaromonas sp. JS666 and Polaromonas naphthalenivorans CJ2 have two and three paralogous copies of these transporter genes, respectively, where one is a part of the BoxR regulon, while others are likely regulated by IclR-family TFs (not studied here) whose genes are adjacent to the transporter genes. Moreover, in Cupriavidus taiwanensis and Ralstonia eutropha H16, one copy of boxR is associated with this benzoate ABC transporter, while the second one, with benK. In Azoarcus sp. EbN1 both these transporters are in the same operon with bzd genes (Figure 7).
The box gene clusters described so far lack genes encoding 3-hydroxyacyl-CoA hydro-lyase and 3-hydroxyadipyl-CoA dehydrogenase to complete the pathway and connect it to the TCA cycle (Gescher et al., 2002). Genes encoding COG1024/COG447 enoyl-CoA hydratase/isomerase and COG1250 3-hydroxyacyl-CoA dehydrogenase in BoxR-regulated operons in Burkholderia xenovorans LB400 and in several Comamonadaceae from the selected genome set likely perform these functions (Figure 1).
BoxR regulons of several studied bacteria include a gene encoding COG2030 acyl dehydratase (Figure 6, 7 and Supplementary Table S2). The function of this protein in the benzoate degradation remains unknown, but its sequence is similar to enzymes catalyzing the hydrolytic ring cleavage and aldehyde oxidation during the aerobic phenylacetate metabolism via CoA thioesters (Ferrández et al., 1998; Gescher et al., 2002; Teufel et al., 2011) and enoyl-CoA conversion into 3-hydroxyacyl-CoA (Park and Lee, 2003). Thus, this protein could be involved in the reactions carried out by BoxC and BoxD, or in the further steps of β-oxidation.
The BoxR regulon in Azoarcus sp. EbN1 might also include COG824 acyl-CoA thioesterase and formate dehydrogenase FdhBA, since the intergenic distance between these genes and the lactonase gene from the box genes is very short and thus they could be co-transcribed. Thioesterase may be required to hydrolyze CoA thioester intermediates of the benzoate degradation pathway if enzymes downstream of benzoyl-CoA become limiting, and thus release CoA preventing its deprivation (Gescher et al., 2002). Formate dehydrogenase may be involved in the oxidation of formate generated by BoxC, providing reducing equivalents (Gescher et al., 2006). In Azoarcus sp. BH72, these genes are also localized near the box operon, but the intergenic distance is too large to imply co-transcription.
Benzoate metabolic gene clusters (regulated either by BenR and/or AphS, or BoxR, Figure 5, 7 and Supplementary Table S2) in studied Rhodocyclales and Rhodoferax ferrireducens DSM 15236 also include genes encoding COG3945 protein with the hemerythrin/HHE cation-binding motif. Its role in the benzoate metabolism is not clear (Gescher et al., 2002), possibly, it could protect the cell from toxic compounds and/or act as an oxygen store/scavenger during the oxygen concentration changes. Indeed, it has been shown that the metabolism of aromatic compounds may cause oxidative stress response (Chávez et al., 2004; Denef et al., 2006; Carmona et al., 2009).
Moreover, we predict that Tmz1t_3130 encoding enamine deaminase (COG251, YjgF/RidA family) in Thauera sp. MZ1T also belongs to the BoxR regulon. Proteins of this family quench reactive enamine/imine intermediates that can inactivate a number of enzymes by covalent binding to their active sites (Lambrecht et al., 2012; Flynn et al., 2013; Flynn and Downs, 2013). In particular, some YjgF-like proteins are known to deaminate short-lived enamines 2-aminomuconate to 4-oxalocrotonate during the degradation of nitrobenzene (He and Spain, 1998; Park and Kim, 2000) and intermediate 2-amino-5-carboxymuconic 6-semialdehyde into 2-hydroxy-5-carboxymuconic 6-semialdehyde in the meta-cleavage pathway of 4-amino-3-hydroxybenzoate degradation (Orii et al., 2004; Takenaka et al., 2009). Moreover, chorismatase XanB2 that converts chorismate into 3-hydroxybenzoate and 4-hydroxybenzoate has an YjgF-like domain and is important for the oxidative stress defense (Zhou et al., 2013). Thus, Tmz1t_3130 may indeed play a role in the utilization of a benzoate derivative and stress resistance.
Experimental studies of BoxR and BzdR have demonstrated that they bind TGCA repeats separated by either 1, 6, or 15 nucleotides (Barragán et al., 2005; Durante-Rodríguez et al., 2008, 2010; Valderrama et al., 2012). Here we show that, according to the phylogenetic footprinting data, the main variant of the motif has 6 nt between TGCA, forming the ATGCACTATAGTGCAT palindrome (Figure 2A). The identified sites are almost always multiple, often separated by several DNA turns, mainly 3–5 (Supplementary Table S3).
Regulation of the Catechol Metabolism by CatR and CatM/BenM
The nomenclature of homologous LysR-family TFs that control the catechol and/or benzoate metabolism is not consistent and depends on the species they are characterized in Chugani et al. (1997), Collier et al. (1998), Cowles et al. (2000), Tover et al. (2001), Clark et al. (2004), Ezezika et al. (2006). CatR and CatM/BenM likely substitute each other, completely or partially, but it can be analyzed only experimentally, hence, here we predict the regulation based on their co-localization with respective operons. We use the name CatR for TFs that regulate only cat genes and are co-localized with them; and CatM/BenM for those TFs that control cat and/or ben genes. This distinction is supported by the branching of the phylogenetic tree of these TFs (Figure 8).
FIGURE 8.

Phylogenetic tree of the LysR-family regulators of the aromatic metabolism. The color code is consistent with that in Figure 1: AphT – green, CatR – light blue, CatM/BenM – blue, CatR∗ – dark blue, ClcR and TfdR(S) – magenta, MmlR – violet.
Neither CatR nor CatM/BenM have been found among Rhodocyclales and Comamonadaceae (apart from Polaromonas naphthalenivorans CJ2) in our genome set, while most Burkholderia spp. and Ralstonia spp. have two TFs.
All Burkholderia spp. have both CatR and CatM/BenM, where CatR regulates its own gene and the divergently transcribed catBAC operon, except for Burkholderia glumae BGR1 that has only CatR, which regulates catBCA and possibly also adjacent GTPase genes (see below). CatM/BenM seems to regulate its own gene and divergently transcribed benABCD in B. mallei ATCC 23344, B. phymatum STM815, B. pseudomallei K96243 and B. xenovorans LB400 (Supplementary Figure S4). In those Burkholderia, where catM/benM is not adjacent to the regulated genes (B. cepacia AMMD, Burkholderia sp. 383, B. vietnamiensis G4), it controls the operon comprised of cat(B)(C)A–benABCD, as well as the gene of a COG251 protein (Supplementary Figure S4 and Supplementary Table S2). The possible role of COG251 enamine deaminases (YjgF/RidA family) in the aromatic metabolism was described above in the BoxR section.
Bordetella petrii DSM 12804 also has both CatR and CatM/BenM. CatR regulates its own gene and divergently transcribed catB and the gene of a COG3181 transport protein. The CatM/BenM regulon is comprised of the TF itself and the divergent operon formed by catBA, benABCD, the gene of a COG251 protein, benK, fragments of catI and catF, catDC, the genes of COG3181, COG1012, COG1062, COG2271 proteins, benE, as well as two genes encoding proteins with predicted GTPase activity (Supplementary Figure S5). The fragments of catI and catF are homologous to catI and catF of various Comamonadaceae, where they are regulated by an IclR-family TF (Supplementary Table S2). COG1062 and COG1012 dehydrogenases likely convert benzyl-alcohol through benzaldehyde into benzoate, for further degradation by BenABCD. The COG2271 transporter may be involved in import of hydroxybenzoate or some other benzoate-related compound. The GTPases in the regulon are distantly homologous to each other and to the genes forming operons with catA in A. baylyi ADP1 and catBCA in B. glumae BGR1.
CatR in Ralstonia metallidurans CH34 and both studied Ralstonia eutropha strains regulates its own gene and divergently transcribed catB(C)–COG3181–(catD). Notably, in R. eutropha these genes are positioned close to phenol metabolism genes, while in R. metallidurans CH34, catR, catM/benM and their regulated genes form one cluster. CatM/BenM in these three Ralstonia species regulates catA–benABCD and its own divergent gene, as well as co-transcribed catB in R. eutropha JMP134, and catBCD and COG3246 protein in R. eutropha H16 (Supplementary Figure S5). Homologs of the latter in other bacteria are often associated with genes of protocatechuate 3,4-dioxygenase subunits, and other member of this family include 3-keto-5-aminohexanoate cleavage enzyme that catalyzes condensation of the compound with acetyl-CoA yielding 3-aminobutanoyl-CoA and acetoacetate (Bellinzoni et al., 2011). Thus, this COG3246 protein might be a 3-keto acid (e.g., β-ketoadipate) cleavage enzyme that catalyzes carbon-carbon condensation with CoA esters and is involved in the catechol metabolism.
In R. eutropha JMP134, CatM/BenM also likely controls the expression of separately localized benK and benE. Candidate binding sites conforming to the CatR or CatM/BenM consensus have been also observed upstream of plasmid genes encoding a COG3181 protein (PHG382) and catBC-COG3181 (PHG405-403) in R. eutropha H16, though they might be regulated by other LysR-family TFs, e.g., MmlR (PHG389, see below), PHG401 or PHG406 (Supplementary Table S3).
Ralstonia pickettii 12J has only CatM/BenM, which controls its own gene and the divergent catBCA–benABCDKE operon. In Polaromonas naphthalenivorans CJ2, a similar situationis observed, with catM/benM and benABCDKE regulated by CatM/BenM (Supplementary Table S2).
Regulation of Chlorocatechol and 2,4-Dichlorophenoxyacetic Acid Metabolism by ClcR and TfdR/TfdS
In the analyzed bacteria, the clc or tfd genes of chloroaromatic metabolism have been identified only in Bordetella petrii DSM 12804, Burkholderia xenovorans LB400, and Ralstonia eutropha JMP134 (Figure 8), all of which are prominent in their ability to degrade a remarkable variety of aromatic compounds (Chain et al., 2006; Gross et al., 2008; Lykidis et al., 2010).
The ClcR regulons (Supplementary Table S2) in B. petrii and B. xenovorans are comprised of clcR and the divergently transcribed operon consisting of clcAB, the clcC gene encoding a COG3181 protein [homologous, but not orthologous to the ones in the CatR(M)/BenM, AphS, AphR, AphT regulons, data not shown], clcDE, and possibly also the gene of an AraC-family TF (Bpet1539, Bxe_A1125). The function of the latter is unknown, but it is distantly homologous to the regulators of anthranilate (AntR, AndR) and benzoate (BenR) degradation, thus it might indeed be involved in the regulation of metabolism of some benzoate derivative.
Bordetella petrii also has a second copy of the clc gene cluster that has the same composition and organization as the first one, except for an AraC-family regulator gene. Notably, this clc gene cluster apparently has a specific mode of regulation. It is known that in addition to the AkACC–N5–GGTAT motif (Figure 2B), which is the repression binding site (RBS), both CatR and ClcR bind an adjacent activation binding site (ABS) (Parsek et al., 1994; Chugani et al., 1997, 1998; Tover et al., 2000, 2001). A similar ABS was also identified for the tfd genes, while the second clc gene cluster in B. petrii (Bpet3747; Bpet3748–3752) lacks half of the palindromic RBS, yet retains the ABS (Figure 9).
FIGURE 9.

Multiple alignment of the upstream regions of regulatory genes clcR, tfdR, tfdS, tfdT. ABS is highlighted with green, RBS – with yellow, start codon ATG is marked with red font. Asterisks mark completely conserved positions of the alignment. #Since genes of TFs are divergent to their regulated genes, ABS sequences here are in reverse complement compared to the sequences (ABS in catB, clcA upstream regions) described earlier (Parsek et al., 1994; Chugani et al., 1997, 1998; Tover et al., 2000, 2001).
The TfdR/TfdS regulon (Supplementary Tables S2, S3) in R. eutropha includes several co-localized, divergently oriented pairs of operons from a plasmid (Leveau and van der Meer, 1996; Laemmli et al., 2000; Vedler et al., 2004):
-
simple (i)
tfdT and tfdCDEF (tfdB is adjacent, but is not a part of the tfd operon). TfdT is highly similar toTfdR/TfdS, but is inactive due to an insertion element;
-
simple (ii)
tfdDIICIIEIIFIIBIIK and tfdR, possibly co-transcribed with an adjacent chimerical gene, encoding partially the HTH domain of an IclR-family TF (see below) and partially COG1024 enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase (Reut_D6476). We predict that it may be involved in the conversion of 2,3-didehydroadipyl-CoA to 3-ketoadipyl-CoA;
-
simple (iii)
tfdS and tfdA; tfdS may form an operon with the gene of an IclR-family TF (Reut_D6477) that is distantly homologous to the regulators of the 4-hydroxybenzoate and protocatechuate utilization, PobR and PcaR, and modulates expression of the tfd genes (Trefault et al., 2009). The adjacent genes encoding acyl-CoA synthetase COG318, thiolase COG183, and a COG1545 protein are possibly co-transcribed with tfdA or, at least, are involved in the lower 2,4-D metabolism. The COG318 protein likely acts on the β-ketoadipate produced by TfdF, while COG183 is likely a β-ketoadipyl-CoA thiolase that further degrades it into acetyl-CoA and succinyl-CoA, thus channeling the 2,4-D metabolism into the TCA cycle. The exact function of the COG1545 protein remains unknown, but it is distantly related to acyl dehydratases and is likely functionally connected to β-ketoadipyl-CoA thiolase, as the genomic context shows that various COG1545 and COG183 proteins are often associated.
Regulation of 4-Methylmuconolactone Metabolism by MmlR
MmlR and 4-methylmuconolactone metabolism genes were found only in R. eutropha H16 and R. eutropha JMP134 (Figure 8 and Supplementary Table S2). Notably, in R. eutropha H16 these genes are localized on a plasmid, while in R. eutropha JMP134 they are chromosomal.
A candidate MmlR binding site has been identified upstream of the mmlLRFGHIJ operon based on its conservation in these two species and its high similarity to the binding motifs of the homologous TFs ClcR/TfdR/TfdS and CatR(M)/BenM (Figure 2B, Supplementary Figure S6, and Supplementary Table S3). The similarity of the motifs may result in the cross-regulation by the respective TFs.
A Novel LysR-Family Regulator of Aromatic Metabolism, CatR∗
In five Commamonadaceae species we have identified a TF homologous to CatR, ClcR, and CynR (Figure 8). The phylogenetic footprinting has revealed a possible binding motif (Figure 2B), an odd palindrome conforming to the general LysR-family consensus (T–N11–A). Similarly to CatR, this TF is predicted to regulate diverse combinations of genes (Supplementary Figure S7) encoding CatA, CatB, CatC homologs and COG3181 proteins (all of them form branches on the phylogenetic trees that are separate from those controlled by CatR, data not shown; Supplementary Table S2), and its own divergently transcribed gene (except in Delftia acidovorans SPH-1). Notably, all studied bacteria that have this TF lack CatR(M)/BenM, except for Polaromonas naphthalenivorans CJ2, where both these regulators belong to the same gene cluster, with ben genes under the CatM/BenM control. Thus, this TF either substitutes CatR(M)/BenM, or shares functions with it.
Discussion
This comparative study showed complexity and variability in the regulation of aromatic metabolism in Betaproteobacteria. The aromatic metabolism is organized by key substrates, which are the merging points of various upper metabolic pathways and are further degraded by the lower pathways flowing into the central metabolism. The upper parts of pathways, rare pathway variants, and degradative pathways of exotic and complex, in particular, xenobiotic compounds are generally controlled by a single TF each (e.g., the bph, box, mml, or tfd genes). The regulation of more common and/or central parts of the metabolism (e.g., the aph, ben, or cat genes) may vary even between close species and could involve several TFs with diverse modes of interaction (Figure 1 and Supplementary Table S2). In particular, TFs may simultaneously control certain operons (dual regulation), the regulation of a pathway may be divided between different TFs (shared regulation), or a TF may control expression of another TF (cascade regulation). For example, in Acidovorax sp. JS42 and L. cholodnii SP-6, the aph genes are under shared and/or dual regulation by AphS, AphR and AphT (Figure 4), and in D. aromatica RCB and Thauera sp. MZ1T there is cascade regulation of BenR by AphS (Figure 5).
These observations are likely not restricted to Betaproteobacteria and can be propagated to other classes, e.g., Gammaproteobacteria. Most aromatic metabolism pathways and respective TFs studied here have been identified in Gammaproteobacteria as well, and are similarly regulated. Some examples are ben and cat genes regulated by homologous TFs BenM and CatM, respectively, in Acinetobacter baylyi ADP1, and ben genes regulated by BenR in Pseudomonas putida (Collier et al., 1998; Cowles et al., 2000; Ezezika et al., 2006; Li et al., 2010). An interesting direction of research could be the analysis of horizontal gene transfer between and within Beta- and Gammaproteobacteria involving both pathway operons and respective TFs. One specific question could be whether operons always travel with their regulators, or there are cases when a newly arrived operon falls under regulation by a pre-existing TF. As more genomes become available, one might hope to reconstruct the evolutionary history of the metabolic genes and their regulators in considerable detail.
Among the studied genomes, D. aromatica RCB and Thauera sp. MZ1T have the most complex interaction of studied aromatic regulatory proteins, and the most frequent and, at the same time, variable connection is between AphS, AphR, AphT, and BenR (Figure 10 and Supplementary Table S2).
FIGURE 10.
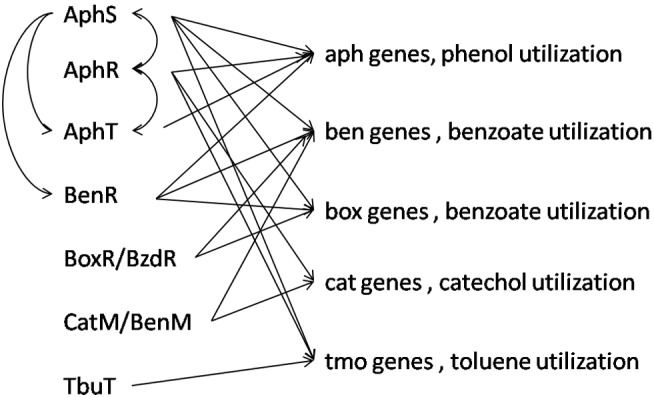
Integrated scheme of interactions between aromatic metabolism TFs and regulated genes in all studied genomes.
We have identified a novel LysR-family TF, CatR∗, that regulates the metabolism of catechol (or some catechol derivative) and either substitutes CatR(M)/BenM, or shares functions with it.
We have also predicted several new members of aromatic regulons, with some COGs belonging to several regulons (Table 1), the most notable example being COG3181 transporter proteins: in different species, genes from this COG have been assigned to AphS, AphR, AphT, ClcR, CatR(M)/BenM, and CatR∗ regulons. Validation of predicted metabolic functions may be an object of further experimental study.
Table 1.
Predicted functions of the main new members of aromatic regulons.
| COG or other ID | Predicted function | Regulated by TFs |
|---|---|---|
| COG115 | 4-Amino-4-deoxychorismate lyase | AphR |
| COG183 | β-ketoadipyl -CoA thiolase | TfdR(S) |
| COG251 | Enamine deaminase involved in utilization of benzoate derivative and stress resistance | CatM/BenM, BoxR |
| COG318 | β-ketoadipyl -CoA synthetase, involved in the lower 2,4-D metabolism | TfdR(S) |
| COG1012 | Benzaldehyde dehydrogenase | CatM/BenM |
| COG1024 | Enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase, possibly involved in the conversion of 2,3-didehydroadipyl-CoA to 3-ketoadipyl-CoA | TfdR(S) |
| COG1024/ COG447 | 3-Hydroxyacyl-CoA hydro-lyase | BoxR |
| COG1062 | Benzyl-alcohol dehydrogenase | CatM/BenM |
| COG1250 | 3-Hydroxyadipyl-CoA dehydrogenase | BoxR |
| COG1545 | Distantly related to acyl dehydratases, functionally connected to β-ketoadipyl-CoA thiolase | TfdR(S) |
| COG1894, COG3383 | Formate dehydrogenase possibly involved in the oxidation of formate generated by BoxC | BoxR |
| COG2271 | Hydroxybenzoate or some benzoate-related compound transporter | CatM/BenM |
| COG3181 | Aromatic transporter family receptor | AphS, AphR, AphT, ClcR, CatR(M)/BenM, CatR∗ |
| COG3246 | 3-Keto acid (possibly, β-ketoadipate) cleavage enzyme involved in the catechol metabolism | CatM/BenM |
| COG3485 | Dioxygenase of some catechol derivative, possibly 3,4-dihydroxybenzoate or 3,4-dihydroxyphenylacetate | AphT |
| COG3945 | Protein with hemerythrin/HHE cation-binding motif, possibly protecting the cell from toxic compounds and/or acting as an oxygen store/scavenger | AphS, BenR, BoxR |
| COG3971 | 2-Oxo-hepta-3-ene-1,7-dioic acid hydratase | AphT |
| COG4231 | 2-Ketoacid ferredoxin oxidoreductase, distantly homologous to arylpyruvate ferredoxin oxidoreductases | AphS, BenR |
| COG4447, COG1033, PF07044, DUF1302 | Proteins possibly involved in coping with toxic concentrations of aromatic compounds and with the solvent-stress response | AphS, AphR, AphT |
In addition to the structure of regulatory networks and regulon content, we have also made some observations about the possible mechanism of regulation. In particular, AphR belongs to the XylR/DmpR group of regulators, which also includes TbuT, HbpR, and other TFs that control metabolism of aromatic compounds (Arai et al., 1999; Jaspers et al., 2001; Tropel and van der Meer, 2002). These TFs are known to bind pairs of palindromic sequences, approximately 16–20 nt in length and with a 29–42 nt distance between their centers, which are usually localized 100–200 nt upstream of the –12/–24 target promoters (Jaspers et al., 2001; Tropel and van der Meer, 2002). The correct spacing between two binding sites is necessary for the cooperative binding shown for XylR/DmpR TFs: both sites should face the same side of the DNA helix, e.g., the centers of binding sites are separated by three complete DNA helical turns in the case of XylR and DmpR, and by three or four turns, in the case of HbpR (Jaspers et al., 2001; Tropel and van der Meer, 2002). We have observed that almost all genes predicted to be under regulation of AphR and its homologs in the studied bacteria are preceded by multiple sites, separated by three (or rarely by one) DNA turns (Supplementary Table S3).
Our study also supports previous observations that genes of aromatic metabolism are often organized in very long supraoperonic clusters and are associated with mobile DNA elements that facilitate their transfer between genomes. For example, transposases and/or integrases flank the aph genes in B. vietnamiensis G4 and M. petroleiphilum PM1; bph, box, and clc genes in B. xenovorans LB400; cat and clc genes in B. petrii DSM 12804, etc (Figure 3, 4, 6 and Supplementary Figure S5). These features allow for biochemical diversity of bacteria and their rapid adaptation for various compounds, including xenobiotics.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
IS and MG conceived the study. IS designed and performed the comparative analysis, and visualized results. IS and MG participated in writing and revising the manuscript, read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This study was supported by the Russian Science Foundation under grant 18-14-00358.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00642/full#supplementary-material
References
- Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., et al. (1997). Gapped BLAST and PSIBLAST: a new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. PMID: 9254694 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai H., Akahira S., Ohishi T., Kudo T. (1999). Adaptation of Comamonas testosteroni TA441 to utilization of phenol by spontaneous mutation of the gene for a trans-acting factor. Mol. Microbiol. 33 1132–1140. 10.1046/j.1365-2958.1999.01554.x [DOI] [PubMed] [Google Scholar]
- Arai H., Ohishi T., Chang M. Y., Kudo T. (2000). Arrangement and regulation of the genes for meta-pathway enzymes required for degradation of phenol in Comamonas testosteroni TA441. Microbiology 146(Pt 7) 1707–1715. 10.1099/00221287-146-7-1707 [DOI] [PubMed] [Google Scholar]
- Barragán M. J., Blázquez B., Zamarro M. T., Mancheño J. M., García J. L., Díaz E., et al. (2005). BzdR, a repressor that controls the anaerobic catabolism of benzoate in Azoarcus sp. CIB, is the first member of a new subfamily of transcriptional regulators. J. Biol. Chem. 280 10683–10694. 10.1074/jbc.M412259200 [DOI] [PubMed] [Google Scholar]
- Bellinzoni M., Bastard K., Perret A., Zaparucha A., Perchat N., Vergne C., et al. (2011). 3-Keto-5-aminohexanoate cleavage enzyme: a common fold for an uncommon Claisen-type condensation. J. Biol. Chem. 286 27399–27405. 10.1074/jbc.M111.253260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D. A., Boguski M. S., Lipman D. J., Ostell J., Ouellette B. F., Rapp B. A., et al. (1999). GenBank. Nucleic Acids Res. 27 12–17. 10.1093/nar/27.1.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büsing I., Kant M., Dörries M., Wöhlbrand L., Rabus R. (2015). The predicted σ54-dependent regulator EtpR is essential for expression of genes for an aerobic p-ethylphenol and p-hydroxyacetophenone degradation in “Aromatoleum aromaticum” EbN1. BMC Microbiol. 15:251. 10.1186/s12866-015-0571-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne A. M., Kukor J. J., Olsen R. H. (1995). Sequence analysis of the gene cluster encoding toluene-3-monooxygenase from Pseudomonas pickettii PKO1. Gene 154 65–70. 10.1016/0378-1119(94)00844-I [DOI] [PubMed] [Google Scholar]
- Byrne A. M., Olsen R. H. (1996). Cascade regulation of the toluene-3-monooxygenase operon (tbuA1UBVA2C) of Burkholderia pickettii PKO1: role of the tbuA1 promoter (PtbuA1) in the expression of its cognate activator, TbuT. J. Bacteriol. 178 6327–6337. 10.1128/jb.178.21.6327-6337.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona M., Zamarro M. T., Blázquez B., Durante-Rodríguez G., Juárez J. F., Valderrama J. A., et al. (2009). Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol. Mol. Biol. Rev. 73 71–133. 10.1128/MMBR.00021-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain P. S., Denef V. J., Konstantinidis K. T., Vergez L. M., Agulló L., Reyes V. L., et al. (2006). Burkholderia xenovorans LB400 harbors a multi-replicon, 9.73-Mbp genome shaped for versatility. Proc. Natl. Acad. Sci. U.S.A. 103 15280–15287. 10.1073/pnas.0606924103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez F. P., Lünsdorf H., Jerez C. A. (2004). Growth of polychlorinated-biphenyl-degrading bacteria in the presence of biphenyl and chlorobiphenyls generates oxidative stress and massive accumulation of inorganic polyphosphate. Appl. Environ. Microbiol. 70 3064–3072. 10.1128/AEM.70.5.3064-3072.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. W., Zhang Y., Jiang C. Y., Liu S. J. (2014). Benzoate metabolism intermediate benzoyl coenzyme A affects gentisate pathway regulation in Comamonas testosteroni. Appl. Environ. Microbiol. 80 4051–4062. 10.1128/AEM.01146-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugani S. A., Parsek M. R., Chakrabarty A. M. (1998). Transcriptional repression mediated by LysR-Type regulator CatR bound at multiple binding sites. J. Bacteriol. 180 2367–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugani S. A., Parsek M. R., Hershberger C. D., Murakami K., Ishihama A., Chakrabarty A. M. (1997). Activation of the catBCA promoter: probing the interaction of CatR and RNA polymerase through in vitro transcription. J. Bacteriol. 179 2221–2227. 10.1128/jb.179.7.2221-2227.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T. J., Phillips R. S., Bundy B. M., Momany C., Neidle E. L. (2004). Benzoate decreases the binding of cis,cis-muconate to the BenM regulator despite the synergistic effect of both compounds on transcriptional activation. J. Bacteriol. 186 1200–1204. 10.1128/JB.186.4.1200-1204.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier L. S., Gaines G. L., III, Neidle E. L. (1998). Regulation of benzoate degradation in Acinetobacter sp. strain ADP1 by BenM, a LysR-type transcriptional activator. J. Bacteriol. 180 2493–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles C. E., Nichols N. N., Harwood C. S. (2000). BenR, a XylS homologue, regulates three different pathways of aromatic acid degradation in Pseudomonas putida. J. Bacteriol. 182 6339–6346. 10.1128/JB.182.22.6339-6346.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14 1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denef V. J., Klappenbach J. A., Patrauchan M. A., Florizone C., Rodrigues J. L., Tsoi T. V., et al. (2006). Genetic and genomic insights into the role of benzoate-catabolic pathway redundancy in Burkholderia xenovorans LB400. Appl. Environ. Microbiol. 72 585–595. 10.1128/AEM.72.1.585-595.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36 W465–W469. 10.1093/nar/gkn180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durante-Rodríguez G., Valderrama J. A., Mancheño J. M., Rivas G., Alfonso C., Arias-Palomo E., et al. (2010). Biochemical characterization of the transcriptional regulator BzdR from Azoarcus sp. CIB. J. Biol. Chem. 285 35694–35705. 10.1074/jbc.M110.143503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durante-Rodríguez G., Zamarro M. T., García J. L., Díaz E., Carmona M. (2008). New insights into the BzdR-mediated transcriptional regulation of the anaerobic catabolism of benzoate in Azoarcus sp. CIB. Microbiology 154(Pt. 1) 306–316. 10.1099/mic.0.2007/011361-0 [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. 10.1186/1471-2105-5-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezezika O. C., Collier-Hyams L. S., Dale H. A., Burk A. C., Neidle E. L. (2006). CatM regulation of the benABCDE operon: functional divergence of two LysR-type paralogs in Acinetobacter baylyi ADP1. Appl. Environ. Microbiol. 72 1749–1758. 10.1128/AEM.72.3.1749-1758.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrández A., Miñambres B., García B., Olivera E. R., Luengo J. M., García J. L., et al. (1998). Catabolism of phenylacetic acid in Escherichia coli. Characterization of a new aerobic hybrid pathway. J. Biol. Chem. 273 25974–25986. 10.1074/jbc.273.40.25974 [DOI] [PubMed] [Google Scholar]
- Flynn J. M., Christopherson M. R., Downs D. M. (2013). Decreased coenzyme A levels in ridA mutant strains of Salmonella enterica result from inactivated serine hydroxymethyltransferase. Mol. Microbiol. 89 751–759. 10.1111/mmi.12313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J. M., Downs D. M. (2013). In the absence of RidA, endogenous 2-aminoacrylate inactivates alanine racemases by modifying the pyridoxal 5’-phosphate cofactor. J. Bacteriol. 195 3603–3609. 10.1128/JB.00463-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler J. F., Harwood C. S., Gibson J. (1988). Purification and properties of benzoate-coenzyme A ligase, a Rhodopseudomonas palustris enzyme involved in the anaerobic degradation of benzoate. J. Bacteriol. 170 1709–1714. 10.1128/jb.170.4.1709-1714.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand M. S., Koonin E. V., Mironov A. A. (2000). Prediction of transcription regulatory sites in Archaea by a comparative genomic approach. Nucleic Acids Res. 28 695–705. 10.1093/nar/28.3.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gescher J., Eisenreich W., Wörth J., Bacher A., Fuchs G. (2005). Aerobic benzoyl-CoA catabolic pathway in Azoarcus evansii: studies on the non-oxygenolytic ring cleavage enzyme. Mol. Microbiol. 56 1586–1600. 10.1111/j.1365-2958.2005.04637.x [DOI] [PubMed] [Google Scholar]
- Gescher J., Ismail W., Olgeschläger E., Eisenreich W., Wörth J., Fuchs G. (2006). Aerobic benzoyl coenzyme A (CoA) catabolic pathway in Azoarcus evansii: conversion of ring cleavage product by 3,4-dehydroadipyl-CoA semialdehyde dehydrogenase. J. Bacteriol. 188 2919–2927. 10.1128/JB.188.8.2919-2927.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gescher J., Zaar A., Mohamed M., Schägger H., Fuchs G. (2002). Genes coding for a new pathway of aerobic benzoate metabolism in Azoarcus evansii. J. Bacteriol. 184 6301–6315. 10.1128/JB.184.22.6301-6315.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross R., Guzman C. A., Sebaihia M., dos Santos V. A., Pieper D. H., Koebnik R., et al. (2008). The missing link: Bordetella petrii is endowed with both the metabolic versatility of environmental bacteria and virulence traits of pathogenic Bordetellae. BMC Genomics 9:449. 10.1186/1471-2164-9-449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z., Spain J. C. (1998). A novel 2-aminomuconate deaminase in the nitrobenzene degradation pathway of Pseudomonas pseudoalcaligenes JS45. J. Bacteriol. 180 2502–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D. H., Richter D. C., Rausch C., Dezulian T., Franz M., Rupp R. (2007). Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics 8:460. 10.1186/1471-2105-8-460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers M. C., Schmid A., Sturme M. H., Goslings D. A., Kohler H. P., Van Der Meer J. R. (2001). Transcriptional organization and dynamic expression of the hbpCAD genes, which encode the first three enzymes for 2-hydroxybiphenyl degradation in Pseudomonas azelaica HBP1. J. Bacteriol. 183 270–279. 10.1128/JB.183-1.270-279.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J. J., Kim J. H., Kim C. K., Hwang I., Lee K. (2003). 3- and 4-alkylphenol degradation pathway in Pseudomonas sp. strain KL28: genetic organization of the lap gene cluster and substrate specificities of phenol hydroxylase and catechol 2,3-dioxygenase. Microbiology 149(Pt 11),3265–3277. [DOI] [PubMed] [Google Scholar]
- Kahng H. Y., Byrne A. M., Olsen R. H., Kukor J. J. (2000). Characterization and role of tbuX in utilization of toluene by Ralstonia pickettii PKO1. J. Bacteriol. 182 1232–1242. 10.1128/JB.182.5.1232-1242.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemba M., Jakobs B., Wittich R. M., Pieper D. (2000). Chromosomal integration of tcb chlorocatechol degradation pathway genes as a means of expanding the growth substrate range of bacteria to include haloaromatics. Appl. Environ. Microbiol. 66 3255–3261. 10.1128/AEM.66.8.3255-3261.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J., Fuchs G. (1992). Enzymatic reduction of benzoyl-CoA to alicyclic compounds, a key reaction in anaerobic aromatic metabolism. Eur. J. Biochem. 205 195–202. 10.1111/j.1432-1033.1992.tb16768.x [DOI] [PubMed] [Google Scholar]
- Laemmli C. M., Leveau J. H., Zehnder A. J., van der Meer J. R. (2000). Characterization of a second tfd gene cluster for chlorophenol and chlorocatechol metabolism on plasmid pJP4 in Ralstonia eutropha JMP134 (pJP4). J. Bacteriol. 182 4165–4172. 10.1128/JB.182.15.4165-4172.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht J. A., Flynn J. M., Downs D. M. (2012). Conserved YjgF protein family deaminates reactive enamine/imine intermediates of pyridoxal 5’-phosphate (PLP)-dependent enzyme reactions. J. Biol. Chem. 287 3454–3461. 10.1074/jbc.M111.304477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy J. G., Johnson G. R., Olsen R. H. (1997). Cross-regulation of toluene monooxygenases by the transcriptional activators TbmR and TbuT. Appl. Environ. Microbiol. 63 3736–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner M., Schmitt K., Bauer S., Hot D., Hubans C., Levillain E., et al. (2009). Genomic island excisions in Bordetella petrii. BMC Microbiol. 9:141. 10.1186/1471-2180-9-141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveau J. H., van der Meer J. R. (1996). The tfdR gene product can successfully take over the role of the insertion element-inactivated TfdT protein as a transcriptional activator of the tfdCDEF gene cluster, which encodes chlorocatechol degradation in Ralstonia eutropha JMP134 (pJP4). J. Bacteriol. 178 6824–6832. 10.1128/jb.178.23.6824-6832.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Yan Y., Ping S., Chen M., Zhang W., Li L., et al. (2010). Genome-wide investigation and functional characterization of the b-ketoadipate pathway in the nitrogen-fixing and root-associated bacterium Pseudomonas stutzeri A1501. BMC Microbiol. 10:36. 10.1186/1471-2180-10-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López Barragán M. J., Carmona M., Zamarro M. T., Thiele B., Boll M., Fuchs G., et al. (2004). The bzd gene cluster, coding for anaerobic benzoate catabolism, in Azoarcus sp. strain CIB. J. Bacteriol. 186 5762–5774. 10.1128/JB.186.17.5762-5774.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykidis A., Pérez-Pantoja D., Ledger T., Mavromatis K., Anderson I. J., Ivanova N. N., et al. (2010). The complete multipartite genome sequence of Cupriavidus necator JMP134, a versatile pollutant degrader. PLoS One 5:e9729. 10.1371/journal.pone.0009729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín M., Pérez-Pantoja D., Donoso R., Wray V., González B., Pieper D. H. (2010). Modified 3-oxoadipate pathway for the biodegradation of methylaromatics in Pseudomonas reinekei MT1. J. Bacteriol. 192 1543–1552. 10.1128/JB.01208-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFall S. M., Klem T. J., Fujita N., Ishihama A., Chakrabarty A. M. (1997a). DNase I footprinting, DNA bending and in vitro transcription analyses of ClcR and CatR interactions with the clcABD promoter: evidence of a conserved transcriptional activation mechanism. Mol. Microbiol. 24 965–976. [DOI] [PubMed] [Google Scholar]
- McFall S. M., Parsek M. R., Chakrabarty A. M. (1997b). 2-chloromuconate and ClcR-mediated activation of the clcABD operon: in vitro transcriptional and DNase I footprint analyses. J. Bacteriol. 179 3655–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov A. A., Vinokurova N. P., Gel’fand M. S. (2000). Software for analyzing bacterial genomes. Mol. Biol. 34 253–262. 10.1007/BF02759643 [DOI] [PubMed] [Google Scholar]
- Miyazaki R., Yano H., Sentchilo V., van der Meer J. R. (2018). Physiological and transcriptome changes induced by Pseudomonas putida acquisition of an integrative and conjugative element. Sci. Rep. 8:5550. 10.1038/s41598-018-23858-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novichkov P. S., Rodionov D. A., Stavrovskaya E. D., Novichkova E. S., Kazakov A. E., Gelfand M. S., et al. (2010). RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 38 W299–W307. 10.1093/nar/gkq531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo Y., Delawary M., Kimbara K., Takagi M., Ohta A., Nagata Y. (2001). BphS, a key transcriptional regulator of bph genes involved in polychlorinated biphenyl/biphenyl degradation in Pseudomonas sp. KKS102. J Biol Chem. 276 36146–36154. 10.1074/jbc.M100302200 [DOI] [PubMed] [Google Scholar]
- Orii C., Takenaka S., Murakami S., Aoki K. (2004). A novel coupled enzyme assay reveals an enzyme responsible for the deamination of a chemically unstable intermediate in the metabolic pathway of 4-amino-3-hydroxybenzoic acid in Bordetella sp. strain 10d. Eur. J. Biochem. 271 3248–3254. 10.1111/j.1432-1033.2004.04258.x [DOI] [PubMed] [Google Scholar]
- Park H. S., Kim H. S. (2000). Identification and characterization of the nitrobenzene catabolic plasmids pNB1 and pNB2 in Pseudomonas putida HS12. J. Bacteriol. 182 573–580. 10.1128/JB.182.3.573-580.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. J., Lee S. Y. (2003). Identification and characterization of a new enoyl coenzyme A hydratase involved in biosynthesis of medium-chain-length polyhydroxyalkanoates in recombinant Escherichia coli. J. Bacteriol. 185 5391–5397. 10.1128/JB.185.18.5391-5397.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsek M. R., McFall S. M., Shinabarger D. L., Chakrabarty A. M. (1994). Interaction of two LysR-type regulatory proteins CatR and ClcR with heterologous promoters: functional and evolutionary implications. Proc. Natl. Acad. Sci. U.S.A. 91 12393–12397. 10.1073/pnas.91.26.12393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov D. A. (2007). Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem. Rev. 107 3467–3497. 10.1021/cr068309+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda H., Yamada A., Miyauchi K., Masai E., Fukuda M. (2004). Characterization of transcriptional regulatory genes for biphenyl degradation in Rhodococcus sp. strain RHA1. J. Bacteriol. 186 2134–2146. 10.1128/JB.186.7.2134-2146.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka S., Sato T., Koshiya J., Murakami S., Aoki K. (2009). Gene cloning and characterization of a deaminase from the 4-amino-3-hydroxybenzoate-assimilating Bordetella sp. strain 10d. FEMS Microbiol. Lett. 298 93–98. 10.1111/j.1574-6968.2009.01699.x [DOI] [PubMed] [Google Scholar]
- Teramoto M., Harayama S., Watanabe K. (2001). PhcS represses gratuitous expression of phenol-metabolizing enzymes in Comamonas testosteroni R5. J. Bacteriol. 183 4227–4234. 10.1128/JB.183.14.4227-4234.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teufel R., Gantert C., Voss M., Eisenreich W., Haehnel W., Fuchs G. (2011). Studies on the mechanism of ring hydrolysis in phenylacetate degradation: a metabolic branching point. J. Biol. Chem. 286 11021–11034. 10.1074/jbc.M110.196667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tover A., Ojangu E. L., Kivisaar M. (2001). Growth medium composition-determined regulatory mechanisms are superimposed on CatR-mediated transcription from the pheBA and catBCA promoters in Pseudomonas putida. Microbiology 147(Pt. 8) 2149–2156. 10.1099/00221287-147-8-2149 [DOI] [PubMed] [Google Scholar]
- Tover A., Zernant J., Chugani S. A., Chakrabarty A. M., Kivisaar M. (2000). Critical nucleotides in the interaction of CatR with the pheBA promoter: conservation of the CatR-mediated regulation mechanisms between the pheBA and catBCA operons. Microbiology 146(Pt. 1) 173–183. 10.1099/00221287-146-1-173 [DOI] [PubMed] [Google Scholar]
- Trefault N., Guzmán L., Pérez H., Godoy M., González B. (2009). Involvement of several transcriptional regulators in the differential expression of tfd genes in Cupriavidus necator JMP134. Int. Microbiol. 12 97–106. [PubMed] [Google Scholar]
- Tropel D., van der Meer J. R. (2002). Identification and physical characterization of the HbpR binding sites of the hbpC and hbpD promoters. J. Bacteriol. 184 2914–2924. 10.1128/JB.184.11.2914-2924.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valderrama J. A., Durante-Rodríguez G., Blázquez B., García J. L., Carmona M., Díaz E. (2012). Bacterial degradation of benzoate: cross-regulation between aerobic and anaerobic pathways. J. Biol. Chem. 287 10494–10508. 10.1074/jbc.M111.309005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg B., Bhamidimarri S. P., Winterhalter M. (2015). Crystal structure of a COG4313 outer membrane channel. Sci. Rep. 5:11927. 10.1038/srep11927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedler E., Vahter M., Heinaru A. (2004). The completely sequenced plasmid pEST4011 contains a novel IncP1 backbone and a catabolic transposon harboring tfd genes for 2,4-dichlorophenoxyacetic acid degradation. J. Bacteriol. 186 7161–7174. 10.1128/JB.186.21.7161-7174.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wöhlbrand L., Wilkes H., Halder T., Rabus R. (2008). An aerobic degradation of p-ethylphenol by “Aromatoleum aromaticum” strain EbN1: pathway, regulation, and involved proteins. J. Bacteriol. 190 5699–5709. 10.1128/JB.00409-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K. M., Karl M. R., Blatt L. M., Simon M. J., Winter R. B., Fausset P. R., et al. (1991). Cloning and characterization of a Pseudomonas mendocina KR1 gene cluster encoding toluene-4-monooxygenase. J. Bacteriol. 173 5315–5327. 10.1128/jb.173.17.5315-5327.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaar A., Eisenreich W., Bacher A., Fuchs G. (2001). A novel pathway of aerobic benzoate catabolism in the bacteria Azoarcus evansii and Bacillus stearothermophilus. J. Biol. Chem. 276 24997–25004. 10.1074/jbc.M100291200 [DOI] [PubMed] [Google Scholar]
- Zamarro M. T., Martín-Moldes Z., Díaz E. (2016). The ICEXTD of Azoarcus sp. CIB, an integrative and conjugative element with aerobic and anaerobic catabolic properties. Environ. Microbiol. 18 5018–5031. 10.1111/1462-2920.13465 [DOI] [PubMed] [Google Scholar]
- Zhou L., Wang J. Y., Wu J., Wang J., Poplawsky A., Lin S., et al. (2013). The diffusible factor synthase XanB2 is a bifunctional chorismatase that links the shikimate pathway to ubiquinone and xanthomonadins biosynthetic pathways. Mol. Microbiol. 87 80–93. 10.1111/mmi.12084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.