

Summary
The dicistrovirus, Cricket paralysis virus (CrPV) encodes an RNA interference (RNAi) suppressor, 1A, which modulates viral virulence. Using the Drosophila model, we combined structural, biochemical, and virological approaches to elucidate the strategies by which CrPV-1A restricts RNAi immunity. The atomic resolution structure of CrPV-1A uncovered a flexible loop that interacts with Argonaute 2 (Ago-2), thereby inhibiting Ago-2 endonuclease-dependent immunity. Mutations disrupting Ago-2-binding attenuates viral pathogenesis in wild-type but not Ago-2-deficient flies. CrPV-1A also contains a BC-box motif that enables the virus to hijack a host Cul2-Rbx1-EloBC ubiquitin ligase complex, which promotes Ago-2 degradation and virus replication. Our study uncovers a viral-based dual regulatory program that restricts antiviral immunity by direct interaction with and modulation of host proteins. While the direct inhibition of Ago-2 activity provides an efficient mechanism to establish infection, the recruitment of a ubiquitin ligase complex, enables CrPV-1A to amplify Ago-2 inactivation to restrict further antiviral RNAi immunity.
Keywords: Insects, CrPV, antiviral immunity, RNAi, Ago-2, RNAi suppressor, E3 ligase, Ago-2 degradation
Introduction
The conflict between viral pathogens and their hosts drives the coevolution of both host defenses and viral suppression mechanisms that inhibit them (tenOever, 2016). In many species, including insects, RNA interference (RNAi) serves as a system of adaptive antiviral immunity. The importance of RNAi-mediated immunity and viral evasion strategies is exemplified by insects that transmit a wide variety of viral infectious agents to humans including sandfly fever, Dengue, Chikungunya, and Zika virus (Blair, 2011; Nayak et al., 2013; Varjak et al., 2017). The insect antiviral RNAi response requires the Dicer-2 (Dcr2) and the Argonaute 2 (Ago-2) enzymes, which cleaves double-stranded (ds) viral RNA replication intermediates and single-stranded viral RNA genomes, respectively, to limit virus replication (Aliyari et al., 2008; Marques et al., 2013). To block this antiviral response, many insect viruses encode suppressors that inhibit host RNAi pathways (Galiana-Arnoux et al., 2006; Li et al., 2002; Nayak et al., 2010; van Cleef et al., 2014; van Mierlo et al., 2012; van Rij et al., 2006). One common strategy employed by insect virus RNAi suppressors, including Flock house virus (FHV) B2, Drosophila C Virus (DCV) 1A, and Culex Y Virus (CYV) VP3 proteins, is to encode a dsRNA-binding motif; this serves to simultaneously prevent cleavage of the viral dsRNA by Dcr2 and segregate viral derived siRNA from incorporation into Ago-2 containing effector complexes (Aliyari et al., 2008; van Cleef et al., 2014; van Rij et al., 2006). In contrast, Cricket paralysis virus (CrPV) encoded RNAi suppressor CrPV-1A and Nora virus RNAi suppressor protein VP1 do not bind dsRNA, but inhibits Ago-2 directly (Nayak et al., 2010; van Mierlo et al., 2012).
CrPV infects diverse types of insects (Bonning and Miller, 2010). We reasoned that the broad host range of CrPV may give insights into the general molecular and cellular mechanisms of insect antiviral immunity. In addition, CrPV also replicates efficiently in vitro both in Drosophila S2 cells and Aedes aegypti-derived Aag2 cells, leading to cell death and thus enabling the molecular dissection of RNAi suppressor function. Previously we showed that CrPV-1A plays an essential role in mediating CrPV infection in Drosophila cells by targeting Ago-2 (Nayak et al., 2010). Moreover, expression of CrPV-1A in honeybee and gypsy moth cell lines also compromises host immunity and promotes efficient virus replication, suggesting a general mechanism for CrPV-1A in promoting virus replication and disease (Carrillo-Tripp et al., 2016; 2014).
To understand how CrPV-1A counters insect immunity, we determined the crystal structure of CrPV-1A, an Argonaute-interacting RNAi suppressor protein, to 2.6 Å resolution. This structure allowed for the identification of the Argonaute interacting site, which we show is essential for its antiviral function. We also used affinity-purification mass spectrometry to characterize the CrPV-1A interactome, which identified an additional function that is dependent on an Elongin B-Elongin C interacting BC-box motif (Kobayashi et al., 2005). Through this BC-box, CrPV-1A recruits a cellular ubiquitin ligase complex that promotes Ago-2 degradation and facilitates viral replication. In summary, our findings uncover a broad-spectrum mechanism of immune evasion mediated by an RNAi suppressor protein relying on direct enzymatic inactivation of the Ago-2 and an amplification mechanism through degradative clearance of Ago-2 protein.
Results
The crystal structure of CrPV-1A RNAi suppressor has no structural homolog
To gain insight into the molecular mechanism of Ago-2 inhibition, we sought to characterize CrPV-1A structure and function. There are no structures available for other Ago-2 inhibitors and CrPV-1A has no significant amino acid sequence similarity to any known protein. Size exclusion chromatography (SEC) coupled with multi-angle light scattering (MALS) showed that purified CrPV-1A recombinant protein exists as monomer with a minor fraction of the protein in equilibrium with a dimeric form (Fig S1A). The 1H-15N heteronuclear single quantum coherence (HSQC) nuclear magnetic resonance (NMR) techniques suggested the presence of folded CrPV-1A protein. However, the HSQC-spectrum also contained incomplete peak dispersion and resonance broadening, indicating that the protein is dynamic and may contain flexible regions (Fig S1B). We crystallized CrPV-1A159 (residues 1–159) and solved the structure at 2.6 Å resolution (Table S1). The final model contained density for residues 14–154 of CrPV-1A.
The CrPV-1A monomer has no structural homolog in the DALI server (Holm and Sander, 1995), and consists of a parallel beta sheet (β1 and β3) flanked by six alpha helices (α1- α6) and one beta-strand (β2) (Fig 1A, 1B). Its surface model has a tooth-shaped structure, in which two loops form the root (Fig 1C). One of these loops, spanning residues 103–116 (103NYCPEHRYGSTFGN116) is located between α3 and β3 and is poorly resolved in the electron density. Limited trypsin proteolysis followed by electrospray ionization-time of flight (ESI-TOF) mass spectrometry analysis suggested that this loop is flexible (Fig S1C). The other loop is cysteine-rich and spans residues 56–66 (56CYC58MDDFD64CGC66) connecting α2 and β2. Each asymmetric unit of the CrPV-1A crystal contains two identical monomers that are superposed with a root-mean-square deviation of 1.3 Å for 141 Cα positions (Fig 1D). Analysis of the CrPV-1A crystal packaging by PISA (Proteins, Interfaces, Surfaces, and Assemblies) (Krissinel and Henrick, 2007) showed the presence of a major interface (Interface I) burying a solvent-accessible area of 1253 Å2. Notably, symmetric hydrogen bonds between E55 and Y57 mediate the formation of a cross-beta sheet (two β2 strands). PISA also identified a second interface (Interface II) that buries a solvent-accessible area of 535 Å2 formed by salt bridges and hydrogen bonds (Fig 1D).
Fig 1: Overview of CrPV-1A structure.

(A, B) Side and top view of CrPV-1A monomer (C) Representation of the surface electrostatic potential in the CrPV-1A with negatively charged regions colored in red and corresponding positively charged regions is in blue. (D) Two adjacent monomers [monomer 1(cyan) and monomer 2(orange)] create two contact interfaces. Interface I is formed by hydrogen bonding between pairs of glutamic acids (Glu55) and tyrosine residues (Tyr57) on antiparallel beta strands. Interface II forms due to crystal contacts and is driven by hydrogen bonding between arginine 71 (Arg71) and glutamate 74 (Glu74) and electrostatic interactions (ES) between lysine 77 (Lys77) and glutamate 92 (Glu92).
CrPV-1A contains an Ago-2-recognition element that mediates RNAi suppression by blocking endonuclease activity
The CrPV-1A structure guided our identification of features critical for Ago-2 inhibition. We expressed either wild-type (WT) CrPV-1A148 (the N-terminal 1–148 residues) or alanine substitutions in CrPV-1A148 in Drosophila S2 cells and assessed their effect on Ago-2 activity using a luciferase-based RNAi assay (Fig 2A). The WT CrPV-1A148 completely blocked Ago-2 mediated silencing (Fig 2B, lower panel). To test the role of crystal interfaces on CrPV-1A activity, we mutated both interface I (green) and interface II (red) residues, and found they lacked any effect on RNAi suppression. Similarly, alanine substitutions in either surface-exposed residues in this region (black) or the cysteine-rich loop (magenta) did not affect CrPV-1A RNAi suppression (Fig 2B). We next examined the role of the flexible loop in RNAi suppression. We replaced the 14-residue loop with a flexible linker (GS loop) containing alternating glycine-serine residues. This CrPV-1A(GS) construct expressed well in the S2 cells but did not suppress RNAi (Fig 2B, compare to mock), indicating that this flexible loop contributes to RNAi suppression. We next used alanine scanning to identify the residue(s) in this loop mediating RNAi inhibition (Fig 2C). Only two mutations, P106A and F114A, caused significantly defective RNAi inhibition (Fig 2C). Thus, we concluded that P106 and F114, which map to opposite sides of the loop, are required for Ago-2 inhibition and loss of RNA silencing, with minor contributions from E107 and Y110. This type of Ago-2-inhibiting element has not been previously observed and we herein refer to this element as Targeting Argonaute for Loss Of Silencing or TALOS.
Fig 2: A flexible loop in CrPV-1A inhibits RNAi response in Drosophila cells.

(A) Schematic of a dual-luciferase reporter assay to screen for RNAi suppression (B) The CrPV-1A crystal structure showing amino acid residues selected for mutagenesis (yellow sticks) (top panel). The ability of these mutants to inhibit the RNAi response in S2 cells was tested in the RNAi reporter assay (bottom panel). (C) Residues (F114 and P106) in the flexible loop showing strongest inhibition of RNA silencing are highlighted in bold. The RNAi suppression data in (B) and (C) represent mean (±SD) of at least three independent experiments (n = 3) for each condition. (D) The affinity of CrPV-1A proteins for interactions with Ago-2 in S2 cells was probed by Flag-IP and Western blot analysis using antibodies directed against Drosophila Ago-2 protein. Expression levels of CrPV-1A proteins were probed with an anti-Flag antibody. One of three representative experiments is shown (n = 3). (E) S2 cell extracts were incubated with Fluc siRNA, radio-labeled capped Fluc RNA substrate, and purified CrPV-1A or variants. Slicing of the mRNA substrate was detected by denaturing PAGE and autoradiography. One of three representative experiments is shown (n = 3).
We next tested if CrPV-1A acts by binding to Ago-2. WT or mutants of Flag-tagged CrPV-1A were expressed in S2 cells, and interactions with Ago-2 were tested by immunoprecipitation (IP; Fig. 2D). We found that CrPV-1A interacted with Ago-2 efficiently, but mutations in the flexible loop containing the TALOS element abrogated or reduced binding. Replacement of the flexible loop of CrPV-1A with a GS loop or the F114A mutation abrogated Ago-2 binding (Fig. 2D), which correlates with an increase in silencing as detected by the RNAi reporter assay (Figs. 2B, 2C). Mutations that reduced CrPV-1A mediated RNAi suppression, such as P106A, Y110A, and E107A, also showed reduced Ago-2 binding, suggesting that Ago-2 inhibition is mediated by the CrPV-1A TALOS element interaction. Indeed, mutations such as Y104A of CrPV-1A suppressed RNAi to WT levels and maintained interactions with Ago-2. These data indicate that the TALOS element responsible for Ago-2 inhibition, also mediates interaction with Ago-2 (Fig 2D).
We further tested if the TALOS element is needed for inhibition of Ago-2 endonuclease activity in a cell-free slicing assay (Nayak et al., 2010). Purified recombinant CrPV-1A or CrPV-1A variants with mutations in the TALOS element were added to Drosophila cell-free extracts and Ago-2-directed cleavage of a radiolabelled luciferase capped RNA target by a cognate luciferase siRNA was assessed (Fig 2E). WT CrPV-1A prevented Ago-2-mediated luciferase RNA target degradation. Notably, the inhibitory effect of recombinant purified TALOS mutants on in vitro slicing activity of Ago-2 correlated well with their ability to co-IP Ago-2 and suppress RNAi in S2 cells. Indeed, purified CrPV-1A(F114A) and CrPV-1A(GS) had no effect on Ago-2 nuclease activity, while addition of CrPV-1A(P106A) showed modest effects and CrPV-1A(Y110A) had a very minor effect suppressing Ago-2 endonuclease activity. CrPV-1A(E107A), which has only marginal effects in suppressing RNAi in S2 cells, had no effect in the in vitro slicing assay. Together, our data shows that the TALOS element in CrPV-1A contains an opposing proline-phenylalanine pair separated by seven amino acids that mediates Ago-2 binding, inhibition of slicing activity and suppression of RNAi in Drosophila cells.
Bipartite residues of the TALOS element and neighboring sequences determine the RNAi inhibition specificity
We next examined the determinants in the TALOS element needed for Ago-2 inhibition. Our mutagenesis implicated the opposing P106 and F114 residues as crucial for Ago-2 interaction. To better understand if the spacing and positioning between these residues is critical for activity, we introduced single and double Ala insertions at two positions in the loop: between His108 and Arg109 (H108-A-R109 or H108-AA-R109 respectively) and between Ser112 and Thr113 (S112-A-T113 or S112-AA-T113 respectively) (Fig 3A). Of note, Ala replacements of either H108; R109; S112 or T113 had no effect on CrPV-1A inhibition of Ago-2 (Fig 2B). A single Ala insertion, however, in H108-A-R109 reduced CrPV-1A suppressor activity to a level comparable to F114A; whereas the double Ala insertion H108-AA-R109 completely abrogated CrPV-1A function (Fig 3B). By contrast, equivalent Ala insertions in the other position of the loop, S112-A-T113 and S112-AA-T113 had no effect on CrPV1A activity. Homology modeling of the H108-AA-R109 mutant suggests that introduction of two Ala residues increases the helical propensity of this region, which may disrupt residue spacing or flexible loop dynamics (Fig 3B, Fig S2A). No such effects were predicted for the S112-A/AA-T113 insertion, which may explain the lack of phenotype for this mutant. We also reduced the loop length by either deleting residue H108 (H108∆) or both H108 and R109 (H108/R109∆∆) (Fig 3A). The H108∆ mutation had a modest effect on activity while H108/R109∆∆ had a more severe effect. Importantly, the RNAi inhibition of the mutants in the loop correlated with their ability to bind Ago-2 (Fig 3B, lower panel). These experiments suggest that spacing between P106 and F114 or the structural dynamics of the flexible loop are important for Ago-2 binding and CrPV-1A function.
Fig 3: The TALOS element in CrPV-1A shows relaxed specificity for interaction with Ago-2.

(A) List of amino acid insertions and deletions introduced into the CrPV-1A flexible loop. (B) Effects of insertions and deletions on TALOS element-mediated RNAi suppression and Ago-2 interaction were measured using the RNAi reporter assay and Western blot analysis, respectively. (C) Effects of mutation of F114 and P106 for their ability to suppress RNAi and interact with Ago-2 were probed by RNAi-reporter assay and Western blot analysis, respectively. RNAi suppression data in (B) and (C) represent mean (±SD) of at least three independent experiments (n = 3) for each condition. Western blots analysis in (B) and (C) shows one of three representative experiments (n = 3).
See also Fig S2
Since our mutagenesis analyses point to key roles for P106 and F114, we next examined if these positions in the loop can tolerate additional amino acid substitutions. Proline is the only cyclic amino acid and plays unique roles in protein folding, structure stabilization and protein interactions. Interestingly, proline can participate in proline-aromatic residue interactions that can stabilize the structure of short peptides (Zondlo, 2013). To test if these properties were relevant to the P106 - F114 pair in the CrPV-1A loop, we replaced P106 with Gly (P106G), Tyr (P106Y), Phe (P106F) or Trp (P106W). None of these mutations supported the CrPV-1A RNAi inhibition activity (Fig 3C), suggesting that P106 is indispensable at this position, perhaps by having a structural role in positioning F114 for Ago 2 binding and/or inhibition. Next, we tested the role of F114 by mutagenesis followed by RNAi suppression and Ago-2 binding assays. F114 was mutated to either non-aromatic or aromatic amino acid residues. CrPV-1A retained function when F114 was replaced by aromatic amino acids Trp and Tyr, as well as by hydrophobic amino acids Leu and Cys. However, F114 substitution with Ala or with either Gly or Pro, with charged Arg or Asp or with polar Asn all led to defective RNA silencing inhibition and reduced Ago-2 binding. The strongest reduction in CrPV-1A activity, equivalent to that of replacing the entire loop with GS linker, was observed by F114 replacement with negatively charged Asp (F114D) followed by F114G, F114P, F114R and F114N (Fig 3C). By using higher input IP sample and longer exposure, we could detect a minor interaction of F114A suggesting that this mutation still has residual Ago-2 binding activity. F114G further reduced in affinity for Ago-2 as did F114P and F114R. F114D fully abrogated Ago-2 binding. Notably, F114 replacement with Ser or Thr retained CrPV-1A function and Ago-2 binding, albeit to a lesser extent than WT. These experiments reveal relaxed specificity of F114 in TALOS element for Ago-2 interaction. Since Ae. aegypti mosquito-derived cells, such as Aag2 are also susceptible to CrPV infection, we tested whether CrPV-1A also interacts with mosquito Ago-2. Indeed Flag-tagged CrPV1-A efficiently associated with Ago-2 in Aag2 cells, as judged by IP followed by mass spectrometry. By contrast, CrPV-1A(F114A), which served as a negative control, did not bind to mosquito Ago-2 (Fig S2B). This indicates that the TALOS element in CrPV-1A can broadly interact with Dipteran Ago-2 from Drosophila and mosquito and that these interactions rely on residue F114.
The CrPV-1A TALOS element is essential for virus replication and pathogenesis
To determine if the TALOS element is important for CrPV replication and pathogenesis, we introduced Ala mutations (Fig 4A) in the CrPV3 infectious cDNA clone (Kerr et al., 2015). These include P106A and F114A, which abrogate Ago-2 binding and E107A and Y110A, which do not significantly affect RNAi suppression and served as controls. We compared the replication and pathogenesis of viruses with these mutations with both the WT and an infection-incompetent viral clone with a premature stop codon in the non-structural protein (CrPV3-Stop). Initially, infectious in vitro synthesized vRNAs from these clones were transfected into S2 cells and their replication measured via viral titer (Fig. 4B) and production of viral protein 3CD (Fig. 4B, bottom panel). As expected, mock-transfected and cells transfected with CrPV3-STOP vRNA did not produce any virus. Cells transfected with vRNAs for CrPV3-P106A, CrPV3-F114A RNA produced 3-logs unit less virus than WT CrPV vRNA, when assayed 48 hours post transfection (Fig 4B). Importantly, vRNA for CrPV3-E107A or CrPV3-Y110A produced WT levels of virus titers. Immunoblot analysis of viral non-structural protein 3CD in cells transfected with CrPV3, CrPV3-E107A, CrPV3-Y110A viruses mirrored the effects observed for viral titers (Fig 4B, bottom panel). Of note, a CrPV-1A mutant that fully abrogates Ago-2 binding such as F114G, has a lethal viral replication phenotype (data not shown). To test if the effects of mutations in the TALOS element on viral replication relate to their association with Ago-2, we compared viral replication in control or Ago-2-depleted cells. Ago-2 was depleted from S2 cells by dsRNA soaking and viral replication was examined by viral titers and immunoblot analysis for viral protein 3CD (Fig 4C). The lack of Ago-2 enabled the CrPV3-P106A and CrPV3-F114A to replicate at WT levels, but not CrPV3-Stop thus establishing that the CrPV-1A TALOS element counteracts the antiviral effect of Ago-2 (Fig 4C).
Fig 4: Importance of the TALOS element in virus pathogenesis.

(A) Schematic representation of the positions (small triangles) at which amino acid mutations were introduced into the CrPV3 infectious cDNA clone. (B) Viral titer by CrPV3 or CrPV3 variant RNA in S2 cells was measured by FFU assay. The synthesis of a viral protein and loading controls were visualized by a Western blot analysis using an antibody raised against CrPV 3CD peptides and tubulin, respectively. (C) Ago-2-depleted S2 cells were transfected with CrPV3, CrPV3-P106A, or CrPV3-F114A RNAs. Viral titer and expression of 3CD protein was measured by FFU assay and Western blot, respectively. Titer value in (B) and (C) represents the mean (±SD) of at three replicate experiments (n = 3). ns, not significant; *p<0.05 (Unpaired t test). The statistical significance represents measurement compared to CrPV3. One of two representative experiments (n = 2) is shown for Western blot analysis. (D) CrPV3 or CrPV3-F114A virus, or PBS were injected into flies (n = 10) of either WT or Ago-2 knockout background. Survival data represents mean of three independent experiments (n = 3). ns, not significant; *p<0.05; ****p<0.0001 (Log-rank test). (E) Viral RNA production in injected flies (panel D) was measured by qPCR. Each data point represents the mean (±SD) of three independent qPCR measurements (n = 3) using infected flies (n = 10). ns, not significant; *p<0.05; ****p<0.0001 (Generalized estimating equation test).
We then examined how the CrPV-1A TALOS element mutation F114A affects infectivity of CrPV in WT or heterozygous Ago-2-knockout (Ago-2−/−) flies. For this, 500 fluorescence forming unit (FFU) of CrPV3 or CrPV3-F114A virus was injected intra-thoracically into either WT or Ago-2−/− flies. WT CrPV3 caused 100% fly mortality by day 7 (Fig. 4D, lightgreen) while depletion of Ago-2 sensitized flies to infection with WT virus, with 100% mortality by day 6 (Fig. 4D, salmon). The CrPV3-F114A virus was attenuated in WT flies with 70% mortality by 7 (Fig. 4D, blue). However, in Ago-2−/− flies, CrPV3-F114A was as virulent as WT CrPV3 (Fig 4D, gold). Similar results were obtained when Ago-2−/− flies were infected using lower virus loads, i.e. either 5 or 50 FFU of virus (Fig. S3). We also examined the effect of F114A mutation on viral RNA replication in WT and Ago-2−/− flies by qPCR measurements using CrPV3 specific primers (Table S2). In WT flies, CrPV3-F114A replication was impaired, with a reduction of 2.5 orders of magnitude in viral RNA accumulation compared to WT CrPV3 (Fig 4E). By contrast, in Ago-2−/− flies there was no significant difference (~1.5 fold) between these viruses (Fig. 4E) suggesting that the pathogenicity of the WT and mutant virus correlated with the virus replication rate. These experiments show that loss of replication efficiency and virulence of CrPV3-F114 virus is directly linked to its antiviral action on Ago-2.
CrPV-1A recruits components of a Drosophila Cullin-Ring E3 ubiquitin ligase
To further investigate if the interaction of CrPV-1A TALOS element and Ago-2 is sufficient to regulate the RNAi response during virus infection, we characterized its CrPV-1A interactome in Drosophila S2 cells (Fig S4). An inducible Flag-CrPV-1A (encoding 166 amino acids from the N-terminus) stably expressed in S2 cells was subjected to affinity purification and mass spectrometry (AP-MS); Flag-GFP and an un-induced CrPV-1A cell line served as controls. We did at least 3 replicates of each bait purification and determined the enrichment of co-purifying proteins relative to 5 control purifications. Using an established statistical model (SAINT express) (Teo et al., 2014), we inferred the likelihood that proteins detected by AP-MS are true interactors of the bait protein. We identified 51 host proteins that co-immunoprecipiated with CrPV-1A from S2 cells, including, Ago-2 (Fig 5A, Table S3). Analyses of these interactors via the String database (Szklarczyk et al., 2017) grouped the interactions into functionally associated pathways and protein complexes (Fig 5B). Among the most significantinteractors, were the known subunits of E3 ubiquitin ligase complex including Cullin 2 (Cul2), Elongin C (EloC), Elongin B (EloB), and Ring-box protein 1 (Rbx1) (marked with asterisk in Fig 5A and highlighted in yellow in Fig 5B), which together form a Cul2-E3 ubiquitin ligase (Nguyen et al., 2017).
Fig 5: The interactomes of CrPV-1A and Ago-2 protein in S2 cells.

(A) For AP-MS experiments, a SaintScore threshold of 0.9, corresponding to a Bayesian False Discovery Rate (BFDR) of < 0.01, was used for reporting specific interactions. Heat map plots of most and least frequently observed interactors for a given bait protein in red and black, respectively. AP-MS experiment represents at least 3 biological replicates per bait protein (n = 3–5) (B) Networks for CrPV-1A, CrPV-1A+Infection, and Ago-2 interacting proteins depicting functionally associated protein clusters were generated using the String database (String Score Threshold = 0.9), and a layout was created using Cytoscape. The yellow dotted circle highlights the most prominent functionally associated protein interactions for CrPV-1A.
To test if these CrPV-1A interactors are recruited through their association with Ago-2, we did AP-MS analysis of Flag-Ago-2 in S2 cells. We identified 74 Ago-2 interactors, confirming reported interactions with heat shock proteins such as DroJ2 (Hsp40), Hsp60 and Hsc70 (Iwasaki et al., 2010; Nakanishi, 2016). Fifteen Ago-2 interactors were also associated with CrPV-1A including components of the chaperonin TRiC and the proteasome as well as proteins involved in TCA cycle, metabolism, and translation. Importantly, the Cul2-E3 ligase components were only found in the CrPV-1A purification (Fig 5A, 5B, Table S3). Of note AP-MS analysis of flag-tagged DCV-1A (encoding 97 amino acids from the N-terminus), a dsRNA binding RNAi suppressor (van Rij et al., 2006) identified 15 interactors including siRNA pathway proteins (i.e. Dcr2, loqs, and blanks), but no E3 ligase components were identified, providing additional confidence for the association of the E3 ligase machinery to CrPV-1A.
We next tested if the Cul2-E3 complex also binds to CrPV-1A in the context of viral infection by infecting S2 cells that express the Flag-CrPV-1A with CrPV for 7 hours. By AP-MS analysis of these infected cells, we identified 318 CrPV-1A interactors during infection. Most interestingly, 49 out of 51 protein interactors identified in the CrPV-1A overexpression experiment, were also detected in the context of infection. In both cases, E3 ligase components were the most prominent interactors (Fig 5A, Table S3). These experiments show additional infection-dependent CrPV-1A interactors including components of the cellular transcription, DNA replication, splicing, and ribosome biogenesis (Fig. 5A, 5B, Table S3). Given the prominence of the Cul2-E3 ligase interaction, we next assessed its relevance to CrPV replication.
A BC-box motif in CrPV-1A mediates viral E3 ligase assembly
E3 ubiquitin ligase complexes containing Cul2, EloB+EloC (herein EloBC), and Rbx1, recruit their substrates through exchangeable substrate receptors. The substrate recognition proteins share a helical EloBC recognition motif termed a “BC-box”. We hypothesized that the Cul2/EloBC/Rbx1 complex interacts with CrPV-1A to assemble a viral E3 ligase. Flag-CrPV-1A complexes from S2 cells were subjected to glycerol gradient ultracentrifugation to determine if CrPV-1A forms a stable complex with Cul2/EloBC/Rbx1. Analysis of fractions by immunobloting, silver staining and correlation with mass-spectrometry showed that CrPV-1A co-fractionates with EloBC, Rbx1, Cul2 as well as with Ago-2 (Fraction 5–11, Fig 6A, S5A). This raised the possibility that CrPV-1A is a component of the viral Cul2-E3 ligase that targets Ago-2 for degradation. While an amino acid BLAST search did not identify a BC-box motif in CrPV-1A, inspection of the CrPV-1A sequence showed a consensus motif (SL)xxx(A)xxx(L) at the N-terminus (Fig 6B), similar to that previously reported for HIV and BIV Vif proteins that bind EloBC (Kobayashi et al., 2005; Yu et al., 2003; W. Zhang et al., 2014). To determine if the putative BC-box motif in CrPV-1A is a bona fide EloBC interacting domain, we co-expressed His-tagged CrPV-1A with untagged EloB, and EloC in E. coli and purified CrPV-1A protein by nickel affinity chromatography (NAC) followed by SDS-PAGE and Coomassie Blue staining. Indeed, we observed the formation of a recombinant CrPV-1A complex with EloBC. Furthermore, CrPV-1A mutations in the BC-box motif (CrPV-1A(BCmut)) completely abolished the interaction (Fig 6C, left panel). These results show that CrPV-1A has an exposed helical motif that functions as a BC-box to promote E3-ligase recruitment. Drosophila Cul2 shares a high degree of amino acid sequence similarity with human Cul2 (43.78%) (Fig S5B). Since human Cul2 interacts with BC-box and EloC (Nguyen et al., 2015), we next incubated the purified and untagged CrPV-1A-EloBC complex (Fig 6C, Step-I) with E.coli cell lysates expressing a His-tagged N-terminal domain of human Cul2 (hCul2-NTD). By NAC, we observed co-purification (Fig 6C, right panel) of CrPV-1A-EloBC with His-tagged hCul2-NTD. Formation of the CrPV-1A-EloBC ternary complex (Fig 6D, left panel) and CrPV-1A-EloBC-Cul2 quaternary complexes (Fig 6D, right panel) was further assessed by gel filtration chromatography. Thus, Cul2 is recruited to the CrPV-1A-EloBC complex by a similar mechanism to that reported for human VHL-EloBC (Cardote et al., 2017, Nguyen et al., 2015).
Fig 6: CrPV-1A hijacks cellular E3 ligase components.
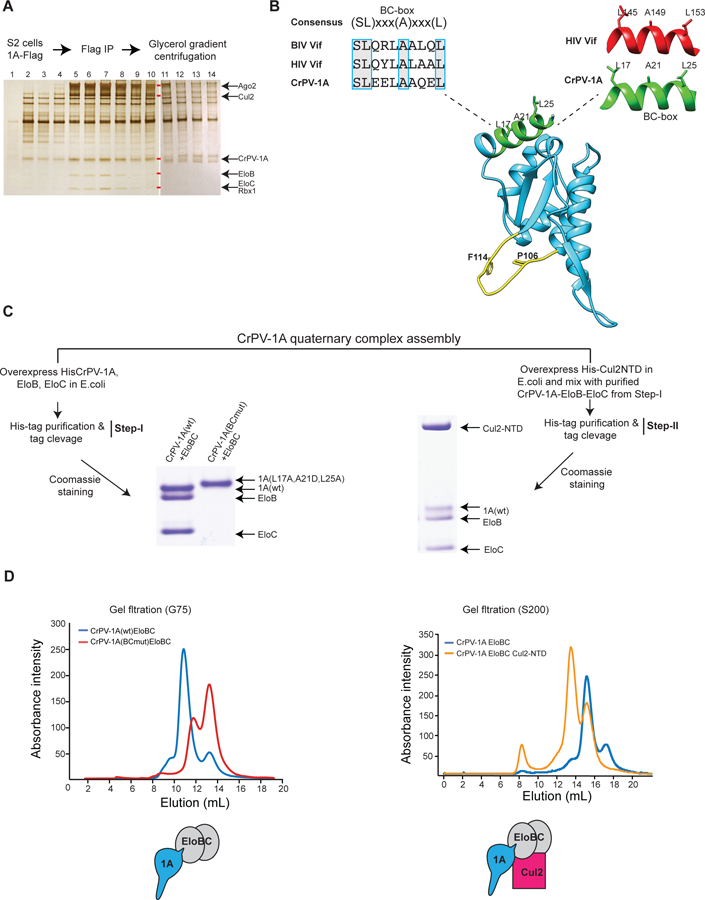
(A) Glycerol gradient fractions containing CrPV-1A associated proteins were analyzed using SDS-PAGE and silver staining. Gel bands corresponding to Ago-2, Cul2, EloB, EloC, and Rbx1 are indicated with red rectangles, and their identities were verified by mass spectrometry. After fraction 11, HSP27 was observed to run with similar electrophoretic mobility to CrPV-1A as confirmed by mass spectrometry. One of two representative silver stain gels is shown (n = 2). (B) Blue boxes highlight the consensus BC-box amino acid sequences of CrPV-1A, HIV Vif, and BIV Vif. Structural conservation of the BC-box motif for CrPV1A and HIV Vif is shown. The TALOS element residues (P106 and F114) and the BC-box motif are highlighted in yellow and green, respectively. (C) Assembly of CrPV-1A+EloB+EloC ternary and CrPV-1A+EloB+EloC+Cul2 quaternary complexes were examined by overexpression the proteins in E.coli, His-tag pull-down, and Coomassie blue staining. Elution patterns of the complexes was examined by Superdex-G75 and Superdex-200 column chromatography, respectively.
See also Fig S5
We next confirmed that this E3 ligase assembly mode is preserved in S2 cells. AP-MS analysis of stable S2 cell lines expressing CrPV-1A(BCmut) showed that the BC-box mutation completely abrogated interaction with EloBC and Cul2 in vivo (Fig 5A). Importantly, as expected CrPV-1A(BCmut) still interacted efficiently with Ago-2 since the TALOS element mediating Ago-2 binding and inhibition is on a interface of CrPV-1A distal to the BC-box motif (Fig 6B). Taken together, we concluded that CrPV-1A recruits EloBC/Cul2/Rbx1 in a BC-box dependent manner to assemble a virus hijacked E3 ligase and disrupting this interaction does not affect interaction with Ago-2.
CrPV-1A ubiquitin ligase promotes virus replication
To test if CrPV-1A acts as a specificity factor to create a viral ubiquitin E3 ligase to target Ago-2 for degradation, we expressed Ago-2-flag in CrPV-1A or CrPV-1A(BCmut) expressing S2 cells and monitored Ago-2 expression in the presence and absence of the proteasome inhibitor MG132 (Fig 7A, 7B). Ago-2 expression was only detected in the presence of MG132 or when co-expressed in CrPV-1A(BCmut). Of note, mock S2 cell lines with no expression of the CrPV-1A protein did not degrade Ago-2 protein (Fig S6A). These experiments suggest that Ago-2 degradation is promoted by CrPV-1A in an EloBC-Cul2 dependent manner. To assess if the E3 ligase function is required for RNAi suppression, we compared the effect of mutations in the BC-box of CrPV-1A with those in the TALOS element (F114A). Notably, our RNAi assay showed that a functional BC-box is dispensable for RNAi suppression by CrPV-1A protein (Fig 7C). Conversely, the TALOS element was dispensable for formation of the Cul2-E3 ligase complex, since CrPV-1A(F114A) can efficiently interact with EloBC (Fig S6B). Indeed, gel filtration analysis showed similar elution patterns for the CrPV-1A(F114A)/EloBC and CrPV-1A/EloBC complexes (Fig S6C). We conclude that Ago-2 binding to CrPV-1A is independent of its interaction with EloBC.
Fig 7: CrPV-1A-hijacked E3 ligase contributes to virus replication.

(A, B) The stability of Ago-2 in CrPV-1A or CrPV-1A(BC-mut) expressing S2 cells at indicated time points (h) in the presence or absence of MG132 was visualized by Western blot. A lane between 2 and 3 as shown in Fig B has been removed from the original blot for clarity. Western blots analysis are representative of three independent experiments (n = 3) (C) Effects of a single point or combined mutations in the BC-box of the CrPV-1A on RNAi suppression using the RNAi reporter assay. RNAi suppression data represent mean (±SD) of at least three independent experiments (n = 3) for each condition. (D) Red dots represent the ubiquitin sites on Ago-2 derived from S2 cells or S2 cells expressing CrPV-1A protein by UbiScan analysis. The Ubiquitin sites reported are representative of three independent experiments (n = 3). Approximate boundaries of the domains in Drosophila Ago-2 were derived from homology-based modeling using the structure of human Ago-2. (E) S2 cells expressing Ago-2-flag were depleted for Cul2 and EloB followed by infection with CrPV at MOI of 2. Twenty hours post infection, Ago-2 expression in S2 cells and loading controls were analyzed by Western blot using flag and actin antibodies, respectively. Western blots analysis is representative of two independent experiments (n = 2). (F) Cul2-depleted (dsCul2) or Luc-depleted (dsLuc) S2 cells were monitored by infection of CrPV at MOI of 2, and viral titer was measured by FFU assay. Data shows one of three representative experiments with the mean (±SD) of three replicates (n = 3). *p<0.05; ***p<0.001 (Multiple t test). (G) Ago-2-depleted S2 cells were transfected with CrPV3 RNA or CrPV3 variant RNA and viral titer was measured by FFU assay. Each titer value represents the mean (±SD) of at least three replicate experiments (n = 3). The statistical significance represents measurement compared to CrPV3. ns, not significant; *p<0.05 (Unpaired t test) (H) A model showing the mechanism of immune restriction against CrPV (top) and the mechanism deployed by CrPV to counteract this host immune response (bottom) in Drosophila cells.
Next, we examined if CrPV-1A affects the ubiquitination status of Ago-2. A proteome-wide UbiScan using an anti-diglycine remnant (K-ε-GG) antibody identified ubiquitination sites in S2 cells or S2 cells expressing CrPV-1A. In total, we identified 23 ubiquitination sites on endogenous Drosophila Ago-2 spanning all protein domains except for the PolyQ repeat region in both of these conditions (Fig 7D, Table S4). Thus, endogenous function and/or levels of Ago-2 are likely regulated by ubiquitination even in uninfected cells. Of note, expression of CrPV-1A enhanced ubiquitination at several sites on Ago-2 upon treatment with proteasome inhibitor MG132 including K406, K515, K547, K690, K735, K879, K916, K935, and K1092 (Fig S7). We then examined Ago-2 degradation during CrPV infection. We depleted Cul2 and EloB from S2 cells by dsRNA soaking prior to infection with CrPV. Compared to control cells, knockdown of either Cul2 or EloB stabilized Ago-2 protein levels by CrPV infection (Fig 7E). These data suggest that a CrPV-1A-mediated assembly of the E3 ligase promotes Ago-2 degradation during infection in Drosophila cells.
To see if the CrPV-1A E3 ligase has a role during virus replication, we tested whether Cul2 depletion affects viral replication. Cul2-depleted S2 cells were infected with CrPV for 8, 12, and 14h. Cul2 knockdown reduced viral replication to 50% by 14h (Fig 7F), suggesting that the viral E3 ligase is important for efficient CrPV replication. We engineered BC-box mutations in CrPV-1A in the CrPV3 infectious cDNA and monitored virus replication in S2 cells. Mutations in the BC-box, L17A, A21D or the combined L17A+A21D+L25A/(BC-mut) led to drastic decreases in virus replication, with over a 3-log reduction in viral titer for the BC-mut mutations. By contrast, L25A alone did not affect virus production (Fig 7G). The drastic reduction in titer of BC-mutant viruses compared to CrPV infection in Cul2-knockdown condition (Fig. 7F) could possibly due to the incomplete depletion of Cul2 by dsRNA soaking (~80%, data not shown). Importantly, when Ago-2 was depleted from S2 cells by dsRNA soaking, the defective replication phenotype of BC-mutant viruses was significantly rescued albeit not to the same titer obtained with WT viruses (Fig 7G). Of note, the rescue of the BC-mutant viruses was not observed in the Cul2-depleted S2 cells indicating that degradation of Ago-2 is required for sustained infection (Fig S6D). The lack of complete rescue of the BC-mutant viruses could be due to incomplete Ago-2 depletion or to additional functions of the CrPV-1A-Cul2/E3 ligase during replication. In sum, these data indicate that the E3 ligase activity of CrPV-1A plays a key role in viral replication that is linked to Ago-2 degradation.
Discussion
In insect and plant cells, the host RNAi machinery plays an essential antiviral function (Nayak et al., 2013; Szittya and Burgyán, 2013). Thus, many viruses target different components of the antiviral RNAi pathway, including the core effector Ago-2 (Csorba et al., 2015; Gammon and Mello, 2015). In the present study, structural and functional characterization of the Ago-2 suppressor, CrPV-1A, uncovered an unusual mechanism of action. The residues responsible for blocking Ago-2 catalytic activity map to a 14-mer-element in a flexible loop region within CrPV-1A, which we term TALOS(Fig 1, Fig 2C). The function of the TALOS element depends on the identity of two residues, P106 and F114, precisely spaced to face each other in the loop. Changing their spacing or nearby local structure by deletion or insertion in the loop interferes with CrPV-1A mediated RNAi inhibition and Ago-2 interaction (Fig 3A, Fig 3B). Within the loop, the proline residue is essential, conformationally rigid, and likely acts as a pivot that promotes the proper orientation of F114 for Ago-2 interaction. Indeed, proline substitution with either a flexible glycine or bulky aromatic amino acids (Tyr, Trp, and Phe) causes loss of RNAi inhibition activity (Fig 3C). The requirement of F114 for Ago-2 binding and inhibition is less stringent since a range of hydrophobic and aromatic amino acids can maintain binding and inhibitory activity. It is thought that CrPV-1A blocks Ago-2 by preventing base pairing between the Ago-2-bound guide RNA and a target (e.g. viral) RNA (Watanabe et al., 2017). Based on our data we hypothesize that Ago-2 inhibition may require the residue at position 114 to prevent the Ago-2-bound guide RNA from engaging with the target. Indeed, in vitro experiments show that alanine substitution at either P106 or F114 abolish the capacity of recombinant CrPV-1A proteins to inhibit Ago-2-directed hydrolysis of the target mRNA (Fig 2E).
Our mutagenesis analysis of the TALOS element showed significant plasticity in the sequence requirement for Ago-2 inhibition. From an evolutionary and pathogenesis point of view this may increase virus evolvability in the context of a virus-host arms race, which drives rapid evolution of both antiviral RNAi genes (e.g., Dicer-2, and Ago-2) and suppressors (Obbard et al., 2006). Judging from the plasticity of the TALOS element, we anticipate that equivalent Ago-2-interacting determinants may have evolved in other insects and plant viruses to modulate Argonaute-silencing. Argonaute proteins across kingdoms show remarkable structural similarities and share a conserved mechanism of RNA silencing (Schirle and MacRae, 2012; Swarts et al., 2014). Thus, a TALOS-like element may be a functionally flexible, broadly acting eukaryotic motif employed by viruses to neutralize Ago-2-dependent host immunity. Indeed, F114 residue in CrPV-1A interacts with Aedes Ago-2 suggesting that the TALOS element could target a conserved region of the dipteran Ago-2. Moreover, since CrPV infects diverse insects, it is possible that the TALOS element plasticity provides a context-dependent adaptive advantage, where distinct residues at position 114 can modulate Ago-2 suppression to different degrees and in distinct insect species. Importantly, depletion or elimination of Ago-2 protein in both S2 cells and adult flies, respectively, rescues F114 mutant virus replication directly linking the TALOS element action to Ago-2 (Fig 4C, 4D, 4E).
CrPV-1A has an additional sequence element, the BC-box, which serves to recruit the Drosophila E3-ligase adaptor protein EloBC (Fig 5A, Fig 5B) and this interaction drives viral E3 ligase assembly (Fig 6). Consequently, CrPV-1A not only inhibits RNAi in the host, but also targets Ago-2 for degradation (Fig 7A, 7B) thus enabling optimal viral replication (Fig 7E, 7F, 7G). Interestingly, Ago-2 is ubiquitylated in Drosophila cells and CrPV-1A regulates the ubiquitination status of Ago-2 in a proteasome- dependent manner (Fig 7D, S7). In principle, CrPV-1A may also target other host proteins for degradation affecting cellular pathways during infection. For example, CrPV-1A can also inhibit stress granule formation in Drosophila cells (Khong et al., 2017); however, how these functions are related remains to be determined. Altogether, we demonstrate how viral evolutionary plasticity can create protein-interaction motifs, which in turn lead to disruption of host pathways (Fig 7H).
Of note, other viral proteins in plants and animals exploit the ubiquitin-proteasome pathway to target host proteins for degradation. For example, the plant polerovirus encodes P0 RNA silencing suppressor that targets Argonaute 1 for degradation through an F-box motif (Baumberger et al., 2007; Csorba et al., 2010; Pazhouhandeh et al., 2006). In animal viruses HIV and BIV, a specialized BC-box protein Vif mediates degradation of APOBEC3 proteins (Desimmie et al., 2014). Unlike these cases, CrPV-1A combines the activities of a direct competitive inhibitor to block Ago-2 endonuclease activity with that of an E3-ubiquitin ligase for the catalytic degradation of Ago-2.
We hypothesize that the dual acquisition of TALOS and BC-box sequence elements, each conferring binding specificity to a distinct host protein allowed CrPV to combine and amplify immune-suppressive activities. Importantly, both activities contribute to viral pathogenesis. Acquisition of E3 ligase activity to inactivate Ago-2 may be relevant under conditions where CrPV establishes a persistent and asymptomatic infection as reported in field population of crickets and honeybees(Anderson and Gibbs, 1988; Scotti et al., 1981). Here, lower CrPV-1A expression levels may be insufficient to block Ago-2 activity effectively and thus Ago-2 degradation could be critical for effective long-term replication of the virus. It is possible that distinct CrPV-1A functions are required for different stages of virus replication. For example, the TALOS element may antagonize the active form of Ago-2, associated with the RISC complex, while the BC-box may affect the homeostatic regulation of Ago-2 ubiquitination to prevent active de novo synthesized Ago-2 from participating in antiviral immunity. Overall, our study uncovers a two-step mechanism to regulate RNAi antiviral immunity and suggests that the evolutionary plasticity built into CrPV-1A may enable fine tuning of the virus-insect interactions to ensure successful virus replication. In addition, these findings test critical aspects of the structure and functionality of a multifunctional protein from the perspective of a host-virus arms race. The increasing relevance of insect-virus interactions on global health, driven by spread of Dengue, Chikungunya, and Zika viruses, highlight the importance of understanding how viruses modulate the RNAi antiviral response in insects.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Raul Andino (raul.andino@ucsf.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila melanogaster
Wolbachia-free fly stocks listed in Key Resource Table were used for survival assays. Flies were maintained at 25°C and 70% humidity on standard cornmeal food. 6-day-old female flies were used for experiments.
Drosophila Schneider 2 cells
S2 cells (Thermo Fisher Scientific) derived from Oregon R late embryonic stage male fly were cultured without CO2 in either Shield’s and Sang media (Sigma) or Schneider’s medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, 1X Penicillin-Streptomycin-Glutamine at 25°C.
Aedes aegypti Aag2 cells
Aag2 cells (Peleg et al., 1968) derived from Aedes aegypti mosquito (Sex unspecified) were cultured at 28°C without CO2 in Schneider’s Drosophila medium (Thermo Fisher Scientific), supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1X non-essential amino acids (NEAA, 10 mM each of Glycine, L-Alanine, L-Asparagine, L-Aspartic Acid, L-Glutamic Acid, L-Proline, and L-Serine in de-ionized water), and 1X Penicillin-Streptomycin-Glutamine (Thermo Fisher Scientific).
E. coli cells
For in vitro biochemistry experiments and crystallization of native CrPV-1A, protein expression was carried out using BL21(DE3) cells grown at 37°C in Luria-Bertani (LB) medium. For NMR studies, CrPV-1A protein expression was carried out using BL21(DE3) cells grown at 37°C using SBMX minimal media (Daugherty et al., 2010). For phase determination of Sel-Met derivative of CrPV-1A, protein expression was carried out using B834(DE3) grown at 30°C in PASM-5052 media (Studier et al., 2005).
Cricket Paralysis Virus
CrPV-3 was grown and propagated in Drosophila S2 cells using Shield’s and Sang media (Sigma) at 25°C as described (Kerr et al., 2015).
METHOD DETAILS
Purification of native CrPV-1A159
DNA fragment encoding the N-terminal 159 amino acids of CrPV-1A (CrPV-1A159) was cloned in pHis-Gb1-II vector (Nayak et al., 2010) and the resultant construct (pHis-Gb1-II-CrPV-1A159) was expressed as an N-terminal His-tag fusion protein using Escherichia coli BL21(DE3) cells in Luria-Bertani (LB) medium. Purifications of the His-tagged protein was carried out using Ni-NTA agarose (QIAGEN) in a buffer containing 20 mM Hepes pH 7.4, 150 mM KCl, 150 mM Na2SO4, 10 mM beta-mercaptoethanol, 10% glycerol containing cOmplete Mini EDTA-free protease inhibitor tablets (Roche). The N-terminal His-tag from the fusion protein was cleaved by in-house purified His-tagged TEV protease. The cleaved His-Tag and the TEV protease were subsequently removed by Ni-NTA agarose chromatography. The flow-through containing the untagged protein was further purified over a monoQ column equilibrated with buffer A (30 mM Hepes pH 7.9, 150 mM KCl, 10 mM beta-mercaptoethanol). Elution of the protein was carried out in buffer A containing 700 mM KCl. The purified sample was dialyzed to bring down the salt concentration to 150 mM KCl and further analyzed over a Superdex 75 gel filtration column equilibrated with buffer A.
Purification of Sel-Met derivative of CrPV-1A159
Methionine auxotroph B834(DE3) cells (Novagen) with pHis-Gb1-II-CrPV-1A159 plasmid was grown in 50 mL standard LB medium until the OD600 = 0.6. Cells were harvested and re-suspended in 1 L fresh PASM-5052 media containing methionine, Selenomethionine, and Vitamin B12 at concentration 10 mg/L, 125 mg/L, 100 nM respectively (Studier et al., 2005). The culture was grown at 37°C till the OD600 = 0.6, induced with IPTG to a final concentration of 0.1 mM, and grown for an additional 5 hours at 30°C. Although PASM-5052 is an auto-inducing medium, IPTG was used for the induction of the CrPV-1A159 protein. Incorporation of Selenomethionine was verified by electrospray ionization-time of flight (ESI-TOF) mass spectrometric analysis using a LCT Premier LC/electrospray ionization-MS instrument (Waters Corporation) and MassLynx software. Selenomethionine-labeled CrPV-1A159 was purified as described above for the native protein.
Purification of CrPV-1A159+EloBC+Cul2 complex
Untagged Drosophila Elongin B and Elongin C were cloned using pACYCDuet-1 vector (Novagen). Co-expression of the His-CrPV-1A, untagged Elongin B and Elongin C proteins was achieved by transforming pHis-Gb1-II-CrPV-1A159, pACYCDuet-1-Elongin B-Elongin C plasmids in BL21(DE3) cells, growing cells in LB and inducing with 0.5 mM IPTG for 4 hours at 30°C. Purifications of the His-tagged protein complex was carried out using Ni-NTA agarose in a buffer containing 20 mM Hepes pH 7.4, 150 mM KCl, 150 mM Na2SO4, 10 mM beta-mercaptoethanol, 10% glycerol containing cOmplete Mini EDTA-free protease inhibitor tablets (Roche). The N-terminal His-tag from the CrPV-1A159 fusion protein was cleaved by in-house purified His-tagged TEV protease. The cleaved His-Tag and TEV protease were subsequently removed by Ni-NTA agarose chromatography. The flow-through contained the untagged purified CrPV-1A159+Elongin BC complex. For expression of human Cul2, a plasmid (pNIC-Cul2-NTD) (Thomas et al., 2013) containing His-tagged N-terminal half domain of Cul2 (Cul2NTD) was expressed in BL21(DE3) cells as described above. Subsequently, the untagged purified CrPV-1A159+Elongin BC complex was added to the E. coli cell lysates expressing His-Cul2-NTD. Purifications of the His-Cul2NTD was carried out using Ni-NTA agarose in a buffer containing 20 mM Hepes pH 7.4, 300 mM KCl, 150 mM Na2SO4, 10 mM beta-mercaptoethanol, 10% glycerol containing protease inhibitor cocktail (Roche). The N-terminal His-tag from the His-Cul2NTD was removed as described above.
Multiangle Light Scattering
The purified CrPV-1A159 protein sample (5 μg/μL in 50 μL volume) was applied over a KW803 HPLC column (Shodex) equilibrated in 30 mM Hepes pH 7.9, 150 mM KCl, 10 mM DTT at a flow rate of 0.35 mL/min. The column was coupled with multi-angle light scattering equipment, and scattered light intensity (refractive index) of the column eluate (elution volume) was recorded using a DAWN-HELIOS laser photometer (Wyatt Technology). The molecular mass of the eluent was computed using the ASTRA software (Wyatt Technology). All the measurements were performed at 4°C.
NMR Spectroscopy
For CrPV-1A159 1H-15N HSQC NMR spectroscopy, protein expression and purification were performed as described above for CrPV-1A159 native protein except BL21(DE3) cells were grown in SBMX, a phosphate-based minimal media (Each liter of SBMX media contains 40 mL “25X Salts” (32.3 g KH2PO4, 175 g K2HPO4, 36.5 g NaCl, 1.2 g K2SO4 in 1 L H2O), 4 g glucose, 1 g NH4Cl, 1 mL 0.1% (w/v) Thiamine-HCl, 2 ml “O solution” (500 mL total solution containing 26.8 g of MgCl2.6 H2O + 10 ml of a solution containing 92 ml H2O, 8 ml concentrated HCl, 5 g FeC12.4 H2O, 184 mg CaC12.2 H2O, 64 mg H3BO3, 40 mg MnCl2.4 H2O, 18 mg CoCl2.6 H2O, 4 mg CuC12.2 H2O, 340 mg ZnCl2, and 605 mg Na2NoO4.2 H2O), 1 mL ‘Vitamin Solution’ (2.2 mg biotin, 2.2 mg folic acid, 220 mg p-aminobenzoic acid, 220 mg riboflavin, 440 mg pantothenic acid, 440 mg niacinamide, 440 mg pyridoxine-HCl in 1 L 50% (v/v) EtOH) and ampicillin to 100 ug/ml as described previously (Daugherty et al., 2010). The purified protein was buffer exchanged in 20 mM Hepes pH 7.5, 100 mM KCl, 100 mM Na2SO4, 10 mM DTT and HSQC spectrums were acquired using Bruker 800 MHz spectrometer. Spectra were processed with NMRPIPE (Delaglio et al., 1995) and analyzed with SPARKY (Goddard and Kneller, 2007).
Structure determination of CrPV-1A159
Crystals were grown at room temperature using hanging drop diffusion by mixing protein (7.7 mg/mL) and reservoir solution (17% PEG 3350 and 0.2 M LiCl) at 0.5:1 ratio. Crystals were gradually transferred to reservoir solution containing 20% and 25% PEG3350 and flash frozen in liquid nitrogen. Data were collected at an advanced light source, Lawrence Berkeley National Laboratory, Berkeley on Beamline 8.3.1. The native CrPV-1A159 crystals diffracted to 2.6 Å resolution. Two-wavelength anomalous dispersion data of selenium derivatives were collected at peak and remote wavelengths. The collected data were integrated and scaled using HKL2000 (Otwinowski and Minor, 1997). Multi-wavelength anomalous dispersion method was used for phase calculation. Five selenium sites were found by SOLVE program (Terwilliger et al., 1999) and the density modification was performed using RESOLVE (Terwilliger et al., 2003). The initial model was manually built by using COOT program (Emsley and Cowtan, 2004). Several cycles of rigid-body annealing, positional refinement, simulated annealing and B-factor refinement and model rebuilding were conducted using CNS (Brunger, 2007) and COOT programs. The dimeric model of CrPV-1A159 containing two identical monomeric units was generated using REFMAC (Murshudov et al., 2011) with R/Rfree of 22.7/28.5 %. The figures were generated using UCSF Chimera (Pettersen et al., 2004).
Limited trypsin proteolysis
Purified CrPV-1A159 protein (2.35 μg) was incubated with sequencing grade trypsin (2.35 ng) (Promega) in a buffer containing 50 mM Tris pH8.0, 75 mM KCl, 5 mM beta-mercaptoethanol for 30 min at room temperature. The reaction was stopped by adding SDS sample loading buffer, and the extent of proteolysis was analyzed by Coomassie blue staining. The mass of the fragments was analyzed by electrospray ionization-time of flight (ESI-TOF) mass spectrometry using a LCT Premier LC/electrospray ionization-MS instrument (Waters Corporation) and MassLynx software.
Site-directed mutagenesis
For RNAi reporter assay in S2 cells, CrPV-1A148 was cloned in vector pAc5.1/V5-HisA (Thermo Fisher Scientific). For expression in Aag2 cells, CrPV-1A148 was cloned in a vector (pUb) under the control of polyubiquitin promoter (Gift from Zach Adelman). Amino acid mutations into CrPV-1A or in the CrPV infectious clone were introduced by PCR using Phusion High Fidelity DNA Polymerase enzyme (NEB) and complementary phosphorylated primers. The DpnI digestion, plasmid transformation, and plasmid isolation were carried out by standard molecular biology techniques. Transformations and propagations of plasmid constructs were carried out using SURE 2 Supercompetent Cells (Agilent Technologies). Desired mutations in CrPV-1A and the infectious CrPV clone were verified by Sanger sequencing. For infectious clones entire genomic region was sequenced to ensure the integrity of the viral genome.
Generation of stable S2 cell lines
Transgenes encoding CrPV-1A, DCV-1A, and Argonaute 2 were cloned in frame with C-terminal 3xFLAG and 6xHis tags in an inducible expression vector pMT/V5-HisA (Thermo Fisher Scientific). Stable S2 cell lines were generated by cotransfecting pMT plasmids (19 μg) and pCoBlast selection plasmid (1 μg) using Effectene transfection reagent (QIAGEN) and propagating the S2 cells in the presence of 25 μM Blasticidin for three weeks as described by the manufacturer (Thermo Fisher Scientific). Stable S2 cell lines were maintained in Schneider’s medium supplemented with 25 μM Blasticidin. Expression of proteins was induced by adding 500 μM copper sulfate to the medium for desired time periods as indicated.
Dual luciferase reporter Assay
Drosophila S2 cells were grown on 96 well plates (approximately 30,000 cells/well) for 24 hours before transfection. Plasmids encoding firefly luciferase (pMT-Fluc) (12 ng), Renilla luciferase (pMT-Rluc) (3 ng) and CrPV-1A148 (pAc5.1-CrPV-1A148) (50 ng) were co-transfected with specific dsRNA targeting firefly luciferase (Fluc dsRNA) (10 ng) or non-specific dsRNA targeting green fluorescence protein (GFP dsRNA) (10 ng) using Effectene transfection reagent (QIAGEN). Twenty-four hours post-transfection, expressions of firefly and Renilla luciferases under the control of metallothionine (MT) promoter were induced overnight by adding 500 μM CuS04 to the medium. S2 cells were lysed in 100µl of 1X lysis buffer, and measurement of firefly and Renilla-luciferase were carried out using Dual-Luciferase Reporter Assay kit according to the manufacture’s instruction (Promega). Renilla expression was measured for normalization purpose. The fold luciferase silencing was obtained by dividing the normalized firefly luciferase signal resulting from treatment with non-silencing control GFP dsRNA by specific Fluc dsRNA. Each experiment was preformed at least in 3 replicates.
Immunoprecipitation and Western blotting
pAc5.1/V5-HisA or pUb plasmid constructs (1–2 µg) expressing Flag-tagged CrPV-1A148 were transfected into S2 cells or Aag2 cells in 10 cm dishes format using Effectene (QIAGEN) and TransIT-2020 (Mirus Bio) transfection reagents respectively. At 20 hours post transfections, cells were harvested and washed 2X with PBS at 4°C, and the pellet was stored at −70°C for further analysis. After thawing, cells were kept in ice and re-suspended in IP buffer [30mM Hepes pH 7.4, 150mM KCl, 2mM MgOAc, 2mM DTT, 0.1% Triton-X-100] supplemented with cOmplete Mini EDTA-free protease inhibitor tablet (Roche) for 15 minutes. The extracts were centrifuged at 14,000 rpm for 30 minutes, and the supernatants were either probed using Western blot analysis or added to Dynabeads M-270 Epoxy (Thermo Fisher Scientific) conjugated with anti-Flag M2 antibody (Sigma) for 1hr at 4°C for immunoprecipitation analysis. Beads were washed 5X with IP buffer, and bound proteins were eluted from the beads in SDS sample buffer by boiling at 95°C for 3 minutes. With exception, immunoprecipitated samples from Aag2 cells were eluted using 3X flag peptides (Sigma) and stained using Pierce Silver Stain Kit (Thermo Fisher Scientific). The immunoprecipitated samples from S2 cells were run on 4–20% gradient SDS-PAGE gel and transferred to PVDF membrane (Millipore). The membrane was blotted with either for Drosophila Ago-2 (Abcam) or anti-Flag M2 antibody (Sigma) and signals were developed using ECL plus chemiluminescence substrate (Thermo Fisher Scientific). Loading controls were visualized by using antibody raised against either tubulin or actin. Western blotting analyses were performed using at least two independent experiments.
Ago-2 slicing assay
The Ago-2 slicing assays were performed as described previously (Nayak et al., 2011). A firefly luciferase (Fluc) DNA template containing a T7 promoter was amplified using primers T7-Luc For and Luc Rev. Primers are listed in Table S2. The Fluc mRNA targets (592 bp in length) were synthesized using the Fluc DNA template by in vitro transcription at 37°C. The Urea-PAGE gel-purified mRNA transcripts were radio-labeled with 5’ G-cap using Vaccinia Capping System (New England Biolabs). Cell-free extracts were prepared by resuspending Drosophila S2 cells in Lysis buffer [30 mM Hepes-KOH (pH 7.4), 100 mM KOAc, 2 mM MgCl2] supplemented with cOmplete Mini EDTA-free protease inhibitor tablet (Roche) and freezing the resuspended cells at −80°C for 24 hours. The frozen cells were thawed and kept on ice for 1 hour and centrifuged at 13,000xg for 20 min. The supernatant contained S2 cell-free extract and was incubated with a specific siRNA targeting the Fluc mRNA substrate for 30 minutes followed by addition recombinant CrPV-1A159 for an additional 10 minutes. The target cleavage reactions were initiated by incubating the radiolabeled Fluc mRNA substrate for 3hr at 25°C. A 100nt 5’ cleaved Fluc mRNA product was analyzed using 6% denaturing polyacrylamide gel electrophoresis and autoradiography. Slicing assay analyses were performed using at least three independent experiments.
Sequence alignment and Homology modeling
The N-terminal 384 amino acids of human Cullin 2 (hCul2) and Drosophila Cullin 2(dCul2) were aligned using Clustal Omega (Goujon et al., 2010). Modeling of the CrPV-1A flexible loop with insertion and deletion of amino acids were performed using SWISS-MODEL(Bordoli et al., 2008) (Biasini et al., 2014). The CrPV-1A PDB file together with CrPV-1A protein sequences (containing either insertions or deletions) was submitted using “User Template” option for model building. Homology-based structural modeling of the Drosophila Ago-2 was performed using Chimera interface to MODELLER (Pettersen et al., 2004; Webb and Sali, 2016) using a human Ago-2 structure (PDB ID: 4OLA) as a template (Schirle and MacRae, 2012).
Fluorescence Foci Forming Units assay
Determination of CrPV viral titres by Fluorescence Foci Forming Units assay were performed as previously described (Garrey et al., 2010). S2 cells (1.5 ×106) were incubated with 10 µL of serial dilutions of virus for 30 minutes, then resuspended in 500 µL of Shield’s and Sang media (Sigma). Of this, 100 µL was aliquoted into a 96-well plate coated with concavilin A (0.5 mg/mL; Calbiochem) and incubated at 25°C for 8 h. Cells were then washed with PBS before being fixed with 3% paraformaldehyde for 15 min followed by methanol for 10 min. The fixed cells were washed with PBS and then incubated with CrPV anti-VP2 antibody (1:250 dilution in 5% bovine serum albumin in PBS) for 1 h at room temperature. Subsequently, cells were washed three times with PBS and incubated with a Texas Red IgG anti-rabbit (1:500 dilution in 5% bovine serum albumin in PBS; Invitrogen) for 1 h at room temperature. Finally, cells were washed with PBS and stained with Hoechst dye (0.5 μg/mL) in PBS. The number of infected cells was quantified after plates were analyzed with a Cellomics Arrayscan HCS instrument. Each titre is the result of at least three replicate experiments.
dsRNA knockdowns and viral rescue experiments
To generate dsRNA for AGO-2 and Cul2, total cellular RNA was isolated from S2 cells using TRIzol (Thermo Fisher Scientific). Following this, cDNA was generated via RT-PCR using random hexamer primers and Superscript III reverse transcriptase (Invitrogen). Sequences to make dsRNA were amplified using primers containing a T7 promoter flanking either side of the amplicon. Primers AGO-2-for and AGO-2-rev were used to amplify the AGO-2 sequence, while primers Cul2-for and Cul2-rev were used to amplify the Cul2 sequence. GFP was amplified from a GFP plasmid construct with primers GFP-for and GFP-rev. Firefly luciferase was amplified from a Luciferase construct with primers Luc-for and Luc-rev. Primers are listed in Table S2. After purification, 1 µg of each amplicon was used for in vitro transcription at 37°C. RNAs were subsequently purified via RNeasy kit (QIAGEN). For dsRNA-mediated knockdowns, 6.5 × 106 S2 cells were seeded into a T25 flask (Corning) and allowed to settle for 1 h at 25°C. Media was aspirated and cells were treated with 85 µg of dsRNA in 1.65 mL of serum-free media for 1 h while rocking at 25°C. 3.35 mL of complete media (with 10% FBS) was then added and cells were incubated for 72 h at 25°C. After 72 h, cells were pelleted, subjected to a second dose of dsRNA (85 µg) in the same manner as above and then incubated for a further 48 h. Following dsRNA treatment, 1.5 × 106 S2 cells were seeded onto each well of a 12-well plate and allowed to settle for 1 h. For the transfection of in vitro transcribed RNA derived from the respective CrPV-3 mutants, 1.5 µg of each RNA was incubated with 125 µL of OptiMEM (Invitrogen) serum-free media for 5 min at room temperature. Simultaneously, 1.25 µL of Lipofectamine 2000 (Invitrogen) was incubated in 125 µL of OptiMEM for 5 min at room temperature. The RNA and Lipofectamine was then mized and incubated for 15 min at room temperature. Media was aspirated from cells and the RNA:Lipid mixture was added to cells and was incubated at 25°C while rocking for 15 min. Finally, 1 mL of complete Shield’s and Sang media (Sigma) was added to the cell and were allowed to incubate for 48h at 25°C before harvesting the cell pellets. Viral titers measurements were performed in three replicate experiments and yield on harvested cells was determined by Fluorescence Foci Forming Units assay.
Fly survival assay
Both fly strains, y*w* and Ago-2414 were anesthetized with CO2 and injected with 50 nL of appropriate CrPV variants dilutions made in PBS. Nanoject II (Drummond Scientific) nanoinjector, equipped with fine glass needles was used. Flies were carefully placed into new food vials and scored for one week. Flies were moved to new food vials every 3 days. Fly survival assay was analyzed using three independent fly injection experiments. For virus RNA loads, 3–4 days post injection (time at which flies started showing mortality) flies were anesthetized, frozen in dry ice and crushed in TRIzol (Thermo Fisher Scientific). Total RNA isolation was performed according to the manufacturer instructions. 10 individuals were typically used per data point. RNA samples were treated with DNAse I (New England Biolabs) and reverse transcribed with Maxima Reverse Transcriptase (Thermo Fisher Scientific) both according to the manufacturer’s recommendations. qPCR analysis of three independent replicates was performed with SensiFAST SYBR no ROX kit (BIOLINE). Relative quantification of CrPV RNA was carried out by using qPCR CrPV Fwd and qPCR CrPV Rev primers and normalization to internal control RpL12 using qPCR RpL12 Fwd and qPCR RpL12 Rev primers. Primers are listed in Table S2. Mortality curve and viral RNA yield were generated using ggplot2 in R (Wickham et al., 2009).
Affinity Purification Mass-spectrometry
Each AP-MS experiment was repeated with 3 to 5 biological replicates per bait protein. Stable S2 cells expressing Flag-His tagged bait proteins were harvested at 4°C and washed twice with PBS. Cell pellets were re-suspended in lysis buffer (30 mM Hepes pH 7.4, 150 mM potassium-chloride, 2 mM magnesium-chloride, 0.5% Triton-X-100, supplemented with cOmplete, Mini-EDTA-free protease inhibitor tablets (Roche) and passed five times through a 27-gauge needle syringe. Cell lysates were centrifuged at 15,000 rpm for 30 minutes and supernatants were then pre-incubated with EZview Red Protein A gel beads (Sigma) for 2 hours at 4°C followed by mixing with EZview Red ANTI-FLAG M2 Affinity gel beads for 2 hours at 4°C. The beads were washed five times with lysis buffer and bound proteins were eluted with lysis buffer containing 400 μg/ml 3x FLAG peptide (Sigma) for 60 minutes at 4°C with slight agitation. Eluted samples were further incubated with Dyanbeads for His-Tag protein isolation (Thermo Fisher Scientific) for 60 minutes at 4°C. Beads were washed three times with wash buffer (lysis buffer without Triton-X-100) and eluted with wash buffer containing 250 mM imidazole and 0.1% NP-40. Twenty microliters of protein samples were run by 4–12% Bis-Tris gel (Thermo Fisher Scientific) and protein bands were visualized by Coomassie staining.
Six bands of approximately equal area were excised from each gel lane and processed separately. Cysteine residues were reduced with 10 mM dithiothreitol (Sigma-Aldrich) in 25 mM NH4HCO3 at 56°C for 1h, followed by alkylating the thiol groups with 55 mM iodoacetamide (Sigma) at room temperature for 45min. The samples were then incubated with 100ng sequencing-grade trypsin (Promega) overnight at 37°C. Peptides were extracted from the gel slices twice using 60% acetronitrile and 5% formic acid, dried, desalted with µC18 Ziptips (Millipore), and then re-suspended in 10 µL of 0.1% formic acid in water for MS analysis. LC-MS analyses were conducted using either a QExactive Plus (QE) or an LTQ-Orbitrap Velos (Velos) mass spectrometer (Thermo Scientific) coupled online to a nanoAcquity UPLC system (Waters Corporation) through an EASY-Spray nanoESI ion source (Thermo). Peptides were loaded onto an EASY-Spray column (75 µm x 15 cm column packed with 3µm, 100 Å PepMap C18 resin) 2% B (mobile phase A: 0.1% formic acid in water; mobile phase B: 0.1% formic acid in acetonitrile) for 20 min at a flow rate of 600nl/min. Peptides were separated at 400nL/min using a gradient from 2% to 25% B over 48 min followed by a second gradient from 25% to 37% B over 6 minutes and then a column wash at 75% B and reequilibration at 2% B.
Precursor scans were acquired in the Orbitrap analyzer (QE: 350–1500 m/z, resolution: 70,000@200 m/z, AGC target: 3e6; Velos: 300–1800 m/z, resolution: 30,000@400 m/z, AGC target: 2e6. The top 10 or 6 (QE or Velos) most intense, doubly charged or higher ions were isolated (4 m/z isolation window), subjected to high-energy collisional dissociation (QE: 25 NCE; Velos: 30 NCE), and the product ions measured in the Orbitrap analyzer (QEplus resolution: 17,500@200 m/z, AGC target: 5e4; Velos resolution: 7,500@400 m/z, AGC target: 9e4). Each baits was analyzed with similar numbers of experimental replicates performed on the Velos and the QEplus to avoid introducing systematic biases due to the improved performance of the QEplus. Peaklists were generated using Proteome Discoverer v1.4 (Thermo) and searched using Protein Prospector v 5.18.21 (UCSF). The data were first searched against the UniProtKB Drosophila melanogaster reference proteome (downloaded Nov 21, 2014) concatenated with the sequences of the FLAG-His tagged bait proteins as well as 40 common contaminating proteins from other species (20,089 entries total). An equal-sized randomized decoy database was also appended to the search. The search parameters allowed for tryptic specificity with 2 missed cleavages, 6 ppm precursor and 25 ppm product ion tolerance, carbamidomethylation of cysteine as a fixed modification, and methionine oxidation, peptide N-terminal glutamine to pyroglutamic acid conversion, and protein N-terminal methionine loss and/or acetylation as variable modifications (up to two per peptide). Merged results were initially reported at score thresholds of 20/20 and expectation value thresholds of 0.0001/0.01 (protein/peptide) using the option to report only “interesting” protein variants. These initial results (protein FDR: <1%) were manually curated to select only isoforms/variants that were identified by two unique peptides and to select variants consistently across experiments. The peaklists were then re-searched against this restricted database of 3450 sequences using otherwise the same parameters as in the initial search. Results were reported per gel band and then collated into gel lane level results by summing the spectral counts to a reported protein across gel lanes.
All bait proteins contained several peptides in common originating from the Flag-tag linker sequence: EFSGLRSYDLGDTIERGR. To account for differences in bait expression levels and instrument performance, the number of spectra matching this common sequence in each enrichment was used to calculate normalized spectral counts (NSC) across experiments. Spectral counts matching each protein were divided by the counts matching the common bait-sequence in a given purification and multiplied by the mean bait-sequence counts across all experiments. Alternately, the areas of the extracted ion chromatograms matching these same peptide sequences were calculated using the MS1 filtering functionality of Skyline. Comparing the two methods gave essentially identical results. Control experiment from uninduced S2 cells and from eGFP-Flag-His pulldowns were normalized instead by total spectral counts per gel lane to the mean of total spectral counts across all experiments. Control experiments were normalized different because: a) uninduced cells lack the common bait-sequence used for normalization and b) to achieve higher stringency by avoiding transformations that would reduce control counts. The statistical significance of the affinity captured prey proteins was modeled using SAINT express v2.5 (Teo et al., 2014). The normalized spectral count tables were manipulated into SAINT format, appended with gene names (downloaded from UniProtKB) and protein lengths. SAINT express was run in spectral count mode with explicit control purifications. Two replicates of uninduced S2 cells and three replicates of eGFP-Flag-His pulldowns were used as the control samples. The SAINT option to choose the three control replicates with highest spectral counts for each prey was invoked. Interactions were considered specific at a SAINT score of 0.9, corresponding to a Bayesian FDR of ≤ 0.01.
The results were further analyzed by hierarchical clustering. NSC values from the prey proteins surpassing the SAINT threshold were averaged across replicates and log2 transformed (to create a normal distribution). These values were then scaled within each pulldown. Dendrograms were calculated using a Euclidean distance metric and ward.D agglomeration and visualized with the gplots R library. For visualization purposes, the dendrogram was omitted. Network visualizations were created in Cytoscape v3.6.1 using stringApp v1.3.2 to import interactions from the String database v10.5 at a score threshold of 0.9 (Szklarczyk et al., 2017). Unless otherwise noted, all data processing and analysis was performed in R v3.3.1.
Ago-2 degradation assay
Inducible S2 cells stably expressing CrPV-1A or CrPV-1A(BC-mut) were transfected with Ago-2-flag plasmids. At desired post hour induction cells were harvested, washed 2X with PBS at 4°C, and the pellet was stored at −70°C. The frozen S2 cells were thawed on ice and re-suspended in lysis buffer [30mM Hepes pH 7.4, 150mM KCl, 2mM MgOAc, 2mM DTT, 0.1% Triton-X-100] supplemented with cOmplete Mini EDTA-free protease inhibitor tablet (Roche). The extracts were centrifuged at 14,000 rpm for 30 minutes, and the supernatants were probed for Ago-2 by Western blot analysis using anti-flag antibody. For measuring Ago-2 degradation in CrPV infected cells, 106 uninduced stable S2 cells containing Ago-2-flag transgene were treated with 12.5 µg of either Cullin 2 or Elongin B or Luciferase dsRNA in 0.5 ml serum-free Schneider’s Drosophila media (Thermo Fisher Scientific). One hour post dsRNA treatment, 1 ml complete Schneider’s Drosophila media containing 10% FBS was added, and cells were incubated for 72 h at 25°C. After 72 h cells were counted and CrPV was added at MOI 2 for 1 h. Subsequently, Ago-2 expression was induced by adding CuSO4 to the medium and infection was carried out for 20 hours. S2 cells were harvested, washed 2X with PBS at 4°C, and the pellets were resuspended in RIPA buffer (50 mM Tris pH (8.0), 150 mM NaCl, 0.1% SDS, 0.5% Sodium deoxycholate, 1% Triton-X 100, 1.0 mM EDTA) supplemented with cOmplete protease inhibitor tablet (Roche). The extracts were centrifuged at 14,000 rpm for 30 minutes, and the supernatants were probed for Ago-2 by Western blot analysis using anti-flag antibody. Ago-2 degradation was analyzed using at least two independent experiments.
Glycerol gradient fractionation
The Flag-His tag purified CrPV-1A sample as described in Affinity Purification Mass-spectrometry experiment above was overlaid on a 4.8 ml 10–40% glycerol gradient and spun at 45,000 rpm for 16 hours using a SW-55Ti rotor in a Beckman ultracentrifuge. The 200μL fractions were collected from top, protein bands were analyzed using a 4–12% Bis-Tris gel and visualized using Pierce Silver Stain Kit (Thermo Fisher Scientific).
Ubiquitin Site Analysis
UbiScan analysis was performed as per manufacturer’s recommendation using PTMScan technology (Cell Signaling). In brief, S2 cells control and S2 cells stably expressing CrPV-1A treated with MG132 were harvested by centrifugation and washed twice with cold 1X PBS. Cell pellets were resuspended in freshly prepared room temperature Urea lysis buffer and stored at −80°C. Frozen pellets were thawed and sonicated for 15 sec three times, keeping on ice for 1 min between cycles. Sonicated samples were centrifuged at 10,000xg for 30 min at 15°C and the supernatant was harvested. After Bradford protein quantification assay, 10 mg of lysates were subsequently reduced and alkylated in the presence of DTT and Iodoacetic acid. The final urea concentration was diluted to 2M and treated with sequencing grade Trypsin-TPCK overnight at room temperature. Trypsin pre-digested and post-digested lysates were analyzed using a Tricine gel. The fully digested lysates were subsequently acidified by adding formic acid and centrifuged at 1780xg for 15 min to remove any precipitate. The supernatant was harvested, and peptide purification was carried out using a Sep-Pak C18 solid phase extraction cartridge (Waters). The eluted peptide sample was frozen in liquid nitrogen and lyophilized. The lyophilized peptides were subjected to immunoaffinity purification using a Ubiquitin remnant motif (K-ε-GG) antibody (Cell Signaling). The eluted samples were concentrated and purified using ZipTip (EMD Millipore) and brought up in 0.1% formic acid for mass spectrometry analysis.
LC-MS analysis of the in-solution digested peptides was generally performed as detailed in the AP-MS methods section using the LTQ-Orbitrap Velos system with the following changes: after loading onto the EASY-Spray column, peptides were eluted with a gradient of 2%−25% B over 108 minutes. Database searches were conducted using Protein Prospector using the D. melanogaster reference proteome noted previously. The search parameters allowed for tryptic peptides with 2 missed cleavage, 20 ppm precursor and 30 ppm product ion tolerance, carboxymethylation of cysteine as a fixed modification, and the variable modifications were the same as previously noted with the addition of the GlyGly remnant on uncleaved Lys residues. Results are reported at score thresholds of 20/20 and expectation value thresholds of 0.0001/0.0001 (protein/peptide). The SLIP score, for site-localization of PTMs, was set to 6. All spectra that matched ubiquitylated Ago-2 peptides were manually inspected for correct precursor monoisotopic selection, a long consecutive series of product ions, and that most of the high-intenisty peaks matched the sequence.
A spectral library of Ago-2 ubiquitylated peptides was exported to Skyline 4.1 in blib format, and the MS1 filtering based quantitation was performed against the RAW data files. The first three isotopes of the precursor ion were extracted and added for quantitation. All extracted ion chromatograms were manually inspected and adjusted to choose the correct peaks based on retention time, profile, and isotope distribution. Peak areas were averaged when multiple peptides or charge states identified the same ubiquitylated site. Data were presented to show the increase in peak area induced by MG132 treatment as a percentage of the area of the corresponding peak in S2 cells with MG132 treatment. That is, the red and blue bars in Figure S7 represent: (Pk.Area(S2+MG132) – Pk.Area(S2-MG132)) / Pk.Area(S2+MG132) and (Pk.Area(CrPV-1A+MG132) – Pk.Area(CrPV-1A-MG132)) / Pk.Area(S2+MG132), respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
AP-MS
The statistical significance of the AP-MS results was assessed using the Saint Express v 2.5 software package (Teo et al., 2014). These data represent 3–5 biological replicates (n = 3–5) for each affinity purified bait protein, and 5 negative control purifications. Saint Express performed statistical inference based on the fold-enrichment of normalized spectral counts matching each identified protein in the experimental conditions over the spectral counts observed in the three highest negative controls. Prey proteins with a SAINT score greater than or equal to 0.9 were considered significant. This corresponded to a BFDR (Bayesian False Discovery Rate) of less than 0.01. Heatmaps of the results were generated in R using the gplots package, and network visualizations were generated using Cytoscape. More information is provided in the Method Details section.
Fly survival assay
Three independent fly injection experiments (n = 3) were performed and fly survival data were tested for statistical significance using the log-rank test. For each infection condition at least 10 flies (n = 10) were used. For virus RNA loads measurement, qPCR analysis of three independent replicates (n = 3), and 10 individuals flies (n = 10) were typically used per data point. Statistical analysis was performed using generalized estimating equations to adjust for correlation of measurements within subject. A model was fitted with viral titer as the dependent variable and regressed on time post-infection, experimental condition, and their interaction, to look for differences in trends in viral titer over time across experimental groups. All experimental groups were compared to WT + CrPV3.
CrPV3 Titer in S2 cells
For viral titer measurement in S2 cells, the unpaired t test was used for statistical significance. Each Viral titers measurement was performed using three replicate experiments (n = 3).
All error bars represent standard deviation (SD). R and Prism software was used for statistical analysis.
Supplementary Material
Table S3. Results of AP-MS experiments, Related to Fig 5.
Table S4.. UbiScan mass spectrometry analysis, Related to Fig 7.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Mouse monoclonal ANTI-FLAG M2 | Sigma Life Science | Cat# F3165; RRID:AB_259529 |
| Rabbit polyclonal Anti-Argonaute-2 (Drosophila) | Abcam | Cat# ab5072; RRID:AB_2096294 |
| Mouse monoclonal Anti-beta Actin | Abcam | Cat# ab8224; RRID:AB_449644 |
| Ubiquitin Remnant Motif (K-ε-GG), Academic license | Cell Signaling Technology | Cat #5562 |
| Mouse monoclonal Anti-alpha Tubulin | Santa Cruz | Cat #sc-8035; RRID:AB_628408 |
| Rabbit polyclonal CrPV anti-3CD | Garrey et al., 2010 | N/A |
| Rabbit polyclonal CrPV anti-VP2 | Garrey et al., 2010 | N/A |
| Goat Anti-Rabbit IgG, Texas Red | Thermo Fisher Scientific | Cat #T6391; RRID:AB_10374713 |
| Bacterial and Virus Strains | ||
| CrPV-3 | Kerr et al., 2015 | N/A |
| Escherichia coli BL21(DE3) | John D Gross Lab | N/A |
| Escherichia coli B834(DE3) | Novagen | Cat# 69041 |
| SURE 2 Supercompetent Cells | Agilent Technologies | Cat# 200152 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| L-Selenomethionine | Anatrace | Cat# S2000 |
| Sequencing Grade Modified Trypsin, Frozen | Promega | Cat# V5113 |
| Phusion High Fidelity DNA Polymerase enzyme | New England Biolabs | Cat# M0530L |
| Dynabeads M-270 Epoxy | Thermo Fisher | Cat# 14301 |
| EZview Red ANTI-FLAG M2 Affinity Gel | Sigma-Aldrich | Cat# F2426 |
| EZview Red Protein A Affinity Gel | Sigma-Aldrich | Cat# P6486 |
| MG-132 proteasome inhibitor | Sigma-Aldrich | Cat# C2211 |
| cOmplete Mini EDTA-free Protease Inhibitor tablets | Roche | Cat# 11836170001 |
| Ni-NTA Agarose | QIAGEN | Cat# 30210 |
| Dynabeads His-Tag Isolation and Pulldown | Thermo Fisher Scientific | Cat# 10103D |
| 3X FLAG Peptides | Sigma-Aldrich | Cat# F4799 |
| Critical Commercial Assays | ||
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1910 |
| Vaccinia Capping System | New England Biolabs | Cat# M2080S |
| SensiFAST SYBR No-ROX Kit | BIOLINE | Cat# BIO-98005 |
| Pierce Silver Stain Kit | Thermo Fisher Scientific | Cat# 24612 |
Acknowledgements
We thank J. Holton and G. Meigs at LBNL, ALS beamline 8.3.1 for X-ray data collection, and Pascal Egea for assistance with crystallography. We thank Z. Whitfield, M. Tioni, and M.G. Knight members of the RA laboratory for critical reading of the manuscript and Cinthia Blat for help with the statistical analysis. We thank K. Ehmsen, J. Binning, and B. Jayaraman for their helpful comments. We thank Zach Adelman for pUb plasmid. This work was financially supported by NIH NIGMS 841GM103481, NIH 1S10D016229, and Howard Hughes Medical Institute grant to ALB, NIH P50 GM082250 to JDG, CIHR Projects Grant (PJT-148761) to EJ, NIH 1R01 AI12744701 to JF, DARPA HR0011–11-C-0094 and NIH R01 AI091575 and AI137471 to RA.
Footnotes
Declaration of Interests: The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
132 Thermo RAW files, corresponding to mass spectrometry analysis of each gel slice from the AP-MS experiments, have been deposited at the MassIVE repository (http://massive.ucsd.edu) under the accession code: MSV000082752. The accession number for the CrPV-1A159 coordinates reported in this paper is PDB ID: 6C3R.
References
- Aliyari R, Wu Q, Li H-W, Wang X-H, Li F, Green LD, Han CS, Li W-X, and Ding S-W (2008). Mechanism of Induction and Suppression of Antiviral Immunity Directed by Virus-Derived Small RNAs in Drosophila. Cell Host Microbe 4, 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DL, and Gibbs AJ (1988). Inapparent Virus Infections and their Interactions in Pupae of the Honey Bee (Apis mellifera Linnaeus) in Australia. J Gen Virol 69, 1617–1625. [Google Scholar]
- Baumberger N, Tsai C-H, Lie M, Havecker E, and Baulcombe DC (2007). The Polerovirus silencing suppressor P0 targets ARGONAUTE proteins for degradation. Curr Biol 17, 1609–1614. [DOI] [PubMed] [Google Scholar]
- Biasini M, Bienert S, and Waterhouse A (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic acids Res 42, W252–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair CD (2011). Mosquito RNAi is the major innate immune pathway controlling arbovirus infection and transmission. Future Microbiol 6, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonning BC, and Miller WA (2010). Dicistroviruses. Annu Rev Entomol 55, 129–150. [DOI] [PubMed] [Google Scholar]
- Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, and Schwede T (2008). Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc 4, 1–13. [DOI] [PubMed] [Google Scholar]
- Brunger AT (2007). Version 1.2 of the Crystallography and NMR system. Nat Protoc 2, 2728–2733. [DOI] [PubMed] [Google Scholar]
- Cardote TAF, Gadd MS, and Ciulli A (2017). Crystal Structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Ligase Complex. Structure 25, 901–911.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Tripp J, Dolezal AG, Goblirsch MJ, Miller WA, Toth AL, and Bonning BC (2016). In vivo and in vitro infection dynamics of honey bee viruses. Sci Rep 6, 22265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Tripp J, Krueger EN, Harrison RL, Toth AL, Miller WA, and Bonning BC (2014). Lymantria dispar iflavirus 1 (LdIV1), a new model to study iflaviral persistence in lepidopterans. J Gen Virol 95, 2285–2296. [DOI] [PubMed] [Google Scholar]
- Csorba T, Kontra L, and Burgyán J (2015). viral silencing suppressors: Tools forged to fine-tune host-pathogen coexistence. Virol J 479-480, 85–103. [DOI] [PubMed] [Google Scholar]
- Csorba T, Lózsa R, Hutvagner G, and Burgyán J (2010). Polerovirus protein P0 prevents the assembly of small RNA-containing RISC complexes and leads to degradation of ARGONAUTE1. Plant J 62, 463–472. [DOI] [PubMed] [Google Scholar]
- Daugherty MD, Liu B, and Frankel AD (2010). Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat Struct Mol Biol 17, 1337–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Desimmie BA, Delviks-Frankenberrry KA, Burdick RC, Qi D, Izumi T, and Pathak VK (2014). Multiple APOBEC3 Restriction Factors for HIV-1 and One Vif to Rule Them All. J Mol Biol 426, 1220–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Galiana-Arnoux D, Dostert C, Schneemann A, Hoffmann JA, and Imler J-L (2006). Essential function in vivo for Dicer-2 in host defense against RNA viruses in drosophila. Nat Immunol 7, 590–597. [DOI] [PubMed] [Google Scholar]
- Gammon DB, and Mello CC (2015). RNA interference-mediated antiviral defense in insects. Curr Opin Insect Sci 8, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrey JL, Lee Y-Y, Au HHT, Bushell M, and Jan E (2010). Host and viral translational mechanisms during cricket paralysis virus infection. J Virol 84, 1124–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, and Kneller DG (2007). SPARKY 3.112 Vol. 3 University of California San Francisco. [Google Scholar]
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, and Lopez R (2010). A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 38, W695–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, and Sander C (1995). Dali: a network tool for protein structure comparison. Trends Biochem Sci 20, 478–480. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Kobayashi M, Yoda M, Sakaguchi Y, Katsuma S, Suzuki T, and Tomari Y (2010). Hsc70/Hsp90 Chaperone Machinery Mediates ATP-Dependent RISC Loading of Small RNA Duplexes. Mol Cell 39, 292–299. [DOI] [PubMed] [Google Scholar]
- Kerr CH, Wang QS, Keatings K, Khong A, Allan D, Yip CK, Foster LJ, and Jan E (2015). The 5’ untranslated region of a novel infectious molecular clone of the dicistrovirus cricket paralysis virus modulates infection. J Virol 89, 5919–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong A, Kerr CH, Yeung CHL, Keatings K, Nayak A, Allan DW, and Jan E (2017). Disruption of Stress Granule Formation by the Multifunctional Cricket Paralysis Virus 1A Protein. J Virol 91, e01779–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, and Uchiyama T (2005). Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem 280, 18573–18578. [DOI] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Li H, Li W-X, and Ding S-W (2002). Induction and Suppression of RNA Silencing by an Animal Virus. Sci Signal 296, 1319. [DOI] [PubMed] [Google Scholar]
- Marques JT, Wang J-P, Wang X, de Oliveira KPV, Gao C, Aguiar ERGR, Jafari N, and Carthew RW (2013). Functional Specialization of the Small Interfering RNA Pathway in Response to Virus Infection. PLoS Pathog 9, e1003579–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA, IUCr (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K (2016). Anatomy of RISC: how do small RNAs and chaperones activate Argonaute proteins? Wiley Interdisciplinary Reviews: RNA 7, 637–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak A, and Andino R (2011). Slicer Activity in Drosophila melanogaster S2 Extract. Methods Mol Biol (Clifton, NJ) 721, 231–244 [DOI] [PubMed] [Google Scholar]
- Nayak A, Berry B, Tassetto M, Kunitomi M, Acevedo A, Deng C, Krutchinsky A, Gross J, Antoniewski C, and Andino R (2010). Cricket paralysis virus antagonizes Argonaute 2 to modulate antiviral defense in Drosophila. Nat Struct Mol Biol 17, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak A, Tassetto M, Kunitomi M, and Andino R (2013). RNA Interference-Mediated Intrinsic Antiviral Immunity in Invertebrates. Curr Top Microbiol Immunol 371, 183–200. [DOI] [PubMed] [Google Scholar]
- Nguyen HC, Wang W, and Xiong Y (2017). Cullin-RING E3 Ubiquitin Ligases: Bridges to Destruction. Subcell. Biochem 83, 323–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HC, Yang H, Fribourgh JL, Wolfe LS, and Xiong Y (2015). Insights into Cullin-RING E3 Ubiquitin Ligase Recruitment: Structure of the VHL-EloBC-Cul2 Complex. Structure 23, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obbard DJ, Jiggins FM, Halligan DL, and Little TJ (2006). Natural selection drives extremely rapid evolution in antiviral RNAi genes. Curr Biol 16, 580–585. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pazhouhandeh M, Dieterle M, Marrocco K, Lechner E, Berry B, Brault V, Hemmer O, Kretsch T, Richards KE, Genschik P, and Ziegler-Graff V (2006). F-box-like domain in the polerovirus protein P0 is required for silencing suppressor function. P Natl Acad Sci Usa 103, 1994–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg J (1968). Growth of arboviruses in monolayers from subcultured mosquito embryo cells. Virology 35, 617–619. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Schirle NT, and MacRae IJ (2012). The crystal structure of human Argonaute2. Science 336, 1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti PD, Longworth JF, Plus N, Croizier G, and Reinganum C (1981). The Biology and Ecology of Strains of an Insect Small RNA Virus Complex. Adv Virus Res 26 117–143. [DOI] [PubMed] [Google Scholar]
- Swarts DC, Makarova K, Wang Y, Nakanishi K, Ketting RF, Koonin EV, Patel DJ, and van der Oost J (2014). The evolutionary journey of Argonaute proteins. Nat Struct Mol Biol 21, 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szittya G, and Burgyán J (2013). RNA Interference-Mediated Intrinsic Antiviral Immunity in Plants. Curr Top Microbiol Immunol 371, 153–181. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, and Mering von C (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45, D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tenOever BR (2016). The Evolution of Antiviral Defense Systems. Cell Host Microbe 19, 142–149. [DOI] [PubMed] [Google Scholar]
- Teo G, Liu G, Zhang J, Nesvizhskii AI, Gingras A-C, and Choi H (2014). SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J Proteomics 100, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, and Berendzen J, IUCr. (1999). Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, IUCr. (2003). Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr 59, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JC, Matak-Vinkovic D, Van Molle I, and Ciulli A (2013). Multimeric Complexes among Ankyrin-Repeat and SOCS-box Protein 9 (ASB9), Elongin BC, and Cullin 5: Insights into the Structure and Assembly of ECS-type Cullin-RING E3 Ubiquitin Ligases. Biochemistry 52, 5236–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Cleef KWR, van Mierlo JT, Miesen P, Overheul GJ, Fros JJ, Schuster S, Marklewitz M, Pijlman GP, Junglen S, and van Rij RP (2014). Mosquito and Drosophila entomobirnaviruses suppress dsRNA- and siRNA-induced RNAi. Nucleic Acids Res 42, 8732–8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mierlo JT, Bronkhorst AW, Overheul GJ, Sadanandan SA, Ekström J-O, Heestermans M, Hultmark D, Antoniewski C, and van Rij RP (2012). Convergent evolution of argonaute-2 slicer antagonism in two distinct insect RNA viruses. PLoS Pathog 8, e1002872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rij RP, Saleh M-C, Berry B, Foo C, Houk A, Antoniewski C, and Andino R (2006). The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Genes Dev 20, 2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varjak M, Donald CL, Mottram TJ, Sreenu VB, Merits A, Maringer K, Schnettler E, and Kohl A (2017). Characterization of the Zika virus induced small RNA response in Aedes aegypti cells. PLoS Negl Trop Dis 11, e0006010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Iwakawa H-O, Tadakuma H, and Tomari Y (2017). Biochemical and single-molecule analyses of the RNA silencing suppressing activity of CrPV-1A. Nucleic Acids Res 1–8. [DOI] [PMC free article] [PubMed]
- Webb B, and Sali A (2016). Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Protein Sci 86, 2.9.1–2.9.37. [DOI] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag)
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, and Yu X-F (2003). Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302, 1056–1060. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang H, Li Z, Liu X, Liu G, Harris RS, and Yu X-F (2014). Cellular requirements for bovine immunodeficiency virus Vif-mediated inactivation of bovine APOBEC3 proteins. J Virol 88, 12528–12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zondlo NJ (2013). Aromatic-proline interactions: electronically tunable CH/π interactions. Acc Chem Res 46, 1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. Results of AP-MS experiments, Related to Fig 5.
Table S4.. UbiScan mass spectrometry analysis, Related to Fig 7.