

Abstract
Lynch syndrome (LS) is an autosomal dominant inherited disorder that is associated with an increased predisposition to certain cancers caused by loss-of-function mutations in one of four DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, or PMS2). The diagnosis of LS is often challenged by the identification of missense mutations where the functional effects are not known. These are termed variants of uncertain significance (VUSs) and account for 20–30% of noncoding and missense mutations. VUSs cause ambiguity during clinical diagnosis and hinder implementation of appropriate medical management. In the current study, we focus on the functional and biological consequences of two nonsynonymous VUSs in PMS2. These variants, c.620G>A and c.123_131delGTTAGTAGA, result in the alteration of glycine 207 to glutamate (p.Gly207Glu) and the deletion of amino acid residues 42–44 (p.Leu42_Glu44del), respectively. While the PMS2 p.Gly207Glu variant retains in vitro MMR and ATPase activities, PMS2 p.Leu42_Glu44del appears to lack such capabilities. Structural and biophysical characterization using circular dichroism, small angle X-ray scattering, and X-ray crystallography of the N-terminal domain of the PMS2 variants indicate that the p.Gly207Glu variant is properly folded similar to the wild-type enzyme, whereas, p.Leu42_Glu44del is disordered and prone to aggregation.
Keywords: PMS2, Lynch syndrome, CMMRD, variants of uncertain significance, DNA mismatch repair, structural analysis
Introduction
One necessary category in the five-tier genetic variant classification system is variants of uncertain significance (VUSs) (Richards, et al., 2015). This tier must be utilized when there is a lack of information that would otherwise classify the variant as either pathogenic (i.e. disease causing) or benign (i.e. does not result in disease). A VUS result can often hinder appropriate medical management of patients undergoing genetic testing for hereditary cancer syndromes (Hoffman-Andrews, 2017; Macklin, et al., 2018). Lynch syndrome (LS) is an autosomal dominant cancer syndrome that predisposes individuals to a higher lifetime risk for developing colorectal (CRC), endometrial, gastric, biliary tract, and other cancers (Lynch, et al., 2015; Sehgal, et al., 2014). LS results from heterozygous mutations in one of four mismatch repair (MMR) genes (MSH2, MSH6, MLH1, and PMS2) as well as more recently in the EPCAM gene, which is located approximately 17-kb upstream of MSH2 (Tutlewska, et al., 2013). While a majority of pathogenic mutations found in LS patients often result from premature truncation or deletion of the MMR protein product, approximately 20–30% of missense mutations are classified as VUSs and require further characterization (Heinen, 2016).
The MMR pathway is an evolutionarily conserved, post-replicative DNA repair mechanism that corrects errors such as base mismatches, and insertion/deletion loops (IDLs) that are caused by the slippage of replicative DNA polymerases at DNA repeat sequences (microsatellites) (Jiricny, 2013; Modrich, 2006; Modrich, 2016). Therefore, the MMR pathway serves to increase the fidelity of replication by 2–3 orders of magnitude (Kunkel and Erie, 2015). The heterodimeric MutS complexes, MutSα (MSH2-MSH6) or MutSβ (MSH2-MSH3), are ATPases that initiate the first step of MMR. MutSα recognizes base-base mismatches and short (1–2 nt) IDLs, while MutSβ recognizes longer (>2 nt) IDLs. After mismatch recognition, MutSα (or β) recruits MutLα, a heterodimeric complex formed between MLH1 and PMS2 (Hsieh and Zhang, 2017). MLH1 and PMS2 harbor conserved ATP-binding domains located in the N-termini of the enzymes and their C-terminal domains are involved in heterodimerization, which is necessary for the stability of PMS2 (Guarne, 2012; Hinrichsen, et al., 2017). The C-terminal domain of PMS2 also possesses an endonuclease domain that preferentially nicks the discontinuous strand of mismatched DNA, an activity that appears to be stimulated via interactions with the proliferating cell nuclear antigen (PCNA) (Genschel, et al., 2017; Kadyrova and Kadyrov, 2016).
With the advent of panel testing, variants (pathogenic, benign, or VUSs) in the MMR genes are being identified much more rapidly (Espenschied, et al., 2017). However, the availability of experimental evidence such as biochemical, structural, and functional studies to accurately characterize VUSs as pathogenic or benign, is still lacking. This is particularly observed for variants in PMS2 that have not been studied as rigorously as variants in the other MMR genes (Blount and Prakash, 2018). Approximately 26% of the variants that have been identified in PMS2 remain categorized as VUSs.
In this study, we scrutinized two VUSs in the PMS2 gene, c.620G>A (rs374704824) and c.123_131delGTTAGTAGA (rs863224676), that result in the mutation of glycine 207 to glutamate (p.Gly207Glu) and the deletion of three amino acids (aa) 42–44 (p.Leu42_Glu44del) in the N-terminal domain of the enzyme, respectively (Table 1, Supp. Figure S1). The ClinVar database lists the p.Gly207Glu variant as being either a likely benign variant or a VUS with conflicting interpretations of pathogenicity among submitters, while the p.Leu42_Glu44del variant is classified as being either a likely pathogenic variant or a VUS, also with conflicting interpretations. In the current manuscript, we use clinically available data combined with in vitro biochemical and structural tools to characterize these VUSs. Our goal is to provide additional evidence to aid in variant classification and assist with downstream clinical decisions regarding cancer risk assessment and reduction in LS families with these PMS2 variants.
Table 1:
PMS2 variants: Location, Sequence Conservation, and Database Classification
| Variant Alteration (gDNA) | Mutation in Protein | Allele Frequency | Sequence Conservation (Align-GVGD) | Location | Database Classification | ||
|---|---|---|---|---|---|---|---|
| ClinVar | InSiGHT Class | CLIA approved lab | |||||
| c.620G>A | p.Gly207Glu | .00045 (ExAC) Highest in South Asians at 0.0022 | Intermediate conservation (9/14) | ATPase Domain | Conflicting: likely benign/ VUS | Not classified | VUS → Likely benign per Invitae |
| c.123_131delGTTAGTAGA | p.Leu42_Glu44del | None noted | Highly Conserved (L(V/I/L)E) | ATPase Domain | Conflicting: likely pathogenic/ VUS | Not in database | VUS → Likely pathogenic per GeneDx |
Methods
Pedigree Analysis
For the purposes of this study, we chose two VUSs in the PMS2 gene identified in patients seen at the University of South Alabama Mitchell Cancer Institute, Mobile, AL. We selected these variants due to the availability of detailed three-generation pedigrees and their location within the ATPase domain of the PMS2 protein. Pedigree analysis was performed as part of genetic risk assessment in a clinical setting (Fig. 1). For each of the variants, the proband’s personal health history and reported family histories were further assessed after obtaining approval from the University of South Alabama Institutional Review Board (IRB Approval #18–275).
Figure 1.
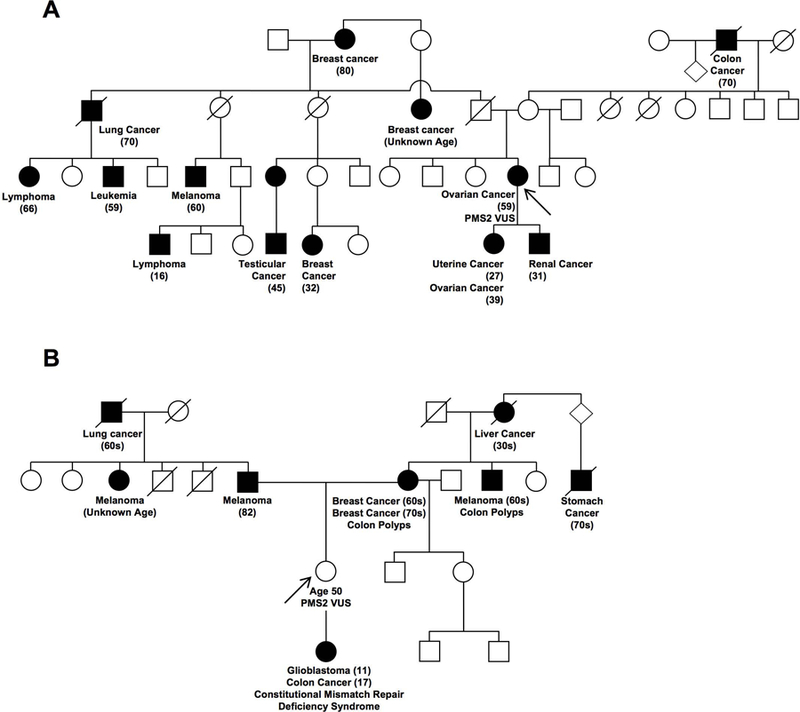
Three-generation pedigrees of two LS families with VUSs in PMS2. A) Family pedigree with a c.620G>A variant that results in the alteration of glycine 207 to glutamate (p.Gly207Glu). No other family members have undergone genetic testing, despite IRB-approved efforts to gather further genetic information. B) Family pedigree with a c.123_131delGTTAGTAGA VUS that results in the deletion of amino acids 42–44. The proband (arrow), tested positive for the PMS2 VUS and her offspring was clinically diagnosed with constitutive mismatch repair deficiency syndrome (CMMRD). The CMMRD patient inherited a known PMS2 pathogenic mutation (from father) and the PMS2 VUS (from mother) in trans. Circles represent females and squares represent males. Black squares/circles indicate individuals diagnosed with cancer and a strike-through represents individuals who are deceased. The arrow points to the proband (person who underwent genetic counseling/testing).
Plasmids, Protein Expression, and Purification
The pET15b-PMS2 construct expresses N-terminal residues 1–365 of PMS2 and was kindly provided by Dr. Wei Yang (National Institute of Health). The Quikchange II XL site-directed mutagenesis kit (Agilent Technologies) was used to generate the PMS2 p.Gly207Glu and PMS2 p.Leu42_Glu44del variants following the manufacturer’s protocol and propagated in XL10 Gold E. coli. Primer sets to generate the variants were as follows: p.Gly207Glu forward primer 5’ – gctgtcgttttccttgttcaagctgattggtgcaa-3’ and reverse primer 5’-ttgcaccaatcagcttgaacaaggaaaacgacagc-3’; p.Leu42_Glu44del forward primer 5’-cactgcggtaaaggagaacagtctggatgctg-3’ and reverse primer 5’-cagcatccagactgttctcctttaccgcagtg-3’. The nucleotide sequences of wild type (WT) and variant constructs were verified by DNA sequencing (Eurofins). The 6x histidine (His)-tagged WT and variant forms of PMS2 were expressed in E. coli Rosetta 2 (DE3) cells (Novagen) following induction by isopropyl-β-D-thiogalactopyranoside (IPTG) at 18 °C for 18 hours. Cells were lysed using a chilled French press with a high pressure setting of 18,000 psi with a Thermo IEC French press cell disruptor in the presence of 1 mM phenylmethylsulfonyl fluoride (PMSF). The crude lysate was subjected to centrifugation at 13,500 rpm for 1 hour at 4 °C in a Sorvall Instruments RC5C with an SS34 rotor. Protein was purified on a HiTrap TALON crude column (GE Healthcare, 1.6 × 2.5 cm) in 10 mM potassium phosphate pH 6.3, 150 mM potassium chloride, 5% glycerol, 20 mM imidazole and 2 mM β-mercaptoethanol with a 10-column volume (CV) imidazole gradient (up to 500 mM imidazole) followed by a HiTrap Q FF column (GE Healthcare, 1.6 × 2.5 cm). When indicated, thrombin was used to cleave the His tag from the protein. Removal of thrombin and the cleaved His tag was achieved by flowing the protein through a HiTrap SPFF column (GE Healthcare, 1.6 × 2.5 cm). Buffers for QFF and SPFF columns consisted of buffer A: 20 mM HEPES, pH 6.3, 150 mM potassium chloride, 5% glycerol, and 1 mM DTT; and buffer B contained the same components with the addition of 1 M potassium chloride. For protein crystallization and downstream structural studies, the protein was further purified using a Superdex 200 10/300 GL column (GE Healthcare). The PMS2 p.Leu42_Glu44del variant was highly prone to aggregation and consistently found in the pellet following centrifugation of the crude lysate. Initial protein preparations were successfully obtained by dissolving the pellet with 8 M urea followed by centrifugation at 13,500 rpm for 1 hour. The unfolded PMS2 p.Leu42_Glu44del variant protein was found in the soluble fraction and isolated following incubation and elution from TALON metal affinity resin (TaKaRa). The protein was refolded by stepwise dialysis from 8 to 0 M urea. The dialyzed protein was highly prone to precipitation, and thus the His-tag was left uncleaved and concentrated following collection from the HiTrap QFF column using an Amicon Ultra-15 10 kDa cutoff centrifugal filter (Millipore). A second GST tagged construct of the PMS2 p.Leu42_Glu44del variant was produced by PCR amplification using Thermo Scientific PCR Supermix. Forward and reverse primers were designed with Gateway Technology attachment sites (attB1 and attB2, respectively). Forward primer included the nucleotide sequence for a tobacco etch virus recognition site (TEV site) for removal of 5’-tags from the fusion construct. Primer sets used to generate the construct are: PMS2 p.Leu42_Glu44del forward PCR primer 5’-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAAACCTGTATTTTCAGGGAATGGAGCGAGCTGAGAGCT-3’ and reverse PCR primer 5’-GGGGACCACTTTGTACAAGAAAGCTGGGTCTCAACTATCAAACATTCCTATCAAAGAG-3’. Following PCR amplification, PMS2 p.Leu42_Glu44del was propagated to pDest15 following manufacturer’s standard protocol for BP and LR reactions (Thermo Fisher Scientific) and expressed as described above as a GST fusion protein. To isolate the GST-PMS2 p.Leu42_Glu44del fusion protein, the pellet was suspended in 10 mM potassium phosphate, pH 8.0, 150 mM potassium chloride, 5% glycerol, 2 mM β-mercaptoethanol. Lysate was loaded onto a GSTrap FF column (GE Healthcare, 1.6 × 2.5 cm) and eluted using a 10-column volume gradient of 10 mM Tris pH 9.0, 150 mM potassium chloride, 5% glycerol, 2 mM β-mercaptoethanol, and 30 mM glutathione.
ATPase Activity Assay
ATPase activity was assessed using a malachite green assay (Biomol Green reagent, Enzo Life Sciences) as described in (Rule, et al., 2016). Reactions were assembled with 20 μM enzyme in 20 mM HEPES pH 8.5, 13 mM NaCl, and 1% glycerol in the presence of 5 mM ATP and 5 mM MgCl2 totaling 30 μL volume. Reactions were initiated by the addition of ATP and MgCl2 and 5 μL aliquots were removed at time 0, 15, 30, 45, and 60 min. The reactions were incubated in a water bath at 37 °C and quenched by diluting aliquots into 245 μL 1x HNG buffer followed by flash freezing in liquid nitrogen. Samples were thawed and two 50 μL aliquots from each time point were pipetted into flat bottom 96 well clear plates (Fisher). To determine amounts of orthophosphate released, 100 μL of Biomol green reagent was added to each well, and allowed to incubate at 25 °C for 25 min. Following incubation, plates were read at 620 nm using a Tecan Infinite M1000 Pro multimode plate reader. The mean ± SEM was derived from the cumulative data generated from three separate experiments.
Cell Culture and Transfection
HCT116 cells were acquired from ATCC (CCL-247) and cultured in HyClone McCoy’s 5a medium modified (GE Healthcare Life Sciences) supplemented with 10% fetal bovine serum (Atlanta Biologicals), 50 U penicillin, and 50 μg/mL streptomycin (Gibco). Cultures were maintained at 37 °C in a 5% CO2 humidified atmosphere. Cells were co-transfected using pSG5 PMS2-WT (Addgene Plasmid #16475), p.Gly207Glu variant, or p.Leu42_Glu44del variant concomitantly with pCEP9 MLH1 (Addgene plasmid #16458). The pSG5 PMS2-WT plasmid acquired from Addgene contained a missense mutation at lysine 541 (K541E). The K541E mutation was reverted to the consensus sequence using the Quikchange II XL site directed mutagenesis kit with forward primer 5’-aaaagagtcgtcagttttaggcgctttctcctgag-3’ and reverse primer 5’-ctcaggagaaagcgcctaaaactgacgactctttt-3’. Using the modified pSG5 PMS2-E541K plasmid, both the p.Gly207Glu and p.Leu42_Glu44del variants plasmids were produced using primer sets described above. HCT116 cells were seeded (eight 150 mm culture dishes per construct for MMR assays with nuclear extracts, one 150 mm culture dish per construct for western blots of nuclear extracts, or one 100 mm culture dish for whole cell extracts) and grown to 70% confluency prior to transient transfection with the PMS2 and MLH1 vectors. Cells were transfected with 2.5 μg (100 mm dish) or 7 μg (150 mm dish) of each plasmid with a 1:6 ratio of PEI (1 mg/mL; Polysciences, MW 25,000, Linear) and cultured for an additional 48 hours prior to collection.
Preparation of Nuclear Extracts for Mismatch Repair Assays
Nuclear extracts were prepared as previously described (Geng, et al., 2011) with minor modifications. HCT116 cells (2.5 × 108) were washed with 5 mL cold PBS, detached using cell lifters in 2 mL PBS, and collected by centrifugation at 3000 x g for 5 min at 4 °C. Cells were washed with 20 mL cold hypotonic buffer (20 mM HEPES, pH 7.5, 5 mM KCl, 0.5 mM MgCl2, 0.2 M sucrose, 2 mM DTT, and 0.1% PMSF, and 1X HALT protease inhibitor cocktail) and collected by centrifugation at 3000 x g for 5 min. The cell pellet was suspended in 5 mL hypotonic buffer without sucrose (20 mM HEPES, pH 7.5, 0.5 mM MgCl2, 2 mM DTT, and 0.1% PMSF, and 1X HALT protease inhibitor cocktail) and incubated on ice for 10 min. Cells were lysed using 10 strokes with a dounce homogenizer (pestle A). Nuclei were collected by centrifugation at 2000 x g for 5 min and resuspended in 1.25 mL extract buffer (50 mM HEPES, pH 7.5, 10% Sucrose, 2 mM DTT, and 0.1% PMSF, and 1X HALT protease inhibitor cocktail) supplemented with 0.03 volumes of 5 M NaCl and mixed on a rotator for 1 hour at 4 °C. Nuclear debris was removed by centrifugation at 15,000 x g for 30 min. The supernatant was concentrated to 7–10 mg/ml with an Amicon Ultra-15 10 kDa cutoff centrifugal filter (Millipore), snap frozen in liquid nitrogen, and stored at −80 °C.
SDS-PAGE and Western Blot
For preparation of whole cell extracts, HCT116 cells were washed twice with 5 mL cold PBS, and collected by scraping in 250 μL CelLyticM (Sigma). Cells were lysed by agitation on a rotator for 15 min at room temperature. Cell debris was pelleted by centrifugation at 16,900 x g for 15 min at 4 °C. Protein concentration was determined using the DC Protein assay kit (BioRad) with bovine serum albumin as a standard. For western blot analysis, 50 μg of protein from whole cell extracts and nuclear extracts were loaded and separated by SDS-PAGE on a 4–20% polyacrylamide gel (BioRad). After transfer, the membrane was probed with PMS2 mouse monoclonal antibody (1:1000; Santa Cruz Biotechnology, Cat # sc-25315) or MLH1 mouse monoclonal antibody (1:1000; Santa Cruz Biotechnology, Cat # sc-133228 X). PCNA (D3H8P) XP rabbit monoclonal antibody (1:1000; Cell Signaling, Cat #13110S) was used as a loading control. An ECL anti-mouse IgG secondary antibody conjugated to horseradish peroxidase (HRP) (1:10,000, GE Healthcare NA931VS, from sheep) or anti-rabbit IgG secondary antibody conjugated to HRP (1:10,000, GE Healthcare NA934V, from donkey) and Advansta Inc Westernbright Sirius - femtogram substrate were used to visualize antibody binding using a BioRad ChemiDoc imager. Relative PMS2 expression was quantified and normalized to PCNA using ImageLab 5.2.1 software (BioRad). The mean ± SEM was derived from the cumulative data generated from three separate experiments. Statistical analysis was performed in GraphPad Prism 7 using an unpaired t test with Welch’s correction where equal standard deviations are not assumed.
In Vitro Mismatch Repair Assays
Mismatch repair (MMR) assays were performed as previously described in (Geng, et al., 2011) with minor modifications in a 40 μL volume containing 100 μg of nuclear extract, 76.7 fmol (100 ng) of pUC19HXB DNA substrate, 0.1 mM of dNTPs in the standard MMR buffer (20 mM Tris-HCl, pH 7.6, 1.5 mM ATP; 1 mM glutathione; 5 mM MgCl2, and 50 μg/mL BSA) and incubated at 37 °C for 1 hour. The reaction was terminated by the addition of 80 μL of stop solution (25 mM EDTA, 0.67% sodium dodecyl sulfate, and 90 μg/mL proteinase K) and incubated for an additional 30 minutes at 37 °C. DNA was isolated by extraction (1:1 v/v) twice with phenol, twice with phenol/chloroform/iso-amyl alcohol (25:24:1), and twice with chloroform. To minimize organic solvent carry over, phase lock tubes (VWR) were used for each extraction. The DNA was ethanol precipitated and digested with 10 and 20 units of AseI and XhoI restriction enzymes, respectively, and 10 μg of RNAseA (VWR) for 1 hour at 37 °C. DNA was separated by electrophoresis on a 0.8% agarose gel in 1X TAE buffer at 125 V for 1 hour. Agarose gels were stained with ethidium bromide (0.001 mg/mL) for 20 minutes and destained in 1X TAE prior to being imaged with a BioRad ChemiDoc imager. ImageLab 5.2.1 software (BioRad) was used to assess repair yield. Repair yield equals the ratio of the summed intensities of the 759 bp and 1,216 bp fragments to the total intensities of the 759 bp, 1,216 bp, and 1,975 bp bands. Repair assays were conducted in triplicate and graphed as the mean ± SEM. Statistical analysis was performed in GraphPad Prism 7 using an unpaired t test with Welch’s correction where equal standard deviations are not assumed.
Small Angle X-ray Scattering (SAXS)
SAXS data were collected at beamline 18-ID (BioCAT) of the Advanced Photon Source (APS) at Argonne National Laboratory (Fischetti, et al., 2004) for PMS2 WT, p.Gly207Glu, as well as p.Leu42_Glu44del. Additional replicates of PMS2 WT and p.Gly207Glu were collected at SIBYLS beamline 12.3.1 at the Advanced Light Source at Lawrence Berkeley National Laboratory (Classen, et al., 2013; Hura, et al., 2009). The data presented here represents the X-ray scattering recorded at the SIBYLS beamline for three concentrations of PMS2 WT (1.1, 2.5, and 5.1 mg/ml) and three concentrations of PMS2 p.Gly207Glu (1.0, 2.5, and 5.1 mg/ml). X-ray scattering data for PMS2 p.Leu42_Glu44del was recorded at the BioCAT beamline at two concentrations (0.31 and 0.42 mg/ml). Concentrations were determined based on extinction coefficients and MW for individual proteins measuring the absorbance at λ 280 nm with a NanoDrop One spectrophotometer. For PMS2 WT and p.Gly207Glu proteins, all measurements were performed in a buffer containing 20 mM HEPES, pH 6.3, 150 mM KCl, 5% glycerol, and 1 mM DTT. For GST-PMS2 p.Leu42_Glu44del, all measurements were performed in a buffer containing 10 mM Tris, pH 9.0, 150 mM KCl, 5% glycerol, and 1 mM DTT. Representative data from the two beamlines are shown in Table 2 up to a momentum transfer of 0.4 Å-1. For data collected at SIBYLS, samples were exposed to λ 1.127 Å for 10 seconds and framed at 0.3 second intervals. For each concentration series, the 34 buffer subtracted data files were merged to increase the signal to noise ratio using FrameSlice (SIBYLS). For data collected at BioCAT, samples were exposed to λ 1.04 Å at 0.5 second intervals every 3 seconds. Radial scattering measurements from each exposure were integrated followed by averaging of the exposures without any radiation damage. Averaged scattering curves from these exposures were buffer subtracted, scaled, and superimposed to evaluate concentration dependency using Scatter 3.0 (Förster, et al., 2010). Guinier approximation based forward scattering (I0) was determined using PRIMUS (Konarev, et al., 2003) and radius of gyration (Rg) were determined using Scatter. The scaled scattering intensity curves, normalized pairwise interatomic distance distribution P[r] function, and Kratky analysis curves were generated in Scatter and the raw data was plotted in GraphPad Prism 7.
Table 2:
SAXS Table
| (A) Sample details | |||
|---|---|---|---|
| PMS2 WT | PMS2 p.Gly207Glu | PMS2 p.Leu42_Glu44del | |
| Organism | Homo sapiens | Homo sapiens | Homo sapiens |
| Source | E. coli | E. coli | E. coli |
| UniProt sequence ID (residues in constructs) |
P54278 (1–365) |
P54278 (Gly 207 → Glu) | P54278 (Deletion of aa 42 – 44) |
| Affinity Tag | - | - | GST |
| Theoretical MW | 40.88 kDa | 40.95 kDa | 68.73 kDa |
| Extinction coefficient/1000 [M−1 cm−1] | 13.41 | 13.41 | 64.75 |
| (B) Data collection parameters | |||
| Beamline | SIBYLS | SIBYLS | APS (BioCAT 18-ID) |
| Wavelength (Å) | 1.127 | 1.127 | 1.04 |
| Q Range (Å−1) | 0.0091–0.5881 | 0.0091–0.5881 | 0.0049–0.3829 |
| Temperature (°C) | 25 | 25 | 25 |
| (C) Software used for data reduction, merging, analysis, and interpretation | |||
| SAXS data reduction | PRIMUS (ATSAS 2.8.1), SCATTER (Bioisis) | ||
| SAXS data merging | FrameSlice | ||
| Extinction coefficient estimate | ProtParam (Expasy) | ||
| Basic analyses: Guinier and P(r), | PRIMUS (ATSAS 2.8.1), SCATTER (Bioisis) | ||
| (D) Structural parameters | |||
| I(0) from P(r) | 146.7 ± 3.89 | 146.0 ± 2.90 | NA |
| Rg (Å) from P(r) | 25.71 ± 0.34 | 25.27 ± 0.20 | NA |
| I(0) from Guinier | 171.7 ± 0.49 | 172.9 ± 0.68 | NA |
| Rg (Å) from Guinier | 27.43 ± 0.41 | 28.13 ± 0.58 | NA |
| Dmax (Å) | 78.5 | 77.5 | NA |
Protein Crystallization and Structure Determination
Crystallization and Data Collection.
Crystals of the PMS2 p.Gly207Glu protein were obtained using hanging drop vapor diffusion methods at 16 °C in conditions containing 4% tacsimate pH 5 (v/v) and 25% PEG 3350 (w/v). The concentrated PMS2 p.Gly207Glu protein (7 mg/mL) was combined with 0.1 μl of an additive (0.1 M calcium chloride dehydrate) prior to the addition of crystallization reagent in a 1:1 ratio. Crystals were obtained after ~7 days, mounted onto MiTeGen micro loops, and flash cooled by plunging into liquid nitrogen. The data were collected at the Stanford Synchrotron Radiation Light Source (SSRL) beamline BL9–2 with a Dectris Pilatus 6M PAD detector (Cohen, et al., 2002; Gonzalez, et al., 2008; McPhillips, et al., 2002; Russi, et al., 2016). Data (0.20 sec per exposure) were collected to 2.6 Å with a crystal-to-detector distance of 300 mm and an image width of 0.2° per frame. The data were processed and reduced using the PROTEUM III program suite (Bruker, AXS). Data collection statistics are summarized in Table 3.
TABLE 3.
Data collection, phasing, and refinement statistics for PMS2 p.Gly207Glu
| PDB Code | 6MFQ |
|---|---|
| Beamline | SSRL (BL9–2) |
| Wavelength (Å) | 0.97946 |
| Space group | P 21 21 21 |
| Unit-cell parameters (Å, °) | a = 74.989 b = 74.962 c = 134.399 |
| α = β = γ = 90 | |
| Molecules per asymmetric unit | 2 |
| Data collection statistics | |
| Resolution range (Å) | 38.46 – 2.6 (2.693 – 2.6) |
| Unique reflections | 23960 (2361) |
| Redundancy | 14.8 (15.1) |
| Rmerge | 0.182 (1.567) |
| Rmeas | 0.189 (1.62) |
| Rpim | 0.049 (0.416) |
| CC1/2 | 0.998 (0.584) |
| Overall I/σ | 12.8 (1.6) |
| Completeness (%) | 100 (100) |
| MR phasing statistics | |
| Top LLG | 2382.145 |
| Top TFZ | 42.8 |
| Refinement statistics | |
| Twin Law | k,h,-l |
| Reflections used in refinement | 23937 (2353) |
| Reflections used for R-free | 2010 (197) |
| Rwork (%) | 0.2306 (0.3326) |
| Rfree (%) | 0.2424 (0.3304) |
| Number of non-hydrogen atoms | 4511 |
| Macromolecules | 4478 |
| Solvent | 262 |
| r.m.s.d values | |
| Bond length (Å) | 0.002 |
| Bond angles (°) | 0.50 |
| B-factor (Å2) | |
| Wilson B | 45.26 |
| Protein | 50.28 |
| Water | 47.72 |
| Ramachandran plot | |
| Ramachandran favored (%) | 97.98 |
| Ramachandran allowed (%) | 2.02 |
| Ramachandran outliers (%) | 0.00 |
| Clashscore | 5.49 |
, where is the average intensity from multiple observations of symmetry-related reflections. Rwork and , where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. Rfree was calculated with 10% of the reflections not used in refinement. MR, molecular replacement. Values for the highest resolution shell are shown in parentheses.
Structure Determination and Refinement.
Orthorhombic pseudo-merohedral twinned crystals of PMS2 p.Gly207Glu protein belonging to the P212121 space group contained 2 molecules per asymmetric unit with a calculated solvent content of 47.14%. The structure of the PMS2 p.Gly207Glu variant was solved by molecular replacement with Phaser-MR (Adams, et al., 2010) using the crystal structure of WT N-terminal domain of PMS2 (PDB: 1H7S) as a starting model (Guarne, et al., 2001). Water molecules were updated during refinement and manually checked. Refinement was performed with Phenix.Refine (Afonine, et al., 2012) using the twin law operator k,h,-l. Model building and map fitting were performed in COOT (Emsley, et al., 2010). The final model for the structure was found to exhibit good geometry, as determined using MolProbity (PHENIX; (Chen, et al., 2010)). Refinement statistics are shown in Table 3. All structure figures were prepared using PyMOL (The PyMOL Molecular Graphics System, Version 1.7.6 Schrödinger, LLC).
Protein Data Bank Accession Code.
Atomic coordinates and structure factor amplitudes have been deposited with the protein data bank (http://www.pdb.org) and are accessible under accession code 6MFQ.
Results
Genetic Risk Assessment
In the current manuscript, we evaluated two VUSs identified within the PMS2 gene, c.620G>A and c.123_131delGTTAGTAGA, which result in the alteration of a glycine to a glutamate at position 207 (p.Gly207Glu) and the deletion of amino acid residues 42–44 (p.Leu42_Glu44del), respectively. For each variant, a detailed three-generation pedigree analysis was performed (Fig. 1). The first family (Fig. 1A; i.e. with the p.Gly207Glu variant) presented with multiple cancers, particularly a serous ovarian cancer in the proband, which was confirmed by pathology. We note that no other tumors, beyond the proband’s, were confirmed by pathology or other clinical documentation. A panel test, analyzing 46 cancer-predisposition genes, was performed by Invitae and revealed the presence of the c.620G>A PMS2 VUS. No other pathogenic mutations or additional VUSs were identified in the 46 genes analyzed. Following the identification of the PMS2 variant, a sample of the ovarian tumor tissue was analyzed by immunohistochemistry (IHC) and revealed expression of all four MMR proteins (MLH1, MSH2, MSH6 and PMS2; Supp. Figure S2). Further analysis for microsatellite instability (MSI) was not performed on the ovarian tumor due to lack of sufficient tumor tissue.
The second family (Fig. 1B) presented with an individual who has a clinical diagnosis of Constitutional Mismatch Repair Deficiency syndrome (CMMRD). CMMRD is an autosomal recessive disorder, resulting from biallelic pathogenic mutations in any one of the four MMR genes associated with LS. This rare condition is characterized by the onset of childhood cancers including glioblastoma, CRC, hematologic malignancies and often the presence of café au lait macules (Ramachandra, et al., 2014). Genetic testing of the individual affected with CMMRD (a colorectal cancer panel analyzing 19 genes and performed at GeneDx) revealed two PMS2 variants; a VUS (c.123_131delGTTAGTAGA) that causes the deletion of aa 42–44 (p.Leu42_Glu44del) as well as a known PMS2 pathogenic mutation (c.2404C>T), which results in the mutation of Arg802 to a premature stop codon (p.Arg802*) (De Vos, et al., 2006). This testing also revealed a VUS in AXIN2 identified as c.1294G>A, but no further analysis was performed clinically or for the purposes of this study. It was determined via parental testing that the proband (Fig. 1B, arrow) carries the PMS2 VUS while the other parent carries the PMS2 pathogenic mutation, confirming that these two variants were inherited in trans. Here, we present biochemical and structural data for the VUSs identified in these families that are located within the N-terminal domain of the PMS2 protein.
Expression of Variant PMS2 in Human Cells
Initial studies assessing the relative expression and stability of PMS2 WT, p.Gly207Glu, and p.Leu42_Glu44del were performed using in vitro studies in HCT116 cells. In these cells, the expression of MLH1 is ablated due to hypermethylation of the MLH1 promoter (Chang, et al., 2000). As a consequence, PMS2 protein expression is lost as its stability is dependent upon its ability to dimerize with MLH1, an interaction that is mediated via the C-terminal domains of both proteins (Guarne, 2012; Kosinski, et al., 2010; Li and Modrich, 1995). We transiently co-transfected identical amounts of plasmids encoding full-length PMS2 WT, p.Gly207Glu, or p.Leu42_Glu44del concomitantly with WT MLH1 and assessed relative protein expression after 48 hours. Cells were harvested, and relative expression of PMS2 WT, p.Gly207Glu, or p.Leu42_Glu44del, and MLH1 from whole cell and nuclear extracts were assessed by immunoblotting against both PMS2 and MLH1 (Fig. 2A). Quantification of the relative PMS2 WT and variant expression levels revealed a consistent, statistically significant reduction in the expression of p.Leu42_Glu44del in both whole cell (p<0.001) and nuclear extracts (p<0.01) compared to the WT and the p.Gly207Glu variant (Fig. 2B). While it is documented that the stability of the PMS2 protein is reliant upon the expression of MLH1 (Dudley, et al., 2015; Mohd, et al., 2006; Rosty, et al., 2016), we consistently observed lower levels of MLH1 when it is co-expressed with the PMS2 p.Leu42_Glu44del variant but not with PMS2 WT or p.Gly207Glu (Supp. Figure S3). This result could be a consequence of reduced stability of the MutLα complex that leads to decreased protein levels of both PMS2 p.Leu42_Glu44del and MLH1.
Figure 2.

Transient expression and mismatch repair capacity of PMS2 p.Leu42_Glu44del (Δ42–44) is reduced compared to the PMS2 WT and PMS2 p.Gly207Glu (G207E) variant. HCT116 cells were co-transfected with full-length MLH1 WT and full-length PMS2 WT, p.Gly207Glu, or p.Leu42_Glu44del plasmids. Following 48 hours of incubation, whole cell and nuclear extracts were produced and relative expression levels were assessed by immunoblotting. A) Western blot analysis showing expression levels of PMS2 (non-transfected, WT, p.Gly207Glu, or p.Leu42_Glu44del), MLH1 WT, and PCNA from HCT116 whole cell and nuclear extracts B) Quantification of PMS2 WT and variant protein expression levels normalized to PCNA signal. C) DNA agarose gel showing mismatch repair capacity of nuclear extracts from non-transfected, PMS2 WT, p.Gly207Glu, or p.Leu42_Glu44del transfected HCT116 cells incubated with pUC19HXB (100 ng) for 1 hour. Positive control was produced by incubating nuclear extracts from non-transfected cells with pUC19XB and digesting with AseI and XhoI, as described in the methods. D) Quantification of repair was assessed by measuring the relative signal of digested products to total DNA. The mean ± SEM was derived from the cumulative data generated from three separate experiments; ns = not significant, * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Mismatch repair efficiency of PMS2 WT and variant enzymes
We assessed the relative MMR capacity of nuclear extracts expressing WT and variant PMS2 enzymes using an in vitro assay that was described and implemented previously by several groups (Corrette-Bennett and Lahue, 1999; Drost, et al., 2013; Geng, et al., 2011; Heinen and Juel Rasmussen, 2012; Hinrichsen, et al., 2015; Wang and Hays, 2000). Nuclear extracts were prepared from HCT116 cells co-transfected with full-length WT MLH1 and full-length PMS2 WT, p.Gly207Glu, or p.Leu42_Glu44del. The extracts were incubated with a 1,975 bp mismatch repair substrate containing a G-T mismatch 144-bp away from an Nt.BstNBI 3’ nickase site (Supp. Figures S4 and S5) located on the sense strand. The Nt.BstNBI nickase site directs the MMR machinery to the appropriate strand for mismatch correction. Based on the position of the nick, correction of the G-T mismatch to a canonical G-C base pair restores the XhoI restriction enzyme site, thereby allowing for analysis of repair efficiency. The isolated DNA substrate was digested with AseI (a control restriction site distal to the mismatch, Supp. Figure S5) and XhoI allowing for the quantification of relative mismatch repair capacity of the nuclear extracts expressing either PMS2 WT or variant proteins (Fig. 2C & D). Assessment of repair yield revealed similar repair activity between WT and the p.Gly207Glu variant. However, the repair capacity obtained for the p.Leu42_Glu44del variant was dramatically reduced compared to the WT (p<0.01) and was similar to the control nuclear extracts from non-transfected (NT) cells . This result was not unexpected, given the reduced stability and expression levels observed for the MutLα complex when this variant is present (as described above). The pUC19XB plasmid containing the intact XhoI site was used as a positive control (+C) to indicate the size of the digested bands and serves as an indicator of 100% repair efficiency.
Expression, Purification, and Impact of p.Gly207Glu and p.Leu42_Glu44del Mutations on ATPase Function
To further scrutinize how the p.Gly207Glu and p.Leu42_Glu44del variants affect protein structure, function, and stability of the ATPase domain of PMS2, we acquired a bacterial construct that expresses the N-terminal domain of PMS2 (residues 1 – 365, (Guarne, et al., 2001)). Via site-directed mutagenesis, we generated plasmids to recombinantly express the p.Gly207Glu and p.Leu42_Glu44del variant proteins. Both the WT and p.Gly207Glu variant were soluble and were purified to near homogeneity using published chromatography procedures (Fig. 3A, (Guarne, et al., 2001)). In contrast, the p.Leu42_Glu44del variant was found to be highly insoluble and was consistently found in the bacterial pellet following centrifugation of the crude E. coli lysate. Various attempts to purify soluble PMS2 p.Leu42_Glu44del included co-expression with fusion partners such as the maltose binding protein (MBP-tag) and the His-Patch Thioredoxin (data not shown). These attempts did not improve the solubility of the protein. Furthermore, a separate construct substituting the deleted amino acids (L42, V43, E44) with alanines (A42, A43, A44) and fused to either an N-terminal histidine tag or MBP-tag also did not provide us with soluble protein required for in vitro biochemical assays and structural characterization (data not shown). Small quantities of the His-tagged PMS2 p.Leu42_Glu44del was ultimately isolated by dissolving the bacterial pellet containing the insoluble p.Leu42_Glu44del under urea-based denaturing conditions and gently refolding the protein using a step-wise dialysis procedure as described previously (Cabrita and Bottomley, 2004). For improved solubility of this construct, the His-tag was retained and protein purity was assessed by SDS-PAGE analysis following Coomassie staining (Fig. 3A).
Figure 3.
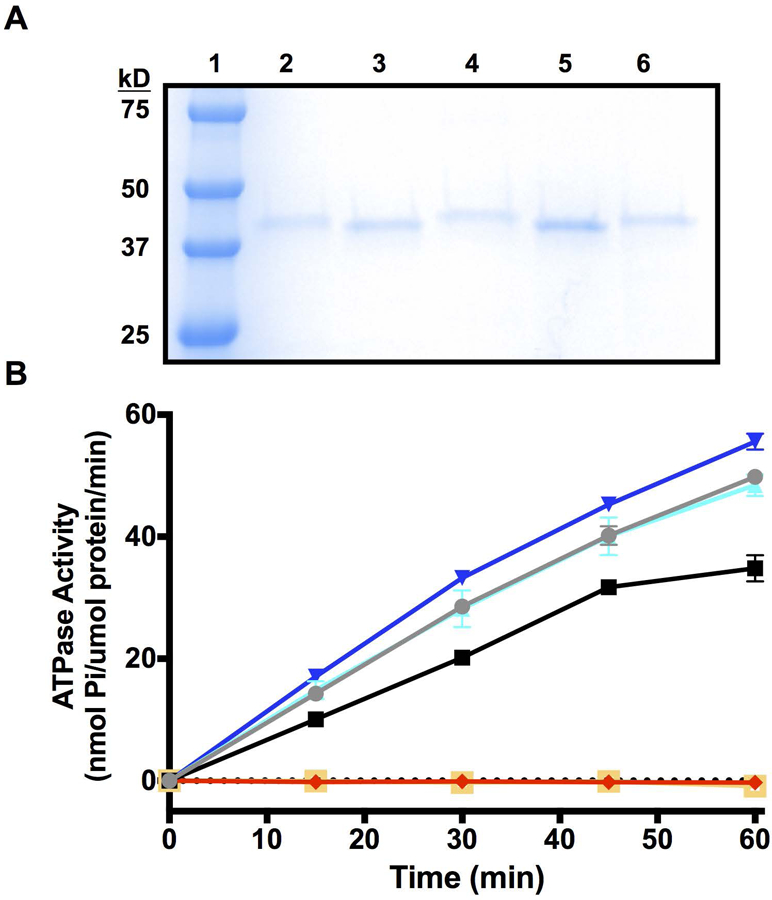
Purification and ATPase activity assessment of PMS2 WT and variant proteins. A) Coomassie stained SDS-PAGE of His-tagged and cleaved PMS2 WT (lanes 2 and 3), p.Gly207Glu (lanes 4 and 5), and His-tagged p.Leu42_Glu44del (lane 6) protein preparations. The molecular weight standard marker is in lane 1. B) ATPase activity of His-tagged and cleaved PMS2 WT, p.Gly207Glu, and p.Leu42_Glu44del protein preparations determined by a molybdate/malachite green-based assay (Enzo Life sciences, BIOMOL Green). The mean ± SEM was derived from the cumulative data generated from three separate experiments. Blue: p.Gly207Glu; Cyan: p.Gly207Glu, His; Gray: WT, His; Black: WT; Red: p.Leu42_Glu44del; Yellow: BSA.
Next, we compared the ATPase activities of the recombinantly expressed PMS2 WT and variant proteins using an in vitro ATPase activity assay. The N-terminal domain of PMS2 has previously been shown to hydrolyze the gamma phosphate from ATP to form ADP and orthophosphate (Pi) (Guarne, et al., 2001). To assess the relative ATPase activity of the WT and variant proteins, a malachite green assay (Rule, et al., 2016) was employed to measure the release of Pi from ATP (Fig. 3B). Both the WT and p.Gly207Glu proteins demonstrated a comparable time-dependent release of Pi. Since the p.Leu42_Glu44del variant was unstable without the His-tag, tagged WT and p.Gly207Glu variant domains were also evaluated to determine whether the fusion tag interfered with ATPase activity. The malachite green assay confirmed the tagged proteins retained similar ATPase activity to the cleaved constructs. In contrast to the WT protein, we observed that the ATPase activity of the p.Leu42_Glu44del variant was completely ablated (similar to BSA control). Therefore, while the p.Gly207Glu variant does not interfere with ATPase activity, our data confirms that the deletion of L42, V43, and E44 from the alpha helix within the active site, compromises the enzyme’s ability to hydrolyze ATP.
Structural Evaluation of the PMS2 Variants
Circular Dichroism
Initial evaluation of secondary structural elements for the purified PMS2 WT and variant N-terminal domains was performed using circular dichroism (CD). For the WT and the p.Gly207Glu variant, the spectra were nearly identical with negative bands at 208 nm and 222 nm that are characteristic of intact α-helices present within the proteins (Greenfield, 2006). Similar α-helical content was observed for both WT and p.Gly207Glu when the spectra were deconvoluted using the CDSSTR algorithm (Dichroweb, (Sreerama and Woody, 2000; Whitmore and Wallace, 2008) (Supp. Figure S6). In contrast, deconvolution of the spectra collected for the p.Leu42_Glu44del variant indicated little to no alpha-helical content that was accompanied by a high degree of disorder (Supp. Figure S6). In addition to the variant enzyme being insoluble and challenging to purify, the CD data obtained was an indication that the deletion of the three amino acids had a large impact on the structural integrity of this variant. The strong denaturing conditions used to purify the p.Leu42_Glu44del variant for this experiment may have resulted in the misfolding of the protein leading us to strategize a means to purify this protein without denaturation for downstream structural studies.
Characterization of PMS2 WT, p.Gly207Glu, and p.Leu42_Glu44del via Small Angle X-Ray Scattering
In order to assess aggregation state and degree of disorder, we employed in solution small angle X-ray scattering (SAXS) methods to evaluate the structural effects caused by the PMS2 variants using recombinant PMS2 WT, p.Gly207Glu, and p.Leu42_Glu44del proteins. We were able to obtain limited amounts (<< 1 mg yield) of soluble p.Leu42_Glu44del as a glutathione S-transferase (GST) fusion protein (GST-p.Leu42_Glu44del) without the need for denaturation with urea (data not shown). A prerequisite to acquiring SAXS data is obtaining purified, monodisperse protein samples (Grishaev, 2012). For this purpose, all proteins were first characterized by size exclusion chromatography. Both WT and p.Gly207Glu demonstrated identical retention volumes (Vr ~15.5 mL) on a Superdex 200 10/300 GL column (Supp. Figure S7). However, the GST-p.Leu42_Glu44del variant demonstrated a drastically different retention volume (Vr = 8.2 mL) compared to the WT or p.Gly207Glu variant proteins, and eluted in the void volume (~8.8 mL) of the size exclusion column suggesting that the protein was likely aggregated. The GST-p.Leu42_Glu44del protein has a theoretical molecular weight of 68.7 kDa, which is well within the resolution limits of the Superdex 200 10/300 GL column (10 kDa – 600 kDa).
Scaled scattering X-ray intensity curves for the WT and p.Gly207Glu variant revealed a lack of concentration-dependent effects (repulsion or aggregation) as indicated by a linear Guinier region (Fig. 4A, Supp. Figures S8A & B). Analysis of the WT and p.Gly207Glu variant curves in the low q region with the Guinier approximation revealed a radius of gyration (Rg) of 27.43 ± 0.41 Å and 28.13 ± 0.58 Å, respectively (Table 2). In contrast, the GST-p.Leu42_Glu44del variant displayed a marked increase in scattering intensity in the low q region, indicating that the protein was highly aggregated (Fig. 4A, Supp. Figure S8C). Comparing the scattering intensity curves of the WT, p.Gly207Glu, and the p.Leu42_Glu44del variant to one another demonstrates that the scattering properties of the latter deviates from the WT and p.Gly207Glu proteins (Fig. 4A). As a consequence, the lack of linearity in the Guinier region excludes the GST-p.Leu42_Glu44del variant from Guinier approximation and pairwise distance distribution analysis. In close agreement to the Rg values derived from the Guinier approximation, the normalized pairwise distance distribution (Pr) curves of the WT and p.Gly207Glu variant exhibit a maximum of 25.71 ± 0.34 Å and 25.27 ± 0.20 Å with an extended tail up to 77.5 Å and 78.5 Å (Dmax), respectively (Table 2). The shape of both the WT and p.Gly207Glu variant Pr curves suggest both proteins have comparable, slightly extended conformations (Fig. 4B). Finally, the Kratky plot of the WT and p.Gly207Glu variant curves have a characteristic bell-shaped appearance with elevated scattering at the higher angles indicating that both samples are mostly ordered, folded, and display a similar degree of flexibility. Whereas, the GST-p.Leu42_Glu44del curve is indicative of a severely compromised protein where Kratky analysis cannot be accurately performed (Fig. 4C). The SAXS data corroborates our observations we obtained throughout our evaluation of the p.Leu42_Glu44del variant.
Figure 4.
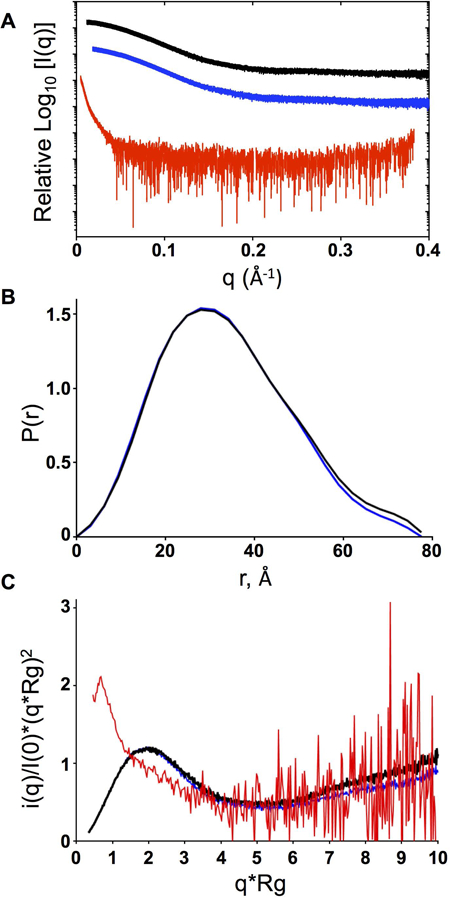
Small angle X-ray scattering (SAXS) analysis of PMS2 WT, p.Gly207Glu, and p.Leu42_Glu44del proteins. A) Scattering intensity curves for PMS2 WT (black), p.Gly207Glu (blue), and p.Leu42_Glu44del (red). B) Normalized pairwise interatomic distance distribution P[r] function for WT and p.Gly207Glu proteins. C) Kratky analysis indicating the degree of disorder for WT, p.Gly207Glu and p.Leu42_Glu44del.
X-ray Crystallography
We were able to grow diffraction-quality crystals of the PMS2 p.Gly207Glu variant, solved the crystal structure to a resolution of 2.6 Å, and refined the model to a final Rwork and Rfree value of 23.06% and 24.24%, respectively (Table 3). As previously reported by (Guarne, et al., 2001), the ATPase domain of PMS2 is composed of two α/β domains. The first of these domains consists of a β-sheet with eight strands packed against five α-helices, whereas, the second α/β domain comprises a five-stranded β-sheet with three α-helices that form a barrel and capped at one end by two β-strands (Guarne, et al., 2001). The p.Gly207Glu structure contained two molecules per asymmetric unit (ASU) similar to the WT structure, where the two molecules are juxtaposed to each other in an antiparallel orientation with crystal packing contacts between the first α/β domain of each molecule. E207 is located in the flexible loop connecting β-strand 7 to β-strand 8 (Fig. 5A). The p.Gly207Glu variant introduces an additional negative charge in a loop comprising a mixture of non-polar, polar, and charged residues (Q205, L206, E207, Q208, G209, and K210) and does not drastically affect the conformation of the β7/ β8 loop (Fig. 5B). Superimposition of the WT (PDB ID: 1H7S) and the p.Gly207Glu structures (PDB ID: 6MFQ) confirms an overall structural similarity between the two proteins with an r.m.s.d. of 0.35 Å calculated on 302 aligned Cα atoms with a maximum deviation of 1.69 Å (Fig. 5A). Thus, the β7/β8 loop in the p.Gly207Glu variant protein is not significantly displaced, and does not appear to cause a significant structural rearrangement.
Figure 5.
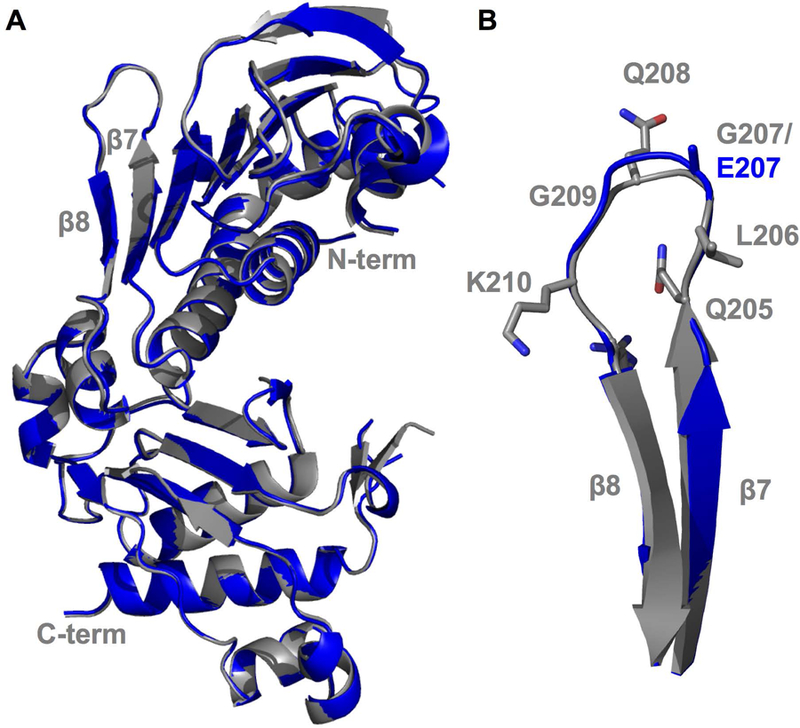
Crystal structure of the N-terminal ATPase domain of the PMS2 p.Gly207Glu variant. A) Superposition of PMS2 WT (Grey; PDB ID: 1H7S) and PMS2 p.Gly207Glu variant (Blue; PDB ID: 6MFQ). Nand C-terminals are notated along with the β7-β8 beta-strands. B) Enlarged view of the β7-β8 flexible loop containing the Glu207 residue superimposed with the PMS2 WT structure.
Discussion
The VUS category is a necessary tier within the genetic variant classification system when there is a lack of supporting data to classify a variant as benign or pathogenic (Morris, et al., 2016). The accurate, evidence-based characterization of VUSs as either pathogenic or benign is clinically necessary to help with the medical management of patients and families (Macklin, et al., 2018). Misclassification of a benign variant as pathogenic could lead to costly and unnecessary screening, chemoprevention, and prophylactic surgeries. Conversely, if a pathogenic mutation is misclassified as benign, risk-reducing medical management options may not be offered to patients with such variants. These false-positive and false-negative results can be avoided using careful consideration of all available evidence during VUS reclassification. Functional studies that elucidate the impact of a VUS on protein function are often needed to assist with the assignment of a VUS as pathogenic or benign (Heinen and Juel Rasmussen, 2012; Rasmussen, et al., 2012). For non-synonymous variants designated as a VUS, activity assays using either extracts from human cells or a cell-free system have been used to evaluate the functional consequences of the variant on MMR (Corrette-Bennett and Lahue, 1999; Drost, et al., 2013; Drost, et al., 2010; Geng, et al., 2011; Heinen and Juel Rasmussen, 2012; Hinrichsen, et al., 2015; Wang and Hays, 2000). For variants in other areas of the gene such as splice sites, in silico predictions and splicing assays have been used to help understand the role of the variant (van der Klift, et al., 2015).
Our initial in silico predictions suggested that the p.Gly207Glu variant (from the NM_000535.6 transcript) is pathogenic with a SIFT score of 0.03 (Deleterious) (Ng and Henikoff, 2003) and a PolyPhen-2 score of 0.92 (Probably Damaging) (Adzhubei, et al., 2013). However, these scores vary between different transcripts that produce this variant and caution is warranted when using such algorithms to predict pathogenicity. Align-GVGD places the p.Gly207Glu variant in class C0 indicating that this missense variant is unlikely to be pathogenic (Tavtigian, et al., 2006). The p.Gly207Glu variant was also previously identified in a BRCA1/2 negative patient with early-onset breast cancer. A clinically validated variant classification algorithm was utilized to determine the pathogenicity of several variants identified in this study and the PMS2 p.Gly207Glu variant was classified as a VUS (Maxwell, et al., 2015). In a separate case, the p.Gly207Glu variant was observed in an Iranian patient diagnosed with CRC, which displayed MSI and loss of PMS2 protein by IHC (MLH1, MSH2 and MSH6 proteins were expressed in the tumor) (Montazer Haghighi, et al., 2009). However, the patient was recruited to the study based on a clinical diagnosis of LS where the MSI and IHC results led to genetic testing, introducing an uncontrolled ascertainment bias when assessing this data. In our proband with this variant, the ovarian tumor showed intact expression of all four MMR proteins via IHC, which indicates that the germline variant does not interfere with PMS2 protein expression (Supp. Figure S2).
The p.Leu42_Glu44del variant was previously identified in an individual suspected to have LS. This individual also had a VUS in the ataxia talangiectasia mutated (ATM) gene; however, no other clinical information or biochemical data was provided (Yurgelun, et al., 2015). Residues 42–44 lie within the same alpha helix located in the ATP-binding active site as a previously reported pathogenic variant, S46I. The S46I variant lacks MMR activity and IHC indicated loss of the PMS2 protein in LS-associated tumors from several individuals (Drost, et al., 2013; Senter, et al., 2008; ten Broeke, et al., 2015).
In our studies, we obtained data using previously established in vitro activity assays and structural studies to evaluate two PMS2 variants (p.Gly207Glu and p.Leu42_Glu44del) by comparing the proteins to the WT enzyme. Extracts from cells expressing the p.Gly207Glu variant demonstrated similar PMS2 expression levels and MMR proficiency as compared to the WT extracts (Fig. 2). Our data corroborates similar results acquired using a cell-free assay that examined the MMR capacity of the p.Gly207Glu variant (Drost, et al., 2013). Biochemical assays with the recombinant N-terminal ATPase domain showed that ATPase activity was not compromised when this variant was introduced (Fig. 3). Furthermore, both in-solution scattering methods and X-ray diffraction data suggest that the p.Gly207Glu variant results in a properly folded enzyme with similar characteristics as the WT enzyme (Figs. 4, 5).
Our studies examining the p.Leu42_Glu44del variant revealed that deletion of residues L42, V43, E44 severely compromised the structure and function of the protein. We observed decreased expression levels of both PMS2 and MLH1 in extracts from cells expressing the p.Leu42_Glu44del variant. Furthermore, the MMR capacity and ATPase activity of the p.Leu42_Glu44del variant were ablated and structural data revealed that the protein product is grossly misfolded and aggregated. We therefore surmise, based on our data and previous reports, that the residues within this highly conserved α-helix (align-GVGD sequence alignment, Supp. Figure S1) are required for PMS2 ATPase activity and stability.
The American College of Medical Genetics and Genomics (ACMG) put forth broad standards and guidelines to aid in variant classification (Richards, et al., 2015). ClinGen working groups are currently refining these guidelines on a per gene basis (ClinGen). However, at the time of manuscript preparation, PMS2 specific guidance was unavailable from ClinGen. Based on the broad 2015 ACMG sequence variant interpretation guidelines, we have summarized our interpretation of the variants discussed in this manuscript in Table 4. The variant reclassifications suggested here are therefore preliminary and await expert consensus, as well as clinical validation of methodologies used. Based on our in vitro functional assays for the p.Gly207Glu variant, we present one strong (criteria BS3) piece of evidence that implicates this variant as Likely Benign. For the p.Leu42_Glu44del VUS, we show one piece of strong evidence (criteria PS3) via established in vitro functional assays (Figs. 2, 3). Furthermore, we suggest several lines of moderate evidence (criteria PM1 – 4) based on the location of this variant at the ATPase active site, low frequency in controls, detected in trans with a known pathogenic variant in a recessive condition (CMMRD), and changes in the length of the protein owing to a deletion of aa 42 – 44. Per the criteria described in Richards et al. (Richards, et al., 2015), this variant is Likely Pathogenic. While the functional assays support a classification of the p.Leu42_Glu44del variant as Likely Pathogenic, caution is warranted while interpreting the clinical significance of this variant. Rather than reclassification of this variant based solely on the data provided herein, we suggest that the data be integrated with other lines of clinical validated evidence such as co-segregation studies. It should be noted here that individuals with Likely Pathogenic variants should be afforded the same medical management plan as those with Pathogenic mutations as per the International Agency for Cancer Research guidelines (Plon, et al., 2008).
Table 4:
Variant classification guideline categories for consideration
| Variant | Possible criteria based on a broad interpretation of the American College of Medical Genetics and Genomics (ACMG) Standards & Guidelines (Richards, et al., 2015) | Pathogenicity | |
|---|---|---|---|
| p.Leu42_Glu44del | Criteria | Implication | Likely Pathogenic (1 strong and 1–2 moderate criteria) |
| PS3 | Well-established in vitro or in vivo studies to support damaging effect | ||
| PM1 | Located in a mutational hot spot and/or critical and well-established functional domain (active site) | ||
| PM2 | Absent from controls (or at extremely low frequency if recessive) | ||
| PM3 | For recessive disorders, detected in trans with a pathogenic variant | ||
| PM4 | Protein length changes as a result of deletion | ||
| p.Gly207Glu | BS3 | Well-established in vitro or in vivo studies show no damaging effect on protein function | Insufficient Data Available |
We acknowledge that variant classification is extremely important to patient medical management and that biochemical and structural analyses, while expensive and time-consuming, are necessary to aid in the variant interpretation process. Therefore, streamlining assays or new high-throughput assay development would greatly assist in this process, making classification of variants on a large scale more feasible (Drost, et al., 2018). For the purposes of understanding LS and MMR variants, we believe that a combination of clinical interpretation, biochemical techniques, and structural methods such as those presented here, can be effectively utilized to study additional VUSs within the MMR genes.
Supplementary Material
Acknowledgements:
The authors would like to recognize the contributions of several individuals. Dr. Wei Yang (National Institutes of Health (NIDDK)) for providing the expression plasmid of the N-terminal domain; Dr. Susan Tsutakawa (Lawrence Berkeley National Laboratory) and Dr. Srinivas Chakravarthy (Advanced Photon Source) for assistance with processing the SAXS data; Dr. Matthew Benning (Bruker) and Dr. Brian Eckenroth (University of Vermont) for advice on solving the crystal structure of p.Gly207Glu; Dr. Julie M. Eggington and Dr. Robert A. Burton (Center for Genomics Interpretation) for helpful suggestions and discussion during variant reclassification; and Dr. Vijay Rangachari and Dr. Gaurav Ghag for assistance with collection of circular dichroism data. Funds for this project were also provided by the University of South Alabama Mitchell Cancer Institute to BD and AP. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. This project was supported by grant 9 P41 GM103622 from the National Institute of General Medical Sciences of the National Institutes of Health. Use of the Pilatus 3 1M detector was provided by grant 1S10OD018090–01 from NIGMS. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institute of General Medical Sciences or the National Institutes of Health. SAXS data was collected at SIBYLS beamline 12.3.1 at the Advanced Light Source (ALS). SAXS data collection at SIBYLS is funded through: DOE BER Integrated Diffraction Analysis Technologies (IDAT) program and NIGMS grant P30 GM124169–01, ALS-ENABLE. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Grant sponsor: BD and AP were supported in part by a grant from the National Institutes of Health (NIEHS grant R00-ES024417 to AP).
References Cited:
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW and others. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66(Pt 2):213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, Sunyaev SR. 2013. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. 2012. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68(Pt 4):352–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount J, Prakash A. 2018. The changing landscape of Lynch syndrome due to PMS2 mutations. Clin Genet 94(1):61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita LD, Bottomley SP. 2004. Protein expression and refolding--a practical guide to getting the most out of inclusion bodies. Biotechnol Annu Rev 10:31–50. [DOI] [PubMed] [Google Scholar]
- Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR. 2000. Steady-state regulation of the human DNA mismatch repair system. J Biol Chem 275(24):18424–31. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66(Pt 1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Classen S, Hura GL, Holton JM, Rambo RP, Rodic I, McGuire PJ, Dyer K, Hammel M, Meigs G, Frankel KA and others. 2013. Implementation and performance of SIBYLS: a dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the Advanced Light Source. J Appl Crystallogr 46(Pt 1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinGen. Clinical Genome Resource https://clinicalgenome.org/working-groups/sequence-variant-interpretation: ClinGen. p Clinical Genome Resource.
- Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. 2002. An automated system to mount cryo-cooled protein crystals on a synchrotron beam line, using compact sample cassettes and a small-scale robot. J Appl Crystallogr 35(6):720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrette-Bennett SE, Lahue RS. 1999. Mismatch repair assay. Methods Mol Biol 113:121–32. [DOI] [PubMed] [Google Scholar]
- De Vos M, Hayward BE, Charlton R, Taylor GR, Glaser AW, Picton S, Cole TR, Maher ER, McKeown CM, Mann JR and others. 2006. PMS2 mutations in childhood cancer. J Natl Cancer Inst 98(5):358–61. [DOI] [PubMed] [Google Scholar]
- Drost M, Koppejan H, de Wind N. 2013. Inactivation of DNA mismatch repair by variants of uncertain significance in the PMS2 gene. Hum Mutat 34(11):1477–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost M, Tiersma Y, Thompson BA, Frederiksen JH, Keijzers G, Glubb D, Kathe S, Osinga J, Westers H, Pappas L and others. 2018. A functional assay-based procedure to classify mismatch repair gene variants in Lynch syndrome. Genet Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost M, Zonneveld J, van Dijk L, Morreau H, Tops CM, Vasen HF, Wijnen JT, de Wind N. 2010. A cell-free assay for the functional analysis of variants of the mismatch repair protein MLH1. Hum Mutat 31(3):247–53. [DOI] [PubMed] [Google Scholar]
- Dudley B, Brand RE, Thull D, Bahary N, Nikiforova MN, Pai RK. 2015. Germline MLH1 Mutations Are Frequently Identified in Lynch Syndrome Patients With Colorectal and Endometrial Carcinoma Demonstrating Isolated Loss of PMS2 Immunohistochemical Expression. Am J Surg Pathol 39(8):1114–20. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66(Pt 4):486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espenschied CR, LaDuca H, Li S, McFarland R, Gau CL, Hampel H. 2017. Multigene Panel Testing Provides a New Perspective on Lynch Syndrome. J Clin Oncol 35(22):2568–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischetti R, Stepanov S, Rosenbaum G, Barrea R, Black E, Gore D, Heurich R, Kondrashkina E, Kropf A, Wang S. 2004. The BioCAT undulator beamline 18ID: a facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the Advanced Photon Source. Journal of synchrotron radiation 11(5):399–405. [DOI] [PubMed] [Google Scholar]
- Förster S, Apostol L, Bras W. 2010. Scatter: software for the analysis of nano-and mesoscale small-angle scattering. Journal of Applied Crystallography 43(3):639–646. [Google Scholar]
- Geng H, Du C, Chen S, Salerno V, Manfredi C, Hsieh P. 2011. In vitro studies of DNA mismatch repair proteins. Anal Biochem 413(2):179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genschel J, Kadyrova LY, Iyer RR, Dahal BK, Kadyrov FA, Modrich P. 2017. Interaction of proliferating cell nuclear antigen with PMS2 is required for MutLalpha activation and function in mismatch repair. Proc Natl Acad Sci U S A 114(19):4930–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A, Moorhead P, McPhillips SE, Song J, Sharp K, Taylor JR, Adams PD, Sauter NK, Soltis SM. 2008. Web-Ice: integrated data collection and analysis for macromolecular crystallography. Journal of Applied Crystallography 41(1):176–184. [Google Scholar]
- Greenfield NJ. 2006. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 1(6):2876–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishaev A 2012. Sample preparation, data collection, and preliminary data analysis in biomolecular solution X-ray scattering. Curr Protoc Protein Sci Chapter 17:Unit17 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarne A 2012. The functions of MutL in mismatch repair: the power of multitasking. Prog Mol Biol Transl Sci 110:41–70. [DOI] [PubMed] [Google Scholar]
- Guarne A, Junop MS, Yang W. 2001. Structure and function of the N-terminal 40 kDa fragment of human PMS2: a monomeric GHL ATPase. EMBO J 20(19):5521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen CD. 2016. Mismatch repair defects and Lynch syndrome: The role of the basic scientist in the battle against cancer. DNA Repair (Amst) 38:127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen CD, Juel Rasmussen L. 2012. Determining the functional significance of mismatch repair gene missense variants using biochemical and cellular assays. Hered Cancer Clin Pract 10(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichsen I, Schafer D, Langer D, Koger N, Wittmann M, Aretz S, Steinke V, Holzapfel S, Trojan J, Konig R and others. 2015. Functional testing strategy for coding genetic variants of unclear significance in MLH1 in Lynch syndrome diagnosis. Carcinogenesis 36(2):202–11. [DOI] [PubMed] [Google Scholar]
- Hinrichsen I, Wessbecher IM, Huhn M, Passmann S, Zeuzem S, Plotz G, Biondi RM, Brieger A. 2017. Phosphorylation-dependent signaling controls degradation of DNA mismatch repair protein PMS2. Mol Carcinog 56(12):2663–2668. [DOI] [PubMed] [Google Scholar]
- Hoffman-Andrews L 2017. The known unknown: the challenges of genetic variants of uncertain significance in clinical practice. J Law Biosci 4(3):648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh P, Zhang Y. 2017. The Devil is in the details for DNA mismatch repair. Proc Natl Acad Sci U S A 114(14):3552–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL 2nd, Tsutakawa SE, Jenney FE Jr., Classen S, Frankel KA, Hopkins RC and others. 2009. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat Methods 6(8):606–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiricny J 2013. Postreplicative mismatch repair. Cold Spring Harb Perspect Biol 5(4):a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyrova LY, Kadyrov FA. 2016. Endonuclease activities of MutLalpha and its homologs in DNA mismatch repair. DNA Repair (Amst) 38:42–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MH, Svergun DI. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of applied crystallography 36(5):1277–1282. [Google Scholar]
- Kosinski J, Hinrichsen I, Bujnicki JM, Friedhoff P, Plotz G. 2010. Identification of Lynch syndrome mutations in the MLH1-PMS2 interface that disturb dimerization and mismatch repair. Hum Mutat 31(8):975–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. 2015. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu Rev Genet 49:291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GM, Modrich P. 1995. Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc Natl Acad Sci U S A 92(6):1950–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. 2015. Milestones of Lynch syndrome: 1895–2015. Nat Rev Cancer 15(3):181–94. [DOI] [PubMed] [Google Scholar]
- Macklin S, Durand N, Atwal P, Hines S. 2018. Observed frequency and challenges of variant reclassification in a hereditary cancer clinic. Genet Med 20(3):346–350. [DOI] [PubMed] [Google Scholar]
- Maxwell KN, Wubbenhorst B, D’Andrea K, Garman B, Long JM, Powers J, Rathbun K, Stopfer JE, Zhu J, Bradbury AR and others. 2015. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet Med 17(8):630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP and others. 2002. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat 9(Pt 6):401–6. [DOI] [PubMed] [Google Scholar]
- Modrich P 2006. Mechanisms in eukaryotic mismatch repair. J Biol Chem 281(41):30305–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrich P 2016. Mechanisms in E. coli and Human Mismatch Repair (Nobel Lecture). Angew Chem Int Ed Engl 55(30):8490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohd AB, Palama B, Nelson SE, Tomer G, Nguyen M, Huo X, Buermeyer AB. 2006. Truncation of the C-terminus of human MLH1 blocks intracellular stabilization of PMS2 and disrupts DNA mismatch repair. DNA Repair (Amst) 5(3):347–61. [DOI] [PubMed] [Google Scholar]
- Montazer Haghighi M, Radpour R, Aghajani K, Zali N, Molaei M, Zali MR. 2009. Four novel germline mutations in the MLH1 and PMS2 mismatch repair genes in patients with hereditary nonpolyposis colorectal cancer. Int J Colorectal Dis 24(8):885–93. [DOI] [PubMed] [Google Scholar]
- Morris B, Hughes E, Rosenthal E, Gutin A, Bowles KR. 2016. Classification of genetic variants in genes associated with Lynch syndrome using a clinical history weighting algorithm. BMC Genet 17(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2003. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FB, Hoogerbrugge N, Spurdle AB, Tavtigian SV and others. 2008. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 29(11):1282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandra C, Challa VR, Shetty R. 2014. Constitutional mismatch repair deficiency syndrome: Do we know it? Indian J Hum Genet 20(2):192–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen LJ, Heinen CD, Royer-Pokora B, Drost M, Tavtigian S, Hofstra RM, de Wind N. 2012. Pathological assessment of mismatch repair gene variants in Lynch syndrome: past, present, and future. Hum Mutat 33(12):1617–25. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E and others. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosty C, Clendenning M, Walsh MD, Eriksen SV, Southey MC, Winship IM, Macrae FA, Boussioutas A, Poplawski NK, Parry S and others. 2016. Germline mutations in PMS2 and MLH1 in individuals with solitary loss of PMS2 expression in colorectal carcinomas from the Colon Cancer Family Registry Cohort. BMJ Open 6(2):e010293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rule CS, Patrick M, Sandkvist M. 2016. Measuring In Vitro ATPase Activity for Enzymatic Characterization. J Vis Exp(114). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russi S, Song J, McPhillips SE, Cohen AE. 2016. The Stanford Automated Mounter: pushing the limits of sample exchange at the SSRL macromolecular crystallography beamlines. J Appl Crystallogr 49(Pt 2):622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal R, Sheahan K, O’Connell PR, Hanly AM, Martin ST, Winter DC. 2014. Lynch syndrome: an updated review. Genes (Basel) 5(3):497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, Lindblom A, Lagerstedt K, Thibodeau SN, Lindor NM and others. 2008. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 135(2):419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIBYLS. SAXS FrameSlice http://sibyls.als.lbl.gov/ran: SIBYLS. p A webapp for merging frame sliced SAXS data.
- Sreerama N, Woody RW. 2000. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem 287(2):252–60. [DOI] [PubMed] [Google Scholar]
- Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A. 2006. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet 43(4):295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Broeke SW, Brohet RM, Tops CM, van der Klift HM, Velthuizen ME, Bernstein I, Capella Munar G, Gomez Garcia E, Hoogerbrugge N, Letteboer TG and others. 2015. Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol 33(4):319–25. [DOI] [PubMed] [Google Scholar]
- Tutlewska K, Lubinski J, Kurzawski G. 2013. Germline deletions in the EPCAM gene as a cause of Lynch syndrome - literature review. Hered Cancer Clin Pract 11(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Klift HM, Jansen AM, van der Steenstraten N, Bik EC, Tops CM, Devilee P, Wijnen JT. 2015. Splicing analysis for exonic and intronic mismatch repair gene variants associated with Lynch syndrome confirms high concordance between minigene assays and patient RNA analyses. Mol Genet Genomic Med 3(4):327–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hays JB. 2000. Preparation of DNA substrates for in vitro mismatch repair. Mol Biotechnol 15(2):97–104. [DOI] [PubMed] [Google Scholar]
- Whitmore L, Wallace BA. 2008. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89(5):392–400. [DOI] [PubMed] [Google Scholar]
- Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, Kaushik P, Roa BB, Wenstrup RJ, Hartman AR, Syngal S. 2015. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 149(3):604–13 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.