

Understanding the mechanism of antibody neutralization/protection against a virus is crucial for antiviral countermeasure development. Tick-borne encephalitis virus (TBEV) and louping ill virus (LIV) are tick-borne flaviviruses (TBFVs) in the family Flaviviridae. They cause encephalomeningitis and encephalitis in humans and other animals. Although vaccines for both viruses are available, infection rates are rising due to low vaccination coverage. In this study, we solved the crystal structures of the LIV envelope protein (E) and a broadly neutralizing/protective TBFV MAb, MAb 4.2, in complex with E from either TBEV or LIV. Key structural features shared by TBFV E proteins were analyzed. The structures of E-antibody complexes showed that MAb 4.2 targets the lateral ridge of both the TBEV and LIV E proteins, a vulnerable site in flaviviruses for other potent neutralizing MAbs. Thus, this site represents a promising target for TBFV antiviral development. Further, these structures provide important information for understanding TBFV antigenicity.
KEYWORDS: louping ill virus (LIV), crystal structure, envelope protein (E), monoclonal neutralizing/protective antibody, tick-borne encephalitis virus (TBEV), tick-borne flaviviruses (TBFVs)
ABSTRACT
Tick-borne encephalitis virus (TBEV) and louping ill virus (LIV) are members of the tick-borne flaviviruses (TBFVs) in the family Flaviviridae which cause encephalomeningitis and encephalitis in humans and other animals. Although vaccines against TBEV and LIV are available, infection rates are rising due to the low vaccination coverage. To date, no specific therapeutics have been licensed. Several neutralizing monoclonal antibodies (MAbs) show promising effectiveness in the control of TBFVs, but the underlying molecular mechanisms are yet to be characterized. Here, we determined the crystal structures of the LIV envelope (E) protein and report the comparative structural analysis of a TBFV broadly neutralizing murine MAb (MAb 4.2) in complex with either the LIV or TBEV E protein. The structures reveal that MAb 4.2 binds to the lateral ridge of domain III of the E protein (EDIII) of LIV or TBEV, an epitope also reported for other potently neutralizing MAbs against mosquito-borne flaviviruses (MBFVs), but adopts a unique binding orientation. Further structural analysis suggested that MAb 4.2 may neutralize flavivirus infection by preventing the structural rearrangement required for membrane fusion during virus entry. These findings extend our understanding of the vulnerability of TBFVs and other flaviviruses (including MBFVs) and provide an avenue for antibody-based TBFV antiviral development.
IMPORTANCE Understanding the mechanism of antibody neutralization/protection against a virus is crucial for antiviral countermeasure development. Tick-borne encephalitis virus (TBEV) and louping ill virus (LIV) are tick-borne flaviviruses (TBFVs) in the family Flaviviridae. They cause encephalomeningitis and encephalitis in humans and other animals. Although vaccines for both viruses are available, infection rates are rising due to low vaccination coverage. In this study, we solved the crystal structures of the LIV envelope protein (E) and a broadly neutralizing/protective TBFV MAb, MAb 4.2, in complex with E from either TBEV or LIV. Key structural features shared by TBFV E proteins were analyzed. The structures of E-antibody complexes showed that MAb 4.2 targets the lateral ridge of both the TBEV and LIV E proteins, a vulnerable site in flaviviruses for other potent neutralizing MAbs. Thus, this site represents a promising target for TBFV antiviral development. Further, these structures provide important information for understanding TBFV antigenicity.
INTRODUCTION
Flaviviruses in the genus Flavivirus of the family Flaviviridae can be divided into three groups, based on the transmission vectors: tick-borne flavivirus (TBFV), mosquito-borne flavivirus (MBFV), and flavivirus with no known vectors (NKVFV) (1, 2). Notorious human pathogens, such as dengue virus (DENV), West Nile virus (WNV), and Zika virus (ZIKV), are all MBFVs (2). Louping ill virus (LIV) and tick-borne encephalitis virus (TBEV) are TBFVs (1–3) belonging to the TBEV serocomplex (4). LIV or TBEV infection can cause encephalitis or encephalomeningitis diseases in the central nervous system (CNS) (2). LIV mainly infects livestock and small animals, including sheep and red grouse. Occasionally, it can also infect humans. LIV was the first tick-borne flavivirus and the only zoonotic flavivirus identified in the British Islands and Norway (3, 5). In contrast, TBEV usually causes encephalitis only in humans, though it can also infect small rodents. TBEV is the most widespread TBFV, and TBEV is believed to annually cause 10,000 to 14,000 cases of infection across the Eurasia continent (6–9). There are three main subtypes of TBEV (the central Europe subtype, the Siberia subtype, and the Far East subtype), from which the rate of mortality varies from 1 to 40% (1, 3, 5, 10–14). Approximately 10% to 20% of survivors suffer long-term sequelae (2). Though efficient vaccines have been commercialized for decades (15), the rate of TBEV infection is still rising due to low vaccination coverage. Currently, no specific therapeutics are available (16). Monoclonal antibody (MAb)-based immunotherapy represents a promising antiviral approach against TBFVs (6), though the use of anti-TBEV immunoglobulins for tick-borne encephalitis (TBE) therapy was discontinued in Europe due to potential antibody-dependent enhancement (ADE) (17–19).
The flavivirus envelope (E) protein is located on the virion surface and is responsible for receptor recognition, virus entry, and subsequent virus-host membrane fusion. E protein is also the main target for neutralizing antibodies (20, 21). Though the structure of TBEV E was solved over 2 decades ago (22), structural information for other TBFV E proteins is lacking (23, 24). The ectodomain of E protein can be divided into three functional domains: domain I (DI), DII, and DIII. DIII is reported to contain epitopes for many potent MAbs, such as E16 for WNV (25), E106 for DENV serotype 1 (DENV1) (26), and ZV67 (27), Z23 (28), Z006 (29), and ZAV190 (30) for ZIKV. Recently, the cryo-electron microscopy (cryo-EM) structures of the TBEV virion and its complex with the neutralizing MAb 19/1786 were determined (6). Further, we previously reported a murine monoclonal antibody (MAb 4.2) that can prophylactically and therapeutically protect mice against LIV challenge (11). It can also neutralize TBEV (the central Europe subtype) (31). The single-chain fragment of the variable region (scFv) of MAb 4.2 Fab expressed in Escherichia coli has a similar virus inhibition effectiveness (31). Previously, the functional features of neutralizing MAbs against TBEV E have been extensively investigated (32–36). Nevertheless, the molecular basis of MAb 4.2 neutralization of TBFV infection is yet elusive.
Here, we present the crystal structures of LIV E as well as MAb 4.2 scFv in complex with the E protein of either LIV or TBEV. Structural analysis revealed that MAb 4.2 recognizes the lateral ridge of DIII on both E proteins, a hot spot for potent neutralizing antibodies against flaviviruses, as previously reported (6, 21, 25–27, 29). Further structural comparison and modeling imply that MAb 4.2 may function by preventing structural rearrangement during the virus-host membrane fusion process.
RESULTS
Overall structure of LIV E.
To obtain soluble LIV E, DNA encoding the ectodomain (residues 1 to 401) was cloned into the pET21a vector, expressed in E. coli cells as inclusion bodies (IB), and subsequently refolded as previously described (37–39). The soluble E protein (sE) mainly existed as a monomer in solution, as shown by size exclusion chromatography (Fig. 1). Crystal screening was performed using the monomeric E protein, and rock-like crystals were obtained after 2 months at 18°C. Diffraction data were collected to a resolution of 3.6 Å. The structure of LIV E was determined in the prefusion conformation, and the E homodimer could be modeled by symmetry operation, similar to other flavivirus E protein structures (22, 38, 40, 41).
FIG 1.

Biochemical characterization of the MAb 4.2 scFv interaction with LIV E/TBEV E. (A) Analytical gel filtration profile of MAb 4.2 scFv and LIV E. MAb 4.2 scFv and LIV E proteins eluted as single monomer peaks in the gel filtration curves. The complex of MAb 4.2 scFv bound to LIV E displayed a shifted complex peak with an extra MAb 4.2 scFv peak. (B) SPR of MAb 4.2 scFv binding to LIV E. (C) Analytical gel filtration profile of MAb 4.2 scFv and TBEV E. (D) SPR of MAb 4.2 scFv bound to TBEV E. All data show that MAb 4.2 binds to both the LIV and TBEV E proteins. In panels A and C, the protein peaks are pointed out with arrows, red rectangles indicate the lanes with the complex peak, and lanes M contain molecular mass markers. mAU, milli-absorbance units; KD, equilibrium dissociation constant.
The overall structure of LIV sE displays the typical class II viral fusion protein architecture, forming head-to-tail homodimers, as seen in all flavivirus E prefusion structures (Fig. 2B). The three domains are arranged in a linear mode: DI has a central β-barrel linked by several loops, DII is an elongated structure that forms the dimerization interface with the fusion loop in the tip region, and DIII is an IgC-like domain (Fig. 2B). The structure of LIV sE is quite similar to that of TBEV sE, with 25 residues in the primary sequence being different (Fig. 2C and Fig. 3).
FIG 2.

Structure of LIV E protein. (A) Schematic diagram of LIV E. Each domain is represented with a different color: DI is in red, DII is in yellow, DIII is in blue, the stem region is in light blue, the transmembrane domain is in dark blue, and the fusion loop is highlighted in lemon. The numbers above the graph represent the starting residue of each segment. (B) Overall view of the LIV E ectodomain dimer. Domains are colored as described in the legend to panel A. Secondary structural elements are labeled in one monomer. The symmetry monomer was generated with PyMOL software. (C) Sequence alignment of LIV E and TBEV E. Secondary structural elements are colored by domains, as described in the legend to panel A. The axis represents the β-strand, a helix means the α-helix, TT is the turning residues of β-strands, and green numbers point out the disulfide bond-forming cysteine. The downward-pointing green arrow represents the potential N-linked glycosylation site on the virion surface, while the downward-pointing red arrows indicate the various residues in the epitope of the two E proteins. The alignment map was generated using the ESPript (version 3.0) program (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi) and manually modified.
FIG 3.

Comparative analyses of all available EDIII structures from different flaviviruses. (A) Sequence alignment of 10 flavivirus EDIIIs. Secondary structural elements follow the LIV E structure. Labels are maintained as described in the legend to Fig. 2C. The alignment map was generated using the ESPript (version 3.0) program (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi) and manually modified. Sequences from all of the following viruses were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/), and GenBank accession numbers are given in parentheses: LIV (NP_044677), TBEV (AGO50945), LGTV (NP_620108), OHFV (NP_878909), DENV2 (AAF18447), WNV (AFO64350), JEV (AAB40664), SLEV (AAP44973), YFV (NP_041726), and ZIKV (AQS26799). (B) Electrostatic surface potential maps of the 10 flavivirus EDIIIs. Black arrows point out the groove in the lateral ridge of TBFV EDIII. The structures of the following viruses were downloaded from PDB (http://www.rcsb.org/), and the PDB accession numbers are given in parentheses: LIV E (6J5C), TBEV E (1SVB), OHFV EDIII (1Z3R), LGTV EDIII (2GG1), DENV2 E (1OAN), WNV E (2HG0), JEV E (3P54), SLEV E (4FG0), YFV E (6IW4), and ZIKV E (5JHM).
In detail, DI is composed of three discrete segments, including residues 1 to 51, 134 to 188, and 279 to 294, which are separated by two portions (residues 52 to 133 and 189 to 278) of DII (Fig. 2A). These three segments fold into 12 β-strands (Fig. 2C), more than the typical 8 strands in the central barrel (22, 42). Residues 8 to 16 and the 150 loop (residue 148 to 159) were not modeled in the final structure due to poor electron density. Therefore, the definitive conformation of the region covering the potential N-linked glycosylation site at N154 is missing (Fig. 2B). Nevertheless, it should be mentioned that since all the proteins were produced in E. coli, there were no posttranslational modifications like those produced in eukaryotic cells, including the potential glycosylation at N154 in LIV E (Fig. 2C). Therefore, the conformations of the residues around N154 might vary from the conformation in the virus particle. The putative dimerization interface within DII is mainly constituted by the αB helix, j strand, and fg loop of each E protomer (Fig. 2B).
DIII (residues 295 to 405), an Ig-like domain, is mainly composed of six strands (strands A, B, C, E, F, and G) and the loops between them (Fig. 2B). Sequence alignment revealed that DIII is the most variable domain among the flavivirus E proteins (Fig. 3A), and thus, antibodies targeting DIII are more likely to be species specific (21). Nine crystal or nuclear magnetic resonance structures of flavivirus E or DIII have been reported, and three of these are from TBFVs (TBEV, Omsk hemorrhagic fever virus [OHFV], and Langat virus [LGTV]) (22–24), and the other six are from MBFVs (DENV, WNV, Japanese encephalitis virus [JEV], St. Louis encephalitis virus [SLEV], yellow fever virus [YFV], and [ZIKV]) (38, 40–46) (Fig. 3). The sequences of these two groups obviously varied: the TBFV DIIIs shared similar residue deletions at positions 335 and 336, 366 and 367, and 386 and 387 but possessed an insertion at position 310 compared to the sequences of the other five MBFVs (Fig. 3A). These variations are thought to be related to host tropism (24). Furthermore, comparison of electrostatic potential maps of DIII revealed that TBFV E proteins are rich in positively charged residues at the lateral ridge and form a crevice due to the residue deletions in the E domain (DE) loop (Fig. 3B).
Complex structures of MAb 4.2 scFv with either LIV DIII or TBEV DIII.
MAb 4.2 can neutralize both LIV and TBEV (31). Neutralizing assays of naturally occurring LIV escape variants suggested that MAb 4.2 engages the 308DKSK311 motif (11), which is relatively conserved within the TBEV E protein (308DKTK311) (Fig. 2C).
To test the binding of MAb 4.2 to E proteins, we performed analytical gel filtration assays. After incubation of MAb 4.2 scFv with TBEV sE or LIV sE, monodisperse peaks for protein complexes eluting before sE and MAb 4.2 scFv were observed (Fig. 1A and C), which indicated the stable binding of MAb 4.2 to both E proteins. We further measured the binding affinities by surface plasmon resonance (SPR) and found that MAb 4.2 scFv binds to LIV sE with an affinity of 45.4 nM but with a lower affinity (390 nM) for TBEV sE (Fig. 1B and D). This difference is consistent with the neutralization efficiencies for these two viruses observed previously (31).
To further analyze the molecular basis of the antibody-E interaction, we determined the crystal structures of MAb 4.2 scFv in complex with the LIV or TBEV E protein. Even though full-length sE proteins were used for crystallization, the electron density for DI and DII could not be resolved in most crystal forms (Fig. 4A and B). Based on cell content analysis, both DI and DII were absent in the crystals, indicating sE degradation during crystallization. The full-length TBEV sE was resolved in only one crystal form, but the density of DI and DII was much weaker than that of DIII, indicating the flexibility of the hinge region connecting DI and DIII (Fig. 4D and Table 1). In all of these structures, MAb 4.2 bound to the E protein in a similar mode involving only DIII. Therefore, further structural analysis was performed using the structures without DI and DII, which were determined with a better resolution.
FIG 4.

Structures of MAb 4.2 in complex with either the LIV or TBEV E protein. (A) Structure of LIV EDIII with MAb 4.2 scFv. The complex structure is colored by chain: LIV EDIII is in blue, the light chain (VL) is in magenta, and the heavy chain (VH) is in cyan. The strands of EDIII and the CDR loops of the antibody that interact with EDIII are labeled. (B) Structure of TBEV EDIII with MAb 4.2 scFv. The corresponding chains are labeled and colored as described in the legend to panel A. (C) Superimposition of the two complex structures. LIV EDIII with MAb 4.2 scFv is in green, and TBEV EDIII with MAb 4.2 scFv is in gray. Labels are maintained as described in the legends to panels A and B. (D) Structure of TBEV E with MAb 4.2 scFv. Domains are colored and labeled as described in the legend to Fig. 2A and panel A. The missing residues are labeled and represented as dotted lines. (E) Comparison of MAb 4.2 scFv bound to TBEV E and TBEV EDIII. The two structures are aligned with the structure of EDIII, and colors and labels are as described in the legend to panels D (MAb 4.2 scFv bound to TBEV E) and B (MAb 4.2 scFv bound to TBEV EDIII). The complex of MAb 4.2 scFv bound to TBEV E is also displayed in transparent surface representation.
TABLE 1.
Data collection and crystallographic statistics
| Parametera | Value(s) forb
: |
|||
|---|---|---|---|---|
| LIV E | LIV EDIII and MAb 4.2 | TBEV EDIII and MAb 4.2 | TBEV E and MAb 4.2 | |
| Data collection statistics | ||||
| Space group | P4132 | I222 | I222 | C121 |
| Cell dimensions | ||||
| a, b, c (Å) | 163.33, 163.33, 163.33 | 93.60, 95.32, 100.92 | 92.67, 95.46, 99.97 | 135.73, 100.69, 140.24 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 100, 90 |
| Resolution (Å) | 50.00–3.60 (3.73–3.60) | 50.00–1.80 (1.86–1.80) | 50.00–1.80 (1.86–1.80) | 50.00–3.30 (3.42–3.30) |
| No. of unique reflections | 9,191 (886) | 42,003 (4,127) | 41,293 (4,076) | 28,405 (2,816) |
| Rmerge | 0.180 (1.539) | 0.101 (0.677) | 0.103 (0.971) | 0.259 (1.499) |
| CC1/2 | 0.999 (0.727) | 0.999 (0.949) | 0.998 (0.807) | 0.979 (0.577) |
| I/σI | 17.7 (1.9) | 16.4 (0.5) | 27.6 (9.4) | 36.2 (6.2) |
| Completeness (%) | 100 (100) | 99.7 (99.1) | 99.9 (99.9) | 99.6 (99.5) |
| Redundancy | 14.6 (15.1) | 13.1 (12.2) | 12.7 (11.2) | 5.3 (5.4) |
| Refinement statistics | ||||
| Resolution (Å) | 47.15–3.59 (3.72–3.59) | 40.26–1.80 (1.86–1.80) | 39.06–1.80 (1.86–1.80) | 44.22–3.29 (3.41–3.29) |
| No. of reflections | 9,070 (818) | 41,383 (3,650) | 38,894 (2,933) | 22,776 (534) |
| Rwork/Rfree | 0.258/0.285 | 0.170/0.190 | 0.179/0.192 | 0.197/0.219 |
| No. of atoms | ||||
| Total | 2,874 | 2,901 | 2,838 | 4,634 |
| Protein | 2,874 | 2,493 | 2,484 | 4,634 |
| Ligand | 0 | 0 | 0 | 0 |
| Water | 0 | 408 | 354 | 0 |
| B factors (Å2) | 73.72 | 25.04 | 24.68 | 105.68 |
| Protein | 73.72 | 23.09 | 23.36 | 105.68 |
| Ligand | 0 | 0 | 0 | 0 |
| Water | 0 | 23.09 | 33.95 | 0 |
| RMSD | ||||
| Bond length (Å) | 0.005 | 0.011 | 0.01 | 0.012 |
| Bond angles (°) | 0.92 | 1.03 | 0.87 | 1.46 |
| Ramachandran plot (%)c | ||||
| Favored | 88.6 | 97.5 | 97.5 | 93.4 |
| Allowed | 10.8 | 2.5 | 2.5 | 5.9 |
| Outliers | 0.5 | 0 | 0 | 0.7 |
Rmerge = ΣiΣhkl|Ii 〈I〉 |/ΣiΣhklIi, where Ii is the observed intensity and 〈I〉 is the average intensity from multiple measurements. CC1/2 is the Pearson correlation coefficient for the two random halves of unique reflections in the unmerged experiment data. h, k, and l are the Miller indices for planes in crystal (Bravais) lattices; they also represent the coordinates in the reciprocal space. Rwork = Σ | |Fo| |Fc | |/Σ|Fo|, where Fo and Fc are the structure factor amplitudes from the data and the model, respectively; Rfree is the R factor for a subset (5%) of reflections that was selected prior to refinement calculations and was not included in the refinement.
Values for the outmost resolution shell are given in parentheses.
Ramachandran plots were generated using the program MolProbity.
The overall structures of the two complexes were quite similar (Fig. 4C), with a root mean square deviation (RMSD) of only 0.18 Å. In general, MAb 4.2 scFv contacted three discrete segments in the lateral ridge of DIII, including the N-terminal loop and the A strand (residues 308 to 313), the BC loop (residues 332 to 336), and the FG loop (residues 387 to 388) (Fig. 5B and C). Reciprocally, CDR2 and CDR3 from the heavy chain variable region (VH) and CDR1 and CDR3 from the light chain variable region (VL) participated in the interaction (Fig. 4A and B). The interacting residues were quite similar in both complexes (Table 2).
FIG 5.

Binding mode comparison of lateral ridge-targeted antibodies. (A) Top view of five antibodies superimposed on their corresponding EDIIIs. Only variable regions of Fab antibodies are shown as a surface representation in different colors: MAb 4.2 is in green, E16 is in yellow, ZV67 is in gray, E106 is in salmon, and 19/1786 is in cyan. Antibody E16 rotates counterclockwise relative to MAb 4.2 approximately 85°, while antibodies ZV67, E106, and 19/1786 span clockwise from MAb 4.2 approximately 50°, 75°, and 170°, respectively. (B to G) Epitopes of MAb 4.2 (B, C), 19/1786 (D), E16 (E), E106 (F), and ZV67 (G). The corresponding EDIIIs are shown in cartoon representation from the top view. Secondary structural elements of each epitope are labeled and colored as described in the legend to panel A. Structures with accession numbers 1ZTX (E16), 4L5F (E106), 5KVG (ZV67), and 5O6V (19/1786) were downloaded from the PDB database.
TABLE 2.
Comparison of amino acid interactions between MAb 4.2 scFv and TBEV E/LIV E
| Residue in MAb 4.2 scFv | LIV E |
TBEV E |
||
|---|---|---|---|---|
| E residue(s) in contact with the MAb 4.2 scFv residue | No. of contacts (no. of hydrogen bonds or salt bridges)a | E residue(s) in contact with the MAb 4.2 scFv residueb | No. of contacts (no. of hydrogen bonds or salt bridges) | |
| H-Y52 | S333, G334, S335 | 1, 7, 21 | S333, G334, T335 | 1, 8, 18 |
| H-S55 | S335, K336 | 4 (1), 4 | T335, K336 | 5 (1), 5 |
| H-T57 | G334, S335, K336 | 4, 5, 7 (1) | G334, T335, K336 | 2, 5, 9 (1) |
| H-Y59 | F332, S333, G334, K336 | 4 (1), 18, 12 (1), 4 | F332, S333, G334, K336 | 4 (1), 19, 12 (1), 4 |
| H-D101 | S335 | 5 | T335 | 7 |
| H-G102 | D308, K309 | 16, 1 | D308, K309 | 17, 1 |
| H-Y103 | D308, K309, S310, E387 | 11, 8, 3, 1 | D308, K309, T310 | 10, 3, 2 |
| H-Y104 | D308, S310, K311 | 5 (1), 1, 8 (1) | D308, T310, K311 | 5 (1), 1, 9 (1) |
| H-I105 | T310 | 2 | ||
| L-Y32 | K309, S310, E387 | 1, 7, 10 (2) | K309, T310, E387 | 2, 7, 9 (2) |
| L-F91 | S310, K311 | 2, 6 (1) | T310, K311 | 3, 6 (1) |
| L-W92 | S310, E387, L388 | 7, 5, 4 | T310, E387, L388 | 9, 6, 3 |
| L-S93 | S310, K311, F312, A313, S333 | 7 (1), 10, 14 (1), 4, 2 | T310, K311, F312, T313, S333 | 7 (1), 9, 14 (1), 5, 2 |
| L-T94 | A313, S333 | 1, 8 | T313, S333 | 3, 3 |
| L-P95 | S333 | 4 | S333 | 5 |
| L-W97 | K311, S333 | 7, 1 | K311, S333 | 8, 2 |
| Total contacts | 250 (11) | 252 (11) | ||
The contacts were defined within a distance of 4.5 Å, and the hydrogen bonds were defined within a distance of 3.5 Å.
Boldface letters represent residues different in TBEV E from LIV E.
For each complex structure, the paratope involves eight or nine residues of VH and seven residues of VL participating in the interaction with DIII and forms a cavity at the interface (Fig. 6B and Table 2). Reciprocally, the footprint in DIII includes residues D308, S310, K311, F312, and S333 and shapes a complementary bulge for the paratope engagement (Fig. 6A). Among the DIII residues, K311 forms hydrogen bonds with both F91 (from VL) and Y104 (from VH), D308 stacks with Y104 (from VH), and S310/T310 and F312 contact S93 (from VL) (Fig. 6C). These interaction networks comprise the main contact interface of the complex. On the other side, residues S55, T57, and Y59 of VH interact with S335/T335, K336, G334, and F332 of DIII, which further stabilizes the binding of scFv to DIII (Fig. 6D).
FIG 6.

Interaction details between MAb 4.2 scFv and LIV EDIII/TBEV EDIII. (A) Amino acids of the footprint on LIV EDIII. DIII is shown in blue surface mode. The residues participating in the interaction with antibodies are labeled and highlighted in green. (B) Amino acids of the paratope on MAb 4.2 scFv. VL and VH are shown in surface mode and colored magenta and cyan, respectively. The residues interacting with antibodies are labeled and printed in blue. The underlined I105 in VL is beyond the distance for contact with LIV EDIII. (C) Major interactions between VL of MAb 4.2 and EDIII. The two complex structures are superimposed and colored similarly: LIV EDIII is in light blue, VL of its complexed MAb 4.2 is in magenta, VH is in cyan, TBEV EDIII is in blue, and VL and VH of MAb 4.2 are in pink and teal, respectively. Residues involved in the interaction are labeled and represented as sticks. Bonds are shown as dotted lines in yellow (LIV) and orange (TBEV). Notably, S310 of LIV is different from T310 in TBEV. (D) Major interactions between residues of VH of MAb 4.2 and EDIII. EDIII and scFv are colored as described in the legend to panel C. (E to G) Surface electrostatic maps of LIV EDIII (E), TBEV EDIII (F), and MAb 4.2 scFv (G) were made using the APBS Electrostatics plug-in in PyMOL software (62). The footprints of the interface on EDIIIs and MAb 4.2 scFv are pointed out with green lines. Of note, compared to TBEV EDIII (F), LIV EDIII exhibits more positive charges in the area highlighted by red dotted circles (E), which is more easily accessible for the mainly negatively charged scFv (G).
As mentioned above, the binding affinities for MAb 4.2 varied between LIV and TBEV. The difference can be interpreted by the available structures. Compared to TBEV E protein DIII (EDIII) (Fig. 6F), the residue differences (Fig. 2C and Table 2) changed the surface electrostatic potential of LIV EDIII to a more positively charged patch around K336 at the interface (Fig. 6E), which enhances the charge complementarity for binding to the negatively charged paratope of MAb 4.2 scFv (Fig. 6G). Therefore, it is likely that the difference in charge complementarity at the interface imbues LIV E with a higher affinity to MAb 4.2 than TBEV E.
Previously, we reported that all MAb 4.2-neutralizing escape variants from murine-adaptive strains encode mutant residues in the epitope region (residues 308 to 311) (47). To interpret the underlying molecular mechanisms of escape, we modeled the EDIII structures of these mutants using the available structure of LIV E and LIV EDIII in complex with MAb 4.2 scFv (Fig. 7B to F). Structural details suggest that the reduced virulence in the mutants with the D308N and S310P mutations is due to the changed surface charges of EDIII (Fig. 7A to C). Meanwhile, all of the mutants with a mutation at position 311 (K311Q, K311N, and K311T) had a longer interaction distance for both F91 (from VL) and Y104 (from VH), which even abolished the interactions (Fig. 7D to F). Therefore, residues 308 to 311 should be the functional residues involved in MAb neutralization, as proven previously (47), while other residues contribute to the tight interaction between EDIII and MAb 4.2 scFv.
FIG 7.

Structural analyses of MAb 4.2 neutralization-resistant mutants. (A) Electrostatic surface potential map of LIV EDIII. The intrinsic residues D308 and S310 are pointed out with black dotted circles. (B and C) Mutants D308N (B) and S310P (C) were modeled by the online server SWISS-MODEL (https://swissmodel.expasy.org/) (63) with the LIV E structure as the template. Residues 308 and 310 are represented as in panel A. Therefore, the surface electrostatic potential changes from negative (A) to positive (B) or neutral (C), which weakens the virulence of mutant LIV strains, as observed in a previous functional study (47). (D to F) Mutants K311Q (D), K311N (E), and K311T (F) were also modeled by use of the SWISS-MODEL, with the LIV EDIII and MAb 4.2 scFv complex structure being used as the template. The complex structure is colored as described in the legend to Fig. 4A, and the modeled EDIIIs are all colored in gray, except for the mutant residue, which is colored by elements; that is, the hydroxyl and amino group are colored in blue and red, respectively. The interactions are represented in yellow (initial complex) or orange (mutant) dotted lines, and the distances were measured by the use of PyMOL software. Therefore, the association between the mutant LIV EDIII and MAb 4.2 is much weaker, even abolishing the interactions, as shown in panel F.
To date, the crystal structures of several lateral ridge-targeted neutralizing MAbs bound to MBFV E proteins have been reported, as exemplified by E16 for WNV, E106 for DENV1, and ZV67 for ZIKV (25–30, 48, 49). In addition, the cryo-EM structure of the TBEV virion in complex with a lateral ridge-targeting MAb, 19/1786, was recently reported (6). Further comparison reveals that the epitope of MAb 4.2 overlaps these MAbs (Fig. 5), including the N-terminal loop and A strand, BC loop, and FG loop. This suggests that the lateral ridge is also a vulnerable site for TBFVs. Notably, superimposition of these structures revealed that MAb 4.2 adopts a unique binding orientation compared to the other MAbs: an 85° clockwise rotation relative to E16 and 50°, 75°, and 170° counterclockwise rotations relative to ZV67, E106, and 19/1786, respectively (Fig. 5A). Unlike the other MAbs, MAb 4.2 does not interact with the DE loop (Fig. 5B to G). Taken together, MAb 4.2 adopts a unique binding mode to interact with the DIII of both LIV E and TBEV E.
Molecular basis for MAb 4.2 neutralization.
As mentioned above, we also solved a complex structure of MAb 4.2 scFv bound to TBEV sE (Fig. 4D). The I0A loop (residues 296 to 303), which links DI and DIII, was not modeled in the final structure (Fig. 4D). The fully exposed flexible I0A loop offers more opportunity for protease cleavage during crystallization, thus resulting in crystals with only DIII bound to MAb 4.2. Of note, the RMSD between TBEV E and TBEV EDIII was very small (0.328 Å) in both complex structures with MAb 4.2 scFv (Fig. 4E), demonstrating the reliable interaction mode of MAb 4.2 scFv with either the LIV or the TBEV E protein, which only involves DIII.
In the structure of full-length TBEV sE in complex with MAb 4.2 scFv, TEBV sE is in its dimeric form bound by two copies of scFv. Therefore, we could superimpose our E-antibody complex structure with the previously reported TBEV E dimer structure (PDB accession number 1SVB) to determine the conformational changes induced by antibody association. Interestingly, compared with its prefusion structure (22), TBEV E was elevated and squeezed toward the dimerization interface upon its association with MAb 4.2 scFv (Fig. 8A), resulting in a 4.6-Å shorter distance between the two αB helices that participate in E dimerization (Fig. 8B). This implies that the binding of MAb 4.2 may restrict TBEV E to retain it in its dimeric form rather than transitioning into the postfusion trimeric form, i.e., preventing domain rearrangement during membrane fusion. Similarly, the WNV antibody E16 also neutralizes viral infection by blocking the conformational changes of E in a step after virus attachment (25). Because the structure of LIV sE is highly similar to that of TBEV sE, we deduce that the restriction effect of MAb 4.2 can be applied to LIV E dimers as well.
FIG 8.
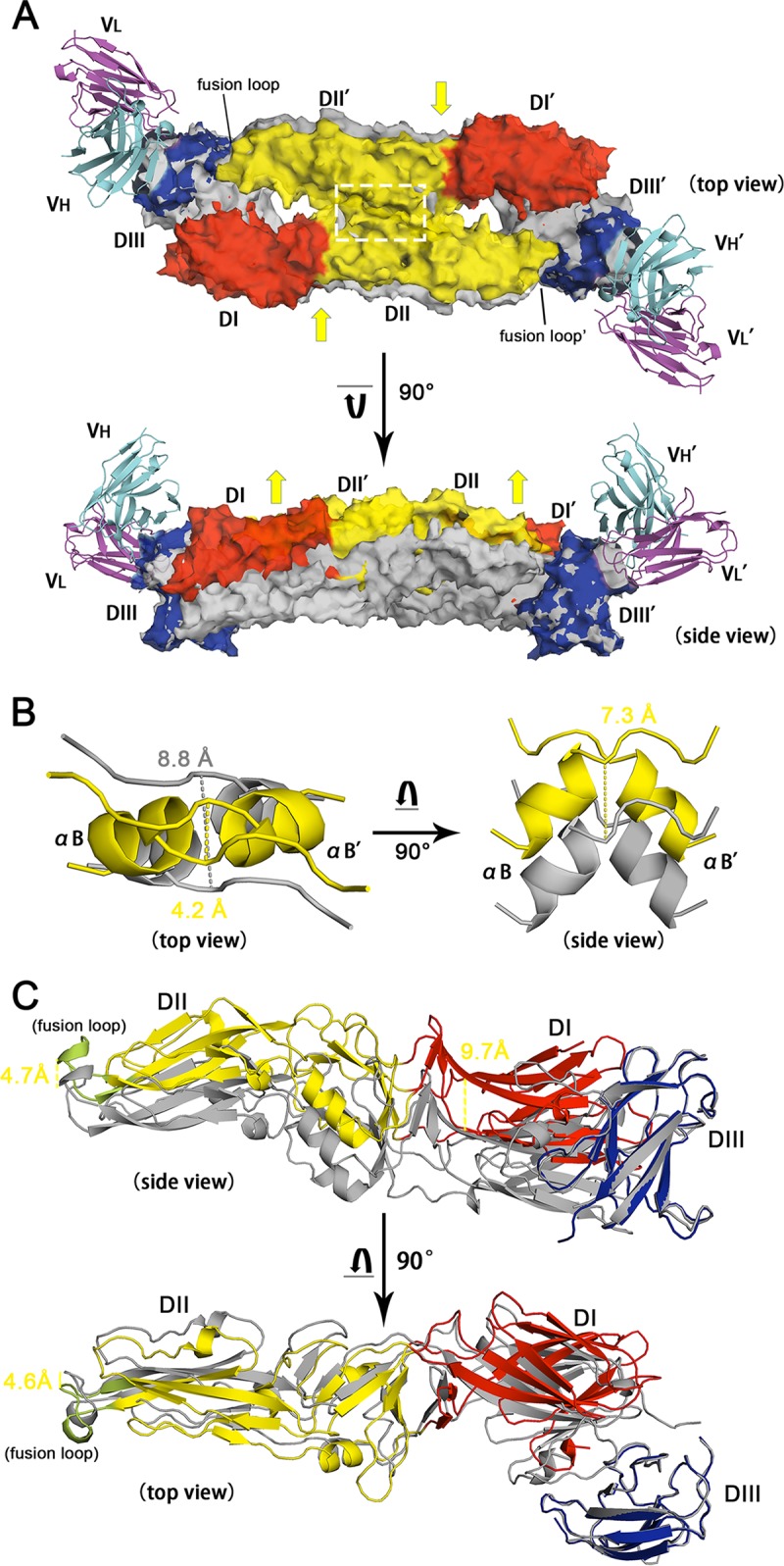
Structural comparison between the free and MAb 4.2 scFv-bound TBEV E homodimers. (A) Superimposition of TBEV E/MAb 4.2 scFv with the TBEV E dimer (PDB accession number 1SVB). The antibody complex is presented with various colors for each domain, as described in the legend to Fig. 4D, while the native structure of the TBEV E dimer is shown in gray. The two structures are aligned with EDIII, leading to the remaining domains being discrete. Both the top view and the side view of the structures are present in cartoon and transparent surface modes. Yellow arrows represent the possible squeezing and elevating of the conformational changes after DIII is associated with MAb 4.2 scFv. (B) The region within the white dashed line in panel A is shown in detail on the left side, while the right side represents the 7.3-Å elevation of the αB helix. The distance between two adjacent αB helices shifts from 8.8 Å to 4.2 Å upon antibody association. (C) Domain movements after binding to MAb 4.2 (not shown in this panel). DI and the fusion loop are elevated by 9.7 and 4.7 Å, respectively. The top view reveals that the fusion loop is squeezed 4.6 Å toward the dimerization interface.
To understand the recognition of DIII by MAb 4.2 in the context of the mature TBEV virion, we superimposed the complex structure of MAb 4.2 scFv/TBEV DIII onto the cryo-EM structure of the TBEV virion (PDB accession number 5O6A) (6). Theoretically, there were three unique positions of EDIII on the TBEV virion, located near the 5-fold and 3-fold axes and the edge of the raft, respectively (Fig. 9A). The DIII locations near the 3-fold axis and the edge of the raft were readily accessible to be bound by the antibody (Fig. 9B). However, when docking MAb 4.2 scFv to any DIII clustered at the 5-fold axis, its light chain would clash with the BC loop of the adjacent DIII (Fig. 9C and D). Therefore, we postulate that a mature TBEV virion could be fully occupied by 120 copies of MAb 4.2 scFv, which is consistent with the cases of antibody E16 bound to WNV virions (25, 50) and antibody 19/1786 bound to TBEV (6). Given the similar binding features, it is quite conceivable that MAb 4.2 shares a similar neutralizing mechanism as E16 for WNV, which inhibits E structural rearrangement at the stage of virus-host membrane fusion.
FIG 9.

Structure of a mature TBEV virion and the possible MAb 4.2 binding mode in the context of the virion. (A) Structure of a TBEV virion (PDB accession number 5O6A) represented in surface mode. Domains are colored as described in the legend to Fig. 7A, and the EDIII epitopes are highlighted in green. The black triangle points out one asymmetric unit on the virion surface, while the black numbers indicate the 2-, 3-, or 5-fold axes. The white dotted line indicates the position of one 5-fold axis on the virion which is shown in detail in panel C. (B) Saturation of the TBEV virion by 120 MAb 4.2 scFvs, which are shown as VH (cyan) and VL (magenta) in surface mode. (C) Epitopes (green) of MAb 4.2 circling the 5-fold axis of the putative TBEV virion particle. (D) Clash of MAb 4.2 scFv with EDIII around the 5-fold axis. The dashed red line points out the possible clash of CDR loops of MAb 4.2 VL with the BC loop of the adjacent EDIII.
DISCUSSION
In this paper, we report the prefusion structure of LIV E, which is quite similar to that of TBEV E. It is the second prefusion E structure and the fourth DIII structure of a TBFV E protein. Sequence alignment and structural comparisons suggest several unique features of TBFV DIII, which exhibits positively charged residues at the lateral ridge and forms a crevice due to residue deletions, distinguishing it from MBFVs. Additionally, we also solved three complex structures of the broadly neutralizing MAb 4.2 bound to LIV EDIII, TBEV EDIII, and TBEV E. Molecular details revealed that MAb 4.2 recognizes epitopes on the lateral ridge of DIII in the two E proteins. Furthermore, the conformation of TBEV E is elevated and squeezed upon binding by MAb 4.2 (Fig. 8A), locking it in the dimeric form. This is because, once the antibody attaches to DIII of the E protein, DI would be elevated up to 9.7 Å due to movement of the flexible I0A loop (missing in the final structure) (Fig. 8C). Then, DII consequently moves upward, shortening the distance between the αB helixes of two E monomers from 8.8 to 4.2 Å (Fig. 8B), which strengthens the binding of two E monomers. Antibody-induced envelope protein rearrangement has been observed with the ZIKV antibody ZKA190, the association of which changes the configuration of E proteins surrounding the 5-fold axis of the mature ZIKV virion (30). Thus, our MAb 4.2 might also have the potential to induce E protein rearrangement on the TBEV virion, which will be an advantage for further applications.
Therapeutic antibodies have shown promising effects in the control of severe infections caused by several MBFVs (21, 51, 52). Antibodies targeting quaternary epitopes or DIII have proven highly potent (21). Neutralizing antibodies, such as Z23 and ZKA190 for ZIKV and 19/1786 for TBEV, all target quaternary epitopes in different domains of the E proteins (6, 28, 30).
As mentioned above, DI and DII are missing in the high-resolution antibody complex structures, which, we speculate, may be due to degradation during the crystallization process. Similar degradation is encountered from time to time in our crystallization studies, e.g., in our previously reported crystal structure of fibronectin-binding protein from Streptococcus suis (PDB accession number 5H3X). We used the full-length protein (552 amino acids) for crystallization trials. However, the crystals unexpectedly contained only the N-terminal 267 amino acids (S2 to K268) (53). Another example is from the crystal structure of the transmembrane protein MCR-2 expressed in E. coli (PDB accession number 6A7W). The full-length MCR-2 was prepared, but the structure-solved crystal contains only the catalytic domain of MCR-2 (54). Here, proteolytic cleavage may have occurred at the proximal upstream portion of EDIII during crystal growth. This is supported by an analysis showing that the proximal upstream sequence of EDIII (LEKLKMKGLT) is a substrate for a panel of proteases. Further, because our E proteins were expressed in E. coli, which lacks the posttranslation modifications common to eukaryotic cell expression systems, they may be more vulnerable to proteases under the crystal conditions.
The MAb 4.2 used in our structural work was functionally investigated by us and our colleagues in the 1990s (11, 31, 47). We found that MAb 4.2 prevents mice against challenge with a lethal dose of LIV (11) and can cross-neutralize the European subtype of TBEV, as well as some other viruses in the TBEV serocomplex (31). Additionally, the single-chain fragment of the variable region (scFv) antibody of MAb 4.2 has a similar virus inhibition effectiveness (31). Previous functional studies suggest that positions 308 and 310 of DIII are pivotal residues that determine the neurovirulence of LIV strains (47), which implies that these residues are located in or adjacent to the putative receptor binding region of LIV E. The corresponding A strand of DENV is also involved in receptor binding (55). Because these two residues are involved in the MAb interaction, MAb 4.2 may potentially inhibit the attachment of LIV to the host cells to exert its neutralizing activity. Finally, a single substitution at residue 311 (lysine to threonine) endows LIV strains with resistance to MAb 4.2 (47), which can be well explained by our complex structure and the modeled EDIII mutants, as discussed above (Fig. 6C and 7F). These findings broaden our understanding of the antigenicity of TBFVs and pave the way toward the development of TBFV-specific therapeutics and vaccines.
MATERIALS AND METHODS
Protein preparation, expression, and purification.
Residues 1 to 401 of the N terminus of LIV (strain 369/T2; GenBank accession number NP_044677) and TBEV (strain HA_2P1-10N_11; GenBank accession number AGO50945) envelope proteins were codon optimized for prokaryotic cell expression and cloned into the bacterial expression vector pET21a with a C-terminal His6 tag. The amino acid sequence of MAb 4.2 was derived from a previous report (47). For expression as a single-chain fragment of the variable region (scFv), the variable region of the light chain and the heavy chain was codon optimized for prokaryotic cell expression and also cloned into pET21a with a (GGGS)3 linker between the two domains. All the recombinant plasmids were transferred into Escherichia coli strain BL21(DE3) and expressed as inclusion bodies (IB) at 37°C. Extraction and refolding of the relevant IB were performed according to previously described methods (37–39) with minor modifications. Briefly, 300 mg IB was diluted dropwise into a 2-liter volume of stirring refolding buffer (100 mM Tris, pH 8.0, 400 mM l-Arg HCl, 2 mM EDTA, 5 mM reduced glutathione, 0.5 mM oxidized glutathione) and further refolded overnight. The refolded protein was then concentrated using an Amicon 400 concentrator with a 10-kDa-cutoff membrane, followed by buffer replacement into the buffer of 20 mM Tris (pH 8.0), 150 mM NaCl. Concentrated proteins were further loaded on a HiLoad 16/60 Superdex 200 PG column (GE Healthcare). Monomeric peaks were collected and concentrated for further use.
Crystallization, data collection, and structure determination.
Before crystal screening, complex proteins were incubated with a molar ratio of 1:1.5 (LIV E or TBEV E/MAb 4.2 scFv) at 4°C for 2 h and subsequently loaded onto a Superdex 200 PG column equilibrated with a buffer of 20 mM Tris (pH 8.0), 50 mM NaCl. LIV E was also recycled for another round of gel filtration before conducting crystal screening. All the proteins were concentrated to 10 mg/ml. The vapor diffusion sitting-drop method was performed with equal volumes of 1 μl protein and 1 μl reservoir solution manually. All the crystals were obtained at 18°C after 2 to 3 months, except for crystals of TBEV E and MAb 4.2 scFv, which were grown at 4°C for more than 3 months. Diffraction-quality crystals were cryoprotected by briefly soaking them in reservoir solution supplemented with 20% (vol/vol) glycerol before flash-cooling in liquid nitrogen. Diffraction data were collected at Shanghai Synchrotron Radiation Facility (SSRF) BL19U (wavelength, 0.97853 Å). In summary, the best crystals were obtained under the following conditions: the LIV E crystal was obtained in 0.1 M citric acid, pH 3.5, 2.0 M ammonium sulfate. The TBEV E and MAb 4.2 scFv complex crystal was obtained in 0.1 M sodium acetate, pH 5, 5% low-molecular-weight polyglutamic acid, 8% polyethylene glycol (PEG) 20000. The TBEV E and MAb 4.2 scFv complex crystal (with only EDIII of TBEV in the final structure) was obtained in 1% (wt/vol) tryptone, 0.05 M HEPES sodium, pH 7.0, 20% (wt/vol) PEG 3350. The LIV E and MAb 4.2 scFv complex crystal was obtained in 0.1 M Na HEPES, pH 7.5, 1.6 M K/Na phosphate.
All the data sets were processed with HKL2000 software (56). The structures were all determined by the molecular replacement method using Phaser software (57), with further molecular adjustments being made manually. For LIV E, the TBEV E prefusion structure (PDB accession number 1SVB) (22) was used as the search model, while MAb 4.2 scFv was solved with our previously reported scFv structure (PDB accession number 5GRJ) (39) as the search model. Then, complex structures were further determined by molecular replacement with the solved monomer structure of LIV-E and MAb 4.2 scFv as the search models, respectively. The atomic models were built by the Coot program (58) and refined with the phenix.refine tool in the Phenix program (59), and the stereochemical qualities of the final models were assessed with MolProbity software (60). Data collection and refinement statistics are summarized in Table 1. All of the structural figures were generated with PyMOL software (61).
Virion modeling.
To analyze the binding mode of antibody MAb 4.2 in the context of the viral particle, the crystal structure of TBEV EDIII bound to MAb 4.2 scFv was superimposed onto each of the three E protomers in an asymmetric unit of the mature TBEV viral particle (PDB accession number 5O6A) by aligning EDIII, which adopts highly similar conformations in the complex and viral particle. The structural model of MAb 4.2 scFv in complex with the TBEV viral particle was obtained by imposing icosahedral symmetry. At the 5-fold vertexes, the epitope is partially shielded by the adjacent E protomers so that the binding of MAb 4.2 scFv is interfered with due to steric hindrance. Therefore, only two copies of the scFv fragment could bind in each asymmetric unit; i.e., one viral particle would be fully occupied by 120 copies of scFv fragment.
SPR assay.
SPR analysis was performed on a BIAcore 3000 machine with CM5 chips (GE Healthcare) at room temperature (25°C). All tested proteins were exchanged to a HEPES buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.005% [vol/vol] Tween 20) via gel filtration. The LIV E/TBEV E proteins were immobilized on the chip to about 1,000 response units (RU). Subsequently, gradient concentrations of MAb 4.2 scFv (0, 1.9, 3.9, 7.8, 15.6, 31.3, 62.5, 125, 250, and 500 nM) were flowed over LIV E on the chip surface, while gradient concentrations (0, 19.5, 39, 78, 156, 312.5, 625, 1,250, 2,500, and 5,000 nM) flowed over TBEV E on the chip surface. The sensor surface was regenerated with treatment with 10 μl of 10 mM NaOH after each cycle of interaction, followed by a flash of HEPES buffers. The binding kinetics were analyzed with the software BIAevaluation (version 4.1) using the 1:1 Langmuir binding model.
Accession number(s).
The atomic coordinates of the LIV E, TBEV E in complex with MAb 4.2 scFv, TBEV EDIII in complex with MAb 4.2 scFv, and LIV EDIII in complex with MAb 4.2 scFv structures have been deposited in the Protein Data Bank (PDB) under accession numbers 6J5C, 6J5G, 6J5F, and 6J5D, respectively.
ACKNOWLEDGMENTS
We thank Zheng Fan of the Institute of Microbiology, Chinese Academy of Sciences, for support with SPR assays and Yan Chai, Chao Su, Dan Lu, Hao Song, and Yi Shi in our group, as well as the staff at the Shanghai Synchrotron Radiation Facility (beamline 19U), for help with data collection, experiments, and manuscript editing.
This study was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29010200), the China National Grand S&T Special Project (2015ZX09102024), and the External Cooperation Program of the Chinese Academy of Sciences (153211KYSB20160001). George F. Gao is a leading principal investigator of the National Natural Science Foundation of China Innovative Research Group (81621091). Lianpan Dai is supported by the Youth Innovation Promotion Association of CAS (grant no. 2015078).
The funding sources had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. We declare no competing financial interests.
G.F.G., L.D., and P.T. contributed to the overall concept and experimental design. X.Y. performed protein purifications and biochemical experiments. X.Y. and J.Q. performed the crystallographic experiments and collected the data. J.Q. and R.P. performed structural determination and modeling. E.A.G. provided the necessary materials and discussion. X.Y., L.D., and G.F.G. analyzed the data and wrote the manuscript.
REFERENCES
- 1.Gould EA, Lamballerie XD, Zanotto PMDA, Holmes EC. 2003. Origins, evolution, and vector/host coadaptations within the genus Flavivirus. Adv Virus Res 59:277–314. doi: 10.1016/S0065-3527(03)59008-X. [DOI] [PubMed] [Google Scholar]
- 2.Pierson TC, Diamond MS. 2013. Flaviviruses, p 747–794. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Gao GF, Zanotto P, Holmes E, Reid H, Gould E. 1997. Molecular variation, evolution and geographical distribution of louping ill virus. Acta Virol 41:259–268. [PubMed] [Google Scholar]
- 4.Gritsun TS, Nuttall PA, Gould EA. 2003. Tick-borne flaviviruses. Adv Virus Res 61:317–371. doi: 10.1016/S0065-3527(03)61008-0. [DOI] [PubMed] [Google Scholar]
- 5.Gao GF, Jiang W, Hussain M, Venugopal K, Gritsun T, Reid H, Gould E. 1993. Sequencing and antigenic studies of a Norwegian virus isolated from encephalomyelitic sheep confirm the existence of louping ill virus outside Great Britain and Ireland. J Gen Virol 74:109–114. doi: 10.1099/0022-1317-74-1-109. [DOI] [PubMed] [Google Scholar]
- 6.Füzik T, Formanová P, Růžek D, Yoshii K, Niedrig M, Plevka P. 2018. Structure of tick-borne encephalitis virus and its neutralization by a monoclonal antibody. Nat Commun 9:436. doi: 10.1038/s41467-018-02882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bogovic P, Strle F. 2015. Tick-borne encephalitis: a review of epidemiology, clinical characteristics, and management. World J Clin Cases 3:430–441. doi: 10.12998/wjcc.v3.i5.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stiasny K, Holzmann H, Heinz FX. 2009. Characteristics of antibody responses in tick-borne encephalitis vaccination breakthroughs. Vaccine 27:7021–7026. doi: 10.1016/j.vaccine.2009.09.069. [DOI] [PubMed] [Google Scholar]
- 9.Gritsun TS, Lashkevich VA, Gould EA. 2003. Tick-borne encephalitis. Antiviral Res 57:129–146. doi: 10.1016/S0166-3542(02)00206-1. [DOI] [PubMed] [Google Scholar]
- 10.Gao GF, Hussain MH, Reid HW, Gould EA. 1993. Classification of a new member of the TBE flavivirus subgroup by its immunological, pathogenetic and molecular characteristics: identification of subgroup-specific pentapeptides. Virus Res 30:129–144. doi: 10.1016/0168-1702(93)90002-5. [DOI] [PubMed] [Google Scholar]
- 11.Gao GF, Hussain M, Reid H, Gould E. 1994. Identification of naturally occurring monoclonal antibody escape variants of louping ill virus. J Gen Virol 75:609–614. doi: 10.1099/0022-1317-75-3-609. [DOI] [PubMed] [Google Scholar]
- 12.Zanotto PMA, Gao GF, Gritsun TS, Marin S, Jiang WR, Venugopal K, Reid HW, Gould EA. 1995. An arbovirus cline across the Northern Hemisphere. Virology 210:152–159. doi: 10.1006/viro.1995.1326. [DOI] [PubMed] [Google Scholar]
- 13.Zanotto PM, Gould EA, Gao GF, Harvey PH, Holmes EC. 1996. Population dynamics of flaviviruses revealed by molecular phylogenies. Proc Natl Acad Sci U S A 93:548–553. doi: 10.1073/pnas.93.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGuire K, Holmes EC, Gao GF, Reid HW, Gould EA. 1998. Tracing the origins of louping ill virus by molecular phylogenetic analysis. J Gen Virol 79:981–988. doi: 10.1099/0022-1317-79-5-981. [DOI] [PubMed] [Google Scholar]
- 15.Brackney DE, Armstrong PM. 2016. Transmission and evolution of tick-borne viruses. Curr Opin Virol 21:67–74. doi: 10.1016/j.coviro.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Boldescu V, Behnam MAM, Vasilakis N, Klein CD. 2017. Broad-spectrum agents for flaviviral infections: dengue, Zika and beyond. Nat Rev Drug Discov 16:565–586. doi: 10.1038/nrd.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kluger G, Schöttler A, Waldvogel K, Nadal D, Hinrichs W, Wündisch GF, Laub MC. 1995. Tickborne encephalitis despite specific immunoglobulin prophylaxis. Lancet 346:1502. doi: 10.1016/S0140-6736(95)92527-9. [DOI] [PubMed] [Google Scholar]
- 18.Waldvogel K, Bossart W, Huisman T, Boltshauser E, Nadal D. 1996. Severe tick-borne encephalitis following passive immunization. Eur J Pediatr 155:775–779. doi: 10.1007/BF02002905. [DOI] [PubMed] [Google Scholar]
- 19.Elsterova J, Palus M, Sirmarova J, Kopecky J, Niller HH, Ruzek D. 2017. Tick-borne encephalitis virus neutralization by high dose intravenous immunoglobulin. Ticks Tick Borne Dis 8:253–258. doi: 10.1016/j.ttbdis.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 20.Dai L, Wang Q, Qi J, Shi Y, Yan J, Gao GF. 2016. Molecular basis of antibody-mediated neutralization and protection against flavivirus. IUBMB Life 68:783–791. doi: 10.1002/iub.1556. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q, Yan J, Gao GF. 2017. Monoclonal antibodies against Zika virus: therapeutics and their implications for vaccine design. J Virol 91:e01049-17. doi: 10.1128/JVI.01049-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature 375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- 23.Mukherjee M, Dutta K, White MA, Cowburn D, Fox RO. 2006. NMR solution structure and backbone dynamics of domain III of the E protein of tick-borne Langat flavivirus suggests a potential site for molecular recognition. Protein Sci 15:1342–1355. doi: 10.1110/ps.051844006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volk DE, Chavez L, Beasley DWC, Barrett ADT, Holbrook MR, Gorenstein DG. 2006. Structure of the envelope protein domain III of Omsk hemorrhagic fever virus. Virology 351:188–195. doi: 10.1016/j.virol.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 25.Nybakken GE, Oliphant T, Johnson S, Burke S, Diamond MS, Fremont DH. 2005. Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature 437:764–769. doi: 10.1038/nature03956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edeling MA, Austin SK, Shrestha B, Dowd KA, Mukherjee S, Nelson CA, Johnson S, Mabila MN, Christian EA, Rucker J, Pierson TC, Diamond MS, Fremont DH. 2014. Potent dengue virus neutralization by a therapeutic antibody with low monovalent affinity requires bivalent engagement. PLoS Pathog 10:e1004072. doi: 10.1371/journal.ppat.1004072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao H, Fernandez E, Dowd KA, Speer SD, Platt DJ, Gorman MJ, Govero J, Nelson CA, Pierson TC, Diamond MS, Fremont DH. 2016. Structural basis of Zika virus-specific antibody protection. Cell 166:1016–1027. doi: 10.1016/j.cell.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Q, Yang H, Liu X, Dai L, Ma T, Qi J, Wong G, Peng R, Liu S, Li J, Li S, Song J, Liu J, He J, Yuan H, Xiong Y, Liao Y, Li J, Yang J, Tong Z, Griffin BD, Bi Y, Liang M, Xu X, Qin C, Cheng G, Zhang X, Wang P, Qiu X, Kobinger G, Shi Y, Yan J, Gao GF. 2016. Molecular determinants of human neutralizing antibodies isolated from a patient infected with Zika virus. Sci Transl Med 8:369ra179. doi: 10.1126/scitranslmed.aai8336. [DOI] [PubMed] [Google Scholar]
- 29.Robbiani DF, Bozzacco L, Keeffe JR, Khouri R, Olsen PC, Gazumyan A, Schaefer-Babajew D, Avila-Rios S, Nogueira L, Patel R, Azzopardi SA, Uhl LFK, Saeed M, Sevilla-Reyes EE, Agudelo M, Yao K-H, Golijanin J, Gristick HB, Lee YE, Hurley A, Caskey M, Pai J, Oliveira T, Wunder EA, Sacramento G, Nery N, Orge C, Costa F, Reis MG, Thomas NM, Eisenreich T, Weinberger DM, de Almeida ARP, West AP, Rice CM, Bjorkman PJ, Reyes-Teran G, Ko AI, MacDonald MR, Nussenzweig MC. 2017. Recurrent potent human neutralizing antibodies to Zika virus in Brazil and Mexico. Cell 169:597–609.e11. doi: 10.1016/j.cell.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Bardelli M, Espinosa DA, Pedotti M, Ng TS, Bianchi S, Simonelli L, Lim EXY, Foglierini M, Zatta F, Jaconi S, Beltramello M, Cameroni E, Fibriansah G, Shi J, Barca T, Pagani I, Rubio A, Broccoli V, Vicenzi E, Graham V, Pullan S, Dowall S, Hewson R, Jurt S, Zerbe O, Stettler K, Lanzavecchia A, Sallusto F, Cavalli A, Harris E, Lok SM, Varani L, Corti D. 2017. A human bi-specific antibody against Zika virus with high therapeutic potential. Cell 171:229–241.e15. doi: 10.1016/j.cell.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang W, Bonnert TP, Venugopal K, Gould EA. 1994. A single chain antibody fragment expressed in bacteria neutralizes tick-borne flaviviruses. Virology 200:21–28. doi: 10.1006/viro.1994.1158. [DOI] [PubMed] [Google Scholar]
- 32.Kiermayr S, Stiasny K, Heinz FX. 2009. Impact of quaternary organization on the antigenic structure of the tick-borne encephalitis virus envelope glycoprotein E. J Virol 83:8482–8491. doi: 10.1128/JVI.00660-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jarmer J, Zlatkovic J, Tsouchnikas G, Vratskikh O, Strauß J, Aberle JH, Chmelik V, Kundi M, Stiasny K, Heinz FX. 2014. Variation of the specificity of the human antibody responses after tick-borne encephalitis virus infection and vaccination. J Virol 88:13845–13857. doi: 10.1128/JVI.02086-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsouchnikas G, Zlatkovic J, Jarmer J, Strauß J, Vratskikh O, Kundi M, Stiasny K, Heinz FX. 2015. Immunization with immune complexes modulates the fine specificity of antibody responses to a flavivirus antigen. J Virol 89:7970–7978. doi: 10.1128/JVI.00938-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heinz FX, Stiasny K. 2017. The antigenic structure of Zika virus and its relation to other flaviviruses: implications for infection and immunoprophylaxis. Microbiol Mol Biol Rev 81:e00055-16. doi: 10.1128/MMBR.00055-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rey FA, Stiasny K, Vaney MC, Dellarole M, Heinz FX. 2018. The bright and the dark side of human antibody responses to flaviviruses: lessons for vaccine design. EMBO Rep 19:206–224. doi: 10.15252/embr.201745302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Zhou M, Han J, Zhu X, Dong T, Gao GF, Tien P. 2005. Generation of murine CTL by a hepatitis B virus-specific peptide and evaluation of the adjuvant effect of heat shock protein glycoprotein 96 and its terminal fragments. J Immunol 174:195–204. doi: 10.4049/jimmunol.174.1.195. [DOI] [PubMed] [Google Scholar]
- 38.Dai L, Song J, Lu X, Deng Y-Q, Musyoki AM, Cheng H, Zhang Y, Yuan Y, Song H, Haywood J, Xiao H, Yan J, Shi Y, Qin C-F, Qi J, Gao GF. 2016. Structures of the Zika virus envelope protein and its complex with a flavivirus broadly protective antibody. Cell Host Microbe 19:696–704. doi: 10.1016/j.chom.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 39.Liu K, Tan S, Chai Y, Chen D, Song H, Zhang CW, Shi Y, Liu J, Tan W, Lyu J, Gao S, Yan J, Qi J, Gao GF. 2017. Structural basis of anti-PD-L1 monoclonal antibody avelumab for tumor therapy. Cell Res 27:151–153. doi: 10.1038/cr.2016.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Modis Y, Ogata S, Clements D, Harrison SC. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A 100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luca VC, AbiMansour J, Nelson CA, Fremont DH. 2012. Crystal structure of the Japanese encephalitis virus envelope protein. J Virol 86:2337–2346. doi: 10.1128/JVI.06072-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nybakken GE, Nelson CA, Chen BR, Diamond MS, Fremont DH. 2006. Crystal structure of the West Nile virus envelope glycoprotein. J Virol 80:11467–11474. doi: 10.1128/JVI.01125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Volk DE, May FJ, Gandham SH, Anderson A, Von Lindern JJ, Beasley DW, Barrett AD, Gorenstein DG. 2009. Structure of yellow fever virus envelope protein domain III. Virology 394:12–18. doi: 10.1016/j.virol.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luca VC, Nelson CA, Fremont DH. 2013. Structure of the St. Louis encephalitis virus postfusion envelope trimer. J Virol 87:818–828. doi: 10.1128/JVI.01950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klitting R, Roth L, Rey FA, de Lamballerie X. 2018. Molecular determinants of yellow fever virus pathogenicity in Syrian Golden hamsters: one mutation away from virulence. Emerg Microbes Infect 7:51. doi: 10.1038/s41426-018-0053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu X, Xiao H, Li S, Pang X, Song J, Liu S, Cheng H, Li Y, Wang X, Huang C, Guo T, ter Meulen J, Daffis S, Yan J, Dai L, Rao Z, Klenk H-D, Qi J, Shi Y, Gao GF. 2019. Double lock of a human neutralizing and protective monoclonal antibody targeting the yellow fever virus envelope. Cell Rep 26:438–446. doi: 10.1016/j.celrep.2018.12.065. [DOI] [PubMed] [Google Scholar]
- 47.Jiang WR, Lowe A, Higgs S, Reid H, Gould EA. 1993. Single amino acid codon changes detected in louping ill virus antibody resistant mutants with reduced neurovirulence. J Gen Virol 74:931–935. doi: 10.1099/0022-1317-74-5-931. [DOI] [PubMed] [Google Scholar]
- 48.Oliphant T, Nybakken GE, Austin SK, Xu Q, Bramson J, Loeb M, Throsby M, Fremont DH, Pierson TC, Diamond MS. 2007. Induction of epitope-specific neutralizing antibodies against West Nile virus. J Virol 81:11828–11839. doi: 10.1128/JVI.00643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stettler K, Beltramello M, Espinosa DA, Graham V, Cassotta A, Bianchi S, Vanzetta F, Minola A, Jaconi S, Mele F, Foglierini M, Pedotti M, Simonelli L, Dowall S, Atkinson B, Percivalle E, Simmons CP, Varani L, Blum J, Baldanti F, Cameroni E, Hewson R, Harris E, Lanzavecchia A, Sallusto F, Corti D. 2016. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353:823–826. doi: 10.1126/science.aaf8505. [DOI] [PubMed] [Google Scholar]
- 50.Kaufmann B, Nybakken GE, Chipman PR, Zhang W, Diamond MS, Fremont DH, Kuhn RJ, Rossmann MG. 2006. West Nile virus in complex with the Fab fragment of a neutralizing monoclonal antibody. Proc Natl Acad Sci U S A 103:12400–12404. doi: 10.1073/pnas.0603488103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi Y, Gao GF. 2017. Structural biology of the Zika virus. Trends Biochem Sci 42:443–456. doi: 10.1016/j.tibs.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Hasan SS, Sevvana M, Kuhn RJ, Rossmann MG. 2018. Structural biology of Zika virus and other flaviviruses. Nat Struct Mol Biol 25:13–20. doi: 10.1038/s41594-017-0010-8. [DOI] [PubMed] [Google Scholar]
- 53.Musyoki AM, Shi Z, Xuan C, Lu G, Qi J, Gao F, Zheng B, Zhang Q, Li Y, Haywood J, Liu C, Yan J, Shi Y, Gao GF. 2016. Structural and functional analysis of an anchorless fibronectin-binding protein FBPS from Gram-positive bacterium Streptococcus suis. Proc Natl Acad Sci U S A 113:13869–13874. doi: 10.1073/pnas.1608406113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X, Lu Q, Qi J, Chai Y, Wang Y, Gao GF. 2018. Structural and functional insights into MCR-2 mediated colistin resistance. Sci China Life Sci 61:1432–1436. doi: 10.1007/s11427-018-9363-4. [DOI] [PubMed] [Google Scholar]
- 55.Cockburn JJ, Navarro Sanchez ME, Fretes N, Urvoas A, Staropoli I, Kikuti CM, Coffey LL, Arenzana Seisdedos F, Bedouelle H, Rey FA. 2012. Mechanism of dengue virus broad cross-neutralization by a monoclonal antibody. Structure 20:303–314. doi: 10.1016/j.str.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 56.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 57.Read RJ. 2001. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr 57:1373–1382. doi: 10.1107/S0907444901012471. [DOI] [PubMed] [Google Scholar]
- 58.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 59.ATLAS Collaboration. 2010. Search for new particles in two-jet final states in 7 TeV proton-proton collisions with the ATLAS detector at the LHC. Phys Rev Lett 105:161801. doi: 10.1103/PhysRevLett.105.161801. [DOI] [PubMed] [Google Scholar]
- 60.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schrodinger LLC. 2015. The PyMOL molecular graphics system, version 1.8. Schrodinger LLC, Cambridge, MA. [Google Scholar]
- 62.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]