

Abstract
Background
The function of proteins is a direct consequence of their three-dimensional structure. The structural classification of proteins describes the ways of folding patterns all proteins could adopt. Although, the protein folds were described in many ways the functional properties of individual folds were not studied.
Results
We have analyzed two β-barrel folds generally adopted by small proteins to be looking similar but have different topology. On the basis of the topology they could be divided into two different folds named SH3-fold and OB-fold. There was no sequence homology between any of the proteins considered. The sequence diversity and loop variability was found to be important for various binding functions.
Conclusions
The function of Oligonucleotide/oligosaccharide-binding (OB) fold proteins was restricted to either DNA/RNA binding or sugar binding whereas the Src homology 3 (SH3) domain like proteins bind to a variety of ligands through loop modulations. A question was raised whether the evolution of these two folds was through DNA shuffling.
Background
The analysis of protein structures as a group in generating and retrieving information is useful in various ways. The structural bioinformatics analysis of protein data bank (PDB) [1] is useful in identifying protein folds [2,3] and identification of unknown protein functions. The analysis of some of the folds illustrated the packing arrangement of the secondary structural elements and features of various non-bonding interactions prevailed in these folds. This in turn helps in identifying active site residues of proteins of unknown functions. For example, the TIM-barrel fold, which is the most frequently observed fold has majority of members as enzymes and the active-site residues are situated on the loops connecting the β-strands to helices or at the C-terminal end of the parallel β-strands of the barrel [4]. Therefore, for any enzyme having a Tim-barrel fold there is a possibility that the active site may be present at the same position consensus with other Tim-barrel fold enzymes.
In depth analysis of a particular fold towards understanding the functional variability with respect to the changes in the fold was not studied much, although there were many reports of fold classifications [2,3]. OB-fold [5] and SH3 domain like folds (which is called as SH3-fold hereafter) were referred interchangeably because they look alike at the first sight. We have attempted to analyze the functional properties of the proteins, which fall into either OB (Fig. 1) or SH3 (Fig. 2) folds. Although there was no overall sequence homology between the proteins of these two classes, we found that the β-strands have certain sequence homology at the residues which are forming the core of the barrel. We also found that OB-fold and SH3-fold slightly differ in their topology due to a possible shuffling of a secondary structural element.
Figure 1.
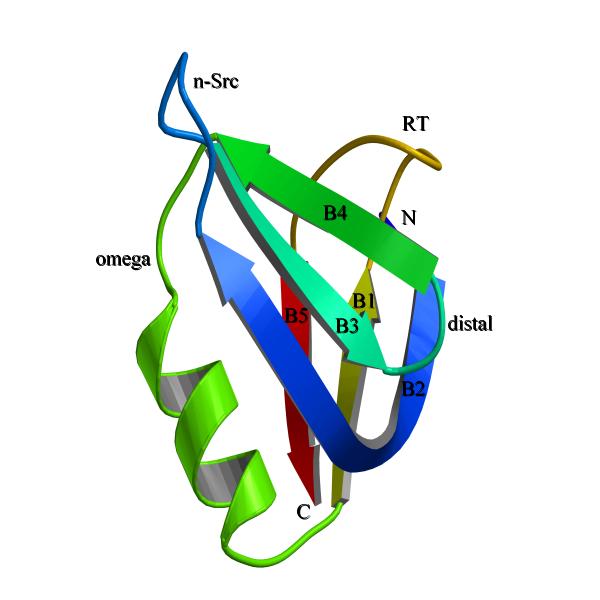
The general OB-fold topology. The β-strands were labeled as B1 to B5. The loops were labeled as RT, n-src, distal and omega. The N- and C-termini were labeled. Note the N-terminal starts with strand B2 and strand B1 was between omega helix and RT-loop. RT-loop was connecting B1 with B5 (see the difference with SH3-fold in Fig. 2). Note the other striking difference between the two folds, the omega helix which was present on majority of OB-fold proteins. This figure was made with bobscript [31] and rendered with povray (http://www.povray.org/).
Figure 2.
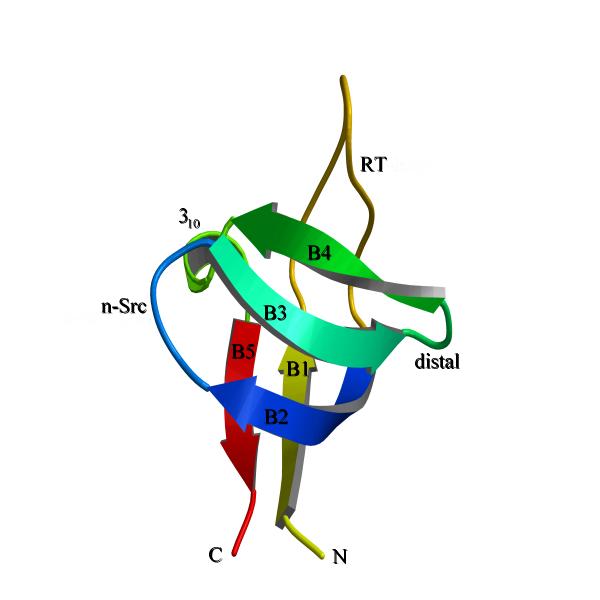
The topology of SH3-fold. The sequential numbering of strands from B1 to B5 could be seen with the connecting loops as RT, n-src, distal. The last loop which was equivalent to omega loop was a short 310-helix in majority of SH3-fold proteins. Although the connectivity of RT-loop was different in OB-fold and SH3-fold, the physical position of RT-loop was approximately equivalent. This figure was made as figure 1.
Results and discussion
Search for SH3-fold and SH3 like folded proteins over various fold classification servers and manual literature search yielded a large number of protein domains. Some of the domains exist as individual proteins and some were part of a multi-domain protein. After superposing the protein domains on each other and through analysis for a common fold architecture we identified two folds, which are common in architecture but differ in topology. Here architecture is defined as immediate apparent similarity in fold irrespective of connectivity and topology is defined as the actual way the secondary structural elements are connected and come together to form a fold. One of the folds is known as OB-fold [5] and the other is SH3-fold. There are at least 30 proteins/domains classified as adopting these two folds [6-10] and the list is increasing. Although, there are more proteins/domains, which could be classified into one of the two folds, they were not included due to too many deviations from a consensus ensemble of structures.
To our surprise we observed that, while OB-fold always binds to either oligonucleotides or oligosaccharides, SH3-fold binds to a wide spectrum of ligands like DNA/RNA (Ribosomal protein L2 [11], Sso7d [12] and HIV Integrase DNA binding domain [13]), peptides (SH3 domains [14]) and folate (dihydrofolate reductase [15]). Although, few enzymes have SH3-folded domains as part of the enzyme, they stabilize the catalytic domain for optimal function (nitrile hydratase [16]) or stabilize the incoming ligand (ferridoxin:thioredoxin reductase [17]).
Both OB and SH3-folds form β-barrels constituted of five β-strands connected by RT, n-src, distal and omega (or a 310-helix in majority of SH3-fold proteins; the loop nomenclature was according to SH3-fold, except omega region which was adopted from OB-fold) loops (Fig. 1,2). When superposed by β-strands alone, the folds align very well with an average root mean square deviation (rmsd) less than 2.0 Å for the β-strands. Although, the strands align well the loops show high positional variability, which was evidenced by having high rmsd (Fig. 3). Nakagawa et al. were the first to identify that OB-fold and SH3-folds were different [11]. But they did not describe the differences in detail. We observed that two of the loops (RT and omega) connect differently amongst the β-strands in the two folds. In SH3-fold RT-loop connects strand B1 to B2 and the 310-helix connects strand B4 to B5 (Fig. 1). In OB-fold, RT-loop connects B1 to B5 and the omega loop connects B4 to B1 (Fig. 2). However, the physical position of RT-loop was retained approximately. The change in the omega loop connectivity results in loop elongation as a α-helix in many OB-folded structures. This feature is the striking difference between SH3-fold and OB-folds.
Figure 3.

Superposition of various OB (left) and SH3-fold (right) structures showing the structural conservation of strands and variations in the loops. The loops are labeled as in Figures 1 and 2. The orientation of the molecules is same as in Figure 1 and 2 for SH3-fold and OB-folds.
Interestingly, none of the proteins in both the folds have any sequence homology with other members. However, when the β-strands alone are considered, they show some homology. This is because the core of the proteins is formed by the interior surface of the β-sheets, which constitutes the β-barrel and the amino acids projecting into the core of the barrel must be hydrophobic (Fig. 4). This is analogous to the earlier observed β-barrel folds [3,18]. We are surprised to note that the ligand-binding region of the proteins under consideration (in both SH3 and OB-fold) is the same: between RT-loop and n-src loop and the sheet (formed by strands B2, B3 and B4) having a RNP motif [19] of the general DNA/RNA binding proteins. For example, in Sac7d which is highly homologous to Sso7d, residues Tyr 8, Lys 9 of RT-loop and Lys 28, Met 29 of n-src loop besides residues Lys 21, Lys 22, Trp 24, Ser 31, Thr 33, Arg 43 of strands B2, B3, B4 are binding the double helix [20]. It is well known that polyproline peptides bind to SH3 domains between the RT-loop and n-src loops [14]. In the C-terminal domain of Nitrile hydratase, which is SH3-folded, Arg 141 of RT-loop is essential for maintaining proper conformation of Cys 113, so that Cys S-gamma bind to Iron or Cobalt ions. The n-src loop residues Trp 161, Pro 162, Pro 164, Ile 167 play an important role in ligand-binding [16]. In the case of OB-fold sometimes the protruding omega loop also participates in ligand binding (For example Aspartyl-tRNA synthase [21] and Heat labile enterotoxin [10]). It could be, therefore inferred that the β-barrel fold creates a base for modulating loops both in length and sequence for a variety of functional binding properties. The variations in loops are necessary for binding to various ligands. It was suggested by Lodi et al[22] that SH3-fold was suitable to graft many different binding properties.
Figure 4.

Structure based sequence alignment of SH3-fold and OB-fold proteins. The alignment was generated by COMPARER [30] server (http://www-cryst.bioc.cam.ac.uk/~robert/cpgs/COMPARER/comparer.html). The proteins were shown with their PDB code. Single letter code for amino acids was used. The proteins aligned for SH3-fold were 1shg: SH3 domain of chicken brain spectrin; 1ihv: the DNA-binding domain of HIV-intergrase; 1d0z: myosin S1 motor domain fragment; 1bia: BirA-biotin operon repressor protein; 1vie: dihydrofolate reductase; 1dj7: ferridoxin-thioredoxin reductase; 1psf: photosystem I protein PsaE; 1whi: ribosomal protein L14. The proteins aligned for OB-fold were 1csp: cold shock protein; 1bov: verotoxin-1; 1ltt: enterotoxin; 1cuk: ruvA protein; 1fjf: ribosomal protein S17; 1asy: aspartyl tRNA-synthetase; 1a0i: T7 DNA-ligase; 1ey0: staphylococcal nuclease. The β-strand regions were marked as 'bbbbb' and the 310-turn as '333'. The 310-turn was conserved in many of the SH3-fold proteins. Highly conserved hydrophobic regions in the strands were marked with * or # depending on the extent of conservation, * being all hydrophobic and # being majority hydrophobic. It is significant that out of 22 residues forming the strands, there were 15 residues have homology.
The architecture of β-strands is similar in both SH3 and OB-folds. However, the subtle differences in both folds are due to changes in connectivity of β-strands. If one looks at the two folds as a one-dimensional chain, it is obvious that the first strand of SH3-fold got inserted between the fourth and fifth strands in OB-fold (Fig. 5). Since the ending of B4 and the beginning of B1 in OB-fold are situated far away in space (Fig. 1), the linker loop (omega loop) should be long and on many occasions it was extended into a α-helix. Because of this extra secondary structure the OB-fold gains further stability. Sometimes the extended omega loop acts as a binding loop giving support to RT- and n-src loops. For example, in Staphylococcal Nuclease, the only enzyme with an OB-fold, the omega loop is extended and the catalytic site is present in between the omega and RT-loops [23]. Amino acids Asp 19, Asp 21 (n-src loop), Arg 35 (B4), Asp 40, Glu 43 (omega loop) and Asp 83, Lys 84, Tyr 85, Arg 87 (RT-loop) participate in catalysis and binding to DNA and a Calcium ion. In summary, this omega loop makes the OB-fold like a molecular clamp to hold a ligand between the four loops (RT, n-src, distal and omega) while the β-sheet formed by strands B2, B3 and B4 make the basic template for a oligonucleotide binding.
Figure 5.

The schematic representation of chain layout of SH3 and OB-folds. The β-strands and loops are marked. Note the insertion of strand B1 between B4 and B5 in OB-fold and consequent changes in the loop positions.
From figure 5 it is clear that the major difference between the two folds is the insertion/deletion of a β-strand, apart from the omega helix in OB-fold. Since the ligand-binding region in both folds is also similar, one could wonder whether these two folds were evolved from a common ancestor. If so, is it a function-driven protein evolution as argued by Fetrow and Godzik [24]? There are both negative as well as positive indicators to support this possibility. The fact that all the proteins considered in this study were not grouped into the same superfamily in the SCOP database [3] indicates that these two folds are not homologous or remotely homologous. The very low sequence homology and classification into different folds in SCOP suggests that they may not be analogous also. However, a simple concept of DNA shuffling, first worked out by Stemmer [25] and later demonstrated by many others, showed that new proteins and folds could be evolved through random fragmentation and reassembly [25-27]. On similar lines, SH3-fold and OB-fold could possibly be evolved from a common ancestor or evolved one from the other, through shuffling of small DNA segments over a large time-scale. Although there is no direct evidence to prove that these two folds are evolved from each other, directed-evolution experiments as demonstrated by Stemmer [25] may be useful to prove or disprove this hypothesis.
Conclusions
The common fold characteristics of both OB-fold and SH3-fold have diversified loops in sequence as well as in length. This feature prompts us to assume that these two folds could be used as a basic fold in designing new proteins with tailored functions. The designing of a chimeric protein with the basic fold of five strands from one protein and loops from another protein with appropriate mutations could be a starting point to test this hypothesis.
Materials and methods
The β-barrel proteins used for the analysis under SH3-fold were SH3 domain of chicken brain spectrin (1SHG), CcdB a topoisomearse poison from E. coli (4VUB), dihydrofolate reductase (1VIE), diphtheria toxin (1BYM), N-terminal domain of eucaryotic translation initiation factor 5a (1EIF), ferridoxin thioredoxin reductase (1DJ7), DNA-binding domain of HIV-1 integrase (1IHV), nitrile hydratase (1AHJ), PsaE from photosystem I protein (1PSF), ribosomal protein L14 (1WHI), C-terminal domain of ribosomal protein L2 (1RL2), Snrnp (1B34), Sso7d (1BF4), tudor domain (1G5V), myosin S1 motor domain (1D0Z) and BirA (1BIA). Under OB-fold the proteins analyzed were cold shock protein (1CSP), aspartyl t-RNA-synthetase (1ASY), heat labile enterotoxin (1LTT), mitochondrial single-stranded DNA-binding protein (3ULL), Rho protein (1A62), replication protein A (1JMC), RuvA (1CUK), ribosomal protein S12, S17 (1FJF), N-terminal domain of ribosomal protein L2 (1RL2), S1 RNA-binding domain (1SRO), staphylococcal nuclease (1EY0), T7 DNA ligase (1A0I), verotoxin-1 (1BOV), C-terminal domain of eukaryotic translation initiation factor 5a (1EIF). The protein data bank code was given in the parenthesis following the name of the protein used in the analysis. For super positioning of proteins programs from CCP4 package [28] were used. For graphical visualization and analysis 'O' program [29] was used. Comparer server [30] was used for structure based sequence alignment.
Acknowledgments
Acknowledgements
V.A. acknowledges a Senior Research Fellowship from Council of Scientific and Industrial Research (CSIR), India.
Contributor Information
Vishal Agrawal, Email: vishal@imtech.res.in.
Radha KV Kishan, Email: kishan@imtech.res.in.
References
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nuc Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH- a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- Conte LL, Ailey B, Hubbard TJP, Brenner SE, Murzin AG, Chothia C. SCOP: a Structural Classification of Proteins database. Nuc Acids Res. 2000;28:257–259. doi: 10.1093/nar/28.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga RK. The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett. 2001;492:193–198. doi: 10.1016/S0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- Murzin AG. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 1993;12:861–867. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkarev A, Pfuetzner RA, Edwards AM, Frappier L. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. 1997;385:176–181. doi: 10.1038/385176a0. [DOI] [PubMed] [Google Scholar]
- Schindelin H, Marahiel MA, Heinemann U. Universal nucleic acid-binding revealed by crystal structure of the B. subtilis major cold-shock protein. Nature. 1993;364:164–168. doi: 10.1038/364164a0. [DOI] [PubMed] [Google Scholar]
- Bycroft M, Hubbard TJP, Proctor M, Freund SMV, Murzin AG. The solution structure of the S1 RNA binding domain: A member of an ancient nucleic acid-binding fold. Cell. 1997;88:235–242. doi: 10.1016/s0092-8674(00)81844-9. [DOI] [PubMed] [Google Scholar]
- Rafferty JB, Sedelnikova SE, Hargreaves D, Artymiuk PJ, Baker PJ, Sharples GJ, Mahdi AA, Lloyd RG, Rice DW. Crystal structure of DNA recombination protein RuvA and a model for its binding to the Holliday junction. Science. 1996;274:415–421. doi: 10.1126/science.274.5286.415. [DOI] [PubMed] [Google Scholar]
- Sixma TK, Pronk SE, Kalk KH, van Zanten BAM, Berghuis AM, Hol WGJ. Lactose binding to heat-labile enterotoxin revealed by X-ray crystallography. Nature. 1992;355:561–564. doi: 10.1038/355561a0. [DOI] [PubMed] [Google Scholar]
- Nakagawa A, Nakashima T, Taniguchi M, Hosaka H, kimura M, Tanaka I. The three-dimensional structure of the RNA-binding domain of ribosomal protein L2; a protein at the peptidyl transferase center of the ribosome. EMBO J. 1999;18:1459–1467. doi: 10.1093/emboj/18.6.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann H, Knapp S, Lundback T, Ladenstein R, Hard T. Solution structure and DNA-binding properties of a thermostable protein from the archaeon Sulfolobus solfataricus. Nature Struct Biol. 1994;1:808–819. doi: 10.1038/nsb1194-808. [DOI] [PubMed] [Google Scholar]
- Eijkelenboom APAM, Lutzke RAP, Boelens R, Plasterk RHA, Kaptein R, Hard K. The DNA-binding domain of HIV-1 integrase has an SH3-like fold. Nature Struct Biol. 1995;2:807–810. doi: 10.1038/nsb0995-807. [DOI] [PubMed] [Google Scholar]
- Lim WA, Richards FM, Fox RO. Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature. 1994;372:375–379. doi: 10.1038/372375a0. [DOI] [PubMed] [Google Scholar]
- Narayana N, Matthews DA, Howell EE, Xuong N-h. A plasmid-encoded dihydrofolate reductase from trimethoprim-resistant bacteria has a novel D2-symmetric active site. Nature Struct Biol. 1995;2:1018–1025. doi: 10.1038/nsb1195-1018. [DOI] [PubMed] [Google Scholar]
- Huang W, Jia J, Cummings J, Nelson M, Schneider G, Lindqvist Y. Crystal structure of nitrile hydratase reveals a novel iron center in a novel fold. Structure. 1997;5:691–699. doi: 10.1016/s0969-2126(97)00223-2. [DOI] [PubMed] [Google Scholar]
- Dai S, Schwendtmayer C, Schurmann P, Ramaswamy S, Eklund H. Redox signaling in chloroplasts: cleavage of disulfides by an iron-sulfur cluster. Science. 2000;287:655–658. doi: 10.1126/science.287.5453.655. [DOI] [PubMed] [Google Scholar]
- Murzin AG, Lesk AM, Chothia C. Beta-trefoil fold. Patterns of structure and sequence in the Kunitz inhibitors interleukins-1 beta and 1 alpha and fibroblast growth factors. J Mol Biol. 1992;223:531–543. doi: 10.1016/0022-2836(92)90668-a. [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Swanson MS, Pinol-Roma S. Heterogeneous nuclear ribonucleoprotein particles and the pathway of mRNA formation. Trends Biochem. 1988;13:86–91. doi: 10.1016/0968-0004(88)90046-1. [DOI] [PubMed] [Google Scholar]
- Robinson H, Gao Y-G, McCray BS, Edmondson SP, Shriver JW, Wang AHJ. The hyperthermophile chromosomal protein Sac7d sharply kinks DNA. Nature. 1998;392:202–205. doi: 10.1038/32455. [DOI] [PubMed] [Google Scholar]
- Cavarelli J, Rees B, Ruff M, Thierry J-C, Moras D. Yeast tRNA(Asp) recognition by its cognate class II aminoacyl-tRNA synthetase. Nature. 1993;362:181–184. doi: 10.1038/362181a0. [DOI] [PubMed] [Google Scholar]
- Lodi PJ, Ernst JA, Kuszewski J, Hickman AB, Engelman A, Craigie R, Clore GM, Gronenborn AM. Solution structure of the DNA binding domain of HIV-1 integrase. Biochemistry. 1995;34:9826–9833. doi: 10.1021/bi00031a002. [DOI] [PubMed] [Google Scholar]
- Judice JK, Gamble TR, Murphy EC, de Vos AM, Schultz PG. Probing the mechanism of Staphylococcal Nuclease with unusual amino acids: Science. 1993;261:1578–1581. doi: 10.1126/science.8103944. [DOI] [PubMed] [Google Scholar]
- Fetrow JS, Godzik A. Function driven protein evolution: A possible proto-protein for the RNA-binding proteins. Pac Symp Biocomput. 1998:485–496. [PubMed] [Google Scholar]
- Stemmer WPC. DNA shuffling by random fragmentation and reassembly: In vitro recombination for molecular evolution. Proc Natl Acad Sci USA. 1994;91:10747–10751. doi: 10.1073/pnas.91.22.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogarad LD, Deem MW. A hierarchical approach to protein molecular evolution. Proc Natl Acad Sci USA. 1999;96:2591–2595. doi: 10.1073/pnas.96.6.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann L, Winter G. Novel folded protein domains generated by combinatorial shuffling of polypeptide segments. Proc Natl Acad Sci USA. 2000;97:10068–10073. doi: 10.1073/pnas.170145497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The CCP4 suite: Programs for protein crystallography Number 4 Collaborative Computational Proteject. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Burke DF, Deane CM, Nagarajaram HA, Campillo N, Martin-Martinez M, Mendes J, Molina F, Perry J, Reddy BV, Soares CM, Steward RE, Williams M, Carrondo MA, Blundell TL, Mizuguchi K. An iterative structure-assisted approach to sequence alignment and comparative modeling. Proteins Suppl, 1999;3:55–60. doi: 10.1002/(SICI)1097-0134(1999)37:3+<55::AID-PROT8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graph. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]