

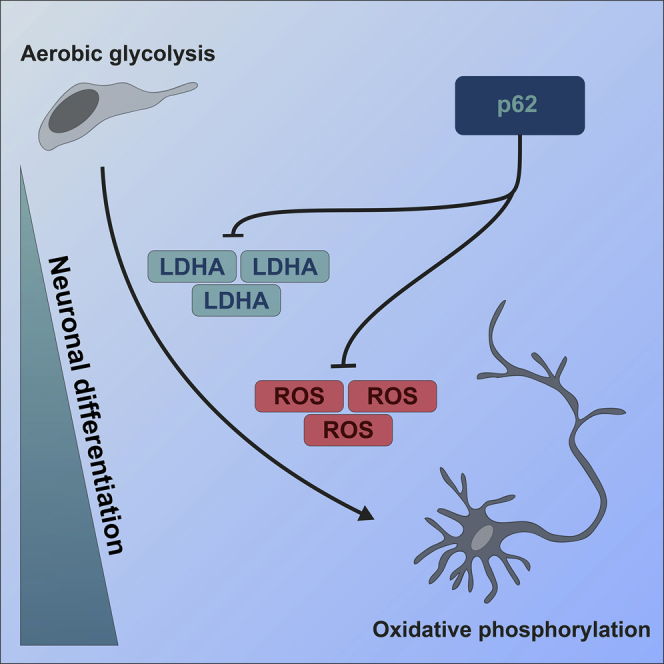
Summary
Neurodegenerative disorders are an increasingly common and irreversible burden on society, often affecting the aging population, but their etiology and disease mechanisms are poorly understood. Studying monogenic neurodegenerative diseases with known genetic cause provides an opportunity to understand cellular mechanisms also affected in more complex disorders. We recently reported that loss-of-function mutations in the autophagy adaptor protein SQSTM1/p62 lead to a slowly progressive neurodegenerative disease presenting in childhood. To further elucidate the neuronal involvement, we studied the cellular consequences of loss of p62 in a neuroepithelial stem cell (NESC) model and differentiated neurons derived from reprogrammed p62 patient cells or by CRISPR/Cas9-directed gene editing in NESCs. Transcriptomic and proteomic analyses suggest that p62 is essential for neuronal differentiation by controlling the metabolic shift from aerobic glycolysis to oxidative phosphorylation required for neuronal maturation. This shift is blocked by the failure to sufficiently downregulate lactate dehydrogenase expression due to the loss of p62, possibly through impaired Hif-1α downregulation and increased sensitivity to oxidative stress. The findings imply an important role for p62 in neuronal energy metabolism and particularly in the regulation of the shift between glycolysis and oxidative phosphorylation required for normal neurodifferentiation.
Keywords: SQSTM1, p62, hypoxia, mitochondria, neurodifferentiation, neuroepithelial-like stem cells, neuronal development, oxidative stress, mitophagy, neurodegeneration
Graphical Abstract
Highlights
-
•
SQSTM1/p62 is dispensable for mitophagy in a human neuronal model
-
•
SQSTM1/p62 is needed for proper neuronal differentiation
-
•
Loss of SQSTM1/p62 leads to increased oxidative stress and poor LDHA downregulation
-
•
SQSTM1/p62 is essential for the shift to OXPHOS during neuronal differentiation
SQSTM1/p62 is a known autophagy adaptor that, if lost, causes childhood-onset neurodegeneration. Data from Wredenberg et al. show that loss of p62 in a neuronal stem cell model does not affect mitophagy but instead leads to impaired differentiation. The authors suggest p62 finely tunes LDHA expression and thus controls the metabolic shift to OXPHOS required for proper differentiation.
Introduction
Brain development is a dynamic, complex, and well-organized process that initiates during the third gestational week and continues throughout development until late adolescence. During this period, neuronal differentiation, maturation, and migration are controlled and tightly regulated by carefully coordinated genetic programs (Paridaen and Huttner, 2014). Small changes in cellular environment and metabolic demands affect cellular fate and are often a requirement for correct neuronal maturation (Kim et al., 2014). Even subtle disturbances during any of these processes can result in a number of neurodegenerative disorders, but the complexity of the brain and cellular metabolism has limited our understanding of neuronal homeostasis in health and disease. A small subgroup of neurodegenerative diseases is caused by inborn errors of metabolism (IEM), defined by the toxic accumulation of metabolic intermediates or the lack of vital substrates. IEM are caused by single gene defects, thus affecting specific biochemical pathways, such as carbohydrate, lipid, amino acid, lysosomal, mitochondrial, and peroxisomal metabolism (El-Hattab, 2015). The molecular and metabolic consequences observed in these diseases therefore offer unique opportunities to study the interplay between the genetic and the metabolic demands of the developing brain, providing vital advances in understanding the changes observed in more complex common neurodegenerative disorders.
We recently reported that the loss of the autophagy adaptor Sequestosome 1 (SQSTM1/p62) in nine individuals from four different families leads to a childhood- or adolescence-onset neurodegenerative disorder, characterized by progressive gait abnormalities, ataxia, dysarthria, dystonia, vertical gaze palsy, and cognitive decline (Haack et al., 2016). Magnetic resonance imaging revealed atrophy of the cerebellum and the vermis cerebelli (Haack et al., 2016). Eleven more patients, with similar clinical presentations, have since been reported (Muto et al., 2018). Nevertheless, there was no clear indication of the pathogenetic mechanism leading to the clinical observations. p62 has been associated with several cellular processes, such as selective autophagy (Lamark et al., 2009) and mitophagy (Park et al., 2014), as well as responses to oxidative stress (Jiang et al., 2015), immune response (Lee et al., 2015), and hypoxia (Rantanen et al., 2013). More recently, p62 was also shown to be an important regulator in cancer metabolism, with increased p62 expression reported in lung (Schlafli et al., 2016), liver, kidney, and breast cancers, as well as glioblastoma (Zeng et al., 2014). On the other hand, loss of p62 has also been suggested to promote prostate cancer progression by affecting the tumor's microenvironment (Valencia et al., 2014).
p62, thus, has been suggested to have both pleiotropic and tissue-specific roles, described in often contradictory reports. For instance, together with the E3-ubiquitin ligase PARKIN and the mitochondrial kinase PINK1, p62 has been widely proposed to regulate the maintenance and turnover of mitochondria by selective autophagy, also termed mitophagy (Geisler et al., 2010). Although the phenomenon of mitophagy under certain experimental conditions is generally accepted, its physiological role and mechanism are more controversial (Grenier et al., 2013), and several studies now dispute the involvement of p62 in this process (Narendra et al., 2010, Okatsu et al., 2010). Likewise, p62 has been proposed to stabilize nuclear factor erythroid 2-related factor 2 (NRF2) transcription factor under oxidative stress conditions, by preventing its KEAP1-mediated degradation and thereby triggering an oxidative stress gene expression response (Copple et al., 2010, Fan et al., 2014, Jain et al., 2010, Komatsu et al., 2010, Lau et al., 2010). However, p62−/− mice were still able to activate NRF2-induced gene responses (Kwon et al., 2012) and no changes in NRF2 and KEAP1 were observed in the absence of p62 in a pancreatic tumor model (Valencia et al., 2014).
To understand the disease mechanisms associated with the loss of p62, we studied the cellular and metabolic consequences in neuronal stem cells and neurons, derived from reprogrammed patient fibroblasts, as well as CRISPR/Cas9-targeted neuroepithelial stem cells (NESCs). Our data suggest that p62 is required for normal neurogenesis by controlling the expression of proteins involved in the metabolic shift from aerobic glycolysis to oxidative phosphorylation (OXPHOS) and protecting from oxidative stress. In particular, lactate dehydrogenase A (LDHA), a protein essential for this shift (Zheng et al., 2016), is upregulated and remains high in p62-knockout cells upon neuronal differentiation.
Results
p62 Is Essential for Neuronal Differentiation
In humans, loss of p62 leads to a childhood-onset neurodegenerative disease with no organs affected other than the central nervous system (Haack et al., 2016, Muto et al., 2018). We therefore sought to study the role of p62 and reprogrammed fibroblasts from two patients carrying a nonsense mutation at the 5′ end of SQSTM1 (p.Arg96∗, patients II:1 and II:4 from family 4 in Haack et al., 2016) to induced pluripotent stem cells (iPSCs), before differentiating them further to neuroepithelial-like stem cells (Pat1NES or Pat2NES) (for details see Experimental Procedures and Falk et al., 2012, Koch et al., 2009). NESCs exhibit an extensive proliferative lifespan and display a high differentiation potential to various neuronal subtypes and glial cells, allowing for a cell-type and patient-specific characterization (Falk et al., 2012, Koch et al., 2009). We confirmed the NESC status of Pat1&2NES cells by immunofluorescence and western blot of Nestin, SOX2, PLZF, and DACH1 (Figures S1A and S1B). To control for potential effects due to differences in genetic background between Pat1&2NES cells and control NESCs, we additionally applied CRISPR/Cas9 gene editing technology to control NESCs, targeting the 5′ terminus of SQSTM1, followed by clonal selection. Loss of p62 was confirmed by western blot and qPCR analysis (Figures 1A and S1C). Two clones with confirmed loss of p62 were chosen as p62-knockout (KO) NESCs (KO1NES and KO2NES), while two clones that retained normal p62 levels were used as CRISPR control (CCo1NES and CCo2NES) cells (see Experimental Procedures for details).
Figure 1.

p62 Is Essential for Neuronal Differentiation
(A–C) (A) Representative western blot analysis of p62 in (top) Co1NES, Pat1NES, and Pat2NES cells and (bottom) CCo1NES, KO1NES, KO2NES, and Pat1NES cells. Tubulin was used as a loading control. n = 3 (n is defined as independent experiments). (B and C) Representative fluorescence images of Co1NEU and Pat1NEU after (B) 6 days differentiation, immunostained against α-tubulin (red) and DAPI (blue) (scale bar, 50 μm; n = 5), or (C) 20 days differentiation, immunostained against neurofilament (green), MAP2A/B (red), and DAPI (blue) or against β-III tubulin (green), HuC/D (red), and DAPI (blue) (scale bar, 100 μm; n = 5).
(D–F) Thirty neuronal bodies of Co1NEU (black), Co2NEU (white), and Co3NEU (gray) (pooled under the name CoNEU); Pat1NEU (red); Pat1NEU + p62 (brown); KO1NEU (yellow); and Pat2NEU (orange) were analyzed for (D) neuronal body area, (E) number of neuronal processes per neuronal body, and (F) width of neuronal processes. Data are expressed as mean ± SD and differences were tested by a two-tailed t test. ∗∗∗p < 0.001, ∗∗p < 0.01; n = 3.
(G) Linear Sholl analysis representation of 20-days-differentiated control neurons (pooled data from Co1NEU, Co2NEU, and Co3NEU, under the name CoNEU, dark gray); Pat1NEU (red); and Pat2NEU (orange). n = 3. Neuronal processes were analyzed from 15 neuronal bodies per line.
(H) Representative western blot analysis of steady-state levels of MAP2A/B, β-III tubulin, HuC/D, SOX2, and p62 in CCo1NES and KO1NES cells undifferentiated and after 14 or 50 days of differentiation. GAPDH was used as a loading control. n = 3.
See also Figures S1 and S2.
Removal of epidermal growth factor and basic fibroblast growth factor from the growth medium induces NESCs to arrest their cell cycle and undergo genomic reprogramming for neuronal differentiation toward neuronal and glial phenotypes. Early steps of this differentiation are marked by the appearance of 1- to 2-μm-thin projections, termed neuronal processes, which will fuse and build a complex network of connected mature neurons with prolonged differentiation. After 6 days of differentiation, control NESCs (CoNEU) presented with the expected formation of neuronal processes, as observed by immunocytochemistry against α-tubulin. In contrast, patient NESCs (Pat1NEU) showed a marked decrease in the formation of these neuronal processes (Figure 1B). After 20 days of differentiation, when morphological maturation of neurons is almost completed, control cells showed a clear organizational structure of thick neuronal processes, while patient-derived neurons showed an aberrant and disorganized morphology along with morphologically undifferentiated cells (Figures 1C and S1D–S1F). Immunostaining, using the neuronal markers Neurofilament, MAP2A/B, HuC/D, and β-III tubulin with subsequent quantification, showed a mild reduction in neuronal body area and a clear reduction in the number and width of mature neuronal processes emerging from these neuronal bodies (Figures 1D–1F). Sholl analysis (Sholl, 1953) demonstrated that patient cells develop interactions within the nearby cell cluster but are unable to connect with more distant cells, having shorter extensions than control cells (Figure 1G) (individual Sholl analyses of the control cells are shown in Figure S1G). This deviant neuronal differentiation could also be observed in CRISPR/Cas9 gene-editing-derived KONES cells (Figures S2A and S2B), as well as in control NESCs, where p62 expression was silenced by short hairpin RNA (Figure S2C). Exogenous expression of wild-type p62 in Pat1NES cells reversed this deficiency (Figures 1D–1F and S2D). Western blot analysis of the neuronal markers β-III tubulin, HuC/D, and MAP2A/B, as well as the stem cell marker SOX2, confirmed that, while control cells were able to undergo neuronal differentiation, cells lacking p62 exhibited a general decrease in the expression of neuronal markers and retained a high degree of stemness, even after 45 days of differentiation (Figure 1H). Elevated levels of SOX2 protein in patient neurons after 25 and 45 days of differentiation were confirmed by immunostaining (Figure S2E). Together, our data show that p62 is required for the formation of neuronal processes and the differentiation into neurons.
Loss of p62 in NESCs Results in Reduced Respiratory Chain Function
Mitochondrial function has recently been shown to be required for neuronal differentiation (Khacho et al., 2017), and we therefore compared mitochondrial respiratory chain function of NES control cells and NESCs lacking p62. In agreement with skeletal muscle and fibroblast samples (Haack et al., 2016), Pat1&2NES cells had a mild reduction in respiratory chain enzyme activities of complexes I and IV (Figure 2A), with reduced basal respiration capacity (Figure 2B), but normal mitochondrial ATP production rates (Figure 2C).
Figure 2.

Loss of p62 in NESCs Results in Reduced Respiratory Chain Function, but p62 Is Dispensable for Mitophagy
(A) Mitochondrial respiratory chain enzyme activities in Co1NES (black), Co2NES (white), Pat1NES (red), and Pat2NES (orange). Complex I (CI; NADH coenzyme Q reductase), complex I + III (CI+III; NADH cytochrome c reductase), complex II (CII; succinate dehydrogenase), complex II + III (CII+III; succinate cytochrome c reductase), and complex IV (CIV; cytochrome c oxidase) are shown. Data are presented as mean ± SD and differences were tested by a two-tailed t -test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3.
(B) Oxygen consumption rates from Co1NES (black), Co2NES (white), Pat1NES (red), and Pat2NES (orange), treated with CCCP or dinitrophenol (DNP). Data are expressed as mean ± SD and differences were tested by a two-tailed t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3.
(C) Mitochondrial ATP production rates in Co1NES (gray) and Pat1NES (red) supplemented with glutamate/succinate (Glu+Succ), pyruvate/L-malate (Pyr+Mal), or palmitoylcarnitine/L-malate (PalCar+Mal). Data are expressed as units per unit of citrate synthase (CS).
(D) Representative confocal images of control fibroblasts transfected with YFP-Parkin (green) and treated with CCCP (20 μM) (top) or O/A (10 μM/1 μM) (bottom) for 2 h. Fibroblasts were immunostained against Tom20 (red) and p62 (blue). n = 3.
(E) Representative confocal images of Co1NES cells treated for 2 h with or without 10 μM CCCP or O/A (5 μM/1 μM) and with or without 50 nM bafilomycin, using antibodies against p62 (green) and TOM20 (red) and DAPI stain (blue). Scale bar: 20 μm. n = 3.
(F) Representative confocal images of 14-days-differentiated neurons treated for 2 h with or without 10 μM CCCP or O/A (5 μM/1 μM) and with or without 50 nM bafilomycin, using antibodies against p62 (green) and TOM20 (red) and DAPI staining (blue). Scale bar: 20 μm. n = 3.
(G and H) Representative western blot analysis of (G) Co1NES and Pat1NES cells treated for 12 h or (H) Co1NEU and Pat1NEU after 20 days of differentiation treated for 2 h with or without 10 μM CCCP or O/A (5 μM/1 μM) and with or without 50 nM bafilomycin for 2 h. n = 3.
See also Figures S2–S4 and Table S2 for p62 primers.
An attractive explanation for the reduced respiratory chain function could be a decreased capacity of the cells to remove damaged mitochondria, since p62, together with the E3-ubiquitin ligase PARKIN and the mitochondrial kinase PINK1, has been reported to be important for this process (Park et al., 2014). Human fibroblasts express low levels of endogenous PARKIN and thus, as previously shown, only the exogenous overexpression of YFP-PARKIN showed the typical co-localization between the mitochondrial outer membrane protein TOM20 and p62 upon treatment with the mitochondrial uncoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP) or with the mitochondrial complex inhibitors oligomycin and antimycin (O/A) (Figures 2D, S2F, and S2G). In contrast, both NESCs and NESCs undergoing neuronal differentiation (14-day neurons) express high levels of endogenous PARKIN and the absence of p62 should therefore have an impact on mitochondrial clearance upon depolarization (Figure S2H). However, both cell types showed very little co-localization of p62 with TOM20 with or without the addition of bafilomycin, an inhibitor of the lysosomal proton pump that blocks lysosomal function (Figures 2E, 2F, and S2G). Furthermore, fibroblasts (Figure S2I), NESCs (Figure 2G), and differentiated neurons (Figure 2H) lacking p62 showed the expected LC3II and PINK1 levels upon CCCP or O/A treatment, suggesting that autophagosome formation and PINK1 stabilization, two classical features of mitophagy, were activated even in the absence of p62. Induction of mitophagy by mitochondrial toxins is associated with the loss of mitochondrial markers (Narendra et al., 2012); however, we observed only a mild reduction of mitochondrial proteins in control and patient fibroblasts, which could be reversed with bafilomycin (Figures S2J and S2K), and no reduction in NESCs (Figure 2G) and neurons (Figure 2H). Co-localization of the autophagosomal marker LC3II or lysosomal marker LAMP2 with the mitochondrial marker TOM20 was not affected in fibroblasts (Figures S2L and S2M), NESCs (Figures S3A and S3B), or differentiated neurons (Figures S4A and S3B) upon loss of p62. Together, these results demonstrate that p62 is not required for the autophagocytic removal of mitochondria upon mitochondrial depolarization in cells with endogenous expression of PARKIN. It also suggests that the observed neuronal phenotypes caused by the loss of p62 are not associated with deficient mitophagy.
Compromised Neuronal Differentiation Can Be Partly Rescued by N-Acetyl-L-cysteine
Oxidative stress and the regulation of reactive oxygen species (ROS) levels have previously been shown to have an important role during neuronal differentiation (Vieira et al., 2011). Mitochondria are the main source of intracellular ROS and a mild mitochondrial defect can lead to increased levels of ROS. Moreover, p62 has been suggested to regulate NRF2, previously shown to regulate genes containing antioxidant response elements (AREs) via KEAP1 (Jiang et al., 2015). KEAP1 degrades NRF2 and thus the loss of p62 would deregulate the KEAP1-NRF2-ARE pathway (Komatsu et al., 2010) and increase sensitivity to oxidative stress (Park et al., 2015). Although patient NESCs undergoing neuronal differentiation did not have increased ROS levels, KO1NEU cells showed a marked increase after 4 days differentiation (Figures 3A and 3B), consistent with the more severe phenotype. In addition, glutathione levels were reduced in the absence of p62 (Figure S4C). Furthermore, NESCs lacking p62 were more sensitive to CCCP treatment in comparison with control cells (Figure 3C). Supplementing growth conditions with the antioxidant N-acetyl-L-cysteine (NAC) allowed p62-deficient NESCs to partially regain their differentiation potential, as indicated by reduced cell death (Figures 3C and 3D) as well as increased axonal projections (Figure 3E), suggesting an important role of p62 in the cellular oxidative stress response. However, we observed no differential response between control cells and cells lacking p62 treated with CCCP in KEAP1 levels (Figure 3F) and antioxidant genes NRF2, γ-glutamylcysteine synthetase (GCLM), and the calcium channel blocker resistance protein (CCBRP, also known as xCT or SLC7A11) (Figures 3G and S4D). Interestingly, p62 protein, mRNA expression levels, and subcellular redistribution to a more punctuated pattern increased in control cells after CCCP and O/A treatment (Figures 2G, 3H, and S4E), suggesting a role of p62 in the antioxidant response. Accordingly, NAC treatment prevented the increase of the antioxidant genes xCT, NRF2, GCLM, and p62 (Figures 3H and 3I). In agreement with increased oxidative stress, several key mitochondrial antioxidant enzymes, such as GPX1, GPX4, NQO1, GSTM4, and GSTP1, were differentially expressed in cells lacking p62 (Figures 3J, 3K, S4F, and S4G), supporting the notion that p62 is essential in protecting neuronal progenitor cells from oxidative damage, independent of the KEAP1-NRF2-ARE pathway.
Figure 3.

Compromised Neuronal Differentiation due to Deficient p62 Can Be Rescued by NAC
(A) Quantification of the average CM-H2DCFDA signal in four control NESCs (Co1NEU, Co2NEU, Co3NEU, CCo1NEU [pooled as CoNEU]), Pat1NES, Pat2NES, and KO1NES.
(B) As in (A), but on cells after 4 days of differentiation. Data are expressed as mean ± SD and differences were tested by a two-tailed t test. ∗p < 0.05, ∗∗p < 0.01; n = 3 with 50 cells analyzed per experiment.
(C) Cell viability assay of CCo1NES, Co1NES, Co2NES, and Co3NES (dark gray, pooled as CoNES), Pat1NES (red), and Pat2NES (orange) after a 15-h treatment with 20 μM CCCP or 20 μM CCCP and 1 mM NAC as indicated. Data are presented as mean ± SD and differences were tested by a two-tailed t test. ∗∗p < 0.01 and ∗∗∗p < 0.001, n = 3.
(D) Representative bright-field images of Co1NES and Pat1NES cells, treated for 15 h with 20 μM CCCP or 20 μM CCCP +1 mM NAC. Scale bar, 100 μm; n = 3.
(E) Representative bright-field images of KO1NES (±1 mM NAC) subjected to differentiation for 6 or 14 days. Scale bar, 100 μm; n = 3.
(F) Representative western blot analysis of Nrf2, KEAP1, and p62 in Co1NES and Pat1NES cells untreated and treated for 12 h with 10 μM CCCP or 10 μM oligomycin and 1 μM antimycin (O/A) as indicated. Tubulin was used as a loading control. n = 3.
(G–I) qRT-PCR analysis in Co1NES (black) and Pat1NES (red) cells, treated (striped) or untreated (solid) for 6 h with 10 μM CCCP and/or 1 mM NAC as indicated for (G) NRF2, GCLM, and xCT (left) and GSS, NQO1, and GSTM4 (right) (n = 4); (H) p62 (n = 3); or (I) NRF2, GCLM, and xCT (n = 3). β-actin was used as an endogenous control. Data are expressed as mean ± SD and differences were tested by a two-tailed t test. ∗p < 0.05 and ∗∗p < 0.01.
(J and K) qRT-PCR analysis of CCo1NES (gray), KO1NES (yellow), and Pat1NES (red) cells for (J) GPX1, GPX4, GSTK1, GSTA1, and GSTP1 or (K) NQO1 and GSTM4 steady-state levels. β-actin was used as an endogenous control. Data are expressed as mean ± SD and differences were tested by a two-tailed t test. ∗p < 0.05, ∗∗p < 0.01, n = 3. See also Figure S4 and Table S2 for primers used.
p62 Regulates Gene Expression Changes Involved in the Metabolic Switch Occurring during Neuronal Differentiation
To identify additional pathways affected by the absence of p62, we studied changes in the global gene expression patterns of NESCs during 4 days of differentiation in the presence or absence of p62. To ensure equal genetic background and stemness status of the NESCs prior to differentiation, we chose to use CRISPR/Cas9-derived KO (KONES) and control (CCoNES) cells. Neuronal development is associated with changes in the expression of several neuronal markers, and as expected, the neuronal markers Pax6, NeuroG1, NeuroD1, and NeuroD2 were upregulated in control cells 4 days after the induction of neuronal differentiation (Table S1). Furthermore, we used WebGestalt to perform gene set enrichment analysis (GSEA) to cluster genes according to gene ontology terms related to biological function, molecular process, or cellular component (Wang et al., 2013). Transcriptomic data revealed a positive enrichment of “central nervous system neuron differentiation” and “neurotransmitter receptor activity” (Figure 4A). A comparison of the 500 most significant genes against the Human Gene Atlas with EnrichR presented fetal brain as the tissue with the most similar gene expression profile (q = 0.002). These data indicate that our neuronal differentiation model indeed represents stages of neuronal development.
Figure 4.

p62 Is Essential for the Metabolic Switch Required during Neuronal Differentiation
(A) Gene ontology (GO) analysis comparing gene expression profiles of CCo1NES and CCo2NES cells after 0 and 4 days of differentiation.
(B) Transcriptomic profiling of CCo1NES, CCo2NES, KO1NES, and KO2NES cells during differentiation. Venn diagram showing the number of significantly differentially expressed genes at p < 0.01 in CCo1NES, CCo2NES, KO1NES, and KO2NES cells during differentiation.
(C) Log2-fold changes of transcripts during differentiation of control CCo1NES and CCo2NES cells against KO1NES and KO2NES cells. In blue is the fitted regression line of a linear model, in gray the 45° line.
(D) Gene set enrichment analysis of RNA sequencing data against four different databases. The dotted line indicates a false discovery rate (FDR) of 0.01.
(E) Comparison of gene expression fold changes of OXPHOS-related gene sets, annotated in three databases, in CCo and KO cells after 4 days differentiation. ∗∗p < 0.01, ∗∗∗p < 0.001, calculated using Welch's t test. Boxplot outlines depict lower and upper quartile and median.
Comparison of differential expression patterns during 4 days of NESC differentiation between p62-KO and control cells identified 964 and 162 unique differentially expressed genes in CCoNES or KONES cells at α = 0.01, respectively (Figures 4B and 4C, Table S1). The expression changes of 120 genes were common between the two genotypes, suggesting that these genes are not related to p62 function. KONES cells had a milder gene response to differentiation, with genes associated with neuronal differentiation induced only in control NESCs (Table S1), corroborating the neuronal differentiation defect observed due to the absence of p62. A pathway-based GSEA revealed a positive enrichment for apoptotic pathways, which is an imprint of increased death of KONES cells under differentiation (Figure 4D). Recent work from Zheng and colleagues suggests that neuronal progenitor cells initiate a metabolic shift from glycolytic to a more OXPHOS metabolism during differentiation (Zheng et al., 2016). In agreement, OXPHOS-related genes increased in control NESCs undergoing differentiation (Figure 4E). However, KONES cells had a significantly lower expression of OXPHOS genes, with a median log2-fold change around 0, thus showing that the loss of p62 prevents NESCs from reprogramming their energy metabolism (Figure 4E).
Loss of p62 Alters Cellular Energy Metabolism
We argued that the loss of p62 altered the intracellular environment already in NESCs, preventing them from fully committing to neuronal differentiation. We therefore compared transcriptomic (Figure 5A) and proteomic (Figure 5B) expression profiles of NESCs lacking p62 with those of control NESCs before differentiation. We found 695 differentially expressed transcripts (5.1% of all accepted transcripts) and 168 differentially expressed proteins (5.5%) at a false discovery rate of <0.05 (Figures 5A and 5B and Table S1). One of the most significant changes observed due to the loss of p62 was the profound upregulation of LDHA, an enzyme that catalyzes the conversion between pyruvate and lactate, in both the transcriptomic and the proteomic datasets (Figures 5A and 5B). We confirmed that this upregulation was a consequence of the loss of p62, as patient and KO cell lines showed the same response by western blotting (Figure S5A) and qRT-PCR (Figure S5B). Interestingly, LDHA levels did not change in control NESCs after 4 days differentiation, while they slightly decreased in the absence of p62, on both protein and transcript levels (Figures 5D and 5E). To our surprise, though, fibroblasts obtained from both patients who were the initial source for the generation of the Pat1 and Pat2 NESCs did not show this increase (Figure S5C), suggesting that this LDHA induction is a tissue-specific response. GSEA against four different pathway libraries further revealed that loss of p62 leads to increased expression of several gene groups involved in pyruvate metabolism and pyruvate dehydrogenase (PDH) regulation (Figures 5A–5C). For instance, the PDH complex was negatively regulated via its inhibitor PDH kinase 3 (PDK3), while the PDH phosphorylase (PDP) was inhibited via the PDP regulatory subunit (PDPR). In addition, gluconeogenesis and fatty acid biosynthesis were downregulated, as reflected by the transcript levels of phosphoenolpyruvate carboxykinase 2 (PCK2) and malic enzymes (ME) 1 and 3, respectively (Figure 5F). Amino acid metabolic pathways appeared upregulated in the absence of p62, probably as a consequence of a dysregulation in pyruvate metabolism (Figure 5C).
Figure 5.

Loss of p62 Alters Cellular Energy Metabolism
(A) High-throughput data analysis of CCo1NES, CCo2NES, KO1NES, and KO2NES cells before differentiation. Volcano plot of differentially transcribed genes, identified by RNA sequencing (n = 13,601 after filtering). FDR, false discovery rate.
(B) Volcano plot of differentially expressed proteins identified by proteomics (n = 3,058 after filtering).
(C) Gene set enrichment analysis of RNA sequencing data against proteomics analysis. Blue- and orange-colored areas indicate increasing confidence.
(D) Representative western blot analysis of LDHA and HK2 in Co1NEU and Pat1NEU after 0, 2, or 4 days of differentiation. Tubulin was used as a loading control. n = 3.
(E) qRT-PCR analysis of LDHA steady-state levels in Co1NEU (dark gray) and Pat1NEU (red) cells after 0 or 4 days of differentiation. β-actin was used as an endogenous control. n = 3.
(F) Expression levels of enzymes involved in pyruvate metabolism in undifferentiated KO1&2NES cells compared with undifferentiated CCo1&2NES cells. Left square of mini-heatmaps, transcriptome; right square, proteome; black dot, data not captured.
(G) Representative western blot analysis of Thr389 phosphorylation of P706SK in Co1NES and Pat1NES cells. Tubulin was used as a loading control. n = 3.
(H) Representative western blot analysis of Myc steady-state levels in CCo1NES, Pat1NES, and KO1NES cells. Tubulin was used as a loading control. n = 3.
LDHA is a key regulator of anaerobic metabolism and its expression is regulated by the Phosphatidylinositol 3-kinase/Akt/Mammalian target of rapamycin (PI3K/Akt/mTOR) pathway, among others (Valvona et al., 2016, Woo et al., 2015). Previous work suggests that p62 regulates mTORC1 pathway activation (Duran et al., 2011), leading to the stabilization of the v-Myc avian myelocytomatosis viral oncogene homolog (c-Myc), an important transcription factor involved in cell cycle progression, apoptosis, cellular transformation, cancer progression (Valencia et al., 2014), and neuronal differentiation (Iavarone and Lasorella, 2014). In agreement, loss of p62 resulted in reduced mTOR activation, as indicated by phosphorylation of Thr389 of P70S6K (Figure 5G), and reduced expression of the LAMTOR proteins LAMTOR1, LAMTOR3, and LAMTOR5 (Figures 5B and 5C). As expected, c-Myc protein levels were reduced in the absence of p62, suggesting reduced mTOR activation (Figure 5H). In stark contrast, transcripts of c-Myc targets, such as genes associated with glucose metabolism (LDHA), DNA replication (MCM2, MCM6), or proteasomal function (PSMA4, PSMA7), were upregulated (Figures 5A–5C), indicating differential gene regulation of c-Myc targets upon loss of p62. Interestingly, the pro-glycolytic modulator hexokinase 2 (HK2) was elevated in NESCs lacking p62, and only slightly reduced during 4 days of differentiation, while it was completely downregulated in control cells (Figure 5D and Table S1), further supporting the notion of a dysregulated energy metabolism.
Physiological Oxygen Concentrations Reveal a Pro-glycolytic Metabolism in the Absence of p62
As described, the increased levels of LDHA could not be explained by c-Myc activation. Exposing mice or cultured cells to oxygen concentrations lower than normoxia (20% oxygen concentration) triggers the activation of anaerobic glycolysis and diminishes mitochondrial aerobic metabolism (Jain et al., 2016). Moreover, transcription of LDHA and other glycolytic genes is activated by Hif-1α under hypoxic conditions (Cui et al., 2017). Results in cancer cells exposed to strict hypoxia (≤1% oxygen concentration) showed hypoxia-activated macroautophagic removal of p62 (Pursiheimo et al., 2009) and p62 regulation of PHD3 activity (Rantanen et al., 2013). Compared with controls, KO1&2NES presented higher levels of LDHA, HK2, and PDK3, a typical pro-glycolytic gene expression profile, already in normoxia, and we therefore hypothesized that p62-deficient NESCs assume a constant hypoxic state when cultured in normoxia. Exposure to physiological oxygen levels would then even further aggravate the abnormal gene expression profile. The exact in vivo oxygen pressure of a tissue is difficult to determine, and we decided to incubate NESCs at 3% oxygen concentration. As previously reported (López-Hernández et al., 2012), Hif-1α expression was low and almost undetectable in control cells in normoxia, but increased profoundly after several hours in hypoxia, before decreasing again after 24 h (Figures 6A, 6B, S6A, and S6B). Hif-1α levels also responded to low oxygen concentrations in NESCs lacking p62 and even remained elevated after 24 h, compared with control NESCs (Figures 6A, 6B, S6A, and S6B). As expected, other factors, such as HK2, PDK1, and PDK3, also had a mild induction under these conditions, while LDHA levels remained increased in patient NESCs (Figures 6C and S6C). We confirmed the induction of Hif-1α by treating NESCs with CoCl2, known to mimic hypoxia (López-Hernández et al., 2015), demonstrating that even in the absence of p62, NESCs responded to hypoxic conditions (Figures 6D and S6D), suggesting that the pronounced pro-glycolytic profile observed in these cells is independent of Hif-1α.
Figure 6.

Physiological Oxygen Concentrations Reveal a Pro-glycolytic Metabolism in the Absence of p62
(A) Representative western blot analysis of Hif-1α and p62 steady-state levels in Co1NES and Pat1NES cells, cultured for 0, 8, or 24 h at 3% O2. Tubulin was used as a loading control (n = 3).
(B) Quantification of (A). Data are expressed as mean ± SD and differences were tested by a two-tailed t test. ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3.
(C) Western blot analysis of HK2, PDK1, PDK3, and LDHA as in (A). Tubulin was used as a loading control. n = 3.
(D) Western blot analysis of Hif-1α expression in Co1NES and Pat1NES cells, cultured for 0, 5, or 24 h in the presence of 200 μM CoCl2. Tubulin was used as loading control. n = 5.
See also Figure S6.
Discussion
More than 130 million people are predicted to suffer from some form of dementia by 2050. With an increasing aging population, neurodegenerative disorders have already become a major burden, not only to the affected. Given the urgency of understanding these diseases, surprisingly few treatment options are available. The importance of particular metabolic networks is increasingly being recognized in disease progression, but studying their interplay has been more difficult. We decided to investigate the molecular consequences of a monogenic neurodegenerative disorder in a brain-appropriate model. The autophagy adaptor protein SQSTM1/p62 has been associated with a number of different functions, including as adaptor for mitochondrial clearance. This association stems from observations made in immortalized cell lines, exogenously overexpressing several components of the proposed process, such as PARKIN or LC3 (Park et al., 2014). We could not confirm this commonly assumed association between p62 and mitophagy in NESCs or neurons, which naturally express high levels of PARKIN.
In contrast, our data indicate that p62 is required for neuronal progenitor cells to change their metabolic microenvironment to a more oxidative metabolism, necessary for neurodifferentiation. Neuronal development is a metabolically dynamic process, during which the interconnection between aerobic glycolysis and OXPHOS is crucial for proper differentiation (Agostini et al., 2016). We observed that the loss of p62 resulted in a significant upregulation of the glycolytic regulators LDHA and HK2 and the inability to promote OXPHOS gene expression. Downregulation of LDHA and HK2 is essential during neuronal differentiation and is blocked by exogenous expression of LDHA (Zheng et al., 2016). Induction of differentiation did lead to the expected decrease in LDHA and HK2 levels in the absence of p62, but levels remained high, suggesting that the initial levels were too high and/or that p62 is involved in their turnover. The consequences, however, are that although the cell is initiating some of the required steps for differentiation, the metabolic environment prevents their execution. Support comes from a recent observation of increased lactate in the ventricular cerebrospinal fluid of a patient with disrupted p62 (Muto et al., 2018). Interestingly, the same study also disrupted p62 expression in a zebrafish model, and reported structural defects in the cerebellum, including reduced axonal connections and complete atrophy. These observations strongly support our observation that p62 is important for correct neuronal function. Several regulators of LDHA have been reported, including c-Myc and Hif-1α (Valvona et al., 2016), and numerous c-Myc targets were upregulated in the absence of p62, including LDHA, but c-Myc itself was downregulated, on both mRNA and protein levels, suggesting alternative transcriptional regulatory mechanisms of its targets. Hif-1α is recognized as the key regulator of systemic and cellular responses to low oxygen concentrations, and p62-KO cells grown under physiological oxygen levels revealed a pronounced Hif-1α stabilization, as well as increased expression of pro-glycolytic genes, such as LDHA, HK2, PDK1, or PDK3. However, in normoxia, where Hif-1α levels are similar, NESCs lacking p62 already present with increased LDHA levels, suggesting that p62 is required for Hif-1α-dependent downregulation during hypoxia but that another factor(s) must be involved in the upregulation of LDHA. Interestingly, fibroblasts from p62−/− patients did not have increased LDHA levels. Recently, two oxygen-sensing cysteine residues in p62 were reported to be important during a p62-dependent autophagic stress response (Carroll et al., 2018), thus suggesting that p62 itself might have oxygen-sensing capacity. Our results indicate that under reduced oxidative stress, either due to reduced oxygen pressure or due to supplementation of antioxidant, neurodifferentiation is improved. Thus, p62 is important during certain stages of development or under stress conditions, which could explain both the normal neuronal development until childhood and the progressive nature of clinical presentation of patients with disrupted p62 (Haack et al., 2016, Muto et al., 2018).
Although mature neurons are thought to primarily rely on OXPHOS for ATP synthesis (Tavernarakis and Palikaras, 2012), a more glycolytic energy metabolism in the brain has been proposed during childhood (Goyal et al., 2014). Such a shift in preference could explain the timing of disease onset and the progressive nature of the clinical presentations in the patients with mutations in p62 (Haack et al., 2016, Muto et al., 2018). The appearance of p62-positive aggregates in Purkinje cells in the cerebellum of old mice also points to an important role of p62 in the brain (Carroll et al., 2018). Increased demands on respiration could also increase oxidative stress, and NESCs lacking p62 displayed increased ROS formation during maximal respiration and differentiation, with several antioxidant pathways induced, including GPX1, GPX4, or ALDH1B1 and decreased levels of NQO1. Alterations in the cellular antioxidant response are in agreement with observations made in p62−/− mice, which also showed reduced NQO1 levels and altered GSH/GSSG ratio (Kwon et al., 2012). Interestingly, NQO1−/− mice showed an unbalanced redox status and accumulation of lactate and decreased glucose levels, suggesting a link between low NQO1 levels and glucose metabolism (Gaikwad et al., 2001). NRF2 is a key regulator of the antioxidant response and the KIR domain of p62 is thought to interact with the NRF2 regulator KEAP1. However, we found no dysfunctional KEAP1-NRF2-ARE pathway regulation upon deletion of p62 in NESCs, which is in agreement with results both in p62−/− mice (Kwon et al., 2012) and in pancreatic tumor cells (Valencia et al., 2014).
p62-KO mice are viable (Durán et al., 2004, Kwon et al., 2012), but die prematurely (Kwon et al., 2012) and present with several neurological phenotypes, ranging from memory loss to behavioral abnormalities to the accumulation of Tau tangles (Ramesh Babu et al., 2008). The patients reported by us also had memory loss and elevated Tau levels (Haack et al., 2016), demonstrating the similarities of the two systems. In addition, p62 was shown to be important for neuronal cell survival and development in rats (Joung et al., 2005). However, p62 ablation rescues the neural stem cell pool in the subventricular zone and dental gyrus of FIP 200-KO mice at p28, suggesting the importance of p62 in the regulation of neuronal development (Wang et al., 2016).
A carefully regulated metabolic environment is increasingly recognized as an important part of neuronal brain development (Agostini et al., 2016, Herrero-Mendez et al., 2009, Schönfeld and Reiser, 2017, Sonntag et al., 2017), and our data clearly show that p62 is required for the metabolic transition during neuronal differentiation.
In conclusion, our findings shed light on the pathogenetic mechanisms behind the childhood onset of ataxia, dystonia, and cognitive decline that result from loss of p62. Importantly, they also indicate that an active promotion of a metabolic shift from glycolysis to OXPHOS is required for neuronal survival beyond the early stages of brain development, and that an inability to promote this shift can lead to neurodegeneration.
Experimental Procedures
Detailed descriptions of experimental procedures can be found in the Supplemental Information.
Ethical Considerations
This study has been approved by the Regional Ethics Committee at Karolinska Institutet in Stockholm, Sweden. Informed consent was obtained from all individuals involved.
Cell Culture
Human primary skin fibroblasts were reprogrammed to iPSCs and subsequently induced to NESCs as described in detail in Supplemental Experimental Procedures.
CRiSPR/Cas9 Gene Editing
Generation of p62-KO cell lines was performed as described in detail in Supplemental Experimental Procedures.
Transcriptomic and Proteomic Analysis
Samples were prepared and analyzed as described in detail in Supplemental Experimental Procedures.
Contact for Details and Resource Sharing
Further information and requests for resources and reagents should be directed to Anna Wredenberg (anna.wredenberg@ki.se).
Author Contributions
Conceptualization, J.C.G., C.M., C.F., A. Wedell, and A. Wredenberg; Software, F.A.S. and H.S.; Methodology, E.U., M.K., N.L., H.B., and A.F.; Formal Analysis, J.C.G., C.M., and F.A.S.; Investigation, J.C.G., C.M., P.C., and F.A.S.; Resources, C.F., A. Wedell, and A. Wredenberg; Data Curation, F.A.S. and H.S.; Writing – Original Draft, J.C.G., C.M., C.F., and A. Wredenberg; Supervision and Funding Acquisition, C.F., A. Wedell, and A. Wredenberg.
Acknowledgments
This study was supported by the Swedish Research Council (A. Wredenberg [2016-02179], A. Wedell [VR521-2013-2341]), Stockholm County Council (A. Wredenberg [K0176-2012] and A. Wedell [20140053]), Swedish Foundation for Strategic Research (A. Wredenberg [ICA 12-0017]), Knut and Alice Wallenberg Foundation (A. Wredenberg and A. Wedell [KAW 2013.0026]), and Swedish Brain Foundation (A. Wedell [FO2015-0146]). A. Wredenberg is a Ragnar Söderberg Fellow in Medicine (M77/13). The authors acknowledge support from the National Genomics Infrastructure funded by Science for Life Laboratory, the Knut and Alice Wallenberg Foundation and the Swedish Research Council, and SNIC/Uppsala Multidisciplinary Centre for Advanced Computational Science for assistance with massively parallel sequencing and access to the UPPMAX computational infrastructure. iPSC derivation was performed at the National iPS Core Facility at Karolinska Institutet, iPS Core, www.ipscore.se. Confocal imaging was performed at the Live Cell Imaging Unit/Nikon Centre of Excellence, Karolinska Institutet, supported by grants from the Knut and Alice Wallenberg Foundation, the Swedish Research Council, the Centre for Innovative Medicine, and the Jonasson donation to the School of Technology and Health, Kungliga Tekniska Högskolan, Sweden. Proteomic analysis was performed at Proteomics Karolinska, http://ki.se/en/mbb/proteomics-karolinska-pkki. The authors would like to thank Dr. T. Pereira for support and help with the hypoxia experiments.
Published: February 28, 2019
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, two tables, and one data file and can be found with this article online at https://doi.org/10.1016/j.stemcr.2019.01.023.
Contributor Information
Christoph Freyer, Email: christoph.freyer@ki.se.
Anna Wredenberg, Email: anna.wredenberg@ki.se.
Supplemental Information
In the table, a list of the complete set of results from RNA sequencing and proteomics analysis is shown.
References
- Agostini M., Romeo F., Inoue S., Niklison-Chirou M.V., Elia A.J., Dinsdale D., Morone N., Knight R.A., Mak T.W., Melino G. Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 2016;23:1502–1514. doi: 10.1038/cdd.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll B., Otten E.G., Manni D., Stefanatos R., Menzies F.M., Smith G.R., Jurk D., Kenneth N., Wilkinson S., Passos J.F. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat. Commun. 2018;9:256. doi: 10.1038/s41467-017-02746-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copple I.M., Lister A., Obeng A.D., Kitteringham N.R., Jenkins R.E., Layfield R., Foster B.J., Goldring C.E., Park B.K. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 2010;285:16782–16788. doi: 10.1074/jbc.M109.096545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X.-G., Han Z.-T., He S.-H., Wu X.-D., Chen T.-R., Shao C.-H., Chen D.-L., Su N., Chen Y.-M., Wang T. HIF1/2α mediates hypoxia-induced LDHA expression in human pancreatic cancer cells. Oncotarget. 2017;8:24840–24852. doi: 10.18632/oncotarget.15266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A., Amanchy R., Linares J.F., Joshi J., Abu-Baker S. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durán A., Serrano M., Leitges M., Flores J.M., Picard S., Brown J.P., Moscat J., Diaz-Meco M.T. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev. Cell. 2004;6:303–309. doi: 10.1016/s1534-5807(03)00403-9. [DOI] [PubMed] [Google Scholar]
- El-Hattab A.W. Inborn errors of metabolism. Clin. Perinatol. 2015;42:413–439. doi: 10.1016/j.clp.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Falk A., Koch P., Kesavan J., Takashima Y., Ladewig J., Alexander M., Wiskow O., Tailor J., Trotter M., Pollard S. Capture of neuroepithelial-like stem cells from pluripotent stem cells provides a versatile system for in vitro production of human neurons. PLoS One. 2012;7:e29597. doi: 10.1371/journal.pone.0029597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W., Tang Z., Chen D., Moughon D., Ding X., Chen S., Zhu M., Zhong Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy. 2014;6:614–621. doi: 10.4161/auto.6.5.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaikwad A., Long D.J., Stringer J.L., Jaiswal A.K. In vivo role of NAD(P)H:quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J. Biol. Chem. 2001;276:22559–22564. doi: 10.1074/jbc.M101053200. [DOI] [PubMed] [Google Scholar]
- Geisler S., Holmström K.M., Skujat D., Fiesel F.C., Rothfuss O.C., Kahle P.J., Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Goyal M.S., Hawrylycz M., Miller J.A., Snyder A.Z., Raichle M.E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014;19:49–57. doi: 10.1016/j.cmet.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier K., McLelland G.-L., Fon E.A. Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front. Neurol. 2013;4:100. doi: 10.3389/fneur.2013.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack T.B., Ignatius E., Calvo-Garrido J., Iuso A., Isohanni P., Maffezzini C., Lönnqvist T., Suomalainen A., Gorza M., Kremer L.S. Absence of the autophagy adaptor SQSTM1/p62 causes childhood-onset neurodegeneration with ataxia, dystonia, and gaze palsy. Am. J. Hum. Genet. 2016;99:735–743. doi: 10.1016/j.ajhg.2016.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Mendez A., Almeida A., Fernández E., Maestre C., Moncada S., Bolaños J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009;11:747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- Iavarone A., Lasorella A. Myc and differentiation: going against the current. EMBO Rep. 2014;15:324–325. doi: 10.1002/embr.201438509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A., Lamark T., Sjottem E., Bowitz Larsen K., Atesoh Awuh J., Øvervatn A., McMahon M., Hayes J.D., Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain I.H., Zazzeron L., Goli R., Alexa K., Schatzman-Bone S., Dhillon H., Goldberger O., Peng J., Shalem O., Sanjana N.E. Hypoxia as a therapy for mitochondrial disease. Science. 2016;352:54–61. doi: 10.1126/science.aad9642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T., Harder B., Rojo de la Vega M., Wong P.K., Chapman E., Zhang D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015;88:199–204. doi: 10.1016/j.freeradbiomed.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung I., Kim H.J., Kwon Y.K. p62 modulates Akt activity via association with PKCzeta in neuronal survival and differentiation. Biochem. Biophys. Res. Commun. 2005;334:654–660. doi: 10.1016/j.bbrc.2005.06.138. [DOI] [PubMed] [Google Scholar]
- Khacho M., Clark A., Svoboda D.S., MacLaurin J.G., Lagace D.C., Park D.S., Slack R.S. Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neurogenesis. Hum. Mol. Genet. 2017;26:3327–3341. doi: 10.1093/hmg/ddx217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.-Y., Rhee I., Paik J. Metabolic circuits in neural stem cells. Cell. Mol. Life Sci. 2014;71:4221–4241. doi: 10.1007/s00018-014-1686-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch P., Opitz T., Steinbeck J.A., Ladewig J., Bruestle O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc. Natl. Acad. Sci. U S A. 2009;106:3225–3230. doi: 10.1073/pnas.0808387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y.-S., Ueno I., Sakamoto A., Tong K.I. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- Kwon J., Han E., Bui C.-B., Shin W., Lee J., Lee S., Choi Y.-B., Lee A.-H., Lee K.-H., Park C. Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 2012;13:150–156. doi: 10.1038/embor.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T., Kirkin V., Dikic I., Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8:1986–1990. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- Lau A., Wang X.J., Zhao F., Villeneuve N.F., Wu T., Jiang T., Sun Z., White E., Zhang D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010;30:3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Sasai M., Ma J.S., Sakaguchi N., Ohshima J., Bando H., Saitoh T., Akira S., Yamamoto M. p62 plays a specific role in interferon-γ-induced presentation of a toxoplasma vacuolar antigen. Cell Rep. 2015;13:223–233. doi: 10.1016/j.celrep.2015.09.005. [DOI] [PubMed] [Google Scholar]
- López-Hernández B., Ceña V., Posadas I. The endoplasmic reticulum stress and the HIF-1 signalling pathways are involved in the neuronal damage caused by chemical hypoxia. Br. J. Pharmacol. 2015;172:2838–2851. doi: 10.1111/bph.13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Hernández B., Posadas I., Podlesniy P., Abad M.A., Trullas R., Ceña V. HIF-1α is neuroprotective during the early phases of mild hypoxia in rat cortical neurons. Exp. Neurol. 2012;233:543–554. doi: 10.1016/j.expneurol.2011.11.040. [DOI] [PubMed] [Google Scholar]
- Muto V., Flex E., Kupchinsky Z., Primiano G., Galehdari H., Dehghani M., Cecchetti S., Carpentieri G., Rizza T., Mazaheri N. Biallelic SQSTM1 mutations in early-onset, variably progressive neurodegeneration. Neurology. 2018;91:e319–e330. doi: 10.1212/WNL.0000000000005869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D., Kane L.A., Hauser D.N., Fearnley I.M., Youle R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D., Walker J.E., Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb. Perspect. Biol. 2012;4:a011338. doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K., Saisho K., Shimanuki M., Nakada K., Shitara H., Sou Y.-S., Kimura M., Sato S., Hattori N., Komatsu M. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paridaen J.T.M.L., Huttner W.B. Neurogenesis during development of the vertebrate central nervous system. EMBO Rep. 2014;15:351–364. doi: 10.1002/embr.201438447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.S., Kang D.H., Bae S.H. p62 prevents carbonyl cyanide m-chlorophenyl hydrazine (CCCP)-induced apoptotic cell death by activating Nrf2. Biochem. Biophys. Res. Commun. 2015;464:1139–1144. doi: 10.1016/j.bbrc.2015.07.093. [DOI] [PubMed] [Google Scholar]
- Park S., Choi S.-G., Yoo S.-M., Son J.H., Jung Y.-K. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy. 2014;10:1906–1920. doi: 10.4161/auto.32177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursiheimo J.P., Rantanen K., Heikkinen P.T., Johansen T., Jaakkola P.M. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene. 2009;28:334–344. doi: 10.1038/onc.2008.392. [DOI] [PubMed] [Google Scholar]
- Ramesh Babu J., Lamar Seibenhener M., Peng J., Strom A.-L., Kemppainen R., Cox N., Zhu H., Wooten M.C., Diaz-Meco M.T., Moscat J., Wooten M.W. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 2008;106:107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- Rantanen K., Pursiheimo J.-P., Högel H., Miikkulainen P., Sundström J., Jaakkola P.M. p62/SQSTM1 regulates cellular oxygen sensing by attenuating PHD3 activity through aggregate sequestration and enhanced degradation. J. Cell Sci. 2013;126:1144–1154. doi: 10.1242/jcs.115667. [DOI] [PubMed] [Google Scholar]
- Schlafli A.M., Adams O., Galvan J.A., Gugger M., Savic S., Bubendorf L., Schmid R.A., Becker K.-F., Tschan M.P., Langer R., Berezowska S. Prognostic value of the autophagy markers LC3 and p62/SQSTM1 in early-stage non-small cell lung cancer. Oncotarget. 2016;7:39544–39555. doi: 10.18632/oncotarget.9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönfeld P., Reiser G. Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem. Int. 2017;109:68–77. doi: 10.1016/j.neuint.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Sholl D.A. Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Sonntag K.-C., Ryu W.-I., Amirault K.M., Healy R.A., Siegel A.J., McPhie D.L., Forester B., Cohen B.M. Late-onset Alzheimer's disease is associated with inherent changes in bioenergetics profiles. Sci. Rep. 2017;7:14038. doi: 10.1038/s41598-017-14420-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavernarakis N., Palikaras K. Mitophagy in neurodegeneration and aging. Front. Cell Neurosci. 2012:1–7. doi: 10.3389/fgene.2012.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia T., Kim J.Y., Abu-Baker S., Moscat-Pardos J. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell. 2014;26:121–135. doi: 10.1016/j.ccr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvona C.J., Fillmore H.L., Nunn P.B., Pilkington G.J. The regulation and function of lactate dehydrogenase A: therapeutic potential in brain tumor. Brain Pathol. 2016;26:3–17. doi: 10.1111/bpa.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira H.L.A., Alves P.M., Vercelli A. Modulation of neuronal stem cell differentiation by hypoxia and reactive oxygen species. Prog. Neurobiol. 2011;93:444–455. doi: 10.1016/j.pneurobio.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Wang C., Chen S., Yeo S., Karsli-Uzunbas G., White E., Mizushima N., Virgin H.W., Guan J.-L. Elevated p62/SQSTM1 determines the fate of autophagy-deficient neural stem cells by increasing superoxide. J. Cell Biol. 2016;212:545–560. doi: 10.1083/jcb.201507023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Duncan D., Shi Z., Zhang B. WEB-based GEne SeT analysis toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo Y.M., Shin Y., Lee E.J., Lee S., Jeong S.H., Kong H.K., Park E.Y., Kim H.K., Han J., Chang M., Park J.-H. Inhibition of aerobic glycolysis represses Akt/mTOR/HIF-1α axis and restores tamoxifen sensitivity in antiestrogen-resistant breast cancer cells. PLoS One. 2015;10:e0132285. doi: 10.1371/journal.pone.0132285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng R.-X., Zhang Y.-B., Fan Y., Wu G.-L. p62/SQSTM1 is involved in caspase-8 associated cell death induced by proteasome inhibitor MG132 in U87MG cells. Cell Biol. Int. 2014;38:1221–1226. doi: 10.1002/cbin.10311. [DOI] [PubMed] [Google Scholar]
- Zheng X., Boyer L., Jin M., Mertens J., Kim Y., Ma L., Hamm M., Gage F.H., Hunter T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife. 2016;5:859. doi: 10.7554/eLife.13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In the table, a list of the complete set of results from RNA sequencing and proteomics analysis is shown.