

Abstract
Hydrogen sulfide (H2S) is an endogenously produced gas that is toxic at high concentrations. It is eliminated by a dedicated mitochondrial sulfide oxidation pathway, which connects to the electron transfer chain at the level of complex III. Direct reduction of cytochrome c (Cyt C) by H2S has been reported previously but not characterized. In this study, we demonstrate that reduction of ferric Cyt C by H2S exhibits hysteretic behavior, which suggests the involvement of reactive sulfur species in the reduction process and is consistent with a reaction stoichiometry of 1.5 mol of Cyt C reduced/mol of H2S oxidized. H2S increases O2 consumption by human cells (HT29 and HepG2) treated with the complex III inhibitor antimycin A, which is consistent with the entry of sulfide-derived electrons at the level of complex IV. Cyt C-dependent H2S oxidation stimulated protein persulfidation in vitro, while silencing of Cyt C expression decreased mitochondrial protein persulfidation in a cell culture. Cyt C released during apoptosis was correlated with persulfidation of procaspase 9 and with loss of its activity. These results reveal a potential role for the electron transfer chain in general, and Cyt C in particular, for potentiating sulfide-based signaling.
Graphical Abstract:
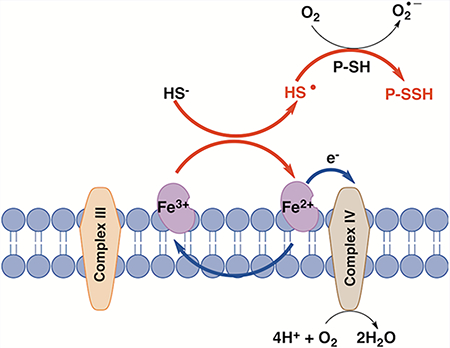
H2S is an energy source for prokaryotes adapted for life in sulfide rich environments, but it is a potent respiratory toxin for other organisms. The targets of H2S poisoning are the metal centers in cytochrome c oxidase, the terminal station in the mitochondrial electron transfer chain (ETC).1,2 At low concentrations, however, H2S modulates physiological processes ranging from inflammation to cardiovascular function and apoptosis.3–5 Cellular H2S levels must therefore be maintained within a nontoxic window by regulating enzymes involved in its biogenesis6–9 and degradation.10,11 The mitochondrial sulfide oxidation pathway transfers electrons from H2S oxidation to complex III via coenzyme Q.12 In red blood cells lacking mitochondria, a noncanonical sulfide oxidation route uses ferric hemoglobin and catalyzes conversion of H2S to thiosulfate.13 Polysulfides that are also produced in this reaction are degraded by reductants such as glutathione.14 Other hemeproteins like myoglobin15 and neuroglobin16 also catalyze ferric heme-dependent H2S oxidation. Unlike hemoglobin and myoglobin, H2S oxidation by neuroglobin, in which the heme has two axial histidine ligands, does not involve direct coordination of sulfide to ferric iron.
Cytochrome c (Cyt C) resides in the mitochondrial intermembrane space and shuttles electrons between complexes III and IV in the ETC. Release of Cyt C into the cytosol triggers apoptotic activation of caspase 9.17 As in neuroglobin, the heme in Cyt C is six-coordinate and exhibits bis-His or His/Met ligation. In principle, electrons from the oxidation of H2S can enter the ETC at the level of complex IV either directly18 or indirectly via initial reduction of ferric Cyt C by sulfide.19 However, the standard redox potential for the S•−, H+/HS− couple is +0.91 V (vs NHE)20 compared to +0.26 V for Cyt C,21 rendering reduction of Cyt C by H2S thermodynamically unfavorable. However, reduction of Cyt C by sulfide has been observed in vitro19,22 and in the ciliated gills of Guenkensia demissa, a mussel that lives in sulfide rich sediments.23 An additional link between H2S and Cyt C was reported in a study on rotenone-induced apoptosis in which H2S exposure preserved mitochondrial function, limited Cyt C release, and inhibited dissipation of the mitochondrial proton membrane potential.24 However, a quantitative analysis of H2S oxidation by Cyt C, identification of the reaction products, and elucidation of its effects on apoptosis and persulfide signaling has been lacking.
In this study, we report the kinetics of Cyt C reduction by H2S, demonstrate formation of thiosulfate as a reaction product, and show stimulation of O2 consumption in human cells treated with antimycin A, consistent with the delivery of electrons from H2S to complex IV via Cyt C. We also demonstrate that Cyt C in the presence of H2S stimulates protein persulfidation, suggesting a potentiating role of the ETC in H2S-dependent signaling in general and in regulation of apoptosis via persulfidation of caspase 9 in particular.
RESULTS AND DISCUSSION
Kinetics of Cyt C Reduction by Sulfide.
Addition of stoichiometric H2S to ferric Cyt C (Soret band at 409 nm) results in its complete conversion to the ferrous form (415 nm) with sharpening of the α/β bands at 550 and 520 nm (Figure 1A). The kinetics of Cyt C reduction by sulfide exhibits a pronounced lag phase (Figure 1B), indicating that the reaction either involves a slow step (e.g., ligand exchange from histidine to HS−) or is accelerated as H2S oxidation products accumulate. Cryo-ESI MS previously used to observe HS− coordination to ferric heme15 did not identify a comparable adduct with Cyt C (Supporting Figure 1). The stoichiometry of the reduction reaction (1.5:1.0 Cyt C:sulfide) was independent of the presence of O2 (Figure 1C). The Cyt C reduction rate showed a bell-shaped dependence on pH with a maximum at 7.5 and inflection points at 6.7 ± 0.1 and 8.5 ± 0.1 (Figure 1D). Phosphate and citrate buffers, previously shown to bind Cyt C and influence its overall charge and redox potential,25 significantly inhibited the rate of ferric Cyt C reduction by sulfide (not shown).
Figure 1.

Reduction of Cyt C by sulfide. (A) The initial spectrum of ferric Cyt C (10 μM) in 100 mM HEPES buffer [pH 7.4 (red)] shifts in the presence of 10 μM Na2S (at 3, 5, 7, 10, 15, and 20 min, black lines) under aerobic conditions at 25 °C. (B) Kinetics of aerobic Cyt C reduction (10 μM) at 3, 5, and 10 μM Na2S in 100 mM HEPES buffer (pH 7.4) at 25 °C. (C) Percent Cyt C [10 μM in 100 mM HEPES buffer (pH 7.4)] reduction as a function of sulfide concentration. The increase in absorbance at 550 nm was monitored after incubation with sulfide at 25 °C for 1 h under aerobic or anaerobic conditions. The red line is a sigmoidal fit to the experimental data with an S0.5 of 2.9 ± 0.1 μM and an n of 2.4 ± 0.2. (D) pH dependence of Cyt C reduction by sulfide. The rate of Cyt C (2.5 μM) reduction by sulfide (500 μM) was determined by anaerobic stopped-flow spectroscopy as described in Methods. The data are the mean ± the standard deviation (SD) of four independent experiments as described in Methods. From the fit, estimates for pKa1 and pKa2 of 6.7 ± 0.1 and 8.5 ± 0.1, respectively, were obtained.
As discussed above, the one-electron reduction of Cyt C with HS− is thermodynamically unfavorable, which could explain the observed lag phase (Figure 1B). In principle, HS•/S•− could dimerize at a diffusion-controlled rate (k = 9 × 109 M−1 s−1)2 giving HS2− (eq 1). However, the probability of HS•/S•− reaching sufficiently high concentrations for the dimerization reaction is likely to be low. Furthermore, formation of HS2− would not provide the driving force for Cyt C reduction, as the reaction (eq 2) is thermoneutral [E°′(Cyt C3+/Cyt C2+) = +0.26 V, and E°′(S2•−, H+/HS2−) = +0.27 V].2,20
| (1) |
| (2) |
Alternatively, O2 could react with the initially formed HS•/S•− at a diffusion-controlled rate (k = 5 × 109 M−1 s−1)2 to produce SO2•−, a strong reducing agent (eqs 3 and 4). However, the extent of Cyt C reduction was independent of O2 (Figure 1C).
| (3) |
| (4) |
An alternative mechanism for explaining the hysteretic behavior is that the initially formed HS•/S•− reacts with HS− (kon = 4 × 109 M−1 s−1, and koff = 5 × 103 M−1 s−1)2,20 forming H2S2•−, which is a strong reducing agent [E°′(HS2−, H+/H2S2•−) = −1.13 V]2,20 and could pull the Cyt C reduction reaction forward. Interestingly, Na2S2 reduced Cyt C more rapidly than sulfide and do so without an observable lag phase (Figure 2A).
Figure 2.

Cyt C reduction kinetics and product analysis. (A) Kinetics of aerobic Cyt C reduction (10 μM) in the presence of 3, 5, and 10 μM Na2S2 in 100 mM HEPES buffer (pH 7.4) at 25 °C. Complete reduction with equimolar Na2S2 was seen within 5−10 min. (B) Kinetics of O2 consumption in the presence of Cyt C and sulfide. Addition of sulfide to Cyt C [50 μM in 100 mM HEPES buffer (pH 7.4)] at 25 °C results in rapid initial consumption of O2 followed by a slower phase. (C) Dependence of the concentration of O2 consumed in the burst phase on sulfide concentration. A hyperbolic fit to the data (mean ± SD of three to five independent experiments) gives a Kact of 0.5 ± 0.1 mM and a maximal O2 consumed of 35 ± 2 μM (red line), yielding a O2 consumed:Cyt C stoichiometry of 0.7:1.0. (D) Consumption of sulfide and accumulation of thiosulfate and sulfite 2 h after addition of 800 μM Na2S to 100 mM HEPES buffer (pH 7.4) without (black bars) or with (gray bars) 100 μM Cyt C at 25 °C under aerobic conditions. The initial levels of sulfide, thiosulfate, and sulfite were 756, 25, and 3.5 μM, respectively. (E) MS detection of sulfur products formed in the reaction of 10 μM Cyt C with 100 μM H2S in ammonium carbonate buffer (pH 7.7) under aerobic conditions. The reaction was monitored for 15 min at room temperature. The observed spectrum is at the top, and the simulated spectra with an isotopic distribution are below.
Degradation of Excess Sulfide in the Presence of Cyt C.
Biphasic O2 consumption kinetics were seen when Cyt C was mixed with excess sulfide (Figure 2B). As the ratio of sulfide to Cyt C concentration was increased, the amplitude of the initial burst phase and the slope of the slow phase increased. These data are consistent with the model in which the burst phase corresponds to the reduction of Cyt C during which the initially formed HS•/S•− is rapidly oxidized, while the slow phase represents subsequent oxidation reactions. The amplitude of the burst phase showed a hyperbolic dependence on sulfide concentration and asymptotically approached 0.7 equiv of Cyt C to O2 consumed (Figure 2C). Biphasic O2 consumption suggested formation of products with higher sulfur oxidation states. SO2 formation (eq 4) would result in accumulation of sulfite, which in the presence of a sulfane sulfur donor (“S”) would form thiosulfate as a final product (eqs 5 and 6).2
| (5) |
| (6) |
We tested for the formation of sulfide oxidation products under aerobic conditions (Figure 2D). Loss of sulfide was paralleled by the accumulation of thiosulfate and, to a lesser degree, sulfite. The recovery of sulfur equivalents in the thiosulfate, which has two sulfur atoms (~280 μM), and sulfite (~40 μM) pools represent ~50% of the sulfide lost. Because of the interference of Cyt C with the cold cyanolysis method, we were unable to evaluate the concentration of sulfane sulfur species in the reaction mixture. ESI-TOF MS in the negative mode confirmed formation of thiosulfate and also revealed the presence of sulfate (Figure 2E). Although sulfite was not seen (possibly because of its low m/z), we detected the sulfur trioxide radical anion SO3•−, the concentration of which increased with time (Supporting Figure 2). Both SO2•− and H2S2•− readily reduce O2 to form superoxide that can disproportionate to give H2O2.2,20 The latter can oxidize ferrous Cyt C, enabling its redox cycling.26,27 Sulfite, peroxide, and heme proteins are known to produce SO3•−, which is a potent oxidant that is proposed to play a role under certain pathological conditions.28–30 SO3•− could also contribute to Cyt C reoxidation to establish a pseudocatalytic cycle for H2S removal.
Cyt C Stimulates in Vitro Protein Persulfidation.
Protein thiols represent a potentially large pool of intracellular HS•/S•− scavengers (eq 7). To test the hypothesis that protein thiols are sinks for HS•/S•−, we first used time-resolved ESI-TOF-MS to monitor a reaction mixture containing Cyt C, H2S, and cysteine. Formation of cysteine persulfide (as [CysSSH + H]+ and [2CysSSH + H]+ ions) and its decay products, CysSSCys (Figure 3A) and CysSSSCys (Supporting Figure 3A), was observed. While formation of CySSH plateaued within 1 min, formation of CysSSCys continued (Figure 3B and Supporting Figure 3B), supporting our previous report of the lability of CysSSH.31
| (7) |
Figure 3.

Cyt C stimulates H2S-dependent persulfidation. (A) Mass spectrum of the reaction mixture (t = 6 min) containing 20 μM Cyt C, 100 μM H2S, and 200 μM cysteine in ammonium carbonate buffer (pH 7.7) at 21 °C and simulated isotopic distributions for some of the identified species: 1, [CysSH + H]+; 2, [CysSSH + H]+; 3, [CysSSCys + H]+; 4, [CysSSCys + Na]+; 5, [2CysSSH + H]+. (B) Kinetics of [CysSSH + H]+ (m/z 151.9) and [CysSSCys + H]+ (m/z 241.0) formation. Data represent means ± SD of three independent experiments. (C) Persulfidation of HSA (10 μM) in phosphate-buffered saline (pH 7.4), incubated for 30 min at room temperature with 500 μM Na2S (lane 1) and 2 μM (lane 2), 20 μM (lane 3), and 200 μM (lane 4) Cyt C. Data represent means ± SD (n = 3; *p < 0.001). (D) Persulfidation of purified (1 μM) EHTE-1 and (10 μM) ATR treated with either 100 μM H2S (lane 1), 10 μM Cyt C (lane 2), or combination of both (lane 3). The results of two separate experiments are shown. Persulfidation (C and D) was detected by the CN-Cy3-based tag switch method (top panels), while equal loading was detected by Coomassie blue staining (bottom panels). The gel was artificially colorized using ImageJ to enhanced visualization of changes, and the fluorescence intensity scale is provided on the right. The persulfidation procedure is described in Supporting Materials and Methods. (E) Aerobic kinetics of Cyt C [20 μM in 100 mM HEPES buffer (pH 7.4) at 25 °C] reduction (monitored at 550 nm) by Na2S (20 μM) with or without untreated or alkylated HSA (20 μM). Sulfide was added at time zero. (F) Kinetics of O2 consumption after addition of 0.2 mM Na2S to ferric Cyt C [50 μM in 100 mM HEPES buffer (pH 7.4)] with or without 50 μM HSA at 25 °C.
Next, we tested Cyt C-dependent persulfidation of a model protein, human serum albumin (HSA). We have previously shown that addition of water-soluble heme iron and H2S triggers protein persulfidation.32 Incubation of HSA with sulfide in the presence of Cyt C also resulted in protein persulfidation (detected using the Cy3-CN-based tag switch method)33 that was proportional to the concentration of Cyt C (Figure 3C). In contrast, when Cyt C was replaced with myoglobin, which oxidizes sulfide to polysulfides and thiosulfate via an inner sphere electron transfer mechanism, an increase in persulfidation was not observed (Supporting Figure 4). The residual persulfidation signals seen in the control lanes result from H2S-induced reduction of the disulfide bond in HSA. We also demonstrated that the human mitochondrial proteins, ETHE-1 and adenosyltransferase (ATR), previously identified as persulfidation targets, were also persulfidated by Cyt C/H2S (Figure 3D).
Interception of reactive sulfur species (HS•/S•−) by cysteine residues on proteins should decrease the rate of Cyt C reduction by H2S and O2 consumption. A substantial decrease in the rate of Cyt C reduction was in fact observed in the presence of equimolar HSA, which was attenuated by prior alkylation of HSA cysteines with iodoacetamide (Figure 3E). HSA also decreased the rate of O2 consumption, impacting both the burst and slow phases of the reaction (Figure 3F). Interestingly, the amplitude of the burst phase was increased in the presence of HSA. The initially formed RSSH•− protein radical anion is expected to be a strong reducing agent in analogy with RSSR•− [E°′(RSSR/RSSR•−) = −1.4 V].34 Because outer sphere electron transfer from the protein radical to Cyt C is likely to be precluded by steric hindrance, O2 would be the only available electron acceptor (eq 8), which could explain the higher amplitude observed for the burst phase (Figure 3F).
| (8) |
Cyt C Silencing Decreases Intracellular Persulfidation.
To further investigate its role in protein persulfidation, Cyt C was silenced in HeLa cells. Under normoxic conditions, Cyt C silencing resulted in a small but statistically significant decrease in endogenous protein persulfidation as detected by the CN-Cy3-based tag switch method (Figure 4A). The mitochondrially targeted H2S donor AP39 caused a large increase in protein persulfidation as seen previously,33 which was reduced by Cyt C siRNA treatment. The perinuclear localization of the persulfidation signal in AP39-treated cells confirms mitochondrial localization as reported for this donor.33,35,36
Figure 4.

Cyt C silencing decreases protein persulfidation. (A) Under normoxic conditions, silencing of Cyt C in HeLa cells (confirmed by Western blot analysis) led to a small but measurable decrease in basal persulfidation levels, which was increased by the mitochondrially targeted H2S donor, AP39 (200 nM). The scale bar is 5 μm. (B) Exposure of HeLa cells to hypoxia for 1 h results in increased endogenous H2S levels as detected by MeRho-Az. The scale bar is 10 μm. (C) Under hypoxic conditions, the levels of protein persulfidation are high but significantly reduced by Cyt C silencing. The scale bar is 10 μm. Representative microscopy images are given on the left, and quantitative image analysis is given on the right. The results represent the average fluorescence intensity (F. I.) detected from ≥40 cells ± the standard error of the mean of five independent images (*p < 0.001).
H2S oxidation is dependent on the presence of a functional ETC, and lower O2 levels lead to H2S accumulation.37 Using MeRho-Az, a bright fluorescence probe for H2S detection,35 we observed a substantial increase in intracellular H2S in HeLa cells exposed to <5% O2 (Figure 4B). A significant increase in protein persulfidation was observed in cells exposed to hypoxic conditions, which was diminished by silencing Cyt C (Figure 4C).
Sulfide Stimulates O2 Consumption in Complex III-Inhibited Cells.
To assess whether electrons from sulfide oxidation can bypass complex III and enter at the level of complex IV (Figure 5A), the O2 consumption rate (OCR) was assessed with human cells in suspension. The OCR decreased to 2.5% (HepG2) and 5% (HT29) of the initial values in the presence of antimycin A, a complex III inhibitor (Figure 5B,C). Addition of sulfide (20 μM) increased the OCR by an average of 30% (HT29) and 95% (HepG2) above the antimycin-treated value. Higher concentrations of sulfide (100 μM) inhibited O2 consumption as expected, presumably because of inhibition of complex IV. Addition of KCN, which poisons complex IV, blocked OCR, confirming that O2 consumption induced by sulfide was linked to mitochondrial respiration.
Figure 5.

Sulfide stimulates O2 consumption by mammalian cells and controls persulfidation of mitochondrial proteins. (A) Cartoon showing electrons from H2S oxidation enter the ETC at the level of complex III via the reduced quinone pool. Entry into complex IV via Cyt C can be monitored in the presence of antimycin A, a complex III inhibitor. (B and C) O2 consumption rates of HT29 and HepG2 cells, respectively, before (control) and after treatment with 2 μg mL−1 antimycin A (AA) followed by 20 μM Na2S. Finally, antimycin A-treated cells were exposed to 5 mM KCN. The data are means ± SD of 12 (B) or 6 (C) independent experiments; the p value shows the statistical significance for the difference between antimycin A with and without sulfide-treated cells. (D) Protein persulfidation in purified mitochondria is affected by the ETC. Functional mitochondria were isolated from S. cerevisiae and preincubated with 2.5 μg mL−1 antimycin A or 10 mM KCN for 10 min at 37 °C or treated with 1 μM H2S for 20 min at 37 °C. Persulfidation was monitored by the improved tag swtich method. The first lane represents the basal persulfidation level in untreated functional mitochondria. The total protein load is shown in the gel on the right.
Next, we assessed how inhibition of complex III and IV affects persulfidation in functional mitochondria that should produce H2S via the action of 3-mercaptopyruvate sulfur-transferase. Purified yeast mitochondria (Saccharomyces cerevisiae) treated with antimycin A showed an increase in intramitochondrial protein persulfidation, an effect that was inhibited by KCN (Figure 5D). This is in accordance with our hypothesis that even when H2S oxidation is inhibited by antimycin A, Cyt C-dependent consumption of H2S can still occur, leading to increased protein persulfidation. Inhibition of cytochrome c oxidase (by KCN), on the other hand, would prevent reoxidation of Cyt C and stop the cycle (Figure 5D).
Collectively, these results suggest that the canonical sulfide oxidation pathway can be bypassed to some extent by direct reduction of Cyt C. The reactive sulfur species formed under these conditions can react with protein thiols increasing persulfidation. We speculate that this situation could be particularly important in ischemia/reperfusion, protecting proteins from ROS-induced hyperoxidation during the reperfusion phase.
Cyt C-Induced Persulfidation Controls Caspase 9 Activity.
In addition to its role in mitochondrial energy metabolism, Cyt C is an important regulator of apoptosis following its release into the cytosol.38,39 In the presence of Cyt C, apoptotic protease activating factors in the apoptosome activate procaspase 9, triggering a protease cascade.40 Because of this functional association with Cyt C, we tested whether caspase 9, a cysteine protease, is persulfidated in the presence of H2S and Cyt C. Incubation with Cyt C and H2S stimulated persulfidation of recombinant murine procaspase 9 (Figure 6A). Next, we assessed caspase 9 activity in HeLa cell extracts using the fluorogenic caspase 9 substrate. Cyt C significantly stimulated caspase 9 activity, which was diminished in the presence of sulfide (Figure 6B).
Figure 6.

Cyt C-mediates procaspase 9 persulfdation and activity. (A) Murine recombinant procaspase 9 (1 μM) was incubated with 5 μM H2S for 30 min at 37 °C in the absence (lane 1) or presence of 0.5 (lane 2) and 2 μM (lane 3) Cyt C. Persulfidation was detected by the CN-Cy3-based tag switch method (top panel), while equal loading was confirmed by Coomassie blue staining (bottom panel). (B) H2S inhibits Cyt C-induced caspase 9 activation in cell lysates. Cyt C (500 nM) was added to freshly prepared HeLa cell lysates, and caspase 9 activity was monitored using a fluorescence caspase 9 kit. Simultaneous addition of Cyt C and H2S (10 μM) inhibited the Cyt C-induced increase in fluorescence. (C) Cyt C-induced caspase 9 activation in Jurkat cells is inhibited by H2S donors. Apoptosis was induced by staurosporine (ST, 2.5 μM), and caspase 9 activity was monitored by flow cytometry using the caspase 9 FITC staining kit 4 and 6 h after induction. Treatment of cells with GYY4137 (100 μM) or ammonium tetrathiomolybdate (ATTM, 200 μM) reduced fluorescence. (D) Inhibition of caspase 3 activation in cells treated with antimycin A and the slow-releasing H2S donor, GYY4137 (100 μM). HeLa cells were incubated with antimycin A (300 μM) overnight to induce the release of Cyt C from mitochondria. GYY4137, when used, was added 2 h prior to antimycin A. Caspase 3 activity was monitored in cell lysates as described for panel B. (E) Persulfidation of procaspase 9 in HeLa cells treated with 2.5 μM ST (1) that were either pretreated for 2 h (2) or treated for the last 2 h (3) with GYY4137 (100 μM): (left) total procaspase 9 levels and (right) persulfidation (PSSH) levels of procaspase 9. PSSH levels were quantified by measuring the difference in fluorescence between DTT-treated and untreated samples and normalizing this to the total procaspase 9 levels. The strategy used for sample processing to detect total versus persulfidated procaspase 9 is explained in the Supporting Information and Supporting Figure 5.
Activity of caspase 9 in intact cells was also assessed by flow cytometry in Jurkat cells treated with staurosporine, an apoptosis inducer.41 Staurosporine induced caspase 9 activity (Figure 6C), which was prevented by the slow-releasing H2S donor, 100 μM GYY4137. In contrast, the faster-releasing H2S donor, ammonium tetrathiomolybdate, only partially inhibited the effect of staurosporine on caspase activation (Figure 6C).
Procaspase 3 is the downstream target of caspase 9, which cleaves it to the active caspase 3 form.42 Induction of apoptosis by antimycin A treatment of HeLa cells for 16 h resulted in the expected increase in caspase 3 activity, which was partially inhibited by preincubating the cells for 2 h with GYY4137 (Figure 6D).
Finally, we checked whether persulfidation of procaspase 9 was affected in HeLa cells treated with staurosporine for 6 h. Cells were either pretreated with GYY4137 prior to staurosporine addition or treated for 2 h after induction of apoptosis by staurosporine (Supporting Figure 5A). GYY4137 treatment decreased total procaspase 9 levels (Figure 6E). Persulfidation of procaspase 9 was however significantly increased when the cells were treated with both staurosporine and the H2S donor (Figure 6E and Supporting Figure 5B).
Several caspases, including caspase 9, are inhibited by S-nitrosation of the active site cysteine.43,44 Several mechanisms have been proposed to explain the antiapoptotic effects of sulfide.45,46 We provide evidence that reactive sulfur species generated by Cyt C-mediated H2S oxidation can tag proteins with the cysteine persulfide modification. We posit that this reactivity might be enhanced under hypoxic conditions when H2S oxidation is inhibited or under apoptotic conditions when Cyt C is released into the cytoplasm and is disconnected from its mitochondrial electron donors, e.g., complex III. If H2S synthesis is upregulated under these conditions, persulfidation of protein targets in the proximity of Cyt C-like procaspase 9 will result. Inhibition of caspase 9 activity by persulfidation raises the possibility of designing peptidomimetic persulfide donors to inhibit its activity, similar to the reported S-nitrosothiol-containing peptide.47
In summary, we propose that the reaction between Cyt C and H2S results in the initial formation of a HS•/S•− radical. In a cellular milieu, HS•/S•− could suffer one of at least two fates: (i) be trapped by proteins, forming protein persulfides and superoxide, or (ii) be trapped by oxygen, forming HSO2• (Figure 7). Dismutation of superoxide generated in scenario (i) would give H2O2, which together with H2S could lead to more persulfidation.48 Formation of HSO2• in scenario (ii) could further facilitate Cyt C reduction, resulting in sulfite and thiosulfate production. Cyt C reduction by H2S can fuel the ETC, bypassing complex III. Oxidation of Cyt C by complex IV would allow it to cycle between sulfide and the ETC, potentially leading to the accumulation of reactive sulfur species. Cyt C-assisted protein persulfidation might represent a previously unrecognized source of reactive sulfur species that mediates the protective effects attributed to protein persulfidation.
Figure 7.

Protein persulfidation catalyzed by the Cyt C/H2S couple.
METHODS
Reagents.
Antimycin A, equine heart ferric Cyt C, HSA, iodoacetamide, and Na2S nonahydrate (99.99%) were purchased from Sigma-Aldrich. Sodium disulfide (Na2S2) was from Dojindo Molecular Technologies, and monobromobimane FluoroPure grade was from Molecular Probes (Grand Island, NY). GYY4137, AP39, and MeRho-Az were synthesized in house as described previously.35,36,49 2-(Methylsulfonyl)-1,3-benzothiazole was from Santa Cruz Biotechnology, and CN-Cy3 was synthesized as reported previously.33 Murine procaspase 9 was from Enzo Life Science. The siRNA for Cyt C was purchased from Santa Cruz Biotechnology. For the anaerobic experiments, buffers were purged with argon. To maintain high purity, Na2S solutions were made in an argon box by dissolving solid Na2S·9H2O into argon-purged water, as recommended.2,19
Cell Culture.
Human colon cancer (HT29), hepatocellular carcinoma (HepG2), cervical cancer (HeLa), and immortalized human T-lymphocyte (Jurkat) cell lines were purchased from ATCC. Cells were cultured in 10 cm plates in EMEM (HepG2) or RPMI 1640 (HT29) supplemented with 10% FBS (Gibco) and a 1% penicillin (10000 units mL−1)/streptomycin (10000 μg mL−1) mixture (Gibco). Jurkat and HeLa cells were cultured in 75 cm flasks in complete RPMI medium and DMEM (Sigma-Aldrich), respectively, supplemented with 10% FBS and a 1% penicillin (10000 units mL−1)/streptomycin (10000 μg mL−1) mixture (Sigma-Aldrich).
A detailed description of the methods is given in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the French State in the frame of the “Investments for the future” Programme IdEx Bordeaux, reference ANR-10-IDEX-03-02, and by an ATIP-AVENIR grant (to M.R.F.), the National Institutes of Health (GM112455 to R.B. and R01GM113030 to M.D.P.), the Medical Research Council, UK (MR/M022706/1 to M.W.), the National Science Foundation (DGE-1309047 to A.K.S.), and the Brian Ridge Scholarship (R.T.). The authors are grateful to M.-F. Giraud for the help with purification of mitochondria.
Footnotes
The authors declare the following competing financial interest(s): M.W. has patents on the therapeutic and agricultural use of mitochondrially targeted, and other, hydrogen sulfide delivery molecules.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00463.
Supporting Figures 1–5, Supporting Materials and Methods, and references (PDF)
REFERENCES
- (1).Keilin D (1929) Cytochrome and Respiratory Enzymes. Proc. R. Soc. London, Ser. B 104, 206–252. [Google Scholar]
- (2).Filipovic MR, Zivanovic J, Alvarez B, and Banerjee R (2018) Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev 118, 1253–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kimura H (2010) Hydrogen Sulfide: From Brain to Gut. Antioxid. Redox Signaling 12, 1111–1123. [DOI] [PubMed] [Google Scholar]
- (4).Kabil O, and Banerjee R (2010) Redox Biochemistry of Hydrogen Sulfide. J. Biol. Chem 285, 21903–21907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kabil O, Motl N, and Banerjee R (2014) H2S and its role in redox signaling. Biochim. Biophys. Acta, Proteins Proteomics 1844, 1355–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Singh S, Padovani D, Leslie RA, Chiku T, and Banerjee R (2009) Relative Contributions of Cystathionine β-Synthase and γ-Cystathionase to H2S Biogenesis via Alternative Trans-sulfuration Reactions. J. Biol. Chem 284, 22457–22466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chiku T, Padovani D, Zhu W, Singh S, Vitvitsky V, and Banerjee R (2009) H2S Biogenesis by Human Cystathionine gamma-Lyase Leads to the Novel Sulfur Metabolites Lanthionine and Homolanthionine and Is Responsive to the Grade of Hyper-homocysteinemia. J. Biol. Chem 284, 11601–11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, and Kimura H (2009) 3-Mercaptopyruvate Sulfur-transferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid. Redox Signaling 11, 703–714. [DOI] [PubMed] [Google Scholar]
- (9).Yadav PK, Yamada K, Chiku T, Koutmos M, and Banerjee R (2013) Structure and Kinetic Analysis of H2S Production by Human Mercaptopyruvate Sulfurtransferase. J. Biol. Chem 288, 20002–20013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kabil O, and Banerjee R (2012) Characterization of Patient Mutations in Human Persulfide Dioxygenase (ETHE1) Involved in H2S Catabolism. J. Biol. Chem 287, 44561–44567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kabil O, Vitvitsky V, and Banerjee R (2014) Sulfur as a Signaling Nutrient Through Hydrogen Sulfide. Annu. Rev. Nutr 34, 171–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hildebrandt TM, and Grieshaber MK (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275, 3352–3361. [DOI] [PubMed] [Google Scholar]
- (13).Vitvitsky V, Yadav PK, Kurthen A, and Banerjee R (2015) Sulfide Oxidation by a Noncanonical Pathway in Red Blood Cells Generates Thiosulfate and Polysulfides. J. Biol. Chem 290, 8310–8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Vitvitsky V, Yadav PK, An S, Seravalli J, Cho U-S, and Banerjee R (2017) Structural and Mechanistic Insights into Hemoglobin-catalyzed Hydrogen Sulfide Oxidation and the Fate of Polysulfide Products. J. Biol. Chem 292, 5584–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bostelaar T, Vitvitsky V, Kumutima J, Lewis BE, Yadav PK, Brunold TC, Filipovic M, Lehnert N, Stemmler TL, and Banerjee R (2016) Hydrogen Sulfide Oxidation by Myoglobin. J. Am. Chem. Soc 138, 8476–8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ruetz M, Kumutima J, Lewis BE, Filipovic MR, Lehnert N, Stemmler TL, and Banerjee R (2017) A distal ligand mutes the interaction of hydrogen sulfide with human neuroglobin. J. Biol. Chem 292, 6512–6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Riedl SJ, and Salvesen GS (2007) The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol 8, 405–413. [DOI] [PubMed] [Google Scholar]
- (18).Nicholls P, and Kim JK (1981) Oxidation of sulphide by cytochrome aa3. Biochim. Biophys. Acta, Bioenerg 637, 312–320. [DOI] [PubMed] [Google Scholar]
- (19).Wedmann R, Bertlein S, Macinkovic I, Böltz S, Miljkovic JL, Muñoz LE, Herrmann M, and Filipovic MR (2014) Working with “H2S”: Facts and apparent artifacts. Nitric Oxide 41, 85–96. [DOI] [PubMed] [Google Scholar]
- (20).Koppenol WH, and Bounds PL (2017) Signaling by sulfur-containing molecules. Quantitative aspects. Arch. Biochem. Biophys 617, 3–8. [DOI] [PubMed] [Google Scholar]
- (21).Battistuzzi G, Borsari M, Cowan JA, Ranieri A, and Sola M (2002) Control of cytochrome c redox potential: Axial ligation and protein environment effects. J. Am. Chem. Soc 124, 5315–5324. [DOI] [PubMed] [Google Scholar]
- (22).Collman JP, Ghosh S, Dey A, and Decreau RA (2009) Using a functional enzyme model to understand the chemistry behind hydrogen sulfide induced hibernation. Proc. Natl. Acad. Sci. U. S. A 106, 22090–22095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Doeller JE, Grieshaber MK, and Kraus DW (2001) Chemolithoheterotrophy in a metazoan tissue: thiosulfate production matches ATP demand in ciliated mussel gills. J. Exp. Biol 204, 3755–3764. [DOI] [PubMed] [Google Scholar]
- (24).Hu L, Lu M, Wu Z, Wong PT-H, and Bian J (2009) Hydrogen sulfide inhibits rotenone-induced apoptosis via preservation of mitochondrial function. Mol. Pharmacol 75, 27–34. [DOI] [PubMed] [Google Scholar]
- (25).Battistuzzi G, Borsari M, Dallari D, Lancellotti I, and Sola M (1996) Anion binding to mitochondrial cytochromes c studied through electrochemistry Effects of the neutralization of surface charges on the redox potential. Eur. J. Biochem 241, 208–214. [DOI] [PubMed] [Google Scholar]
- (26).Radi R, Thomson L, Rubbo H, and Prodanov E (1991) Cytochrome c-catalyzed oxidation of organic molecules by hydrogen peroxide. Arch. Biochem. Biophys 288, 112–117. [DOI] [PubMed] [Google Scholar]
- (27).Alvarez-Paggi D, Hannibal L, Castro MA, Oviedo-Rouco S, Demicheli V, Tórtora V, Tomasina F, Radi R, and Murgida DH (2017) Multifunctional Cytochrome c: Learning New Tricks from an Old Dog. Chem. Rev 117, 13382–13460. [DOI] [PubMed] [Google Scholar]
- (28).Mottley C, Mason RP, Chignell CF, Sivarajah K, and Eling TE (1982) The formation of sulfur trioxide radical anion during the prostaglandin hydroperoxidase-catalyzed oxidation of bisulfite (hydrated sulfur dioxide). J. Biol. Chem 257, 5050–5055. [PubMed] [Google Scholar]
- (29).Neta P, and Huie RE (1985) Free-radical chemistry of sulfite. Environ. Health Perspect 64, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mottley C, Trice TB, and Mason RP (1982) Direct detection of the sulfur trioxide radical anion during the horseradish peroxidase-hydrogen peroxide oxidation of sulfite (aqueous sulfur dioxide). Mol. Pharmacol 22, 732–737. [PubMed] [Google Scholar]
- (31).Yadav PK, Martinov M, Vitvitsky V, Seravalli J, Wedmann R, Filipovic MR, and Banerjee R (2016) Biosynthesis and Reactivity of Cysteine Persulfides in Signaling. J. Am. Chem. Soc 138, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhang D, MacInkovic I, Devarie-Baez NO, Pan J, Park C-M, Carroll KS, Filipovic MR, and Xian M (2014) Detection of protein S-sulfhydration by a tag-switch technique. Angew. Chem., Int. Ed 53, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wedmann R, Onderka C, Wei S, Szijártó IA, Miljkovic JL, Mitrovic A, Lange M, Savitsky S, Yadav PK, Torregrossa R, Harrer EG, Harrer T, Ishii I, Gollasch M, Wood ME, Galardon E, Xian M, Whiteman M, Banerjee R, and Filipovic MR (2016) Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci 7, 3414–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Buettner GR (1993) The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, a-Tocopherol, and Ascorbate. Arch. Biochem. Biophys 300, 535–543. [DOI] [PubMed] [Google Scholar]
- (35).Hammers MD, Taormina MJ, Cerda MM, Montoya LA, Seidenkranz DT, Parthasarathy R, and Pluth MD (2015) A Bright Fluorescent Probe for H2S Enables Analyte-Responsive, 3D Imaging in Live Zebrafish Using Light Sheet Fluorescence Microscopy. J. Am. Chem. Soc 137, 10216–10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Le Trionnaire S, Perry A, Szczesny B, Szabo C, Winyard PG, Whatmore JL, Wood ME, and Whiteman M (2014) The synthesis and functional evaluation of a mitochondria-targeted hydrogen sulfide donor, (10-oxo-10-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)decyl)triphenylphosphonium bromide (AP39). MedChemComm 5, 728–736. [Google Scholar]
- (37).Arndt S, Baeza-Garza CD, Logan A, Rosa T, Wedmann R, Prime TA, Martin JL, Saeb-Parsy K, Krieg T, Filipovic MR, Hartley RC, and Murphy MP (2017) Assessment of H2S in vivo using the newly developed mitochondria-targeted mass spectrometry probe MitoA. J. Biol. Chem 292, 7761–7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, and Kroemer G (2006) Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 13, 1423–1433. [DOI] [PubMed] [Google Scholar]
- (39).Cai J, Yang J, and Jones DP (1998) Mitochondrial control of apoptosis: the role of cytochrome c. Biochim. Biophys. Acta, Bioenerg 1366, 139–149. [DOI] [PubMed] [Google Scholar]
- (40).Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, and Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489. [DOI] [PubMed] [Google Scholar]
- (41).Luetjens CM, Kögel D, Reimertz C, Düßmann H, Renz A, Schulze-Osthoff K, Nieminen A-L, Poppe M, and Prehn JHM (2001) Multiple kinetics of mitochondrial cytochrome c release in drug-induced apoptosis. Mol. Pharmacol 60, 1008–1019. [DOI] [PubMed] [Google Scholar]
- (42).Zou H, Yang R, Hao J, Wang J, Sun C, Fesik SW, Wu JC, Tomaselli KJ, and Armstrong RC (2003) Regulation of the Apaf-1/Caspase 9 apoptosome by Caspase-3 and XIAP. J. Biol. Chem 278, 8091–8098. [DOI] [PubMed] [Google Scholar]
- (43).Kim J-E, and Tannenbaum SR (2004) S-Nitrosation regulates the activation of endogenous procaspase-9 in HT-29 human colon carcinoma cells. J. Biol. Chem 279, 9758–9764. [DOI] [PubMed] [Google Scholar]
- (44).Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, and Gaston B (2001) S-nitrosylation of mitochondrial caspases. J. Cell Biol 154, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sivarajah A, Collino M, Yasin M, Benetti E, Gallicchio M, Mazzon E, Cuzzocrea S, Fantozzi R, and Thiemermann C (2009) Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock 31, 267–274. [DOI] [PubMed] [Google Scholar]
- (46).Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S, and Snyder SH (2012) Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 45, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mitchell DA, Morton SU, and Marletta MA (2006) Design and characterization of an active site selective caspase-3 transnitrosating agent. ACS Chem. Biol 1, 659–665. [DOI] [PubMed] [Google Scholar]
- (48).Cuevasanta E, Lange M, Bonanata J, Coitiño EL, Ferrer-Sueta G, Filipovic MR, and Alvarez B (2015) Reaction of hydrogen sulfide with disulfide and Sulfenic acid to form the strongly Nucleophilic Persulfide. J. Biol. Chem 290, 26866–26880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Alexander BE, Coles SJ, Fox BC, Khan TF, Maliszewski J, Perry A, Pitak MB, Whiteman M, and Wood ME (2015) Investigating the generation of hydrogen sulfide from the phosphonamidodithioate slow-release donor GYY4137. MedChemComm 6, 1649–1655. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.