

Abstract
Long non-coding RNAs (lncRNAs) refer to a class of RNA molecules that are longer than 200 nucleotides and do not encode proteins. Numerous lncRNAs have recently emerged as important regulators of many biological processes in animals and plants, including responses to environmental stress and pathogens. Botryosphaeria dieback is one of the more severe grapevine trunk diseases worldwide. However, how lncRNAs function during Botryosphaeriaceae infection is largely unknown. We performed high-throughput RNA-sequencing (RNA-seq) of susceptible and more tolerant grapevine cultivars infected with Lasiodiplodia theobromae. Overall, we predicted 1826 novel candidate lncRNAs, including long intergenic non-coding RNAs (lincRNAs) and natural antisense transcripts (lncNATs). The data reveal the functions of a set of lncRNAs that were differentially expressed between the resistant cultivar Merlot and the susceptible cultivar Cabernet Franc. Several lncRNAs were predicted to be precursors for grape microRNAs involved in the L theobromae infection. These results provide new insight into the lncRNAs of grapevine that are involved in the response to L theobromae infection.
Keywords: Botryosphaeriaceae, lncRNA, grapevine, plant defence, RNA sequence
Introduction
Non-coding RNAs have been defined as major products transcribed by the eukaryotic genome that differ from mRNA.1 They play important roles in gene regulation without being further translated into polypeptides or proteins. Non-coding RNAs can be grouped based on their expression characteristics: (1) housekeeping non-coding RNAs, which are essential for maintaining the basic functions of cells and generally include rRNAs, tRNAs, small nuclear RNAs (snRNAs), and small nucleolar RNAs (snoRNAs); and (2) regulatory non-coding RNAs, which are specially expressed in specific tissues during developmental stages of organisms or after environment stress.2 Furthermore, on the basis of their length, regulatory non-coding RNA are arbitrarily classified into small (<200 base pairs [bp]) and long non-coding RNAs (lncRNAs; >200 bp). In plants, significant progress has been made towards our understanding of the functions and mechanisms of small non-coding RNAs in the transcriptional and post-transcriptional regulation of gene expression.3–5 However, the biological functions of lncRNAs have yet to be uncovered. In plants, most lncRNAs can be transcribed by RNA polymerase II (Pol II), Pol IV, and Pol V.6 In addition, compared with mRNAs, most lncRNAs are polyadenylated and localized in the nucleus.7,8 In addition, lncRNAs have a low expression level, usually exhibit tissue- or cell-specific expression patterns and show poor conservation among species.9
Through application of whole genome tilling arrays, in silico predictions, and RNA-sequencing (RNA-seq) analysis, thousands of lncRNAs have been identified in Arabidopsis thaliana,10 Zea mays,11 Oryza sativa,12 Triticum aestivum,13 Medicago truncatula,14 and Cucumis sativus,15 and emerging evidence suggests that many of them are responsive to biotic and abiotic stresses.8,16,17 For example, 1212 novel lncRNA candidates were predicted, including 309 differentially expressed lncRNAs under control and Pi starvation conditions in Arabidopsis.18 Using strand-specific RNA-seq, 1113 long intergenic non-coding RNAs (lincRNAs) and 17 defence-related lincTARs responsive to Pectobacterium carotovorum subsp. brasiliense challenge were identified in potato.19 In tomatoes, 1565 lncRNAs that are involved in Tomato yellow leaf curl virus (TYLCV) infection were discovered.20 However, the molecular basis of how lncRNAs regulate responses to environmental stress and pathogens is still poorly understood, with only a few have been functionally investigated. In Arabidopsis, 2 types of lncRNAs, COOLAIR (cold-induced long antisense intragenic RNA) and COLDAIR (cold-assisted intronic non-coding RNA), have been demonstrated to participate in transcript silencing of FLOWERING LOCUS C (FLC) through chromatin modifications during vernalization.21–23 Recently, another antisense lncRNA, ASL, which is not polyadenylated, was identified and plays different roles in FLC silencing.24 Furthermore, Pi starvation induced the lncRNA IPS1 (induced by PHOSPHATE STARVATION 1), which acts as a miR399 target mimic, leading to the reduction of miR399-mediated cleavage of PHO mRNA.25
Grape is one of the most widely cultivated and economically important fruits in the world, both for fruit consumption and for wine production. Grape Botryosphaeriaceous diseases have long been important factors that affect yield and quality, leading to a serious reduction in grape production. To date, Botryosphaeria dieback caused by some members of the Botryosphaeriaceae family is one of the most serious trunk diseases in almost all main grape-growing areas.26–28 The fungi infect grapes through wounds or natural openings, causing serious losses as a result of trunk canker, vascular discolouration, and fruit shrivelling and rot.29–32 Species in the genera Botryosphaeria, Diplodia, Lasiodiplodia, and Neofusicoccum and some others were reported to be associated with Botryosphaeria dieback in grapevine.29,32,33 In China, Li et al34–37 reported morphological and molecular identification of 5 different members of the Botryosphaeriaceae family associated with grapevine trunk disease, including Botryosphaeria dothidea, Diplodia seriata, Lasiodiplodia theobromae, Neofusicoccum parvum, Lasiodiplodia pseudotheobromae, and Neofusicoccum mangiferae. In addition, L theobromae was shown to be the most aggressive Botryosphaeriaceae species on grapevines.31,35 These Botryosphaeriaceous pathogens can cause serious losses in grapevine production. To date, there are no efficient strategies to control this disease. Hence, investigating the interaction between the pathogen and the host is important for designing efficient control strategies. Although emerging evidence suggests that lncRNAs are involved in the response to pathogen attack, whether lncRNAs participate in the Botryosphaeriaceae defence networks in grapevine is still not known.
In this study, to systematically identify and characterize the lncRNAs involved in Botryosphaeriaceae resistance, deep RNA-seq analysis was performed on grape stems in 2 grape cultivars that are susceptible (Cabernet Franc [CF]) and tolerant (Merlot [ML]) with and without L theobromae inoculation.35 In total, 1826 candidate lncRNAs were identified in this analysis. Compared with the mock-inoculated treatments, 782 grape lncRNA candidates have shown significant differential expression patterns in the resistant and susceptible cultivars. Of these, 8 were validated using quantitative real-time polymerase chain reaction (qRT-PCR). In addition, cis and trans roles of lncRNA targeting genes were also examined to annotate lncRNA function. Overall, our results demonstrated that some candidate lncRNAs may play an important role in grape immunity mechanisms, including some acting as miRNA precursors.
Materials and Methods
Plant materials and Lasiodiplodia theobromae inoculation
Dormant branches of 2 grapevine cultivars that are susceptible (CF) and more tolerant (ML)35 to the L theobromae strain CSS-01s on cutting were grown in a greenhouse of Beijing Academy of Agriculture and Forestry Sciences (BAAFS) in Beijing, China. Cuttings from rooting plants were transferred to 25 cm × 25 cm pots and were propagated at a spacing of 20 cm × 20 cm. Cultivation management followed the standard procedures used at BAAFS. Stem inoculations were performed as previous described in Yan et al34,35 with minor modifications. L theobromae CSS-01s was cultured on potato dextrose agar (PDA) medium at 28°C for 2 days prior to inoculation. The semi-lignified current grown shoots were surface-sterilized with 70% alcohol and then were wounded at the middle point using a 4-mm cork borer (2-mm deep). A mycelial agar plug (4 mm in diameter) of L theobromae was placed onto the wound. Controls were mock-inoculated with a plug of sterile PDA medium without L theobromae. These grape pot seedlings were placed under 12 hours of light, at 28°C, and at a relative humidity (RH) of 90% in a plant inoculation room. Shoot phloem within a 0.5- to 2.0-cm range from the wound point was collected at 0 and 24 hours post inoculation (hpi) in 2 biological replicates. The samples were collected and frozen immediately in liquid nitrogen and then stored at −80°C for subsequent use (5 plants were pooled together for each biological replicate).
RNA extraction, library preparation, and sequencing
The total RNA of collected samples was extracted using OminiPlant RNA Kit (CWBIO, Beijing, China) according to the manufacturer’s instruction. The RNA was quantified using Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, Carlsbad, CA, USA). Then, the quality and integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA) with a minimum RNA integrated number (RIN) value of 7.0. The construction of RNA-seq libraries and sequencing were carried at Novogene Bioinformatics Technology Cooperation (Beijing, China). Briefly, RNA samples were treated with Epicentre Ribo-zero™ rRNA Removal Kit (Epicentre, Madison, WI, USA) for rRNA depletion. Whole transcription libraries were prepared using the rRNA-depleted RNA by NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA) following the manufacturer’s recommendations. Then, the libraries were quality checked on the Agilent Bioanalyzer 2100 system. Finally, the resulting libraries were sequenced on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) with paired-end reads of 125 bp. The data for this study have been deposited in National Center for Biotechnology Information (NCBI) Sequence Read Archive Gene with accession number SRP101685.
Bioinformatics pipeline for lncRNAs identification
The grape (Vitis vinifera cv. Pinot Noir) reference genome assembly, PN40024, used throughout this study was downloaded from http://www.genoscope.cns.fr/. Raw reads in FASTQ format were processed through quality trimming and filtering to remove adapter-containing, poly-N containing, and low-quality reads. Each data set of RNA-seq clean reads was aligned to the grape reference genome using TopHat v2.0.9 program38 (TopHat2, –library-type ‘fr-firststrand’ splice-mismatches ‘0’ –min-intron-length ‘70’ –max-intron-length ‘50000’–num-threads ‘6’). The transcripts were assembled using Cufflinks v2.1.139 (Cufflinks2, –num-threads ‘8’ –max-intron-length ‘300000’ –max-mle-iterations ‘5000’ –min-frags-per-transfrag ‘10’ –min-intron-length ‘50’ –minisoform-fraction‘0.1’ –num-importance-samples ‘1000’ –library-type ‘fr-firststrand’) and Scripture40 with default parameters. The transcripts that have 2 or more read coverage were chosen for further analyses. All transcripts less than 200 bp were first sorted out. Compared with the known mRNA and non-coding RNAs using Cuffcompare41(Cuffcompare -o cuffcmp -r genome.grf -s genome.fasta sample1.gtf), the sequences of the remaining transcripts that overlapped with known genes were discarded. Then, transcripts with a FPKM (fragments per kilobase of transcript per million mapped reads) score higher than or equal to 0.5 were retained. The coding potential of the remaining transcripts was searched against CPC (Coding Potential Calculator v2),42 CNCI (Coding-Non-Coding-Index 0.9-r2),43 PfamScan v1.3,44 and phyloCSF v2012102845 programmes by BLASTX (E-value cut-off of 1e–10, coverage > 80%, and identity > 90%) to exclude transcripts with significant homolog to known proteins, respectively. Transcripts predicted with coding potential by any of the 4 tools were filtered out, and those without coding potential were considered as the candidate lncRNAs.
LncRNAs characterization and functional prediction
All identified lncRNAs and mRNAs were aligned to the genome of PN40024 separately to obtain the chromosome distribution. A circular schematic diagram was constructed using Circos46 for comparative visualizations. The full length of all identified lncRNAs was used to align against the whole genome of Arabidopsis and rice with a cut-off E ⩽ 1.0e–10. Potential miRNA precursors were predicted online by subjecting all the lncRNA candidates to Blast search against the miRBase 21 (http://www.mirbase.org)47 and by identifying hits with sequence homology greater than 90%. In addition, candidate lncRNAs targeted by miRNAs were identified using the psRobot software48 with default parameters.
Quantification of gene expression, target gene prediction, and gene ontology enrichment analysis
The FPKMs of both lncRNAs and coding genes were calculated by Cuffdiff v2.1.1 in each sample.41 The transcripts with a P-value < 0.05 were assigned as differentially expressed.
The cis role refers to lncRNA acting on neighbouring target genes. In this study, we searched coding genes 100 kb upstream and downstream of an lncRNA and then analysed their function. The trans role refers to lncRNAs acting on other genes at the expression level. We constructed the co-expression network between lncRNAs and coding RNAs by Pearson’s correlation coefficients with custom scripts (Pearson’s correlation ⩾0.95 or ⩽0.95). Then, gene ontology (GO) enrichment analysis49 of lncRNA target genes was implemented by the GOseq R package, in which gene length bias was corrected. Gene ontology terms with corrected P-values < 0.05 were considered significant functional terms.
qRT-PCR validation of differentially expressed lncRNAs
For qRT-PCR, first-strand cDNA was synthesized from total RNA using the Superscript III First-Strand cDNA Synthesis SuperMix kit (Invitrogen, Carlsbad, CA, USA) and random hexamer primers. Primers were designed using the OligoArchitect™ Online software (Sigma-Aldrich, St. Louis, Mo, USA) and are listed in Supplemental Table S1, and the grape VvEF1-γ (AF176496) gene was used as an internal standard. Polymerase chain reaction (PCR) amplifications were performed in a 7500 real-time system (Applied Biosystems, Foster City, CA, USA) with 15-μL final volumes containing 1.0 μL of cDNA, 0.5 μL of each primer (10 μM), 0.3 μL ROX Reference Dye, 5.2 μL of sterile water, and 7.5 μL of (2×) SYBR® Premix Ex TaqTM II (Tli RNaseH Plus) (Takara, Tokyo, Japan). The conditions for amplification were as follows: 2 minutes of denaturation at 95°C followed by 40 cycles of 95°C for 5 seconds, and 60°C for 35 seconds. Relative gene expression was calculated using the 2−ΔΔCt method. In total, 2 biological replicates and 3 technical replicates were performed for each of the selected lncRNAs.
RT-PCR validation of lncRNAs
First-strand cDNA was reverse transcribed as described above, and PCR amplifications were performed in a C1000 TouchTM Thermal Cycler (Bio-Rad, Hercules, CA, USA). PCR was performed in 25 µL of reaction mixture containing 1 µL of template DNA, 1 µL of 2.5 mM dNTPs, 1 U of LA Taq (Takara), 2.5 µL of 10× LA PCR Buffer II (Mg2+ Plus), and 0.5 µL each of 10-µM forward and reverse primers. Primers were also listed in Supplemental Table S1. Cycling conditions were 5 minutes at 94°C followed by 32 cycles of 94°C for 30 seconds, 60°C for 30 seconds, 72°C for 60 seconds, and a final extension at 72°C for 10 minutes. The PCR products were separated on 1.5% agarose gel.
Results
Deep sequencing and identification of lncRNAs
The grapevine cultivars showed significant differences in tolerance to L theobromae infection, as described in our previous study.35 In this study, the average canker lengths on CF were significantly greater than those on ML after CSS-01s inoculation (Figure 1), indicating that the cultivar ML is much more tolerant to L theobromae than CF.
Figure 1.
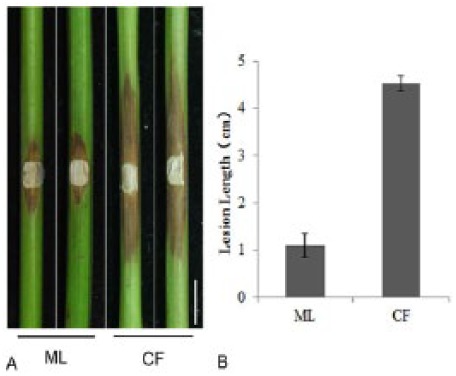
Phenotypic on cultivar ML and CF after inoculating with Lasiodiplodia theobromae strain CSS-01s: (A) photos were taken at 10 days post inoculation (dpi) and (B) canker length was measured and shown as mean values and standard errors from 10 shoots. Bar = 1 cm. CF indicates Cabernet Franc; ML, Merlot.
To identify the lncRNAs involved in mediating the response to L theobromae in grapevine, deep RNA-seq was performed on transcripts that were derived from shoot phloem of grapevine cultivars CF and ML at 0 and 24 hours after CSS-01s inoculation, each with 2 biological replicates. A total of 766 million clean reads were generated from 8 libraries, and approximately 489 million reads (63.79%) were successfully mapped on to the reference PN40024 genome (Table 1). Approximately, 62.12% of the reads were uniquely mapped to a single genomic locus, attesting the reliability of the transcriptome data. A total of 73 657 transcripts were assembled using Cufflinks and Scripture. Identification of grapevine lncRNAs was performed according to the pipeline shown in Figure 2. First, transcript data were filtered according to 4 principles: (1) exon ⩾ 1 and sequencing depth ⩾ 2 reads; (2) transcript length > 200 nt; (3) filter out known non-lncRNA annotations; and (4) FPKM ⩾ 0.5. As a result, 4523 transcript sequences were obtained. Furthermore, the coding potential of the remaining transcripts was subsequently evaluated using CPC, CNCI, PfamScan, and PhyloCSF software, respectively. Finally, 1826 candidate lncRNAs were identified and listed in Supplemental Table S2. Real-time polymerase chain reaction confirmed a total of 13 randomly selected of the RNA-seq identified lncRNAs, thus proving the assembly and identification pipeline (Figure 3).
Table 1.
Summary of RNA-seq data.
| Samples | Raw reads | Clean reads | Total mapped reads | Uniquely mapped reads | Multiple mapped reads |
|---|---|---|---|---|---|
| ML-0 hour Rep1 | 102 942 034 | 99 554 930 | 66 156 046 (66.45%) | 65 039 097 (65.33%) | 1 116 949 (1.12%) |
| ML-0 hour Rep2 | 107 859 336 | 104 413 870 | 69 066 980 (66.15%) | 67 868 457 (65%) | 1 198 523 (1.15%) |
| ML-24 hour Rep1 | 110 089 846 | 104 984 098 | 68 983 852 (65.71%) | 66 629 002 (63.47%) | 2 354 850 (2.24%) |
| ML-24 hour Rep2 | 93 733 566 | 90 051 820 | 55 889 317 (62.06%) | 53 898 495 (59.85%) | 1 990 822 (2.21%) |
| CF-0 hour Rep1 | 99 633 410 | 96 877 110 | 61 577 548 (63.56%) | 60 574 559 (62.53%) | 1 002 989 (1.04%) |
| CF-0 hour Rep2 | 83 588 386 | 81 249 428 | 50 241 614 (61.84%) | 49 446 456 (60.86%) | 795 158 (0.98%) |
| CF-24 hour Rep1 | 99 358 488 | 95 044 372 | 57 759 361 (60.77%) | 55 671 721 (58.57%) | 2 087 640 (2.2%) |
| CF-24 hour Rep2 | 98 390 380 | 93 985 360 | 59 034 378 (62.81%) | 56 811 456 (60.45%) | 2 222 922 (2.37%) |
| Total | 795 595 446 | 766 160 988 | 488 709 096 (63.79%) | 475 939 243 (62.12%) | 12 769 853 (1.67%) |
Abbreviations: CF, Cabernet Franc; ML, Merlot; RNA-seq, RNA-sequencing.
Figure 2.
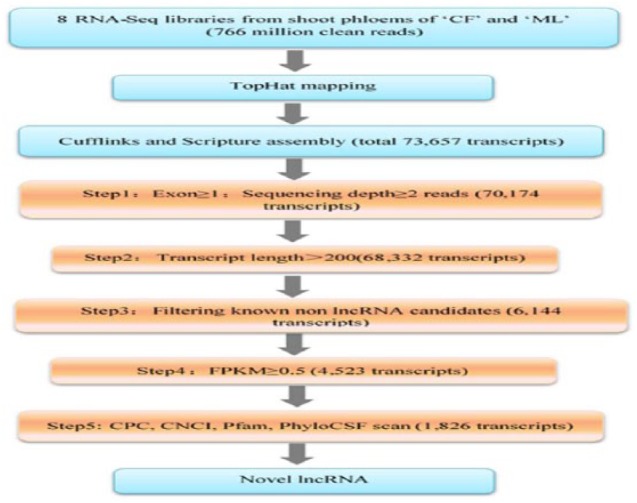
Bioinformatics pipeline for the systematic identification, annotation and classification of grapevine lncRNAs. lncRNA indicates long non-coding RNA.
Figure 3.
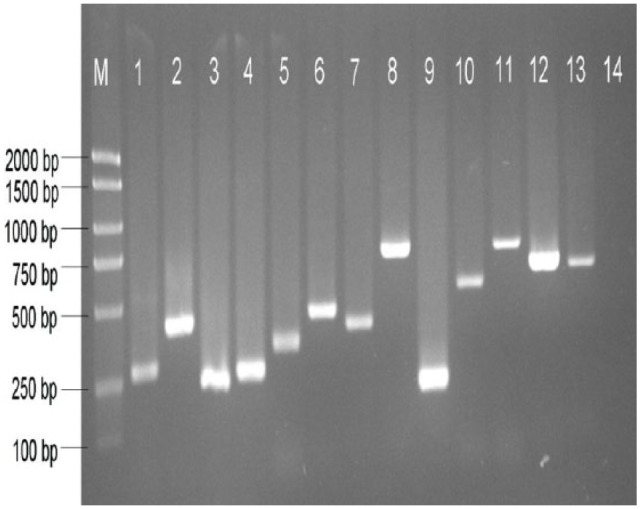
RT-PCR validation of 1 lncRNA transcripts – Lane 1: Lnc36, Lane 2: Lnc367, Lane 3: Lnc440, Lane 4: Lnc485, Lane 5: Lnc625, Lane 6: Lnc1080, Lane 7: Lnc1276, Lane 8: Lnc1327, Lane 9: Lnc1731, Lane 10: Lnc1378, Lane 11: Lnc1388, Lane 12: Lnc1455, Lane 13: Lnc1633, and Lane 14: Negative control. lncRNA indicates long non-coding RNA; RT-PCR, real-time polymerase chain reaction.
Characteristics of grapevine lncRNAs
Based on their genomic location relative to their closest protein-coding genes, the 1826 newly identified grape lncRNAs include 1556 (85%) lincRNAs, 270 (15%) antisense lncRNAs, and no intronic lncRNAs (Figure 4A). Among the intergenic lncRNAs, 449 (24.6%) and 427 (23.4%) are located within 5 kb upstream and downstream of annotated genes, respectively (Supplemental Table S3). The remaining 37% of the intergenic lncRNAs are located at least 5 kb from the closest gene. The lengths of the lncRNAs ranged from 201 to 8661 bp, with more than 60% lncRNAs ranging from 200 to 800 bp (Figure 4B). In addition, full-length lncRNA transcripts (median length of 1001 bp) are shorter than grape mRNA transcripts (median length of 3572 bp). Approximately, 68% of the lncRNAs consist of a single exon, and the rest have multiple exons (Figure 4C). Size distribution of the exons suggested that almost 80% of lncRNAs have sizes ranging from 200 to 800 bp (Supplemental Figure S1A). Most of the lncRNAs (35%) possess short intronic regions (⩽200 bp) and 21% have long intronic regions (>2000 bp) (Supplemental Figure S1B). Moreover, we examined the distribution of lncRNAs on the grape chromosomes and found that the lncRNAs were transcribed from all the 19 chromosomes. Except for those that were not mapped on chromosomes, chromosome 03 has the highest lncRNA density with 4.57 lncRNAs per 1 Mbp of nucleotides, whereas chromosome 11 has the lowest lncRNA density with 2.14 lncRNAs per 1 Mbp of nucleotides (Figure 4D and E).
Figure 4.

Characteristics of grapevine lncRNA: (A) percentage and distribution of lincRNA, antisense lncRNAs, and intronic lncRNAs in grapevine; (B) transcript length distribution of 1826 candidate lncRNAs; (C) number of exons per transcript for all lncRNAs; (D) numbers of lncRNAs on each chromosome; and (E) distribution of lncRNAs along each chromosome. lincRNA indicates long intergenic non-coding RNA; lncRNA, long non-coding RNA.
To evaluate the sequence conservation, the lncRNA sequences were blasted against the genomes of Arabidopsis and rice. Only 56 lncRNAs showed multiple homologous regions with those of A thaliana, whereas 41 lncRNAs were predicted to be highly conserved with the rice genome (Figure 5). In addition, most aligned lncRNAs were conserved with Arabidopsis at both more than 10% and 20% coverage levels. Furthermore, the repeat content was evaluated by RepeatMasker (http://www.repeatmasker.org). The results showed that more than 35% of the putative lncRNAs contain repetitive sequences or transposons (Supplemental Table S4).
Figure 5.

Conservation of grapevine lncRNA: 1826 candidate lncRNAs were blasted with the genomes of (A) Arabidopsis thaliana and (B) Oryza sativa. (C) Number of for conserved lncRNAs with more than 10% or 20% coverage regions. lncRNA indicates long non-coding RNA.
Differentially expressed lncRNAs in response to Lasiodiplodia theobromae infection
To investigate the potential role of lncRNAs in L theobromae inoculation, we performed differentially expressed transcripts analyses in ML and CF at 24 hours after CSS-01s inoculation compared with mock-inoculated samples. Differentially expressed transcripts were analysed by Cuffdiff, and 782 lncRNAs with a P-adjust < 0.05 were assigned as differentially expressed (Figure 6A; Supplemental Table S5). Then, Venn diagrams and heat maps were drawn to show the differentially expressed lncRNAs that were common to both grape cultivars ML and CF or that were specific to either cultivar in response to CSS-01s inoculation (Figure 6B). Of these differentially expressed transcripts, 782 were from intergenic and antisense lncRNAs (704 intergenic and 78 antisense RNAs) (Figure 6B). Among them, 264 differentially expressed lncRNAs (62 up-regulated and 202 down-regulated) were only significantly expressed in ML, 325 were present in both cultivars, and 193 (64 up-regulated and 129 down-regulated) were CF-specific (Figure 6B to D). Among the common lncRNAs, 111 up-regulated and 213 down-regulated were present in both cultivars, and only 1 lncRNA was up-regulated in CF but down-regulated in ML (Figure 6).
Figure 6.

Differential expression of grapevine lncRNA post inoculation with Lasiodiplodia theobromae: (A) heat map plot of differential expressed lncRNAs with q-value < 0.05. (B) Venn plot of differential expressed lncRNAs at 24 hpi. Volcano plot of differential expressed lncRNAs at 24 hpi in (C) ML and (D) CF. CF indicates Cabernet Franc; ML, Merlot; lncRNA, long non-coding RNA.
To confirm the accuracy and reliability of the RNA-seq data, 8 of the differentially expressed lncRNA candidates that were present in both cultivars were randomly selected for qRT-PCR validation. As shown in Figure 7, the qRT-PCR results were in concordance with the RNA-seq data, suggesting that these lncRNAs were likely to play roles in response to L theobromae infection
Figure 7.

qPCR validation of the RNA-seq data using 9 random selected lncRNA. The elongation factor 1-γ (EF1-γ) gene was used as the reference gene. The relative expression level of lncRNAs was calculated relative to its corresponding mock sample. Error bars represented the standard error of 2 biological replicates. lncRNA indicates long non-coding RNA; qPCR, quantitative polymerase chain reaction; RNA-seq, RNA-sequencing.
The cis and trans role of lncRNAs in target genes
To investigate the functions or biological processes that the candidate lncRNAs might be involved in, we predicted the target genes in cis and trans. For the cis analysis of the lncRNAs, we searched coding genes 100 kb upstream and downstream of lncRNAs. The results indicated that 779 differentially expressed lncRNAs, co-localized with 9909 coding genes (Supplemental Table S6). Then, GO enrichment analyses were conducted separately for the neighbouring co-localized genes of these differentially expressed lncRNAs at 24 hpi on cultivars ML and CF. According to the analyses, the GO term ‘structural constituent of cell wall’ (GO: 0005199) was significantly enriched among the co-localized genes of the differentially expressed lncRNAs at 24 hpi on the cultivar ML. However, most of the co-localized genes of differentially expressed lncRNAs on CF were enriched in biological processes and concentrated in ‘response to auxin stimulus’ (GO: 0009733), ‘chitin catabolic process’ (GO: 0006032), ‘response to hormone stimulus’ (GO: 0009725), ‘chitin metabolic process’ (GO: 0006030), and ‘cell wall macromolecule metabolic process’ (GO: 0044036) (Supplemental Table S7).
Based on the expression correlation coefficient (Pearson’s correlation ⩾ 0.95 or ⩽0.95), the co-expression network for the lncRNAs and coding genes were investigated. In total, 393 611 interaction relationships were constructed in trans between 792 lncRNAs and mRNA genes in the V vinifera genome (Supplemental Table S6). Then, GO enrichment analyses were performed separately for the co-expressed genes of these differentially expressed lncRNAs at 24 hpi on cultivars ML and CF. Functional enrichment analysis showed that the trans target genes were significantly enriched in 46 GO terms (Supplemental Table S7).
LncRNAs as potential miRNA precursors
MicroRNAs (miRNAs) are short regulatory RNAs that play an essential role in the regulation of target transcripts at both the transcriptional and post-transcriptional levels in most eukaryotes.50,51 Recent studies suggested that certain lncRNAs could act as miRNA precursors.52 All the lncRNAs were aligned against the miRBase database, and the results had revealed that 36 lncRNAs showed high homology with known miRNA precursors from V vinifera, Arabidopsis lyrata, and Arabidopsis thaliana (Supplemental Table S8). In addition, 2 lncRNAs (LNC_000032 and LNC_001084) were predicted as precursors for miR169, 2 lncRNAs (LNC_000402 and LNC_000403) were precursors for miR156, and another 2 lncRNAs (LNC_001235 and LNC_001272) serve as precursors for miR398.
Discussion
Botryosphaeria dieback causes serious losses to table and grape wine production across the world.32,35 Although fungicides, such as flusilazole, carbendazim, tebuconazole, thiophanate-methyl, and mancozeb, have been reported to inhibit or reduce the infection of Botryosphaeriaceous species,53,54 epidemics still occur. Thus, cultivating disease-tolerant varieties is an efficient way in controlling these diseases. Recently, an increasing number of reports suggested that lncRNAs have been recognized as important regulators of the biotic and abiotic stress responses.16,17 Functional study on lncRNAs has opened up a new field in disease resistance breeding. Recently, 931 differentially expressed lncRNAs responsive to Sclerotinia sclerotiorum infection were identified in Brassica napus.55 Furthermore, 41 lncRNAs were predicted as precursors for fungal phytopathogens responsive miRNAs. In this study, we used an RNA-seq approach to investigate transcriptomic changes in response to L theobromae infection, and we systematically identified 1826 novel lncRNAs in grape. This is the first work to globally identify lncRNAs that respond to L theobromae infection in grape. Hence, this study provides an important resource of grape lncRNAs that can be useful for future research in this direction.
It has been reported that the potential functions of lncRNAs can be predicted by their co-localized and co-expressed transcripts.56,57 The cis analysis showed high relevance with GO terms related to morphological changes, including structural constituent of cell wall, chitin catabolic process, chitin metabolic process, and cell wall macromolecule metabolic process among others. The plant cell wall that is composed of polysaccharides, proteins, and aromatic polymers is the first barrier that plants use to limit pathogen attack.58–60 Emerging evidence of plant–fungal interactions has indicated that perception of fungal chitin by host plants is critical for triggering pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) against fungal pathogens attack.61,62 These results indicated that the expression levels of the target genes involved in cell wall organization and chitin signalling were closely correlated with their lncRNAs on L theobromae infection.
Host endogenous miRNAs and miRNA pathway components play essential roles in plant-immune responses against various pathogens, including bacteria, fungi, oomycetes, and viruses.63 It has been reported that miR156, miR169, miR398, and miR169 were significantly up-regulated by fungal pathogen stress.64,65 In addition, miR482 was reported to guide cleavage of nucleotide-binding site leucine-rich repeat (NBS-LRR) disease resistance genes in Solanaceae and Leguminosae species as well as in Arabidopsis.66,67 Recently, it was shown that miR169 might be involved in bacterial wilt resistance by post-transcriptional regulation of NF-YA transcription factors.68 In addition, miR398 is involved in PTI responses through targeting of 2 copper superoxide dismutase genes, CSD1 and CSD2, and a cytochrome c oxidase gene, COX5b.1.69 In this study, we found that there are 2 lncRNAs that might be precursors for miR156, miR169, and miR398, demonstrating that lncRNA may be important in mediating responses of grape to L theobromae through the interaction with miRNA.
Our current understanding of lncRNA regulation in response to L theobromae infection is still in its infancy. Several approaches including lncRNA silencing and overexpressing or CRISPR in vivo need to be performed to elucidate the specific molecular roles of these candidate lncRNAs and their interaction with other regulatory components involved in L theobromae infection.
Supplemental Material
Supplemental material, Suppplemental_Material for Genome-Wide Identification of Long Non-coding RNAs Responsive to Lasiodiplodia theobromae Infection in Grapevine by Qikai Xing, Wei Zhang, Mei Liu, Lingxian Li, Xinghong Li and Jiye Yan in Evolutionary Bioinformatics
Acknowledgments
The authors are grateful to Xiaoping Tang and Zhigang Dong (Pomology Institute, Shanxi Academy of Agriculture Sciences) for providing the grapevine material.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from CARS-30 and Youth Scientific Fund of BAAFS (QNJJ201609).
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: JY and XL designed the study. QX performed the experiments and analyzed the data. JY and QX wrote the manuscript. WZ, ML and LL contributed reagents and materials. All the authors reviewed and approved the final article.
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rew Genet. 2001;2:919–929. [DOI] [PubMed] [Google Scholar]
- 2. Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. [DOI] [PubMed] [Google Scholar]
- 3. Chen X. Small RNAs in development-insights from plants. Curr Opin Genet Dev. 2012;22:361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cuperus JT, Fahlgren N, Carrington JC. Evolution and functional diversification of MIRNA genes. Plant Cell. 2011;23:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Staiger D, Korneli C, Lummer M, Navarro L. Emerging role for RNA-based regulation in plant immunity. New Phytol. 2013;197:394–404. [DOI] [PubMed] [Google Scholar]
- 6. Wierzbicki AT, Haag JR, Pikaard CS. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell. 2008;135:635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wierzbicki AT. The role of long non-coding RNA in transcriptional gene silencing. Curr Opin Plant Biol. 2012;15:517–522. [DOI] [PubMed] [Google Scholar]
- 8. Zhang YC, Chen YQ. Long noncoding RNAs: new regulators in plant development. Biochem Biophys Res Commun. 2013;436:111–114. [DOI] [PubMed] [Google Scholar]
- 9. Ma L, Bajic VB, Zhang Z. On the classification of long non-coding RNAs. RNA Biol. 2013;10:924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu J, Jung C, Xu J, et al. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell. 2012;24:4333–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li L, Eichten SR, Shimizu R, et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014;15:R40. doi: 10.1186/gb-2014-15-2-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y, Liao J, Li Z, et al. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014;15:512. doi: 10.1186/s13059-014-0512-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xin M, Wang Y, Yao Y, et al. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 2011;11:61. doi: 10.1186/1471-2229-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wen J, Parker BJ, Weiller GF. In Silico identification and characterization of mRNA-like noncoding transcripts in Medicago truncatula. In Silico Biol. 2007;7:485–505 [PubMed] [Google Scholar]
- 15. Hao Z, Fan C, Cheng T, Su Y, Wei Q, Li G. Genome-wide identification, characterization and evolutionary analysis of long intergenic noncoding RNAs in cucumber. PLoS ONE. 2015;10:e0121800. doi: 10.1371/journal.pone.0121800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chekanova JA. Long non-coding RNAs and their functions in plants. Curr Opin Plant Biol. 2015;27:207–216. [DOI] [PubMed] [Google Scholar]
- 17. Shafiq S, Li J, Sun Q. Functions of plants long non-coding RNAs. Biochim Biophys Acta. 2016;1859:155–162. [DOI] [PubMed] [Google Scholar]
- 18. Yuan J, Zhang Y, Dong J, et al. Systematic characterization of novel lncRNAs responding to phosphate starvation in Arabidopsis thaliana. BMC Genomics. 2016;17:655. doi: 10.1186/s12864-016-2929-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kwenda S, Birch PRJ, Moleleki LN. Genome-wide identification of potato long intergenic noncoding RNAs responsive to Pectobacterium carotovorum subspecies brasiliense infection. BMC Genomics. 2016;17:614. doi: 10.1186/s12864-016-2967-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang J, Yu W, Yang Y, et al. Genome-wide analysis of tomato long non-coding RNAs and identification as endogenous target mimic for microRNA in response to TYLCV infection. Sci Rep. 2015;5:16946. doi: 10.1038/srep16946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Swiezewski S, Liu F, Magusin A, Dean C. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature. 2009;462:799–802. [DOI] [PubMed] [Google Scholar]
- 22. Liu F, Marquardt S, Lister C, Swiezewski S, Dean C. Targeted 3′ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science. 2010;327:94–97. [DOI] [PubMed] [Google Scholar]
- 23. Heo JB, Sung S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science. 2011;331:76–79. [DOI] [PubMed] [Google Scholar]
- 24. Shin JH, Chekanova JA. Arabidopsis RRP6L1 and RRP6L2 function in FLOWERING LOCUS C silencing via regulation of antisense RNA synthesis. PLoS Genet. 2014;10:e1004612. doi: 10.1371/journal.pgen.1004612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Franco-Zorrilla JM, Valli A, Todesco M, et al. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet. 2007;39:1033–1037. [DOI] [PubMed] [Google Scholar]
- 26. Bertsch C, Larignon P, Farine S, Clement C, Fontaine F. The spread of grapevine trunk disease. Science. 2009;324:721–721. [DOI] [PubMed] [Google Scholar]
- 27. Dissanayake AJ, Phillips AJL, Li X, Hyde KD. Botryosphaeriaceae: current status of genera and species. Mycosphere. 2016;7:1001–1073. [Google Scholar]
- 28. Chethana KWT, Li X, Zhang W, Hyde KD, Yan J. Trail of decryption of molecular research on Botryosphaeriaceae in woody plants. Phytopathol Mediterr. 2016;55:147–171. [Google Scholar]
- 29. Van Niekerk JM, Fourie PH, Halleen F, Crous PW. Botryosphaeria spp. as grapevine trunk disease pathogens. Phytopathol Mediterr. 2006;45:S43–S54. [Google Scholar]
- 30. Úrbez-Torres JR, Leavitt GM, Voegel TM, Gubler WD. Identification and distribution of Botryosphaeria spp. associated with grapevine cankers in California. Plant Dis. 2006;90:1490–1503. [DOI] [PubMed] [Google Scholar]
- 31. Úrbez-Torres JR, Leavitt GM, Guerrero JC, Guevara J, Gubler WD. Identification and pathogenicity of Lasiodiplodia theobromae and Diplodia seriata, the causal agents of bot canker disease of grapevines in Mexico. Plant Dis. 2008;92:519–529. [DOI] [PubMed] [Google Scholar]
- 32. Úrbez-Torres JR. The status of Botryosphaeriaceae species infecting grapevines. Phytopathol Mediterr. 2011;50:5–45. [Google Scholar]
- 33. Luque J, Martos S, Aroca A, Raposo R, Garcia-Figueres F. Symptoms and fungi associated with declining mature grapevine plants in northeast Spain. J Plant Pathol. 2009;91:381–390. [Google Scholar]
- 34. Yan J, Xie Y, Yao S, Wang Z, Li X. Characterization of Botryosphaeria dothidea, the causal agent of grapevine canker in China. Australas Plant Path. 2012;41:351–357. [Google Scholar]
- 35. Yan J, Xie Y, Zhang W, et al. Species of Botryosphaeriaceae involved in grapevine dieback in China. Fungal Divers. 2013;61:221–236. [Google Scholar]
- 36. Dissanayake AJ, Zhang W, Mei L, et al. Lasiodiplodia pseudotheobromae causes pedicel and peduncle discolouration of grapes in China. Australa Plant Dis Notes. 2015;10:21. [Google Scholar]
- 37. Dissanayake AJ, Zhang W, Li X, et al. First report of Neofusicoccum mangiferae associated with grapevine dieback in China. Phytopathol Mediterr. 2015;54:414. [Google Scholar]
- 38. Kim DG, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: parallel mapping of transcriptomes to detect InDels, gene fusions, and more. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guttman M, Garber M, Levin JZ, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kong L, Zhang Y, Ye Z, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–W349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun L, Luo H, Bu D, et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013;41:e166. doi: 10.1093/nar/gkt646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bateman A, Birney E, Cerruti L, et al. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin M, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–i282. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Griffiths-Jones S, Grocock RJ, Van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu H, Ma YK, Chen T, Wang M, Wang X. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012;40:W22–W28. doi: 10.1093/nar/gks554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Llave C, Xie Z, Kasschau KD, Carrington JC. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002;297:2053–2056. [DOI] [PubMed] [Google Scholar]
- 51. Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, et al. Widespread translational inhibition by plant miRNAs and siRNAs. Science. 2008;320:1185–1190. [DOI] [PubMed] [Google Scholar]
- 52. Wu H, Wang Z, Wang M, Wang XJ. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 2013;161:1875–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bester W, Crous PW, Fourie PH. Evaluation of fungicides as potential grapevine pruning wound protectants against Botryosphaeria species. Australas Plant Path. 2007;36:73–77. [Google Scholar]
- 54. Amponsah NT, Jones E, Ridgway HJ, Jaspers MV. Evaluation of fungicides for the management of Botryosphaeria dieback diseases of grapevines. Pest Manag Sci. 2012;68:676–683. [DOI] [PubMed] [Google Scholar]
- 55. Joshi RK, Megha S, Basu U, Rahman MH, Kav NN. Genome wide identification and functional prediction of long non-coding RNAs responsive to Sclerotinia sclerotiorum infection in Brassica napus. PLoS ONE. 2016;11:e0158784. doi: 10.1371/journal.pone.0158784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Gene Dev. 2009;23:1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liao Q, Liu C, Yuan X, et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011;39:3864–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Underwood W. The plant cell wall: a dynamic barrier against pathogen invasion. Front Plant Sci. 2007;3:85. doi: 10.3389/fpls.2012.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kubicek CP, Starr TL, Glass NL. Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu Rev Phytopathol. 2014;52:427–451. [DOI] [PubMed] [Google Scholar]
- 60. Nafisi M, Fimognari L, Sakuragi Y. Interplays between the cell wall and phytohormones in interaction between plants and necrotrophic pathogens. Phytochemistry. 2015;112:63–71. [DOI] [PubMed] [Google Scholar]
- 61. Kombrink A, Sánchez-Vallet A, Thomma BP. The role of chitin detection in plant–pathogen interactions. Microbes Infect. 2011;13:1168–1176. [DOI] [PubMed] [Google Scholar]
- 62. Shinya T, Nakagawa T, Kaku H, Shibuya N. Chitin-mediated plant–fungal interactions: catching, hiding and handshaking. Curr Opin Plant Biol. 2015;26:64–71. [DOI] [PubMed] [Google Scholar]
- 63. Seo JK, Wu J, Lii Y, Li Y, Jin H. Contribution of small RNA pathway components in plant immunity. Mol Plant Microbe Interact. 2013;26:617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen L, Ren Y, Zhang Y, Xu J, Zhang Z, Wang Y. Genome-wide profiling of novel and conserved Populus microRNAs involved in pathogen stress response by deep sequencing. Planta. 2012;235:873–883. [DOI] [PubMed] [Google Scholar]
- 65. Jin W, Wu F. Characterization of miRNAs associated with Botrytis cinerea infection of tomato leaves. BMC Plant Biol. 2015;15:1. doi: 10.1186/s12870-014-0410-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shivaprasad PV, Chen HM, Patel K, Bond DM, Santos BA, Baulcombe DC. A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell. 2012;24:859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Boccara M, Sarazin A, Thiebeauld O, et al. The Arabidopsis miR472-RDR6 silencing pathway modulates PAMP- and effector-triggered immunity through the post-transcriptional control of disease resistance genes. PLoS Pathog. 2014;10:e1003883. doi: 10.1371/journal.ppat.1003883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hanemian M, Barlet X, Sorin C, et al. Arabidopsis CLAVATA1 and CLAVATA2 receptors contribute to Ralstonia solanacearum pathogenicity through a miR169-dependent pathway. New Phytol. 2016;211:502–515. [DOI] [PubMed] [Google Scholar]
- 69. Li Y, Zhang Q, Zhang J, Wu L, Qi Y, Zhou J. Identification of microRNAs involved in pathogen-associated molecular pattern-triggered plant innate immunity. Plant Physiol. 2010;152:2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Suppplemental_Material for Genome-Wide Identification of Long Non-coding RNAs Responsive to Lasiodiplodia theobromae Infection in Grapevine by Qikai Xing, Wei Zhang, Mei Liu, Lingxian Li, Xinghong Li and Jiye Yan in Evolutionary Bioinformatics