

Abstract
The Chinese forest musk deer (Moschus berezovskii; FMD) is an artiodactyl mammal and is both economically valuable and highly endangered. To investigate the genetic mechanisms of musk secretion and adaptive immunity in FMD, we compared its genome to nine other artiodactyl genomes. Comparative genomics demonstrated that eight positively selected genes (PSGs) in FMD were annotated in three KEGG pathways that were related to metabolic and synthetic activity of musk, similar to previous transcriptome studies. Functional enrichment analysis indicated that many PSGs were involved in the regulation of immune system processes, implying important reorganization of the immune system in FMD. FMD-specific missense mutations were found in two PSGs (MHC class II antigen DRA and ADA) that were classified as deleterious by PolyPhen-2, possibly contributing to immune adaptation to infectious diseases. Functional assessment showed that the FMD-specific mutation enhanced the ADA activity, which was likely to strengthen the immune defense against pathogenic invasion. Single nucleotide polymorphism-based inference showed the recent demographic trajectory for FMD. Our data and findings provide valuable genomic resources not only for studying the genetic mechanisms of musk secretion and adaptive immunity, but also for facilitating more effective management of the captive breeding programs for this endangered species.
Keywords: Moschus berezovskii, musk secretion, adaptive immunity, positive selection, ADA gene, missense mutation
Introduction
The Chinese forest musk deer (Moschus berezovskii; FMD) is primarily found in Southern Asia and is well known for the secretion of musk (Xu et al. 2016). The musk is secreted from the musk gland of sexually mature males. Because of the distinctive fragrance and the significant role in antitumor, anti-inflammation, cardio-cerebral-vascular system, and central nervous system (Cao and Zhou 2007; Feng and Liu 2015), the musk has been a vital component of Chinese traditional medicine and perfume manufacturing (Sheng 1996; Su et al. 2001). Previous studies have explored the mechanisms of musk secretion based on anatomy (Bi et al. 1984; Bi and Shen 1986), mtDNA markers (Chen 2007; Zhao 2009), microsatellite (Guan et al. 2009), microbiota analysis (Li et al. 2016), and transcriptome (Xu et al. 2016), which illustrated various aspects of the secretory mechanisms of musk. However, the genetic mechanisms of musk secretion are still poorly understood.
As the global demand for the musk in the medicine and perfume industries has increased, the population of FMD has declined dramatically in the past five decades, and anthropogenic overexploitation and habitat destruction had placed FMD on the edge of extinction (Yang et al. 2003; Sheng and Liu 2007; He et al. 2014). In order to prevent the FMD from extinction, the Chinese government encouraged enterprises since the early 1950s to take part in captive breeding programs (Sun et al. 2018). However, the population growth of FMD had been limited, in part due to the high susceptibility to infectious diseases, particularly gastrointestinal diseases, respiratory illnesses, abscesses, and parasites (Xu et al. 2014). Previous studies investigating this issue found that the genetic diversity of the major histocompatibility complex (MHC) class II proteins were associated with the resistance or susceptibility of FMD to infectious diseases (Cai et al. 2015; Yao et al. 2015). One recent study based on the blood transcriptomes of purulent and healthy individuals detected that large numbers of the differentially expressed genes were related to the regulation of immune system processes, particularly in parasitic and bacterial infection pathways (Sun et al. 2018). The researchers suggested that abscesses in FMD were likely due to genetic deficiency or pathogenic invasion (Sun et al. 2018). However, the genetic diversity of the immune-related genes at the complete genome level of FMD has largely remained unexplored, thus it could be very useful to understand the mechanism of infectious disease formation in FMD.
Our laboratory sequenced and annotated the first complete genome sequence of one male FMD (Fan et al. 2018), which provided valuable resources to further investigate its genetic mechanisms of musk secretion and immunity. In this study, we compared the FMD genome with nine other artiodactyl species, aiming to address a number of key questions, namely 1) the phylogenetic relationship of artiodactyl species at the genomic level, 2) the genetic mechanisms of the musk secretion of FMD, and 3) the immune adaptation to common diseases of FMD.
Materials and Methods
Genome Data Collection
The full genome sequence of one male FMD was sequenced and annotated by our laboratory (Fan et al. 2018). In order to perform comparative genomic analysis, we collected the genomes of nine other Artiodactyla species from NCBI, including American bison (Bison bison, Bison UMD1.0), wild yak (Bos mutus, BosGru v2.0), water buffalo (Bubalus bubalis, UMD CASPUR WB 2.0), goat (Capra hircus, ARS1), white-tailed deer (Odocoileus virginianus, Ovir.te 1.0), sheep (Ovis aries, Oar v4.0), Tibetan antelope (Pantholops hodgsonii, PHO1.0), wild boar (Sus scrofa, Sscrofa11.1), and wild Bactrian camel (Camelus ferus, CB1).
Gene Family, Phylogeny, and Divergence
We used orthoMCL (Li et al. 2003) to define orthologous genes from the ten Artiodactyla genomes (supplementary table S1, Supplementary Material online). The phylogenetic tree of these ten Artiodactyla species was constructed using the nucleotide sequences of 1:1 orthologous genes. Coding sequences from each 1:1 orthologous family were aligned with PRANK (Löytynoja and Goldman 2010), and were then concatenated to one sequence for each species to build the tree. Modeltest (ver. 3.7) was used to select the best substitution model for the whole concatenated sequence (Posada and Crandall 2005). RAxML (Stamatakis 2014) was applied to reconstruct maximum likelihood (ML) phylogenetic tree with 1,000 bootstrap replicates. Divergence time estimation was performed by the program Mcmctree within PAML (Yang 2007) with several calibration times (5–7 Ma for Cap. hircus/Ov. aries; 2.85–3.89 Ma for Bo. mutus/Bi. bison; 6.85–7.50 Ma for Bo. mutus/Bu. bubalis; 11.5–19.7 Ma for the emergence of Bovinae tribes) (Bai 2015; Tan et al. 2017). Analyses of gene family expansion and contraction were performed by CAFÉ (De Bie et al. 2006). GO enrichment analyses were performed according to previous studies (Huang et al. 2009; Chen et al. 2010).
Positive Selection Analysis
The above alignments of 1:1 orthologous genes and phylogenetic tree were used to estimate the ratio of the rates of nonsynonymous to synonymous substitutions per gene (ω) by ML with the codeml program within PAML under the branch-site model (Yang 2007). We then performed a likelihood ratio test and identified positively selected genes (PSGs) FMD by means of FDR adjustment with Q-values <0.05. We conducted enrichment of the PSGs using KOBAS (Wu et al. 2006; Xie et al. 2011).
FMD-Specific Missense Mutations
We examined the PSGs detected in FMD to identify FMD-specific missense mutations. The FMD-specific missense mutations were validated using three different approaches. Firstly, we downloaded all available protein sequences for each gene from mammals (MHC Class II Antigen DRA, supplementary table S2, Supplementary Material online; MHC Class II Antigen DRB, supplementary table S3, Supplementary Material online; ADA, supplementary table S4; Supplementary Material online), and some other vertebrates from Genbank. Together, these downloaded protein sequences and our study’s identified protein sequences genomes were aligned using MEGA7 (Kumar et al. 2016) for each gene to validate the FMD-specific missense mutations. Secondly, we checked the mutations based on the transcriptomes. The musk gland and heart tissue RNA-seq data (SRA accession: SRR2098995, SRR2098996, and SRR2142357) were downloaded, and then the short reads were de novo assembled into contigs using Trinity (Grabherr et al. 2011). On the basis of trinity results, we identified genes (MHC class II antigen DRA, MHC class II antigen DRB, and ADA) using TBlastN. Lastly, we performed PCR amplification and sequencing with eight FMD muscle samples to verify FMD-specific missense mutation sites in gene MHC class II antigen DRA. None of the FMD muscle samples was used for the genome sequencing, as they were collected from individuals from Miyaluo Farm (Sichuan Province, China). Primers (supplementary table S5, Supplementary Material online) were designed by MHC class II antigen DRA nucleotide sequences of FMD with Primer Premier 5 (Lalitha 2000). The amplification of genes was undertaken using TaKaRa RTaq (TaKaRa Biomedical, Japan) and implemented on a PTC-100 thermal cycler (BioRad, Hercules, CA) in the reaction mixture. The PCR products were sequenced on an ABI PRISM 3730 DNA sequencer at Tsingke Biotechnology Company (Chengdu, Sichuan Province, China) after electrophoresing in 2% agarose gel.
Protein Structure Determination
The crystal structure of the MHC class II antigen DRA and ADA was obtained from SWISS-MODEL (Schwede et al. 2003). We converted the PDB files to PQR format with PDB2PQR server (Unni et al. 2011). The PDB files were further used for visualization of cartoon and surface representations of gene mutants. The visualization of the electrostatic surface potential was conducted using the APBS plugin in PyMOL (DeLano 2002).
Construction of Recombinant pET28a(+)/ADA Expression Vector
The ADA gene synthesis and the pET-28a(+)/ADA construction was performed by Tsingke Biotechnology Company (Chengdu, China). The ADA gene and the vector pET28a (+) plasmid were subjected to digestion with Nco I and EcoR I, and ligated using T4 DNA ligase. The recombinant pET28a(+)/ADA was transformed into Escherichiacoli DH (5α) competent cells for amplification. Positive colonies resistant to ampicillin on a Luria–Bertani (LB) plate were selected and the plasmid pET28a(+)/ADA was confirmed by restriction enzyme mapping and DNA sequencing.
Site-Directed Mutagenesis of ADA Gene
Mutations were generated via PCR using Taq polymerase with mutagenic primers (ADA-Ser-F [forward]: CCTGCCATTGC GGGC TGCCGGGAGG and ADA-Ser-R [reverse]: CATGTAG TAGTTGAACTTGGTCAGG, product size: 6,383 bp) (in which Pro located at position 70 was substituted by Ser by changing the TCT codon to CCT codon), with the recombinant plasmid containing ADA gene template. The amplification was carried out in 25 ml total volume containing 12.5 ml of I-5 2×High-Fidelity Master Mix (TSINGKE, Beijing, China), 1 μl of each primer (10 mM), and ADA gene template. The PCR reactions were performed through the following profile: An initial predenaturation for 5 min at 95 °C followed by 35 cycles at 95 °C—30s, 55 °C—30s, 72 °C—5 min, and a final extension at 72 °C for 10 min. Mutated PCR products were digested with dpnI and then we used the BKL Kit (Takara) to generate annular pET28a(+)/ADA-Pro70Ser plasmid. The recombinant pET28a(+)/ADA-Pro70Ser was transformed into E. coli DH (5α) competent cells for amplification. Positive colonies resistant to ampicillin on a Luria–Bertani (LB) plate were selected and the plasmid pET28a(+)/ADA-Pro70Ser was confirmed by restriction enzyme mapping and DNA sequencing.
Expression of Recombinant Protein
The recombinant plasmids were extracted using a plasmid extraction kit (Tsingke Biotechnology Company, Chengdu, China), and then the recombinant plasmids pET28a(+)/ADA and pET28a(+)/ADA-Pro70Ser were transformed into E. coli BL21(DE3) (TransGen) competent cells for expression. The expression of the proteins was induced by the addition of 0.1 mM isopropyl-β-d-thiogalactoside (IPTG) once the optical density of 600 nm (OD600) of the culture had reached 0.6–0.8. After 4 h of induction, 1 ml culture was centrifuged at 8,000 × g for 5 min. The cell pellet was resuspended in 100 µl phosphate-buffered saline (PBS) and analyzed by SDS-PAGE. The expression level of recombinant ADA was determined by protein bands.
Purification of Recombinant Protein
Following induction, 1 l culture was centrifuged at 8,000 × g for 5 min. The pellet was resuspended in 45 ml PBS and placed in an ice bath for ultrasonic lysis (200 W, 5 s, 5 s). The supernatant was further purified by 80% ammonium sulphate fractionation, dialysis and desalt, filtered using a 0.22 µm filtration membrane and loaded onto a Ni-NTA agarose column (Qiagen, Hilden, Germany). The column was washed with buffer (equilibration buffer) and the protein was eluted with elution buffer (50 mM sodium phosphate; pH 8.0; 0.3 M sodium chloride; and 250 mM imidazole). The eluate was concentrated through a 10 kDa cutoff Centriprep filter (Millipore, Bedford, CA, USA) for SDS-PAGE.
Assay of ADA Activity
We assayed the ADA activity using a commercial kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Single Nucleotide Polymorphism Distribution and Demography from Genome Data
We used SAMtools to detect SNPs between diploid chromosomes for the sequenced FMD (Li et al. 2009). We used the pairwise sequentially markovian coalescent (PSMC) to infer the demographic history of FMD (Li and Durbin 2011). Briefly, the method employed the distribution of heterozygote sites across the FMD genome and a Hidden Markov Model to reconstruct the history of effective population sizes.
Results
Phylogeny, Divergence, and Gene Family
We identified 18,921 gene families for ten Artiodactyla species, of which 8,181 represented 1:1 orthologous gene families. In addition, there were 1,616 gene families specific to FMD (fig. 1a), whereas 5,156 gene families were found in the other nine Artiodactyla species but not in FMD. Furthermore, we identified 11,713 homologous gene families shared by Bo. mutus (Bovidae), Od. virginianus (Cervidae), S.scrofa (Suidae), Cam. ferus (Camelidae), and FMD (Moschidae), and 315 gene families were specific to FMD (fig. 1b). The ML phylogeny constructed based on the above 1:1 orthologous genes indicated that FMD and the Bovidae were within a subclade, which was most likely derived from a common ancestor ∼27.3 Ma. The series relationships within Artiodactyla were recovered as (Camelidae + (Suidae + (Cervidae + (Moschidae + Bovidae)))) (fig. 1c).
Fig. 1.

—Comparative genomics in ten Artiodactyla species studied. (a) Orthologous gene families of ten Artiodactyla species. (b) Comparison of orthologous gene clusters among Bos mutus (Bovidae), Odocoileus virginianus (Cervidae), Sus scrofa (Suidae), Camelus ferus (Camelidae), and FMD (Moschidae). (c) Phylogenetic tree constructed using 1:1 orthologous genes. Branch numbers indicate the number of gene families that have expanded (left) and contracted (right) after the split from the common ancestor. The time lines indicate divergence times among the species.
Compared with other analyzed Artiodactyla species, 592 gene families were expanded, whereas 5,413 gene families were contracted in FMD. The functional analyses of gene families showed the expansions in FMD were enriched to several pathways associated with the immune response (supplementary table S6, Supplementary Material online), suggesting that these functions might be involved in FMD adaptive evolution. However, functional enrichment with contracted gene families also detected several immune response pathways that were possibly related to genetic immunodeficiency. In addition, contracted gene families in FMD were identified to be enriched in keratinization (supplementary table S7, Supplementary Material online), in agreement with FMD lacking horns (Kawtikwar et al. 2010). This enrichment in keratinization was different from some of the other Artiodactyla species.
Positive Selection and Functional Enrichment
We found that 1,561 of the 8,181 1:1 orthologous genes were under positive selection in FMD. The Gene Ontology (GO) annotation classified the PSGs into three categories: Molecular functions, cellular components, and biological processes (supplementary fig. S1, Supplementary Material online). Molecular functions included genes mainly involved in binding (895 genes; GO: 0005488) and catalytic activity (408 genes; GO: 0003824). Genes related to cellular components were primarily cell (1,249 genes; GO: 0005623), cell part (1,247 genes; GO: 0044464), and organelle (1,021 genes; GO: 0043226). Biological process genes were mainly involved in cellular process (1,104 genes; GO: 0009987), biological regulation (787 genes; GO: 0065007), and metabolic process (773 genes; GO: 0008152). The distribution of GO annotations in different functional categories showed a substantial diversity of PSGs.
We identified the biochemical pathways on the basis of the PSGs detected in FMD. The KEGG annotation of the PSGs suggested that they were distributed in 43 pathways related to metabolism (108 genes), genetic information processing (111 genes), environmental information processing (143 genes), cellular processes (126 genes), organismal systems (171 genes), and human diseases (179 genes) (supplementary fig. S2a, Supplementary Material online). Among the identified functional categories, infectious diseases (98 genes) were highly represented (supplementary fig. S2b, Supplementary Material online).
We further performed the KEGG and GO enrichments with all the PSGs. The KEGG terms related to infectious diseases and immune response were overrepresented by PSGs, including primary immunodeficiency (corrected P-value: 0.000423), Cytokine-cytokine receptor interaction (corrected P-value: 0.002556), Measles (corrected P-value: 0.005083), Influenza A (corrected P-value: 0.006335), Malaria (corrected P-value: 0.023315), and Toll-like receptor signaling pathway (corrected P-value: 0.04422) (table 1).
Table 1.
KEGG Enrichment of PSGs in FMD
| Term | Input Number | Background Number | P-value | Corrected P-value |
|---|---|---|---|---|
| Primary immunodeficiency | 10 | 39 | 1.76E–05 | 0.000423 |
| Hematopoietic cell lineage | 14 | 80 | 1.86E–05 | 0.000441 |
| Cytokine-cytokine receptor interaction | 23 | 225 | 0.00014 | 0.002556 |
| Measles | 16 | 135 | 0.00032 | 0.005083 |
| Influenza A | 18 | 168 | 0.000428 | 0.006335 |
| Rheumatoid arthritis | 12 | 89 | 0.000625 | 0.00852 |
| Spliceosome | 15 | 134 | 0.000831 | 0.010626 |
| Cell adhesion molecules (CAMs) | 16 | 152 | 0.001033 | 0.01233 |
| Cytosolic DNA-sensing pathway | 9 | 61 | 0.001665 | 0.01797 |
| Adherens junction | 10 | 74 | 0.001704 | 0.018314 |
| Malaria | 8 | 52 | 0.002354 | 0.023315 |
| Homologous recombination | 6 | 31 | 0.00301 | 0.028138 |
| Cell cycle | 13 | 124 | 0.003032 | 0.028313 |
| Transcriptional misregulation in cancer | 15 | 158 | 0.003631 | 0.031932 |
| Peroxisome | 10 | 83 | 0.003632 | 0.031932 |
| Protein processing in endoplasmic reticulum | 15 | 159 | 0.003833 | 0.033387 |
| African trypanosomiasis | 6 | 34 | 0.004497 | 0.038039 |
| Fatty acid elongation | 5 | 24 | 0.005077 | 0.04097 |
| Graft-versus-host disease | 6 | 35 | 0.005096 | 0.04097 |
| Hepatitis B | 14 | 150 | 0.005562 | 0.043544 |
| Fatty acid metabolism | 7 | 48 | 0.005638 | 0.043544 |
| Toll-like receptor signaling pathway | 11 | 104 | 0.005767 | 0.04422 |
| FoxO signaling pathway | 13 | 135 | 0.00584 | 0.044662 |
| Amyotrophic lateral sclerosis (ALS) | 7 | 50 | 0.006865 | 0.049574 |
GO enrichment identified significant overrepresentation of FMD PSGs involved in environmental adaptation (supplementary table S8, Supplementary Material online). A number of relevant observations from other studies on the activity of these genes hint at their possible functions. For instance, enzyme activator activity (GO: 0008047) could be affected by infection. The expression profile of pig lung tissues postinoculation with Actinobacillus pleuropneumoniae showed a significant representation of genes belonging to this GO term (Zuo et al. 2013). Endoplasmic reticulum (GO: 0005783) plays key roles in important processes like protein transport and energy metabolism. The mRNA expression of GO: 0005783 in mice is altered after heat treatment (Yu et al. 2011).
FMD was a nocturnal mammal and foraged during night or crepuscule (Qi et al. 2011). Currently, it was unclear how FMD evolved to acquire the nocturnal lifestyle. We observed eight PSGs (i.e., GRK7, SLC24A1, SAG, CYP26A1, RDH10, RDH12, DGAT1, and SDR16C5) distributed in phototransduction pathway and retinol metabolism pathways, possibly enhancing adaptation to low-light and night-time environments. In addition, males and females of FMD were visibly very similar, but male individuals were armed with two freakishly long canine teeth in the upper jaw used for fighting with rivals. We also found evidence of strong positive selection in four candidate genes (i.e., KLK4, ACP4, CSF3R, and SSUH2) associated with odontogenesis and amelogenesis.
Genetic Mechanisms of Musk Secretion
We found six PSGs that were closely involved in related pathways (table 2). Firstly, one gene (AKR1D1) involved in steroid metabolism was annotated in steroid hormone biosynthesis (map00140) (supplementary fig. S3, Supplementary Material online). AKR1D1 mainly inactivates the major classes of steroid hormones in steroid hormone metabolism and catalyze key steps of the biosynthetic pathway of bile acids, regulating lipid emulsification and cholesterol homoeostasis (Jin et al. 2014).
Table 2.
KEGG Pathways Related to Musk Secretion
| KEGG Pathway | Map ID | Number of Genes | Positively Selected Genes |
|---|---|---|---|
| Steroid hormone biosynthesis | map00140 | 1 | AKR1D1 |
| Terpenoid backbone biosynthesis | map00900 | 3 | MVD |
| FNTA | |||
| DHDDS | |||
| Aldosterone-regulated sodium reabsorption | map04960 | 2 | SCNN1A |
| FXYD4 |
Furthermore, three PSGs (MVD, FNTA, and DHDDS) were annotated in terpenoid backbone biosynthesis (map00900) (supplementary fig. S4, Supplementary Material online), which are associated with terpenoid metabolism. MVD catalyses the final step in the mevalonate pathway, which is responsible for the biosynthesis of isoprenoids from acetate (Miziorko 2011). This pathway plays a key role in multiple cellular processes by synthesizing sterol isoprenoids, such as cholesterol (Buhaescu and Izzedine 2007; Miziorko 2011), and some cholesterols are converted to steroid hormones (Hinson et al. 1997). DHDDS catalyses the stepwise head-to-tail cis addition of isopentenyl-5-pyrophosphate to farnesyl-pyrophosphate, whereas farnesyl pyrophosphate is a key intermediate in cholesterol and sterol biosynthesis (Welti 2013; Xu et al. 2016). FNTA is another lipid metabolism-related gene (Jung et al. 2013). Additionally, we found two PSGs (SCNN1A, and FXYD4) related to aldosterone metabolism, which were annotated in aldosterone-regulated sodium reabsorption (map04960) (supplementary fig. S5, Supplementary Material online).
Adaptive Immunity
Gastrointestinal disease is the most common disease in FMD (Wang and Meng 2014; Yan et al. 2016). We found that many PSGs were associated with gastrointestinal diseases, and they were distributed in six related pathways (inflammatory bowel disease [IBD], Vibrio cholerae infection, Epithelial cell signaling in Helicobacter pylori infection, pathogenic E. coli infection, Salmonella infection, and Shigellosis) (table 3). Previous studies have demonstrated that infections of V.cholera, pathogenic E. coli, Salmonella, and Shigellosis can cause diarrhoea, dysentery, and some other gastrointestinal disorders (Robins-Browne and Hartland 2002; Alam and Ashraf 2003; Alanis et al. 2005). Infection by H. pylori is likely to result in gastritis with stomach pains and nausea (Butcher 2003; Ryan and Ray 2014).
Table 3.
KEGG Pathways Related to Common Diseases
| Susceptible Diseases | KEGG Pathway | Map ID | Number of PSGs |
|---|---|---|---|
| Gastrointestinal diseases | Inflammatory bowel disease (IBD) | map05321 | 7 |
| Vibrio cholerae infection | map05110 | 2 | |
| Epithelial cell signaling in Helicobacter pylori infection | map05120 | 6 | |
| Pathogenic Escherichia coli infection | map05130 | 2 | |
| Salmonella infection | map05132 | 9 | |
| Shigellosis | map05131 | 4 | |
| Respiratory diseases | Asthma | map05310 | 1 |
| Pertussis | map05133 | 6 | |
| Legionellosis | map05134 | 7 | |
| Tuberculosis | map05152 | 14 | |
| Measles | map05162 | 16 | |
| Influenza A | map05164 | 18 | |
| Abscess disease | Staphylococcus aureus infection | map05150 | 3 |
| Parasitic diseases | Amoebiasis | map05146 | 9 |
| Malaria | map05144 | 8 | |
| Toxoplasmosis | map05145 | 7 | |
| Leishmaniasis | map05140 | 6 | |
| Chagas disease (American trypanosomiasis) | map05142 | 8 | |
| African trypanosomiasis | map05143 | 6 |
Wang and Meng (2014) analyzed 339 dead FMDs, and they found respiratory diseases were the most common cause of mortality (31.27%). In the present study, several genes related to respiratory diseases were found to be under positive selection in FMD, which were distributed in six pathways (Asthma, Pertussis, Legionellosis, Tuberculosis, Measles, and Influenza A). Captive FMD breeding programs are particularly threatened by the prevalence of infectious diseases (Huang et al. 2016), with abscesses occupying 50% of all diagnoses (Luo et al. 2009; Zhao et al. 2011). We detected three PSGs in the Staphylococcus aureus infection pathway. Staphylococcus aureus was reported to be the most frequent cause of human skin or soft tissue abscesses (Lowy 1998; Cheng et al. 2009). Parasitic diseases are also important factors limiting the success of FMD captive breeding programs (Wang and Meng 2014; Yan et al. 2016). We found some PSGs associated with parasitic diseases in FMD, which were distributed in six pathways (Amoebiasis, Malaria, Toxoplasmosis, Leishmaniasis, Chagas disease [American trypanosomiasis], and African trypanosomiasis).
FMD-Specific Missense Mutation
After the examination of the PSGs identified in FMD. We found two PSGs containing FMD-specific missense mutations. The first was at position 123 (Thr) of MHC class II antigen DRA, and the other was located at position 70 (Pro) of ADA.
The mutation at MHC class II antigen DRA (p.Glu123Thr) (fig. 2a) was classified as deleterious by PolyPhen-2 (Adzhubei et al. 2010), and produces a deleterious effect on protein structure (fig. 2b–d). The MHC class II antigen plays a fundamental role in controlling immune responses as well as in autoimmunity, vaccination and infection (Neefjes et al. 2011). MHC class II antigen DRA encodes the alpha subunit of MHC class II antigen DR, and pairs with the beta chain encoded by MHC class II antigen DRB. Therefore, we also examined whether DRB carried any specific mutations. There was one FMD-specific amino acid deletion in MHC class II antigen DRB that was situated at position 94 in humans (fig. 3a). We assessed the impact of FMD-specific substitutions on human proteins for MHC class II antigen DRB by computational predictions: A large proportion (∼50%) of mutations were predicted to be functionally damaging (fig. 3b and c).
Fig. 2.

—Amino-acid sequence alignment and three kinds of visualization of nonmutated and mutated MHC class II antigen DRA. (a) The missense mutation in MHC class II antigen DRA is marked within rectangle. Asterisk means all species have the same amino acid type at this position. Species in red are mammals; species in green are birds; species in blue are reptiles. (b) Altered amino acid at p123 is shown in nonmutant and mutant MHC class II antigen DRA protein models. (c) In the surface of nonmutant and mutant MHC class II antigen DRA, mutation sites of p123 is colored as blue. (d) Electrostatic potential maps on the surface of p123 residues. Compared with nonmutant MHC class II antigen DRA, p123 mutation in mutant MHC class II antigen DRA shows a trend of negatively charged region (blue: positive charges; red: negative charges).
Fig. 3.

—Amino-acid sequence alignment and functional impact prediction of FMD-specific substitutions on human proteins for MHC class II antigen DRB. (a) The missense mutation in MHC class II antigen DRB is marked within rectangle. Asterisk means all species have the same amino acid type at this position. Species in red are mammals; species in green are birds; species in blue are reptiles; species in purple is Amphibia; species in black are Actinopterygii. (b and c) The functional classification and probability of being damaging for FMD-specific missense mutations located in MHC class II antigen DRB, according to the two metrics HumanDiv and HumanVar computed by PolyPhen-2.
We found one FMD-specific missense mutation in the ADA gene (p.Pro70Ser) (fig. 4a) that was predicted to be damaging by PolyPhen-2. ADA is considered as one of the key enzymes of purine metabolism (Glader et al. 1983), and the high degree of amino acid sequence conservation suggests the crucial nature of ADA in the purine salvage pathway (Cristalli et al. 2001). Primarily, ADA in humans is involved in the development and maintenance of the immune system (Wilson et al. 1991).
Fig. 4.

—Amino-acid sequence alignment and functional validation of FMD-specific mutation for ADA. (a) The missense mutation in ADA is marked within rectangle. Asterisk means all species have the same amino acid type at this position. Species in red are mammals; species in green are birds; species in blue are reptiles; species in black is Amphibia. (b) Identification of recombinant plasmid by plasmid polymerase chain reaction (PCR). Lane M1, DNA molecular weight standards; lane 1, PCR product of the wild ADA gene with the recombinant plasmid pET28a-ADA as template; lane 2, PCR product of the mutant ADA gene with the recombinant plasmid pET28a-ADA as template. (c) Expression of pET28a-ADA protein analyzed by SDS-PAGE. Lane M2: Protein molecular mass marker; lane 1: uninduced BL21(DE3)-pET28a-wild ADA (15 °C, 0.1 mM IPTG, 4 h); lane 2: uninduced BL21(DE3)-pET28a-mutant ADA (15 °C, 0.1 mM IPTG, 4 h); lane 3: supernatant of bacterial lysate (wild type, 15 °C, 0.1 mM IPTG, 4 h); lane 4: precipitation of bacterial lysate (wild type, 15 °C, 0.1 mM IPTG, 4 h); lane 5: supernatant of bacterial lysate (mutant type; 15 °C, 0.1 mM IPTG, 4 h); lane 6: precipitation of bacterial lysate (mutant type, 15 °C, 0.1 mM IPTG, 4 h); lane 7: purified pET28a-wild ADA (15 °C, 0.1 mM IPTG, 4 h); lane 8: purified pET28a-mutant ADA (15 °C, 0.1 mM IPTG, 4 h). (d) The ADA activity of mutant type is significantly less than that of wild type.
E scherichia coli Expression by the Missense Mutation Found in ADA of FMD
In order to explore whether the FMD-specific missense mutation (p.Pro70Ser) could affect the ADA activity, we conducted the E. coli expression of wild-type ADA (Ser) and mutated ADA (Pro). We found that the activity of mutated ADA was significantly lower than that of the wild type (supplementary table S9, Supplementary Material online and fig. 4b–d), which suggested that this missense mutation mediated the ADA activity of FMD, and possibly regulated the immune response.
Demography Reconstruction
We identified 7,015,181 heterozygous SNPs in the FMD genome. The distribution in genome-wide SNP density for FMD is shown in figure 5a. On the basis of local SNP densities, we performed PSMC modeling (Li and Durbin 2011) analysis to model the demographic history of FMD. We inferred demographic history between 10 million and 10,000 years ago. PSMC showed that FMD had experienced two bottlenecks (fig. 5b). The effective population size decreased from ∼300,000 individuals 10 Ma to a minimum of 150,000 individuals 2 Ma. Then the FMD populations started to expand, eventually giving rise to 950,000 individuals. However, FMD underwent a second bottleneck ∼400,000 years ago, and the population was reduced to 110,000 individuals.
Fig. 5.
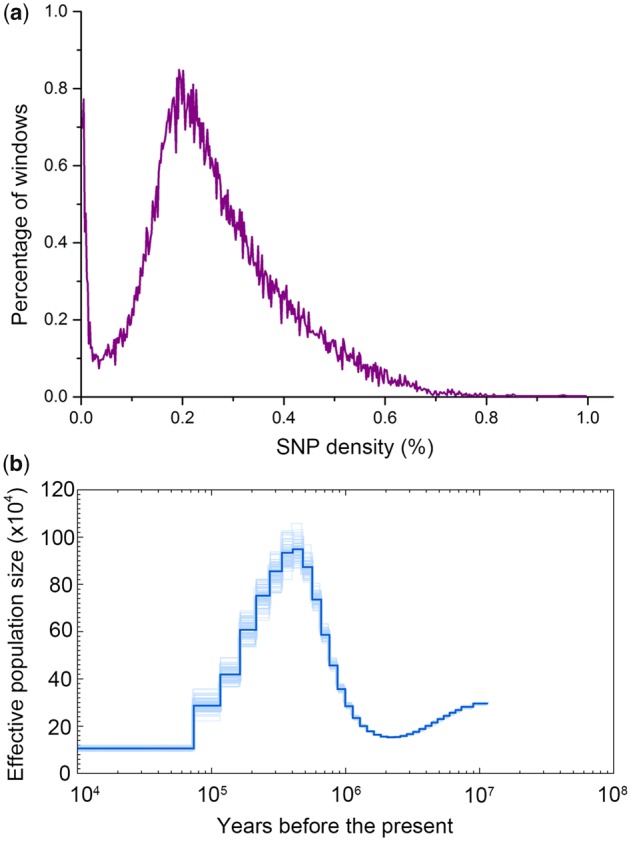
—SNP density distribution and demography reconstruction of FMD. (a) Distribution of SNP density across FMD genome. Heterozygous SNPs between the chromosomes were annotated, and heterozygosity density was observed in nonoverlapping 50-kb windows. (b) PSMC inference of FMD population history on basis of autosomal data. The central bold lines represent inferred population sizes. The 100 thin curves surrounding each line are the PSMC estimates which were generated using 100 sequences randomly resampled from the original sequence. The mutation rate on autosomes used in time scaling, was estimated using Sus scrofa autosome data.
Discussion
Phylogenetic Relationship
Complete genome sequences provided considerable resolution to the phylogenetic relationship and demography of FMD. The genome-wide phylogenetic tree showed that Cervidae (Od. virginianus) and Bovidae (Bi. bison, Bo. mutus, Bu. bubalis, Cap. hircus, Ov. aries, and P. hodgsonii) were more closely grouped, and FMD appeared to be their sister group. This genome-wide phylogenetic tree indicated that Moschidae, Cervidae, and Bovidae were three monophyletic groups. This phylogeny was consistent with other studies that placed Moschidae as an independent family (Li et al. 1998; Peng et al. 2009). Cervidae, Bovidae, and Moschidae originated from a common ancestor and Cervidae diverged earlier than Moschidae and Bovidae. However, previous phylogenetic studies of Artiodactyla species based on mitochondrial data found that Moschidae was the sister group of Cervidae, whereas other work reported that Moschidae was the sister group of Cervidae and Bovidae (Yang et al. 2012).
Genetic Mechanisms of Musk Secretion
To investigate the musk secretion mechanisms in male FMD, it was crucial to understand their metabolic processes and the corresponding pathways and genes. So far, studies from different fields have been performed to explore the mechanisms of musk secretion (Chen 2007; Zhao 2009; Guan et al. 2009; Li et al. 2016; Xu et al. 2016). However, the mechanisms of musk secretion, especially the genetic mechanisms, are still poorly understood. Through comparative genomic analysis, six PSGs were annotated in three KEGG pathways that were related to metabolic and synthetic activity of musk. For example, we found PSGs related to steroid hormone biosynthesis, terpenoid backbone biosynthesis, and aldosterone-regulated sodium reabsorption, which were coincident with the study of Xu et al. (2016). AKR1D1 was annotated in steroid hormone biosynthesis (map00140). AKR1D1 was found to inactivate the major classes of steroid hormones in steroid hormone metabolism and catalyze key steps of the biosynthetic pathway of bile acids, regulating lipid emulsification, and cholesterol homoeostasis (Jin et al. 2014). Three PSGs (MVD, FNTA, and DHDDS) were annotated in terpenoid backbone biosynthesis (map00900) and participated in terpenoid metabolism. MVD catalyzed the final step in the mevalonate pathway, which was responsible for the biosynthesis of isoprenoids from acetate (Miziorko 2011). This pathway played a key role in multiple cellular processes by synthesizing sterol isoprenoids, such as cholesterol (Buhaescu and Izzedine 2007; Miziorko 2011), and some of the cholesterols were converted to steroid hormones (Hinson et al. 1997). DHDDS catalyzed the stepwise head-to-tail cis addition of isopentenyl-5-pyrophosphate to farnesyl-pyrophosphate, whereas farnesyl pyrophosphate was a key intermediate in cholesterol and sterol biosynthesis (Xu et al. 2016). FNTA was another lipid metabolism-related gene (Jung et al. 2013).
Previous studies based on transcriptomes (Xu et al. 2016, 2017) also detected many differentially expression genes (DEGs) in the above two pathways. Our results and theirs did not have overlap candidate genes, but clearly showed the pathways of steroid hormone biosynthesis and terpenoid backbone biosynthesis played a key role in regulating the musk secretion. Indeed, genes implicated in reproduction regulation pathways such as steroid hormone biosynthesis were found in hens with different laying rates (Chen et al. 2007) and Huoyan goose ovaries between the laying period and ceased period (Luan et al. 2014). In order to understand the process of steroid-mediated oocyte maturation, it is necessary to inspect the steroid production and steroid signaling. Previous studies have indicated that steroid production could be induced by gonadotropin, and then steroid can facilitate the maturation of oocyte (Evaul 2009). Steroid metabolism plays an indispensable role in manipulating reproduction, and is closely related to the genital gland, and the metabolism of these compounds is critical to the reproduction. As for the male FMD, the musk gland is situated between naval and genital and is a representative sexual character of this endangered species. The musk gland produces chemical compounds, of which muscone and cholesterol are the primary composition of musk, and then manipulates the composition, formation, and secretion of musk.
Adaptive Immunity and Diseases
Comparative genomics identified many PSGs involved in diseases and the immune response to viral, bacterial and parasitic diseases indicating the susceptibility of FMD to gastrointestinal diseases, respiratory diseases, abscess diseases, and parasitic diseases. In particular, three immune-related genes (MHC class II antigen DRA, MHC class II antigen DRB, and ADA), which had FMD-specific mutations, have been demonstrated to be associated with the immune response (Wilson et al. 1991; Neefjes et al. 2011). The E. coli expression results suggested that the FMD-specific mutation of ADA contributed to the increase of ADA activity, which was likely to enhance the immune response to infectious diseases and could be considered as the evolutionary immune adaptation of FMD (Arredondo-Vega et al. 2002). Moreover, the identified expansions in gene families associated with the immune response, immune system process and defense response in FMD implicated that the immune adaptation had been to the diseases that FMD exhibited the greatest susceptibility. Meanwhile, we found some contracted gene families that were also functionally related to the immune response. These results suggest that some of these common diseases were likely to be caused by genetic deficiency or pathogenic invasion. Further study should focus on these immune-related genes found in the present study, because these genes probably provide us useful clues for further understanding the mechanisms of immune system development in FMD.
Other Adaptive Traits
We observed eight PSGs (GRK7, SLC24A1, SAG, CYP26A1, RDH10, RDH12, DGAT1, and SDR16C5) distributed in phototransduction (map04744) and retinol metabolism pathways (map00830) (Hemond and Vollmer 2015; Wu et al. 2016). Previous studies have reported that mutations in SLC24A1 were implicated in Autosomal-Recessive Congenital Stationary Night Blindness (CSNB) (Riazuddin et al. 2010). SAG was involved in rod vision, which encoded S-arrestin protein, a major soluble photoreceptor protein related to the desensitization of the photoactivated transduction cascade (Shen et al. 2012). Mutations in SAG were also associated with night blindness (Isashiki et al. 1999). RDH12 encoded a photoreceptor cell retinol dehydrogenase, and mutations at this gene were found to be associated with Leber congenital amaurosis and cone-rod dystrophy (Sun et al. 2007). Therefore, these genes may be contributed to the nocturnality of FMD. FMD is largely nocturnal but they can also be active during twilight (Qi et al. 2011). Currently, it is unclear how FMD acquired these pathways/adaptations but being nocturnal may avoid diurnal predators, such as humans and/or environmental conditions.
We found evidence of strong positive selection in four candidate genes (KLK4, ACP4, CSF3R, and SSUH2) that were associated with odontogenesis and amelogenesis. KLK4 (Kallikrein-4) is a glycosylated chymotrypsin-like serine protease and has been identified as essential participants in amelogenesis (Hart et al. 2004). Mutations in KLK4 can lead to autosomal recessive, nonsyndromic enamel malformations in humans and mice (Bartlett and Simmer 2014). ACPT localization has been detected in secretory ameloblasts, odontoblasts, and osteoblasts, and its biallelic mutations could cause nonsyndromic, generalized hypoplastic autosomal-recessive amelogenesis imperfecta in humans (Seymen et al. 2016). After transplantation of pulp stem cells with granulocyte-colony stimulating factor in a dog pulpectomized tooth, the root canal was completely full of regenerated pulp tissue including vasculature and innervation, and the coronal part was filled with regenerated dentin (Iohara et al. 2013). SSUH2 disrupts dental formation and was involved in tooth development (Xiong et al. 2017). Males and females of FMD are generally morphologically similar, but males are armed with two long canine teeth in the upper jaw to fight rivals. Therefore, the strong positive selection of these genes associated with odontogenesis and amelogenesis may enhance the formation of long canine teeth in male FMD.
In summary, this is the first report conducting the comparative genomics of FMD to other Artiodactyla species. Comparative genomics confirmed the PSGs annotated in functional pathways related to steroid compounds metabolism, such as steroid hormone biosynthesis, were present in FMD. The obtained PSGs data provide comprehensive gene information at the genomic level, which can contribute to a better understanding of the genetic mechanisms of musk formation and secretion in FMD. Our identification of the PSGs related to the common susceptible diseases in FMD provide a framework for future studies of these diseases in this species. Furthermore, our work can help enhance the understanding of the pathogenesis and immune system function of FMD-susceptible diseases. The PSGs found in FMD in this study will provide a strong foundation for other functional and comparative genomic studies.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This study was supported by National Key Program of Research and Development, Ministry of Science and Technology (2016YFC0503200) and National Natural Science Foundation of China (31702032). We thank Wujiao Li, Chaochao Yan, Xuhao Song, Jie Jing, Weideng Wei, and Yang Geng for valuable advice.
Author Contributions
C.Z., Z.F., and B.Y. designed and supervised the project. C.Z., Q.W., and J.J. performed the bioinformatics analyses. C.Z., W.Z., J.G., Y.S., G.W., X.J., Y.Z., and X.S. conducted the experiment. C.Z. and Z.F. wrote the manuscript. M.P., J.L., X.Z., Z.F., and B.Y. revised the manuscript. P.B., H.Y., C.P., and Y.S. participated in discussions and provided valuable advice. All authors read and approved the final manuscript.
Literature Cited
- Adzhubei IA, et al. 2010. A method and server for predicting damaging missense mutations. Nat Methods. 74:248.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam NH, Ashraf H.. 2003. Treatment of infectious diarrhea in children. Pediatr Drugs. 53:151–165. [DOI] [PubMed] [Google Scholar]
- Alanis AD, Calzada F, Cervantes JA, Torres J, Ceballos GM.. 2005. Antibacterial properties of some plants used in Mexican traditional medicine for the treatment of gastrointestinal disorders. J Ethnopharmacol. 100(1–2):153–157. [DOI] [PubMed] [Google Scholar]
- Arredondo-Vega FX, et al. 2002. Adenosine deaminase deficiency with mosaicism for a “second-site suppressor” of a splicing mutation: decline in revertant T lymphocytes during enzyme replacement therapy. Blood 993:1005–1013. [DOI] [PubMed] [Google Scholar]
- Bai JL. 2015. Phylotaxonomic position of Tianzhu white yak (Poephagus grunniens) based on nucleotide sequences of multiple subunits of cytochrome c oxidase. J Appl Anim Res. 434:431–438. [Google Scholar]
- Bartlett JD, Simmer JP.. 2014. Kallikrein-related peptidase-4 (KLK4): role in enamel formation and revelations from ablated mice. Front Physiol. 5:240.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi S, Shen Y.. 1986. Study on morphology and chemical communication mechanism in musk gland. Chin J Zool. 0204:11–14. [Google Scholar]
- Bi S, Shen Y, Zhu D.. 1984. Study on ultra-microstructure and musk secretion of musk gland in prosperous secreting stage. Acta Theriol Sin. 02:81–85. [Google Scholar]
- Buhaescu I, Izzedine H.. 2007. Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem. 40(9–10):575–584. [DOI] [PubMed] [Google Scholar]
- Butcher G. 2003. Gastroenterology: an illustrated colour text. 1st ed. Edinburgh; New York: Churchill Livingstone. [Google Scholar]
- Cai R, et al. 2015. Recombination and selection in the major histocompatibility complex of the endangered forest musk deer (Moschus berezovskii). Sci Rep. 5:17285.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Zhou Y.. 2007. Progress on anti-inflammatory effects of musk. China Pharm. 1821:1662–1665. [Google Scholar]
- Chen CF, et al. 2007. Laying traits and underlying transcripts, expressed in the hypothalamus and pituitary gland, that were associated with egg production variability in chickens. Theriogenology 689:1305–1315. [DOI] [PubMed] [Google Scholar]
- Cheng A, et al. 2009. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 2310:3393–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, et al. 2010. De novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PLoS One 512:15633.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. 2007. Studies on the genetic diversity of forest musk deer (Moschus berezovskii) and linkage analysis between the performance of musk productivity and AFLP markers [M. Scie. thesis].
- Cristalli G, et al. 2001. Adenosine deaminase: functional implications and different classes of inhibitors. Med Res Rev. 212:105–128. [DOI] [PubMed] [Google Scholar]
- De Bie T, Cristianini N, Demuth JP, Hahn MW.. 2006. Cafe: a computational tool for the study of gene family evolution. Bioinformatics 2210:1269–1271. [DOI] [PubMed] [Google Scholar]
- DeLano WL. 2002. Pymol: an open-source molecular graphics tool. CCP4 Newsl Protein Crystallogr. 40:82–92. [Google Scholar]
- Evaul KE. 2009. Gonadotropin-induced steroidogenesis and downstream signals leading to oocyte maturation [Doctoral dissertation].
- Fan Z, et al. 2018. The draft genome sequence of forest musk deer (Moschus berezovskii). Gigascience 74:giy038.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Liu J.. 2015. Progress on pharmacological activity of muscone. Food Drug. 17:212–214. [Google Scholar]
- Glader BE, Backer K, Diamond LK.. 1983. Elevated erythrocyte adenosine deaminase activity in congenital hypoplastic anemia. N Engl J Med. 30924:1486–1490. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 297:644.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan T, Zeng B, Peng Q, Yue B, Zou F.. 2009. Microsatellite analysis of the genetic structure of captive forest musk deer populations and its implication for conservation. Biochem Syst Ecol. 373:166–173. [Google Scholar]
- Hart PS, et al. 2004. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet. 417:545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, et al. 2014. Effects of crowding and sex on fecal cortisol levels of captive forest musk deer. Biol Res. 471:48.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemond EM, Vollmer SV.. 2015. Diurnal and nocturnal transcriptomic variation in the Caribbean staghorn coral, Acropora cervicornis. Mol Ecol. 2417:4460–4473. [DOI] [PubMed] [Google Scholar]
- Hinson DD, Chambliss KL, Toth MJ, Tanaka RD, Gibson KM.. 1997. Post-translational regulation of mevalonate kinase by intermediates of the cholesterol and nonsterol isoprene biosynthetic pathways. J Lipid Res. 3811:2216–2223. [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA.. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 371:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, et al. 2016. DNA vaccination based on pyolysin co-immunized with IL-1β enhances host antibacterial immunity against Trueperella pyogenes infection. Vaccine 3430:3469–3477. [DOI] [PubMed] [Google Scholar]
- Iohara K, et al. 2013. A novel combinatorial therapy with pulp stem cells and granulocyte colony‐stimulating factor for total pulp regeneration. Stem Cell Transl Med. 27:521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isashiki Y, et al. 1999. Retinitis pigmentosa with visual fluctuations and arrestin gene mutation. Br J Ophthalmol. 8310:1194–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Chen M, Penning T.. 2014. Rate of steroid double-bond reduction catalysed by the human steroid 5β-reductase (AKR1D1) is sensitive to steroid structure: implications for steroid metabolism and bile acid synthesis. Biochem J. 4621:163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Cho I, Ahn J, Jeon TI, Ha TY.. 2013. Quercetin reduces high‐fat diet‐induced fat accumulation in the liver by regulating lipid metabolism genes. Phytother Res. 271:139–143. [DOI] [PubMed] [Google Scholar]
- Kawtikwar PS, Bhagwat DA, Sakarkar DM.. 2010. Deer antlers-traditional use and future perspectives. Indian J Tradit Know. 92:245–251. [Google Scholar]
- Kumar S, Stecher G, Tamura K.. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 337:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalitha S. 2000. Primer premier 5. Biotech software & internet report: the computer software. J Sci. 16:270–272. [Google Scholar]
- Li D, et al. 2016. The musk chemical composition and microbiota of Chinese forest musk deer males. Sci Rep. 6:18975.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2011. Inference of human population history from individual whole-genome sequences. Nature 4757357:493.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 2516:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS.. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 139:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, et al. 1998. mtDNA difference and molecular phylogeny among musk deer, Chinese water deer, muntjak and deer. Acta Theriol Sin. 183:184–191. [Google Scholar]
- Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med. 3398:520.. [DOI] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N.. 2010. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinformatics 111:579.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan X, et al. 2014. Transcriptome profiling identifies differentially expressed genes in huoyan goose ovaries between the laying period and ceased period. PLoS One 911:113211.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, et al. 2009. Histopathological observations of forest musk deer died in pneumonia and suppurative diseases. Prog Vet Med. 30:122–123. [Google Scholar]
- Miziorko H. 2011. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch Biochem Biophys. 5052:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neefjes J, Jongsma ML, Paul P, Bakke O.. 2011. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 1112:823.. [DOI] [PubMed] [Google Scholar]
- Peng H, et al. 2009. The complete mitochondrial genome and phylogenetic analysis of forest musk deer (Moschus berezovskii). J Nat Hist. 43(19–20):1219–1227. [Google Scholar]
- Posada D, Crandall K.. 2005. Modeltest 3.7. Program and documentation. Available from: http://darwin.uvigo.es. Accessed July 20, 2018.
- Qi W, et al. 2011. The reproductive performance of female Forest musk deer (Moschus berezovskii) in captivity. Theriogenology 765:874–881. [DOI] [PubMed] [Google Scholar]
- Riazuddin SA, et al. 2010. A mutation in SLC24A1 implicated in autosomal-recessive congenital stationary night blindness. Am J Hum Genet. 874:523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins‐Browne RM, Hartland EL.. 2002. Escherichia coli as a cause of diarrhea. J Gastroenterol Hepatol. 174:467–475. [DOI] [PubMed] [Google Scholar]
- Ryan KJ, Ray CG.. 2014. Sherris medical microbiology. New York: McGraw-Hill Education/Medical. [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC.. 2003. Swiss-model: an automated protein homology-modeling server. Nucleic Acids Res. 3113:3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymen F, et al. 2016. Recessive mutations in ACPT, encoding testicular acid phosphatase, cause hypoplastic amelogenesis imperfecta. Am J Hum Genet. 995:1199–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng H. 1996. Protection and utilization of musk deer resources in China. Chin J Wildlife. 91:10–12. [Google Scholar]
- Sheng H, Liu Z.. 2007. The musk deer in China. Shanghai (Chinese): The Shanghai Scientific & Technical Publishers. [Google Scholar]
- Shen YY, et al. 2012. Multiple episodes of convergence in genes of the dim light vision pathway in bats. PLoS One 74:34564.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 309:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B, Wang Y, Wang Q.. 2001. Mitochondrial DNA sequences imply Anhui musk deer a valid species in genus Moschus. Zool Res. 22:169–173. [Google Scholar]
- Sun W, et al. 2007. Novel RDH12 mutations associated with Leber congenital amaurosis and cone-rod dystrophy: biochemical and clinical evaluations. Vis Res. 4715:2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, et al. 2018. Blood transcriptomics of captive forest musk deer (Moschus berezovskii) and possible associations with the immune response to abscesses. Sci Rep. 81:599.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, et al. 2017. Molecular phylogeny and phylogeography of genus Pseudois (Bovidae, Cetartiodactyla): new insights into the contrasting phylogeographic structure. Ecol Evol. 717:7047–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni S, et al. 2011. Web servers and services for electrostatics calculations with APBS and PDB2PQR. J Comput Chem. 327:1488–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Meng X.. 2014. The current status and disease occurrence of captive forest musk deer (Moschus berezovskii) in Shanxi Province. Chin J Ecol. 3310:2720–2726. [Google Scholar]
- Welti M. 2013. Regulation of dolichol-linked glycosylation. Glycoconjugate J. 301:51–56. [DOI] [PubMed] [Google Scholar]
- Xie C, et al. 2011. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39(Web Server issue):W316–W322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, et al. 2014. Research progress in forest musk deer. Heilongjiang Anim Sci Vet Med. 7:147–150. [Google Scholar]
- Xu Z, et al. 2016. De novo assembly of Chinese forest musk deer (Moschus berezovskii) transcriptome from Next-Generation mRNA sequencing. PeerJ PrePrints.
- Xu Z, et al. 2017. Illumina-based de novo transcriptome sequencing and analysis of Chinese forest musk deer. J Genet. 96:1033–1040. [DOI] [PubMed] [Google Scholar]
- Wilson DK, Rudolph FB, Quiocho FA.. 1991. Atomic structure of adenosine deaminase complexed with a transition-state analog: understanding catalysis and immunodeficiency mutations. Science 2525010:1278–1284. [DOI] [PubMed] [Google Scholar]
- Wu J, Mao X, Cai T, Luo J, Wei L.. 2006. KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 34(Web Server):W720–W724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, et al. 2016. Retinal transcriptome sequencing sheds light on the adaptation to nocturnal and diurnal lifestyles in raptors. Sci Rep. 6:33578.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong F, et al. 2017. Mutation in SSUH2 causes autosomal‐dominant dentin dysplasia type I. Hum Mutat. 381:95–104. [DOI] [PubMed] [Google Scholar]
- Yang C, et al. 2012. The complete mitochondrial genome of the Chinese Sika deer (Cervus nippon Temminck, 1838), and phylogenetic analysis among Cervidae, Moschidae and Bovidae. J Nat Hist. 46(27–28):1747–1759. [Google Scholar]
- Yang Q, Meng X, Xia L, Feng Z.. 2003. Conservation status and causes of decline of musk deer (Moschus spp.) in China. Biol Conserv. 1093:333–342. [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 248:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yan M, Yan Q, Yang G.. 2016. The mass diseases of captive musk deer. J Econ Anim. 20:112–117. [Google Scholar]
- Yao G, Zhu Y, Wan Q, Fang S.. 2015. Major histocompatibility complex class II genetic variation in forest musk deer (Moschus berezovskii) in China. Anim Genet. 465:535–543. [DOI] [PubMed] [Google Scholar]
- Yu J, et al. 2011. Integrating miRNA and mRNA expression profiles in response to heat stress-induced injury in rat small intestine. Funct Integr Genomics. 112:203–213. [DOI] [PubMed] [Google Scholar]
- Zhao K, et al. 2011. Isolation and identification on pathogens of Musk-deer abscess disease and antibiotic susceptibility assay. Sichuan J Zool. 30:522–526. [Google Scholar]
- Zhao S. 2009. Assement of genetic diversity in the captive forest musk deer (Moschus berezovskii) and linkage analysis between the performance of musk productivity and DNA molecular markers [D. Scie. thesis].
- Zuo Z, et al. 2013. Transcriptional profiling of swine lung tissue after experimental infection with Actinobacillus pleuropneumoniae. Int J Mol Sci. 145:10626–10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.