

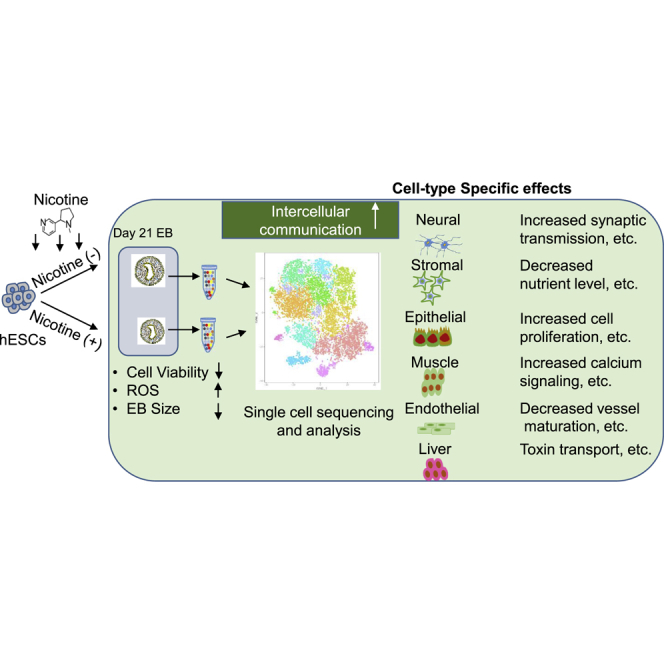
Summary
Nicotine, the main chemical constituent of tobacco, is highly detrimental to the developing fetus by increasing the risk of gestational complications and organ disorders. The effects of nicotine on human embryonic development and related mechanisms, however, remain poorly understood. Here, we performed single-cell RNA sequencing (scRNA-seq) of human embryonic stem cell (hESC)-derived embryoid body (EB) in the presence or absence of nicotine. Nicotine-induced lineage-specific responses and dysregulated cell-to-cell communication in EBs, shedding light on the adverse effects of nicotine on human embryonic development. In addition, nicotine reduced cell viability, increased reactive oxygen species (ROS), and altered cell cycling in EBs. Abnormal Ca2+ signaling was found in muscle cells upon nicotine exposure, as verified in hESC-derived cardiomyocytes. Consequently, our scRNA-seq data suggest direct adverse effects of nicotine on hESC differentiation at the single-cell level and offer a new method for evaluating drug and environmental toxicity on human embryonic development in utero.
Keywords: single-cell RNA sequencing, nicotine, smoking, embryonic stem cells, differentiation, embryonic development
Graphical Abstract
Highlights
-
•
scRNA-seq reveals that nicotine alters transcriptome of hESC differentiation
-
•
Nicotine reduces cell viability, increases ROS level, and changes cell cycle of EBs
-
•
Nicotine disturbs intracellular Ca2+ in hESC-derived cardiomyocytes
Using single-cell RNA sequencing (scRNA-seq), Wu and colleagues elucidate the adverse effects of nicotine exposure on various cell lineages derived from human embryonic stem cells. This study offers new insights into the toxicity of environmental factors on human embryonic development.
Introduction
Maternal smoking during pregnancy is an established risk factor for birth defects such as miscarriage, growth restriction, and premature birth (Jaddoe et al., 2008). It is closely associated with adverse neurobehavioral, cardiovascular, respiratory, endocrine, and metabolic outcomes in the offspring, which can persist into adulthood (Holbrook, 2016). Nicotine, the main chemical constituent of tobacco smoking, is primarily responsible for the elevated risk (Holbrook, 2016). Unfortunately, the introduction and spread of new tobacco products containing nicotine, such as e-cigarettes, is reversing recent progress toward reduction of tobacco use (Bao et al., 2018).
A large body of research has elucidated the negative effects of nicotine in animals, mainly in rodent models. Animal studies have demonstrated that nicotine exposure during pregnancy has detrimental effects on fetal development, such as cellular damage, increased inflammation (Mohsenzadeh et al., 2014), oxidative stress (Lin et al., 2014), endoplasmic reticulum stress (Wong et al., 2016), and impaired cell replication (Repo et al., 2014, Slotkin et al., 1987). The suitability of clinical translation of these studies, however, remains questionable due to interspecies physiological differences and uncertainty over the degree and route of nicotine exposure (Tizabi, 2007, Winzer-Serhan, 2008). To address these issues, some studies have attempted to study the effects of nicotine using human cells. For example, using microarray analysis, Liszewski et al. (2012) demonstrated that tobacco smoke and nicotine have lineage- and stage-specific effects on differentiated human embryonic stem cell (hESCs).
Although the in vitro differentiation of embryonic body (EB) model can be used to mimic early developments from pre-implantation epiblasts to lineage-committed progenitors, conventional bulk RNA sequencing (RNA-seq) analysis has limitations for studying the individual cellular heterogeneity within the EBs. With the recent advent of microdroplet-based single-cell RNA-seq (scRNA-seq) technologies, it is now feasible to analyze transcriptomes at the single-cell level within heterogeneous cell populations (Blakeley et al., 2017, Paik et al., 2018). Here, we used scRNA-seq of EBs to characterize the effects of nicotine on hESC differentiation. We found that nicotine exposure reduced cell viability and increased reactive oxygen species (ROS), resulting in aberrant formation and differentiation of EBs. Nicotine exposure also altered cell cycling in endothelial, stromal, and muscle progenitor cells differentiated from hESCs. Furthermore, nicotine caused lineage-specific effects and dysregulated cell-to-cell communication. We found abnormal Ca2+ signaling pathways in muscle cells upon nicotine exposure that was verified using hESC-derived cardiomyocytes. Taken together, the effects of nicotine exposure on hESC differentiation at the single-cell transcriptomic level offer new insights into mechanisms of nicotine toxicity on early embryonic development, and can provide new tools for optimizing drug toxicity screening.
Results
scRNA-Seq Analysis Reveals Six Major Types of Progenitor Cells
To investigate the effects of nicotine on hESC differentiation, we performed microdroplet-based scRNA-seq to identify unique cell lineages on day 21 control and nicotine-exposed EBs (Figure 1A). We used 10 μM nicotine exposure for 21 days, which is similar to nicotine concentrations found in fetal serum (Luck et al., 1985) and has been used in prior hESC studies (Hirata et al., 2016, Zdravkovic et al., 2008). After dissociation, transcriptomic data of 5,646 single cells from nicotine-exposed EBs and 6,847 single cells from control EBs were acquired. Sequenced data showed high read depth, and were mapped to approximately 3,000 median genes per cell (Figure S1A, left). The percentage of mitochondrial genes present in most cells was less than 10% (Figure S1A, right). We used the Seurat package (Satija et al., 2015) to perform principal-component analysis and t-distributed stochastic neighbor embedding (t-SNE) analysis. Control EBs were divided into 13 clusters, and nicotine-exposed EBs were divided into 12 clusters that exhibited distinct gene expression patterns (Figures S1B and S1C). Control and nicotine-exposed EBs contained similar cell-type markers, without any observed differences in cell types between the two samples (Figure S1B).
Figure 1.

scRNA-Seq Analysis Reveals Cell Lineages in Control and Nicotine-Exposed Embryoid Bodies
(A) Process flow diagram of scRNA-seq analysis on hESC differentiation. Single cells were collected from two independent EB differentiation experiments from day 21 EBs (nicotine-exposed versus control) and were prepared by single-cell barcoded droplets and chemicals from 10× Genomics. Bioinformatics data were processed using Seurat. Cell-type marker, differentially expressed gene, cell communication, and pathway analyses were performed to investigate the effects of nicotine exposure on hESC differentiation.
(B) Separated (left) and combined (middle and right) t-SNE plots of single cells from control and nicotine-exposed EBs. We defined six main types of progenitor cells in day 21 EBs, including muscle progenitor cells (clusters 3 and 13), liver progenitor cells (cluster 5), neural progenitor cells (clusters 3, 4, 8, and 11), stromal progenitor cells (cluster 6), epithelial progenitor cells (clusters 2 and 12), and endothelial progenitor cells (cluster 11). In addition, undifferentiated stem-like cells (USCs) (cluster 1) and undetermined cells (UDCs) (cluster 9) were also identified.
(C) Heatmap showing the expression pattern of top 10 differential genes in each cell type. Representative differential genes for each cell type are listed on the right side. The complete lists of differential genes for each cell type are listed in Table S3.
(D) Violin plots show the expression level distributions of marker genes across cell types.
Next, we performed integrative analysis to compare the cell proportions and gene expression differences in each cell type between nicotine and control EBs. Nicotine exposure induced widespread transcriptomic changes, which were manifested as a shift in the t-SNE projections of singlets (Figure 1B, middle). Previous reports with bulk RNA-seq data also indicated that nicotine affects gene expression in multiple cell lineages (Liszewski et al., 2012). A total of 13 individual clusters were defined from the combined datasets (termed C1 to C13) (Figure 1B, right). Based on differential genes enriched in each cluster, six major types of progenitor cells were identified by Seurat (Figures 1C and 1D). Clusters 3, 4, 8, and 10 were associated with high expression of LHX2 and NR2F1 (de Melo et al., 2016, Tang et al., 2010) and annotated as neural cells. Cluster 5 represented liver progenitor cells with a high expression of FRZB and PTN (Michelotti et al., 2014, Shen et al., 2015). Cluster 6 was annotated as stromal progenitor cells with a high expression of SFRP2 and COL2A1 (Saito et al., 2013, Tabib et al., 2018). Cluster 11 was annotated as endothelial progenitor cells with high expression of GDF15 and DDIT3 (Ahrens et al., 2011, Loinard et al., 2012). Cluster 2 and 12 were annotated as epithelial progenitor cells (EpiPCs) with high expression of IGFBP5 and PODXL (Sugrue et al., 2016, Zhu et al., 2016). Clusters 7 and 13 showed a high expression of HAPLN1 and S100A11, and were annotated as muscle progenitor cells (DeLaughter et al., 2013, Malmstrom et al., 2004). Cluster 1 was enriched for pluripotency genes such as TERF1 and POU5F1, and was annotated as “undifferentiated stem-like cells”. Cluster 9 was enriched for cytoskeletal genes such as ACTB and TUBB, and was annotated as “undetermined cells”.
To further confirm our cluster annotation, we found genes specifically expressed in each cell type that were enriched for the expected appropriate gene ontology (GO) terms. For example, genes that were specifically expressed in muscle progenitor cell clusters were significantly enriched for the cytosolic Ca2+ pathway (p = 2.92 × 10−11) and skeletal system development (p = 3.36 × 10−4). Genes expression in the neural progenitor cell clusters were significantly enriched for nervous system development (p = 1.01 × 10−6) and sensory organ development (p = 5.60 × 10−8). Genes expression in the liver progenitor cell cluster were enriched for liver development (p = 2.52 × 10−4) and response to lipid (p = 1.74 × 10−2). Genes expression in the endothelial progenitor cell cluster were significantly enriched for blood vessel development (p = 1.3 × 10−4) and angiogenesis (p = 1.61 × 10−3). Genes expressions in the EpiPC clusters were enriched for lung development (p = 2.43 × 10−5) and kidney development (p = 5.67 × 10−4) (Figure S1D).
It should be noted that neural, muscle, and epithelial progenitor cells consisted of several sub-clusters. Neural progenitor cells were further divided into four subsets: clusters 3, 4, 8, and 10. Cluster 3 showed a high expression of LHX5/HESX1 that is related to forebrain development (Martynova et al., 2018, Zhao et al., 1999). Cluster 4 was enriched for HMGB2 and PTTG1, which are highly expressed in proliferating neural stem cells (Kimura et al., 2018). Cluster 8 was annotated as neural progenitor cells with an enrichment of HNRNPH1 and PTPRS (Tchetchelnitski et al., 2014, Yazdani et al., 2015), which are related to sensory neurons development. Cluster 10 was enriched for LHX2 and NR2F1 and expressed eye development genes (de Melo et al., 2016, Tang et al., 2010). Muscle progenitor cells were divided into two subsets: clusters 7 and 13. Cluster 7 was annotated as muscle cells for the expression of HAPLN1 and S100A11, which are highly expressed in smooth muscle cells (DeLaughter et al., 2013, Malmstrom et al., 2004). Cluster 13 was enriched for ZFHX3 and NR2F2, which are related to cardiac muscle development (Berry et al., 2001, Pei et al., 2017). EpiPCs were divided into two subsets: clusters 2 and 12. Cluster 2 was enriched for IGFBP5 and HES1, which are related to eye development (Liu et al., 2013, Sugrue et al., 2016). Cluster 12 was enriched for B3GNT7 and PODXL, which are highly expressed in stem-like epithelial cells (Dumont-Lagace et al., 2017) (Figures 1D and S1D).
Overall, six major types of progenitors (neural, liver, stromal, endothelial, epithelial, and muscle) were identified from scRNA-seq data of EBs based on cell markers detected by Seurat. These data may be useful for modeling nicotine exposure on individual organs and cells within the developing fetus.
Nicotine Elicits Cell-Type-Specific Response in Differentiated EBs
Integrated analysis of control and nicotine-exposed EBs at the single-cell level enables us to quantitatively assess cell-type-specific responses to nicotine. Quantification of the cell-type compositional changes showed changes from 5% reduction in epithelial progentior cells to 4% increase in liver progenitor cells following nicotine exposure (Figures 2A and S2A). Next, we performed comparative analysis and calculated the average expression of both the nicotine-exposed and control cells to determine differentially expressed genes (DEGs) in each cell type (Figure 2B). Interestingly, there was a marked difference in the number of DEGs among different cell types, ranging from 5 to 103 genes with a p-value less than 0.01 and a log fold-change more than 0.25. For example, we observed 5 DEGs in liver progenitor cells with nicotine treatment, whereas muscle progenitor cells exhibited the greatest number of 103 DEGs. Among these DEGs, BNIP3, and metallothionein family genes (MT1X, MT1G, MT1E, and MT2A) were uniformly downregulated and RPS10 was upregualted in most of cell types (Figure 2B). BNIP3 gene is an important regulator during long-term nicotine-induced cell death in several cell types (Erkan et al., 2005, Tang et al., 2007). Metallothionein family genes play a role in the protection against metal toxicity and oxidative stress, and have been shown to be suppressed in chronic smokers (Billatos et al., 2018). These genes are involved in apoptosis, ROS generation, mitochondrial function, and response to metal ion pathways (Figure 2C), indicating that EBs have poor cell survival upon nicotine exposure.
Figure 2.

Nicotine Exposure Induces Cell-Type-Specific Response
(A) Cell proportion fluctuation for each cell type with nicotine exposure. Cell proportion fluctuation for each cluster with nicotine exposure is shown in Figure S2A. USCs, undifferentiated stem-like cells; UDCs, undetermined cells; EpiPCs, epithelial progenitor cells; ECs, endothelial progenitor cells.
(B) Plots of the average expression of genes from control and nicotine-exposed EBs for each cell type. Significant differentially expressed genes are labeled in the plots (p < 0.05). The complete list of differentially expressed genes upon nicotine exposure for each cluster is shown in Table S5.
(C) Pathway enrichment analysis of statistically significant gene ontologies following nicotine exposure. The size of the circle represents the significance of gene ontologies, and the darkness of color represents the number of genes involved in the gene ontologies. The complete lists of differentially expressed gene-related pathways upon nicotine exposure for each cluster are listed in Table S6.
(D) Heatmap for differentially expressed genes in scRNA-seq data, public hepatic cell line (HepaRG), human gingival epithelium cell line (HGEC), human smooth muscle cells (HSMCs), and human iPSC-derived endothelial cell (hiPSC-ECs) in terms of fold-change (nicotine-exposed relative to control EBs). Each row represents a single differentially expressed gene identified in (B). The names of cell types are labeled on the left side. Differentially expressed genes in scRNA-seq data corresponding to the public datasets are labeled with a rectangle.
See also Figure S2.
Nicotine also showed cell-type-specific responses. APOE, TUBA1A, and NDUFC2 were significantly upregulated, and H1F0 and SRRM2 were downregulated, in neural progenitor cells (Figures 2B and 2C). Abnormal expression of these genes can lead to β-amyloid formation and increased synaptic transmission (Moreno-Gonzalez et al., 2013), brain malformations (Aiken et al., 2017), and intellectual disability (Tanaka et al., 2018). In muscle progenitor cells, the most upregulated gene following nicotine exposure was HSP90AA1, a myosin chaperone protein gene involved in muscle development and disease (Armant et al., 2016, Etard et al., 2015). Increased expression of HMGB1, known to regulate cardiac excitation-contraction coupling by enhancing the sarcoplasmic reticulum Ca2+ leakage through Toll-like receptor 4 (TLR4)-ROS signaling in cardiac muscle cells, was also observed (Figures 2B, 2C, and S2C).
In stromal progenitor cells, LDHA and DAPL1, known to regulate nutrient levels and amino acid acetylation, were downregulated upon nicotine exposure (Figures 2B and 2C). BINP3, related to lipid metabolism, was downregulated in liver cells (Glick et al., 2012). In epithelial progenitor cells, macrophage migration inhibitory factor (MIF), which is associated with chronic obstructive pulmonary disease in human, was downregulated (Sauler et al., 2015). WDR77, required for proliferation of lung and prostate epithelial cells during development and tumorigenesis (Sheng and Wang, 2016), was upregulated in epithelial progenitor cells in the presence of nicotine (Figures 2B and 2C). In endothelial progenitor cells, LDHA, DDIT3, and IFITM1 were downregulated, and HNRNP2B1 was upregulated upon nicotine exposure (Figures 2B and 2C). Downregulation of LDHA is related to the suppression of glycolysis and endothelial cell dysfunction (Xu et al., 2016). Downregulated DDIT3 exhibits reduced ER stress response upon long-term cigarette smoke exposure (Geraghty et al., 2011). IFITM1 downregulation is related to endothelial lumen formation during angiogenesis (Popson et al., 2014) (Figures 2B and 2C). Likewise, we confirmed cell-type-specific responses to long-term nicotine exposure on EBs using GO pathway enrichment analysis (Figure 2C).
To determine how our analysis correlates with previously reported bulk RNA-seq data from specific cell lines, we examined the expression fold-changes of DEGs in published gene expression data after nicotine exposure, including hepatic cells (HepaRG), human gingival epithelium cells (HGECs), human smooth muscle cells (HSMCs), and human iPSC-derived endothelial cells (hiPSC-ECs) (De Abrew et al., 2016, Gumus et al., 2008, Yoshiyama et al., 2014). Interestingly, the trend in gene expression changes in cluster12 epithelial progenitor cell was similar to that of HGECs, whereas an opposite trend of gene expression changes was observed between cluster-2 epithelial progenitor cells and HGECs. This may be due to HGECs being more similar to stem-like “cluster-12 epithelial cell” in biological identity and more different from “cluster-2 epithelial cell”. In HSMCs, the overall change of DEGs was subtle, but the trends in fold-change expression were consistent with EB-derived muscle cells. The genes downregulated in EB-derived endothelial cells also reduced their expression in hiPSC-ECs upon nicotine exposure (Figure 2D). Taken together, DEG analysis showed cell-type-specific transcriptomic changes upon nicotine exposure, which are consistent with previously reported bulk RNA-seq or microarray analysis in different cell types (De Abrew et al., 2016, Gumus et al., 2008, Yoshiyama et al., 2014). Our data thus provide a novel method for evaluating nicotine toxicity in heterogeneous populations of human EBs at a single-cell level.
Nicotine Dysregulates Viability, ROS Generation, and Cell Cycle in EBs
Infants exposed to nicotine prenatally often exhibit lower birth weights than their peers (Fried and Oconnell, 1987, Slotkin, 1998). Animal studies have shown that nicotine exposure during pregnancy induces cellular damage, oxidative stress, and impaired cell replication (Repo et al., 2014, Slotkin et al., 1987). However, the molecular mechanisms remain poorly understood. Our DEG analysis showed that long-term nicotine exposure induced apoptosis and ROS generation mediated by the downregulation of BNIP3 and metallothionein family genes (Figures 2B and 2C). Therefore, we performed several assays to confirm decreased survival of nicotine-exposed EB. Nicotine-exposed EBs were smaller than control EBs (Figures 3A and 3B), and cell viability was significantly reduced based on quantification of ATP, an indicator of metabolically active cells (Figure 3C). We also found higher levels of ROS in nicotine-exposed EBs compared with control EBs (Figure 3D).
Figure 3.

Nicotine Reduces Cell Viability, Increases ROS Levels, and Changes Cell Cycle in EBs
(A) Representative bright-field images of day 21 control and nicotine-exposed EBs. Scale bar, 100 μm.
(B) Size measurement of control (n = 45) and nicotine-exposed (n = 43) EBs in terms of diameter. EBs were collected from three independent EB differentiation experiments and pooled together for size measurement. ∗∗∗p < 0.001.
(C) Cell viability assay of EBs based on quantitation of the ATP present in control and nicotine-exposed EBs. Cell viability were measured from three independent experiments. ∗p < 0.05.
(D) Reactive oxygen species (ROS) generation in control and nicotine-exposed EBs. ROS was measured from three independent experiments. ∗p < 0.05.
(E) Proportion of cluster 13 muscle cells in G2M, S, or G1 phase.
(F) Distribution of TUBB4B expression in three cell-cycle phases in control (top) and the nicotine-exposed EBs (bottom). Dashed lines represent the center of G2M phase in control and nicotine-exposed EBs.
Clinical and animal studies have shown that nicotine exposure changes the dynamics of cell replication and causes growth restriction (Repo et al., 2014). We thus analyzed cell cycling in the scRNA-seq data to evaluate the growth of EB after nicotine treatment by calculating cell-cycle phase scores based on canonical markers (Nestorowa et al., 2016). Relative to control EBs, nicotine-exposed EBs exhibited a 12% decrease in G1 phase, a 6% increase in G2M phase, and a 5.5% increase of S phase in endothelial progenitor cells. In stromal progenitor cells, we found an 11% decrease in G1 phase and a 12% increase in S phase (Figure S2B). Surprisingly, we found that there was a 20% decrease of cells in the G1 phase, a 5% increase of cells in the G2M phase, and a 15% increase of cells in S phase in the “cluster-13 muscle progenitor cell” (Figure 3E). For example, TUBB4B, a G2M phase marker, was differentially expressed in muscle progenitor cells from nicotine-exposed EBs versus control EBs (Figure 3F). Consequently, nicotine exposure increased ROS production and cell death in EBs and affected the cell cycle of endothelial, stromal, and muscle progenitor cells.
Nicotine Exposure Dysregulates Cell-to-Cell Communication of Differentiated EBs
Smoking and nicotine consumption increase the pathological risk in endocrine, reproductive, respiratory, cardiovascular, and neurologic systems that all rely on intricate and dynamic interactions among multiple functional cell types for homeostasis and function (Kawasaki et al., 2011, Rehan et al., 2009). The effect of nicotine on cell-to-cell communication, however, is not well understood. Recent studies using in vitro co-cultured systems indicate that cell-to-cell communication could be affected by nicotine exposure (Holownia et al., 2015, Larsen et al., 2016, Liu et al., 2017). Our study used a dataset of human ligand-receptor pairs (Ramilowski et al., 2015) to define intercellular communication networks. To examine the effects of nicotine on cell-to-cell communication, we also analyzed ligand-receptor expression differences in nicotine-exposed EBs and control EBs. Overall, we observed increased intercellular communication for each EB cell type upon nicotine exposure (Figures 4A and 4B). For example, the number of ligand-receptor pairs in autocrine circuits from muscle progenitor cell was increased from 51 to 85, and the number of ligand-receptor pairs in muscle-neuron crosstalk was increased from 47 to 71 (Figure 4B).
Figure 4.

Nicotine Increased Cell-to-Cell Communication and Induced HMGB1-TLR4 Pathway among Cell Types in EBs
(A) Intercellular communication analysis among cell types in the control and nicotine-exposed EBs. Line color indicates ligands broadcast by the cell population of the same color (labeled). Lines connect to cell populations where cognate receptors are expressed. Line thickness is proportional to the number of ligands where cognate receptors are present in the recipient cell population. Loops indicate autocrine circuits. Map quantifies potential communication but does not account for anatomic position or boundaries of cell populations. Crosstalk among cell types in control and nicotine-exposed EBs are listed in Table S7.
(B) Detailed view of ligands broadcast by each cell type and those populations expressing cognate receptors primed to receive a signal. Numbers indicate the quantity of ligand-receptor pairs for each inter-population link. Colors of the cell population corresponds to (A) above.
(C) Venn diagrams of ligands present in control or nicotine-exposed EBs and differentially expressed genes between control and nicotine-exposed EBs.
(D) Heatmap of the expression of differentially expressed ligands in control versus nicotine-exposed EBs across different cell types in terms of average fold-change of expression. Seven ligands present in both control and nicotine-exposed EBs (labeled “both”) were selected as differentially expressed ligands. Three ligands were selected as expressed only in nicotine-exposed EBs and labeled as “nicotine.”
(E) Intercellular communication analysis among different cell types shows activated HMGB1-TLR4 pathway upon nicotine exposure. The meaning of the thickness and color of line is explained in (A). USCs, undifferentiated stem-like cells; UDCs, undetermined cells; EpiPCs, epithelial progenitor cells; ECs, endothelial progenitor cells.
Next, we analyzed the expression of ligands in each cell type to identify 61 ligands that were expressed in both nicotine-exposed EBs and control EBs, of which 7 were differentially expressed. Seventy-five ligands were mainly expressed in nicotine-exposed EBs, of which 3 were enriched in certain cell types identified by DEG analysis (Figures 4C and 4D). One extracellular ligand, high-mobility group box 1 (HMGB1), was uniformly upregulated in multiple cell types of nicotine-exposed EBs, but was not presented in control EBs (Figures 4C and 4D). Previous research has shown that HMGB1 regulates Ca2+ handling and cellular contractility by activating its receptor Toll-like receptor 4 (TLR4) in rat cardiomyocytes, which plays an important role in the pathogenesis of cardiac dysfunction in many diseases (Zhang et al., 2014). Here, we found that HMGB1-TLR4 signaling, although not specific, was also activated in muscle progenitor cells (Figure 4E), suggesting that the activated HMGB1-TLR4 pathway may play an important role in cardiac dysfunction upon nicotine exposure.
Disturbance of Intracellular Ca2+ Handling in hESC-Derived Cardiomyocytes by Nicotine Exposure
Our data indicate that nicotine affects the expression of genes associated with intracellular Ca2+ handling via the HMBG1-TLR4 pathway in cardiac muscle cells, and animal studies have shown that nicotine exposure disrupts intracellular Ca2+ homeostasis in cardiac cells (Hu et al., 2013). To investigate whether nicotine affects the Ca2+ handling in cardiac muscle cells, we first checked the expression of HMGB1 and TLR4 in hESC-derived cardiomyocytes. The expression of HMGB1 was increased by 2-fold, and TLR4 increased by 90-fold in hESC-derived cardiomyocytes exposed to nicotine (Figure 5A). We next conducted single-cell Ca2+ measurement using Fura-2 in hESC-derived cardiomyocytes (Figure S3). As shown in Figures 5B–5E, nicotine increased the diastolic Ca2+ (Figures 5B and 5C) and reduced the Ca2+ transient amplitude (Figure 5D), accompanied by prolonged Ca2+ decay (Figure 5E), suggesting compromised intracellular Ca2+ homeostasis. We found that nicotine increased the propensity for arrhythmic Ca2+ release in hESC-derived cardiomyocytes, as indicated by the arrows in Figures 5F and 5G. These data strongly suggest that nicotine increases Ca2+-release abnormalities at the cellular level, predisposing these cells to Ca2+-associated arrhythmia.
Figure 5.

Nicotine Disrupts Intracellular Ca2+ Handling in hESC-Derived Cardiomyocytes
(A) Real-time PCR verification of the expression levels of HMGB1 and TLR4 in hESC-derived cardiomyocytes for nicotine-exposed and control group. Real-time PCR was conducted from three independent differentiation experiments. Nicotine and ethanol were added during cardiomyocyte differentiation process. Ethanol-exposed hESC-derived cardiomyocytes were used as a control. ∗∗∗p < 0.001.
(B–E) Representative single-cell Ca2+ traces with and without treatment of nicotine (1 μM) (B). Cells were paced at 0.5 Hz and recorded at 37°C. Nicotine affects diastolic Ca2+ (C), Ca2+ transient amplitude (D), and Ca2+ uptake (E).
(F) Representative arrhythmic Ca2+ release. The arrhythmic Ca2+ release is indicated by the arrows.
(G) Percentage of cells with normal or arrhythmic Ca2+ release. Numbers indicate the recorded cell number in each condition. Recording was conducted with two independent differentiation. The cell number shows the pooled data.
∗∗p < 0.01, ∗∗∗p < 0.001. Error bars indicate SEM.
Discussion
In this study, we performed scRNA-seq analysis on a total of 12,500 single cells generated from human ESC-derived EBs following 21 days of culture with or without nicotine (Figure 1). Previous studies have demonstrated that nicotine concentrations in fetal serum are much higher than in maternal serum, ranging from 0.3 to 15.4 μM in fetal serum (Luck et al., 1985). Based on these reports, and the concentrations studied in other investigations of nicotine effect on hESCs (0.1–10.0 μM), we decided to use 10 μM of nicotine during hESC differentiation (Hirata et al., 2016, Zdravkovic et al., 2008). We also found that a 6-day exposure to nicotine reduces viability in hESCs (Figure S3D), suggesting that nicotine affects embryo development as early as the pre-implantation stage.
We did not observe cell-type differences between nicotine-exposed EBs and control EBs, although there were minor changes in the cell-type distribution upon nicotine exposure. However, DEG patterns from various progenitor cell populations indicated broad effects on cells derived from all three germ layers (neural, stromal, muscle, endothelial, and epithelial progenitor cells). This is consistent with clinical observations that nicotine-exposed infants have health problems throughout their lives, including impaired function of the endocrine, reproductive, respiratory, cardiovascular, and neurologic systems (Warren et al., 2014). In addition, although the current technology does not allow us to conduct proteomic analyses at a single-cell resolution level, we searched available nicotine-associated proteomic datasets in PubMed and found that our DEGs, HSPA8, BAX, and CKB were also reported to be upregulated in mouse neurons (Matsuura et al., 2016), MIF was downregulated in human endothelial cells (Zhang et al., 2014), and that SLC25A4, REEP5, and ATP5F1 were upregulated in human epithelial cells (Ghosh et al., 2018) (Figure S2E). Moreover, among these DEGs, BNIP3 is uniformly downregulated in most cell types (Figure 2B). We observed downregulated BNIP3 expression in nicotine-exposed EBs compared with the control EBs at the protein level (Figure S2D). These lines of evidence suggest that our scRNA-seq data can reflect protein expression to some extent.
Our scRNA-seq analysis suggests that downregulation of BNIP3 and metallothionein family gene expression in multiple progenitor cell types upon nicotine exposure provides the molecular mechanisms that lead to altered DEG patterns. It has been reported that reduced BNIP3 expression correlates with poor cell survival following long-term nicotine exposure (Tang et al., 2007). Our data indicate that hESC-derived cells adapt to long-term nicotine exposure and cell damage resulting from downregulation of BNIP3 (Figure 2B). This may be analogous to the observed low birth weight, preterm birth, and perinatal death in a developing fetus as a result of maternal smoking during pregnancy (Warren et al., 2014).
Nicotine also displayed cell-type-specific adverse effects, consistent with previous findings in animal and clinical studies (Holbrook, 2016). For example, we found that APOE was upregulated in nicotine-exposed EBs, and it has previously been shown that upregulated APOE leads to brain malformations and intellectual disability (Tanaka et al., 2018). In muscle cells, increased expression of HMGB1 impairs cardiac excitation-contraction (Zhang et al., 2014) and increases nicotine-induced risk for Ca2+-associated arrhythmias (Figure 5). In addition, the DEGs identified by scRNA-seq analysis are similar to bulk transcriptome studies performed in human cell lines (Figure 2D).
Interestingly, we found that nicotine dysregulates the cell cycle of endothelial, stromal, and muscle progenitor cells from G1 phase to S/G2M phases. Previous studies have also shown that nicotine stimulates the cell cycle in aortic smooth muscle cells, epithelial cells, and lung cancer cells (He et al., 2014). One study shows that nicotine enhances proliferation and induces cyclin D1 to stimulate G1 to S/G2 phase transition in human bronchial smooth muscle cells (Hong et al., 2017), which is consistent with our findings in cluster-13 muscle progenitor cells. A possible mechanism is that nicotine activates RAS/MAPK pathway via nicotinic acetylcholine receptors (nAChRs) to trigger a network that positively regulates cell-cycle progression through G1 to S, such as cyclin D (Hong et al., 2017). Here, although no expression difference was observed on cyclin D and RAS/MAPK in our scRNA-seq data, we found that the components of cell-cycle machinery, such as HSP90AA1, TUBB4B, and TUBA1B, which are related to G2M transition (Duggal et al., 2018), and HNRNPH1 and HNRNPA2B1, which are related to G1 to S transition (Duggal et al., 2018), are upregulated in nicotine-exposed muscle cell cluster 3.
As such, scRNA-seq analysis provides a robust tool for investigating cell-to-cell interactions (Kawasaki et al., 2011, Rehan et al., 2009) in development and disease pathobiology. In particular, an activated HMGB1-TLR4 pathway was pronounced in multiple cell types within EBs upon nicotine exposure. High expression of HMGB1 in multiple organs, perhaps induced by the secondary effects of nicotine such as oxidative stress, apoptosis, and inflammatory factors (Loukili et al., 2011, Scaffidi et al., 2002, Kim et al., 2016), and is known to mediate multiple pathological conditions (Ko et al., 2014), has been shown in smokers. For example, upregulated HMGB1 impairs cardiac excitation-contraction coupling by enhancing sarcoplasmic reticulum Ca2+ leakage through TLR4-ROS signaling in cardiomyocytes (Zhang et al., 2014). This indicates that HMGB1 could be a potential drug target for nicotine-induced embryonic defects.
Nicotine has been suggested to mediate its function via an nAChR-dependent or -independent pathway. Nicotinic receptors are expressed in undifferentiated and differentiating cells (Figure S3C). Studies have shown that nAChRs mediate apoptosis, cell proliferation, cell differentiation, regulation of intracellular calcium, oxidative stress, and inflammation by nicotine (Dasgupta and Chellappan, 2006). In addition, nicotine is reported to promote tumor progression by binding to β-adrenergic receptors (Carlisle et al., 2007). In addition, nicotine drives both cell proliferation and cell death via paracrine signaling by cell-cell interaction (Delitto et al., 2014, Scaffidi et al., 2002). Thus, nicotine may induce the adverse effects of EBs through nAChR-dependent and -independent pathways, as well as cell-cell interaction.
Furthermore, scRNA-seq analysis can be used to optimize the treatment manner and period of drug use for patient-specific drug screening/testing. For example, with the increasing availability of commercial human genomic sequencing data, we can evaluate embryonic developmental-specific drug responses with (single nucleotide polymorphism) SNPs, and the response can be confirmed by gene editing with the CRISPR technology (Seeger et al., 2017). Specifically, the SNP (rs141819830) of the GFI1 gene has been reported to be sensitive to maternal smoking in exposed neonates (Gonseth et al., 2016). Thus, we may be able to use patient-specific hiPSC-derived EBs carrying this SNP to evaluate the risk of nicotine toxicity on embryonic development. We anticipate that this platform should help correlate the risk of the GFI1 SNP gene with maternal smoking during pregnancy.
In summary, we used microdroplet-based scRNA-seq to investigate the adverse effects on heterogeneous EBs upon nicotine exposure. Our study offers an effective platform to evaluate the potential effects of nicotine on human embryonic development. Our data provide potential molecular mechanisms for prenatal nicotine toxicity on specific cell populations derived from human ESCs.
Cell Culture and Differentiation
hESC line, H7 (WiCell Research Institute), was seeded on Matrigel (BD Bioscience)-coated plates in Essential 8 Medium (Thermo Fisher Scientific). We generated EBs by clone suspension. EBs were differentiated in DMEM/F12 (Gibco) supplemented with 20% FBS (Gibco), 50 U/mL penicillin/streptomycin (Gibco), 2 mM L-glutamine (Gibco), 1× non-essential amino acids, and 100 μM β-mercaptoethanol (Sigma). At 90% confluency, hESCs were digested using 1 mL Gentle Cell Dissociation Reagent (STEMCELL Technologies) for 5 min. Cell clumps were pipetted into single cells, and 9.0 × 105 cells per well were seeded into AggreWell 800 (STEMCELL Technologies). The day after seeding, EBs/spheroids were harvested from AggreWell 800 plates and transferred into an ultra-low attachment six-well plate (Corning). Nicotine (10 μM; N3876, Sigma) was added into the differentiating medium, and EBs were fed each day for 21 days. The same volume of ethanol (459836, Sigma) was added into the differentiating medium as control.
scRNA-Seq Library Preparation and Analysis
Single cells were collected from two independent EB differentiation experiments from day 21 EBs (control and nicotine-exposed) and dissociated using Accutase (STEMCELL Technologies). They were prepared for the single-cell library separately. In brief, cells were washed with 1× DPBS (Gibco) three times, strainer filtered, and re-suspended in 0.04% BSA. Viable single cells were loaded on to a GemCode Instrument (10× Genomics, Pleasanton, CA) to generate single-cell barcoded droplets (GEMs) using the 10× Single Cell 3′ v.2 chemistry and 10× Chromium system as per the manufacturer's protocol. The quality of the resulting libraries was checked with Bioanalyzer (Agilent Bioanalyzer 2100). Then control and nicotine libraries were combined with equal molar mass and sequenced across two lanes on an Illumina HiSeq machine. Analyses was performed using Seurat R package. Detailed scRNA-seq analysis is available in the Supplemental Experimental Procedures.
EB Diameter Measurement
Bright-field images were captured by an SI8000 Cell Motion Imaging System (Sony Biotechnology) using a 4× objective. Scale was set with a 20-cm-ruler image. The diameter of each EB was measured by drawing a selection line of longest distance in each EB using the Line Selection tool in ImageJ. Analyses were processed with ImageJ and the results were plotted with Prism (GraphPad).
Quantification of ROS Production and ATP
ROS production level and ATP level in EBs were determined using CellTiter-Glo 2.0 (Promega) and ROS-Glo H2O2 (Promega) following the manufacturers' instructions. In brief, EBs were exposed to nicotine in 96-well plates for 21 days. After treatment, H2O2 substrate was added directly to each well of 96-well plates and incubated for 4 h. The resulting supernatant was collected for ROS-Glo H2O2 assay and EBs were subsequently subjected to the CellTiter-Glo 2.0 assay to measure the ATP level. The luminescence intensity was measured using a Synergy HTX multi-mode microplate reader (BioTek).
Intracellular Calcium Imaging
Single hESC-derived cardiomyocytes were plated onto Matrigel-coated coverglass (CS-24/50, Warner Instruments) at a density of ∼10,000 cells per square centimeter. Cells were allowed to recover for 3–4 days and loaded with 5 μM Fura-2 AM (Thermo Fisher Scientific) in Tyrode's solution (140 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 10 mM glucose, 1.8 mM CaCl2, and 10 mM HEPES; pH adjusted to 7.4 with NaOH at room temperature) for 30 min at room temperature. Cells were imaged on a customized Ti-S/L 100 Inverted Microscope-based imaging platform with a 40× oil immersion objective (CFI Super Fluor, NA 1.30 WD 0.22). Bipolar pulse was used to pace cells at 0.5 and 1 HZ. Cells were kept at 37°C while recording. Fura-2 signals were captured in high-frame-rate video recording mode (512 × 512 pixels) at a speed of 50 frames per second. Videos were analyzed with NIS Elements Advanced Research Software (Nikon), and raw ratio-pair data were further processed with a custom-made script based on Interactive Digital Language.
Author Contributions
H.G. designed the study, performed the experiment, analyzed the data, and wrote the manuscript. L.T. analyzed the scRNA-seq data and published bulk RNA-seq data, discussed the results, and wrote the manuscript. J.Z.Z. conducted Ca2+ experiments, discussed the results, and revised the manuscript. T.K. performed the experiments, discussed the results, and revised the manuscript. D.T.P. designed the study, provided advice, and revised the manuscript. W.H.L. performed the experiments, provided advice, and revised the manuscript. J.C.W. designed the study, revised the manuscript, and provided funding support.
Acknowledgments
The authors would like to thank Dhananjay A. Wagh and John A. Coller of Stanford Functional Genomics Facility for assistance with single-cell RNA sequencing. The authors are grateful for critical reading of the manuscript by Yonggang Liu, Amanda J. Chase, and Kitchener D. Wilson. This study was funded by National Institutes of Health (NIH) grants T32 EB009035 (D.T.P.), TRDRP 26FT-0029 (J.Z.Z), Burroughs Wellcome Fund 1015009 (J.C.W.), and R01 HL130020 (J.C.W.), R01 HL113006 (J.C.W.), R01 HL141371 (J.C.W.), R01 HL132875 (J.C.W.), TRDRP 27IR-0012 (J.C.W.).
Published: February 28, 2019
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, three figures, and seven tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2019.01.022.
Accession Numbers
The GEO accession number for single-cell RNA-seq of day 21 control and nicotine-exposed EBs in this paper is GEO: GSE125416.
Supplemental Information
References
- De Abrew K.N., Kainkaryam R.M., Shan Y.Q.K., Overmann G.J., Settivari R.S., Wang X.H., Xu J., Adams R.L., Tiesman J.P., Carney E.W. Grouping 34 chemicals based on mode of action using connectivity mapping. Toxicol. Sci. 2016;151:447–461. doi: 10.1093/toxsci/kfw058. [DOI] [PubMed] [Google Scholar]
- Ahrens I., Domeij H., Topcic D., Haviv I., Merivirta R.M., Agrotis A., Leitner E., Jowett J.B., Bode C., Lappas M. Successful in vitro expansion and differentiation of cord blood derived CD34+ cells into early endothelial progenitor cells reveals highly differential gene expression. PLoS One. 2011;6:e23210. doi: 10.1371/journal.pone.0023210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken J., Buscaglia G., Bates E.A., Moore J.K. The alpha-tubulin gene TUBA1A in brain development: a key ingredient in the neuronal isotype blend. J. Dev. Biol. 2017;5 doi: 10.3390/jdb5030008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armant O., Gourain V., Etard C., Strahle U. Whole transcriptome data analysis of zebrafish mutants affecting muscle development. Data Brief. 2016;8:61–68. doi: 10.1016/j.dib.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W., Xu G.F., Lu J.C., Snetselaar L.G., Wallace R.B. Changes in electronic cigarette use among adults in the United States, 2014–2016. JAMA. 2018;319:2039–2041. doi: 10.1001/jama.2018.4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry F.B., Miura Y., Mihara K., Kaspar P., Sakata N., Hashimoto-Tamaoki T., Tamaoki T. Positive and negative regulation of myogenic differentiation of C2C12 cells by isoforms of the multiple homeodomain zinc finger transcription factor ATBF1. J. Biol. Chem. 2001;276:25057–25065. doi: 10.1074/jbc.M010378200. [DOI] [PubMed] [Google Scholar]
- Billatos E., Faiz A., Gesthalter Y., LeClerc A., Alekseyev Y.O., Xiao X., Liu G., ten Hacken N.H.T., Heijink I.H., Timens W. Impact of acute exposure to cigarette smoke on airway gene expression. Physiol. Genomics. 2018;50:705–713. doi: 10.1152/physiolgenomics.00092.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeley P., Fogarty N., Del Valle I., Wamaitha S., Hu T.X., Elder K., Snell P., Christie L., Robson P., Niakan K. Defining the three cell lineages of the human blastocyst by single-cell RNA-seq. Mech. Dev. 2017;145:S26. doi: 10.1242/dev.131235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle D.L., Liu X., Hopkins T.M., Swick M.C., Dhir R., Siegfried J.M. Nicotine activates cell-signaling pathways through muscle-type and neuronal nicotinic acetylcholine receptors in non-small cell lung cancer cells. Pulm. Pharmacol. Ther. 2007;20:629–641. doi: 10.1016/j.pupt.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Dasgupta P., Chellappan S.P. Nicotine-mediated cell proliferation and angiogenesis: new twists to an old story. Cell Cycle. 2006;5:2324–2328. doi: 10.4161/cc.5.20.3366. [DOI] [PubMed] [Google Scholar]
- DeLaughter D.M., Christodoulou D.C., Robinson J.Y., Seidman C.E., Baldwin H.S., Seidman J.G., Barnett J.V. Spatial transcriptional profile of the chick and mouse endocardial cushions identify novel regulators of endocardial EMT in vitro. J. Mol. Cell. Cardiol. 2013;59:196–204. doi: 10.1016/j.yjmcc.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delitto D., Han S., Hughes S.J., Behrns K.E., Trevino J.G. Nicotine drives pancreatic cancer metastasis through paracrine signaling in the tumor microenvironment. J. Am. Coll. Surgeons. 2014;219:E175. [Google Scholar]
- Duggal S., Jailkhani N., Midha M.K., Agrawal N., Rao K.V.S., Kumar A. Defining the AKT1 interactome and its role in regulating the cell cycle. Sci. Rep. 2018;8:1303. doi: 10.1038/s41598-018-19689-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont-Lagace M., Gerbe H., Daouda T., Laverdure J.P., Brochu S., Lemieux S., Gagnon E., Perreault C. Detection of quiescent radioresistant epithelial progenitors in the adult thymus. Front. Immunol. 2017;8:1717. doi: 10.3389/fimmu.2017.01717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkan M., Kleeff J., Esposito I., Giese T., Ketterer K., Buchler M.W., Giese N.A., Friess H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene. 2005;24:4421–4432. doi: 10.1038/sj.onc.1208642. [DOI] [PubMed] [Google Scholar]
- Etard C., Armant O., Roostalu U., Gourain V., Ferg M., Strahle U. Loss of function of myosin chaperones triggers HSF1-mediated transcriptional response in skeletal muscle cells. Genome Biol. 2015;16:267. doi: 10.1186/s13059-015-0825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried P.A., Oconnell C.M. A comparison of the effects of prenatal exposure to tobacco, alcohol, cannabis and caffeine on birth size and subsequent growth. Neurotoxicol. Teratol. 1987;9:79–85. doi: 10.1016/0892-0362(87)90082-1. [DOI] [PubMed] [Google Scholar]
- Geraghty P., Wallace A., D'Armiento J.M. Induction of the unfolded protein response by cigarette smoke is primarily an activating transcription factor 4-C/EBP homologous protein mediated process. Int. J. Chronic. Obstr. 2011;6:309–319. doi: 10.2147/COPD.S19599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A., Coakley R.C., Mascenik T., Rowell T.R., Davis E.S., Rogers K., Webster M.J., Dang H., Herring L.E., Sassano M.F. Chronic e-cigarette exposure alters the human bronchial epithelial proteome. Am. J. Respir. Crit. Care Med. 2018;198:67–76. doi: 10.1164/rccm.201710-2033OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick D., Zhang W., Beaton M., Marsboom G., Gruber M., Simon M.C., Hart J., Dorn G.W., 2nd, Brady M.J., Macleod K.F. BNIP3 regulates mitochondrial function and lipid metabolism in the liver. Mol. Cell. Biol. 2012;32:2570–2584. doi: 10.1128/MCB.00167-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonseth S., de Smith A.J., Roy R., Zhou M., Lee S.T., Shao X.R., Ohja J., Wrensch M.R., Walsh K.M., Metayer C. Genetic contribution to variation in DNA methylation at maternal smoking-sensitive loci in exposed neonates. Epigenetics. 2016;11:664–673. doi: 10.1080/15592294.2016.1209614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumus Z.H., Du B., Kacker A., Boyle J.O., Bocker J.M., Mukherjee P., Subbaramaiah K., Dannenberg A.J., Weinstein H. Effects of tobacco smoke on gene expression and cellular pathways in a cellular model of oral leukoplakia. Cancer Prev. Res. (Phila). 2008;1:100–111. doi: 10.1158/1940-6207.CAPR-08-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F., Li B., Zhao Z., Zhou Y., Hu G., Zou W., Hong W., Zou Y., Jiang C., Zhao D. The pro-proliferative effects of nicotine and its underlying mechanism on rat airway smooth muscle cells. PLoS One. 2014;9:e93508. doi: 10.1371/journal.pone.0093508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata N., Yamada S., Asanagi M., Sekino Y., Kanda Y. Nicotine induces mitochondrial fission through mitofusin degradation in human multipotent embryonic carcinoma cells. Biochem. Biophys. Res. Commun. 2016;470:300–305. doi: 10.1016/j.bbrc.2016.01.063. [DOI] [PubMed] [Google Scholar]
- Holbrook B.D. The effects of nicotine on human fetal development. Birth Defects Res. C Embryo Today. 2016;108:181–192. doi: 10.1002/bdrc.21128. [DOI] [PubMed] [Google Scholar]
- Holownia A., Wielgat P., Kwolek A., Jackowski K., Braszko J.J. Crosstalk between co-cultured a549 cells and thp1 cells exposed to cigarette smoke. Adv. Exp. Med. Biol. 2015;858:47–55. doi: 10.1007/5584_2015_112. [DOI] [PubMed] [Google Scholar]
- Hong W., Peng G., Hao B., Liao B., Zhao Z., Zhou Y., Peng F., Ye X., Huang L., Zheng M. Nicotine-induced airway smooth muscle cell proliferation involves TRPC6-dependent calcium influx via alpha7 nAChR. Cell Physiol. Biochem. 2017;43:986–1002. doi: 10.1159/000481651. [DOI] [PubMed] [Google Scholar]
- Hu N., Han X.F., Lane E.K., Gao F., Zhang Y.M., Ren J. Cardiac-specific overexpression of metallothionein rescues against cigarette smoking exposure-induced myocardial contractile and mitochondrial damage. PLoS One. 2013;8:e57151. doi: 10.1371/journal.pone.0057151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaddoe V.W.V., Troe E.J.W.M., Hofman A., Mackenbach J.P., Moll H.A., Steegers E.A.P., Witteman J.C.M. Active and passive maternal smoking during pregnancy and the risks of low birthweight and preterm birth: the Generation R Study. Paediatr. Perinat. Epidemiol. 2008;22:162–171. doi: 10.1111/j.1365-3016.2007.00916.x. [DOI] [PubMed] [Google Scholar]
- Kawasaki H., Takatori S., Zamami Y., Koyama T., Goda M., Hirai K., Tangsucharit P., Jin X., Hobara N., Kitamura Y. Paracrine control of mesenteric perivascular axo-axonal interaction. Acta Physiol. (Oxf.) 2011;203:3–11. doi: 10.1111/j.1748-1716.2010.02197.x. [DOI] [PubMed] [Google Scholar]
- Kim C.S., Choi J.S., Joo S.Y., Bae E.H., Ma S.K., Lee J., Kim S.W. Nicotine-induced apoptosis in human renal proximal tubular epithelial cells. PLoS One. 2016;11:e0152591. doi: 10.1371/journal.pone.0152591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A., Matsuda T., Sakai A., Murao N., Nakashima K. HMGB2 expression is associated with transition from a quiescent to an activated state of adult neural stem cells. Dev. Dyn. 2018;247:229–238. doi: 10.1002/dvdy.24559. [DOI] [PubMed] [Google Scholar]
- Ko Y.B., Kim B.R., Nam S.L., Yang J.B., Park S.Y., Rho S.B. High-mobility group box 1 (HMGB1) protein regulates tumor-associated cell migration through the interaction with BTB domain. Cell. Signal. 2014;26:777–783. doi: 10.1016/j.cellsig.2013.12.018. [DOI] [PubMed] [Google Scholar]
- Larsen H.E., Lefkimmiatis K., Paterson D.J. Sympathetic neurons are a powerful driver of myocyte function in cardiovascular disease. Sci. Rep. 2016;6:38898. doi: 10.1038/srep38898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Yon J.M., Hong J.T., Lee J.K., Jeong J., Baek I.J., Lee B.J., Yun Y.W., Nam S.Y. 4-O-Methylhonokiol inhibits serious embryo anomalies caused by nicotine via modulations of oxidative stress, apoptosis, and inflammation. Birth Defects Res. B Dev. Reprod. Toxicol. 2014;101:125–134. doi: 10.1002/bdrb.21092. [DOI] [PubMed] [Google Scholar]
- Liszewski W., Ritner C., Aurigui J., Wong S.S.Y., Hussain N., Krueger W., Oncken C., Bernstein H.S. Developmental effects of tobacco smoke exposure during human embryonic stem cell differentiation are mediated through the transforming growth factor-beta superfamily member, nodal. Differentiation. 2012;83:169–178. doi: 10.1016/j.diff.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.W., Jin G.R., Long C.D., Zhou X., Tang Y., Huang S., Kuang X.L., Wu L.Z., Zhang Q.J., Shen H.X. Blockage of notch signaling inhibits the migration and proliferation of retinal pigment epithelial cells. ScientificWorldJournal. 2013;2013:178708. doi: 10.1155/2013/178708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Wang C.N., Qiu C.Y., Song W., Wang L.F., Liu B. Adipocytes promote nicotine-induced injury of endothelial cells via the NF-κB pathway. Exp. Cell Res. 2017;359:251–256. doi: 10.1016/j.yexcr.2017.07.022. [DOI] [PubMed] [Google Scholar]
- Loinard C., Zouggari Y., Rueda P., Ramkhelawon B., Cochain C., Vilar J., Recalde A., Richart A., Charue D., Duriez M. C/EBP homologous protein-10 (CHOP-10) limits postnatal neovascularization through control of endothelial nitric oxide synthase gene expression. Circulation. 2012;125:1014–U1126. doi: 10.1161/CIRCULATIONAHA.111.041830. [DOI] [PubMed] [Google Scholar]
- Loukili N., Rosenblatt-Velin N., Li J., Clerc S., Pacher P., Feihl F., Waeber B., Liaudet L. Peroxynitrite induces HMGB1 release by cardiac cells in vitro and HMGB1 upregulation in the infarcted myocardium in vivo. Cardiovasc. Res. 2011;89:586–594. doi: 10.1093/cvr/cvq373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck W., Nau H., Hansen R., Steldinger R. Extent of nicotine and cotinine transfer to the human-fetus, placenta and amniotic-fluid of smoking mothers. Dev. Pharmacol. Ther. 1985;8:384–395. doi: 10.1159/000457063. [DOI] [PubMed] [Google Scholar]
- Malmstrom J., Lindberg H., Lindberg C., Bratt C., Wieslander E., Delander E.L., Sarnstrand B., Burns J.S., Mose-Larsen P., Fey S. Transforming growth factor-beta(1) specifically induce proteins involved in the myofibroblast contractile apparatus. Mol. Cell. Proteomics. 2004;3:466–477. doi: 10.1074/mcp.M300108-MCP200. [DOI] [PubMed] [Google Scholar]
- Martynova N.Y., Eroshkin F.M., Orlov E.E., Zaraisky A.G. HMG-Box factor SOXD/SOX15 and homeodomain-containing factor XANF1/HESX1 directly interact and regulate the expression of XANF1/HESX1 during early forebrain development in Xenopus laevis. Gene. 2018;638:52–59. doi: 10.1016/j.gene.2017.09.024. [DOI] [PubMed] [Google Scholar]
- Matsuura K., Otani M., Takano M., Kadoyama K., Matsuyama S. The influence of chronic nicotine treatment on proteins expressed in the mouse hippocampus and cortex. Eur. J. Pharmacol. 2016;780:16–25. doi: 10.1016/j.ejphar.2016.03.025. [DOI] [PubMed] [Google Scholar]
- de Melo J., Zibetti C., Clark B.S., Hwang W., Miranda-Angulo A.L., Qian J., Blackshaw S. LHX2 is an essential factor for retinal gliogenesis and notch signaling. J. Neurosci. 2016;36:2391–2405. doi: 10.1523/JNEUROSCI.3145-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelotti G.A., Tucker A., Machado M.V., Swiderska-Syn M., Choi S.S., Kruger L., Garman K.S., Moylan C.A., Guy C.D., Himburg H. Pleiotrophin regulates the ductular reaction by controlling the migration of cells in liver progenitor niches. Hepatology. 2014;60:408a. doi: 10.1136/gutjnl-2014-308176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohsenzadeh Y., Rahmani A., Cheraghi J., Pyrani M., Asadollahi K. Prenatal exposure to nicotine in pregnant rat increased inflammatory marker in newborn rat. Mediat. Inflamm. 2014;2014:274048. doi: 10.1155/2014/274048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Gonzalez I., Estrada L.D., Sanchez-Mejias E., Soto C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer's disease. Nat. Commun. 2013;4:1495. doi: 10.1038/ncomms2494. [DOI] [PubMed] [Google Scholar]
- Nestorowa S., Hamey F.K., Pijuan Sala B., Diamanti E., Shepherd M., Laurenti E., Wilson N.K., Kent D.G., Gottgens B. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood. 2016;128 doi: 10.1182/blood-2016-05-716480. e20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik D.T., Tian L., Lee J., Sayed N., Chen I.Y., Rhee S., Rhee J.W., Kim Y., Wirka R.C., Buikema J.W. Large-scale single-cell RNA-seq reveals molecular signatures of heterogeneous populations of human induced pluripotent stem cell-derived endothelial cells. Circ. Res. 2018;123:443–450. doi: 10.1161/CIRCRESAHA.118.312913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei F., Jiang J.J., Bai S.Y., Cao H.H., Tian L.Y., Zhao Y., Yang C.X., Dong H.H., Ma Y. Chemical-defined and albumin-free generation of human atrial and ventricular myocytes from human pluripotent stem cells. Stem Cell Res. 2017;19:94–103. doi: 10.1016/j.scr.2017.01.006. [DOI] [PubMed] [Google Scholar]
- Popson S.A., Ziegler M.E., Chen X.F., Holderfield M.T., Shaaban C.I., Fong A.H., Welch-Reardon K.M., Papkoff J., Hughes C.C.W. Interferon-induced transmembrane protein 1 regulates endothelial lumen formation during angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2014;34:1011–1019. doi: 10.1161/ATVBAHA.114.303352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramilowski J.A., Goldberg T., Harshbarger J., Kloppman E., Lizio M., Satagopam V.P., Itoh M., Kawaji H., Carninci P., Rost B. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2015;6:7866. doi: 10.1038/ncomms8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehan V.K., Asotra K., Torday J.S. The effects of smoking on the developing lung: insights from a biologic model for lung development, homeostasis, and repair. Lung. 2009;187:281–289. doi: 10.1007/s00408-009-9158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repo J.K., Pesonen M., Mannelli C., Vahakangas K., Loikkanen J. Exposure to ethanol and nicotine induces stress responses in human placental BeWo cells. Toxicol. Lett. 2014;224:264–271. doi: 10.1016/j.toxlet.2013.10.032. [DOI] [PubMed] [Google Scholar]
- Saito T., Yano F., Mori D., Ohba S., Hojo H., Otsu M., Eto K., Nakauchi H., Tanaka S., Chung U. Generation of COL2A1-EGFP iPS cells for monitoring chondrogenic differentiation. PLoS One. 2013;8:e74137. doi: 10.1371/journal.pone.0074137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauler M., Bucala R., Lee P.J. Role of macrophage migration inhibitory factor in age-related lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2015;309:L1–L10. doi: 10.1152/ajplung.00339.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P., Misteli T., Bianchi M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Seeger T., Porteus M., Wu J.C. Genome editing in cardiovascular biology. Circ. Res. 2017;120:778–780. doi: 10.1161/CIRCRESAHA.116.310197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.P., Zhang F., Lan H.R., Chen K., Zhang Q., Xie G.M., Teng L.S., Jin K.T. Frzb up-regulation is correlated with hepatic metastasis and poor prognosis in colon carcinoma patients with hepatic metastasis. Int. J. Clin. Exp. Pathol. 2015;8:4083–4090. [PMC free article] [PubMed] [Google Scholar]
- Sheng X.M., Wang Z.X. Protein arginine methyltransferase 5 regulates multiple signaling pathways to promote lung cancer cell proliferation. BMC Cancer. 2016;16:567. doi: 10.1186/s12885-016-2632-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin T.A. Fetal nicotine or cocaine exposure: which one is worse? J. Pharmacol. Exp. Ther. 1998;285:931–945. [PubMed] [Google Scholar]
- Slotkin T.A., Cho H., Whitmore W.L. Effects of prenatal nicotine exposure on neuronal development - selective actions on central and peripheral catecholaminergic pathways. Brain Res. Bull. 1987;18:601–611. doi: 10.1016/0361-9230(87)90130-4. [DOI] [PubMed] [Google Scholar]
- Sugrue S.P., Akin D., Newman J., McIntyre L. RNA-seq analysis of impact of PNN on gene expression and alternative splicing in corneal epithelial cells. Invest. Ophth. Vis. Sci. 2016;57:4369. [PMC free article] [PubMed] [Google Scholar]
- Tabib T., Morse C., Wang T., Chen W., Lafyatis R. SFRP2/DPP4 and FMO1/LSP1 define major fibroblast populations in human skin. J. Invest. Dermatol. 2018;138:802–810. doi: 10.1016/j.jid.2017.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H., Kondo K., Chen X., Homma H., Tagawa K., Kerever A., Aoki S., Saito T., Saido T., Muramatsu S.I. The intellectual disability gene PQBP1 rescues Alzheimer's disease pathology. Mol. Psychiatry. 2018;23:2090–2110. doi: 10.1038/s41380-018-0253-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H., Liu Y.J., Liu M., Li X. Establishment and gene analysis of an oxaliplatin-resistant colon cancer cell line thc8307/l-ohp. Anticancer Drugs. 2007;18:633–639. doi: 10.1097/CAD.0b013e3280200428. [DOI] [PubMed] [Google Scholar]
- Tang K., Xie X., Park J.I., Jamrich M., Tsai S., Tsai M.J. COUF-TFS regulate eye development by controlling factors essential for optic vesicle morphogenesis. Development. 2010;137:725–734. doi: 10.1242/dev.040568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchetchelnitski V., van den Eijnden M., Schmidt F., Stoker A.W. Developmental co-expression and functional redundancy of tyrosine phosphatases with neurotrophin receptors in developing sensory neurons. Int. J. Dev. Neurosci. 2014;34:48–59. doi: 10.1016/j.ijdevneu.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Tizabi Y. Nicotine and nicotinic system in hypoglutamatergic models of schizophrenia. Neurotox. Res. 2007;12:233–246. doi: 10.1007/BF03033907. [DOI] [PubMed] [Google Scholar]
- Warren G.W., Alberg A.J., Kraft A.S., Cummings K.M. The 2014 Surgeon General's report: "The health consequences of smoking--50 years of progress": a paradigm shift in cancer care. Cancer. 2014;120:1914–1916. doi: 10.1002/cncr.28695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzer-Serhan U.H. Long-term consequences of maternal smoking and developmental chronic nicotine exposure. Front. Biosci. 2008;13:636–649. doi: 10.2741/2708. [DOI] [PubMed] [Google Scholar]
- Wong M.K., Holloway A.C., Hardy D.B. Nicotine directly induces endoplasmic reticulum stress response in rat placental trophoblast giant cells. Toxicol. Sci. 2016;151:23–34. doi: 10.1093/toxsci/kfw019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R.H., Liu B., Wu J.D., Yan Y.Y., Wang J.N. Mir-143 is involved in endothelial cell dysfunction through suppression of glycolysis and correlated with atherosclerotic plaques formation. Eur. Rev. Med. Pharmacol. 2016;20:4063–4071. [PubMed] [Google Scholar]
- Yazdani N., Parker C.C., Shen Y., Reed E.R., Guido M.A., Kole L.A., Kirkpatrick S.L., Lim J.E., Sokoloff G., Cheng R.Y. HNRNPH1 is a quantitative trait gene for methamphetamine sensitivity. Plos Genet. 2015;11:e1005713. doi: 10.1371/journal.pgen.1005713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama S., Chen Z.Y., Okagaki T., Kohama K., Nasu-Kawaharada R., Izumi T., Ohshima N., Nagai T., Nakamura A. Nicotine exposure alters human vascular smooth muscle cell phenotype from a contractile to a synthetic type. Atherosclerosis. 2014;237:464–470. doi: 10.1016/j.atherosclerosis.2014.10.019. [DOI] [PubMed] [Google Scholar]
- Zdravkovic T., Genbacev O., LaRocque N., McMaster M., Fisher S. Human embryonic stem cells as a model system for studying the effects of smoke exposure on the embryo. Reprod. Toxicol. 2008;26:86–93. doi: 10.1016/j.reprotox.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Zhang C.C., Mo M.H., Ding W.W., Liu W.J., Yan D.W., Deng J.X., Luo X.P., Liu J. High-mobility group box 1 (HMGB1) impaired cardiac excitation-contraction coupling by enhancing the sarcoplasmic reticulum (SR) Ca2+ leak through TLR4-ROS signaling in cardiomyocytes. J. Mol. Cell. Cardiol. 2014;74:260–273. doi: 10.1016/j.yjmcc.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Zhao Y.U., Sheng H.Z., Amini R., Grinberg A., Lee E., Huang S.P., Taira M., Westphal H. Control of hippocampal morphogenesis and neuronal differentiation by the LIM homeobox gene lhx5. Science. 1999;284:1155–1158. doi: 10.1126/science.284.5417.1155. [DOI] [PubMed] [Google Scholar]
- Zhu Z.R., Li Q.V., Lee K., Rosen B.P., Gonzalez F., Soh C.L., Huangfu D.W. Genome editing of lineage determinants in human pluripotent stem cells reveals mechanisms of pancreatic development and diabetes. Cell Stem Cell. 2016;18:755–768. doi: 10.1016/j.stem.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.