

Late (L) genes represent more than one-third of the herpesvirus genome, suggesting that many of these genes are indispensable for the life cycle of the virus. With the exception of BCRF1, BDLF2, and BDLF3, Epstein-Barr virus L genes are transcribed by viral regulators, which are known as the viral preinitiation complex (vPIC) and the host RNA polymerase II complex. Because the vPIC is conserved in beta- and gammaherpesviruses, studying the control of viral L gene expression by the vPIC contributes to the development of drugs that specifically inhibit these processes in beta- and gammaherpesvirus infections/diseases. In this study, we demonstrated that CDK inhibitors induced destabilization of the vPIC component BDLF4, leading to a reduction in L gene expression and subsequent progeny production. Our findings suggest that CDK inhibitors may be a therapeutic option against beta- and gammaherpesviruses in combination with existing inhibitors of herpesvirus lytic replication, such as ganciclovir.
KEYWORDS: BDLF4, CDK2, EBV, phosphorylation, vPIC
ABSTRACT
Temporally controlled gene expression is necessary for the propagation of herpesviruses. To achieve this, herpesviruses encode several transcriptional regulators. In Epstein-Barr virus, BcRF1 associates with five viral proteins (BDLF4, BGLF3, BFRF2, BVLF1, and BDLF3.5) to form the viral late (L) gene regulatory complex, which is called the viral preinitiation complex (vPIC), on TATT-containing promoters. However, regulation of the vPIC has been largely unexplored. In this study, we performed two screens using a kinase inhibitor library and identified a series of cyclin-dependent kinase (CDK) inhibitors that downregulated the expression of L genes without any impact on viral DNA replication through destabilization of the BDLF4 protein. Knockdown of CDK2 by short hairpin RNA (shRNA) and proteasome inhibitor treatment showed that phosphorylation of the BDLF4 protein prevented ubiquitin-mediated degradation. Moreover, we demonstrated that cyclin A- and E-associated CDK2 complexes phosphorylated BDLF4 in vitro, and we identified several serine/threonine phosphorylation sites in BDLF4. Phosphoinactive and phosphomimic mutants revealed that phosphorylation at threonine 91 plays a role in stabilizing BDLF4. Therefore, our findings indicate that S-like-phase CDKs mediate the regulation of L gene expression through stabilization of the BDLF4 protein, which makes the temporal L gene expression system more robust.
IMPORTANCE Late (L) genes represent more than one-third of the herpesvirus genome, suggesting that many of these genes are indispensable for the life cycle of the virus. With the exception of BCRF1, BDLF2, and BDLF3, Epstein-Barr virus L genes are transcribed by viral regulators, which are known as the viral preinitiation complex (vPIC) and the host RNA polymerase II complex. Because the vPIC is conserved in beta- and gammaherpesviruses, studying the control of viral L gene expression by the vPIC contributes to the development of drugs that specifically inhibit these processes in beta- and gammaherpesvirus infections/diseases. In this study, we demonstrated that CDK inhibitors induced destabilization of the vPIC component BDLF4, leading to a reduction in L gene expression and subsequent progeny production. Our findings suggest that CDK inhibitors may be a therapeutic option against beta- and gammaherpesviruses in combination with existing inhibitors of herpesvirus lytic replication, such as ganciclovir.
INTRODUCTION
Epstein-Barr virus (EBV) is a gammaherpesvirus that infects >90% of adults worldwide and is closely linked to the pathogenesis of multiple human malignancies, with 200,000 EBV-associated cancers estimated annually (1). After primary infection, EBV can induce both latent and lytic infections in lymphocytes. Although EBV-associated malignancies are characterized by latent infection, accumulating evidence indicates that lytic viral replication is implicated in the pathogenesis of nasopharyngeal carcinoma (2, 3) and may contribute to the growth of B cell tumors, particularly in patients with immunodeficiency (4, 5), by providing a growth advantage (6, 7) and/or inducing genomic instability (8, 9).
Upon lytic reactivation, EBV genes are expressed in a strictly regulated temporal cascade involving the immediate-early (IE), early (E), and late (L) phases. The IE transcription factors (BZLF1 and BRLF1 proteins) transactivate viral promoters, triggering the EBV lytic cycle. E gene products include proteins involved in viral DNA replication and metabolism (10) and are assembled into replication compartments (11). Replication compartments, in which the EBV genome is amplified 100- to 1,000-fold, become enlarged and fuse to form large globular structures (11). They contain viral DNA, host RNA polymerases, and transcription factors (11–13). Intriguingly, the expression of L genes, which encode mainly structural proteins that encapsulate and mediate the release of infectious virions (14), is tightly linked to viral DNA synthesis (15), because ongoing DNA replication is associated with the recruitment and retention of factors necessary for L gene transcription (16). Recently, cap analysis gene expression sequencing (CAGE-seq) on cells infected with mutant EBV revealed a new EBV kinetics, that of “leaky L genes,” which are active at low levels early and are later upregulated in a DNA replication-dependent manner (17). It should be noted that leaky L and L promoters are transactivated by the viral preinitiation complex (vPIC), with the exception of BCRF1, BDLF2, and BDLF3 (17). At the same time, the virus manipulates cellular signaling pathways to create an optimal environment for productive viral replication (18). High S-phase cyclin-dependent kinase (CDK) activity (19), despite inhibition of cellular DNA replication (20), is known as S-like phase (21). Cyclin A (Cyc A)- and Cyc E-associated CDKs (S-like-phase CDKs) are essential for the transcription of viral IE and E genes, which is likely attributed to transcription factors that are highly expressed during S phase, such as E2F1 and Sp1 (20).
Several studies with beta- and gammaherpesviruses have shed light on the regulation of L gene expression. The L promoters include a TATT element (with T rather than A at the fourth position) instead of the consensus TATA box (22, 23). The EBV-encoding TATA box binding protein homolog, BcRF1, preferentially binds to a TATT element on the L promoters (24). BcRF1 forms the vPIC with five other L gene regulators (BFRF2, BGLF3, BVLF1, BDLF4, and BDLF3.5) (15, 25, 26). The vPIC mediates the recruitment of the RNA polymerase II complex (RNAP II) to L promoters (27) and transactivates the expression of many L genes that encode viral structural proteins (28). The vPIC is conserved in both beta- and gammaherpesviruses (23). EBV BDLF4 and its homologues in human cytomegalovirus (HCMV) (pUL92), murine cytomegalovirus (MCMV) (pM92), and Kaposi's sarcoma-associated herpesvirus (KSHV) (ORF31) play crucial roles in L gene expression (25, 29–31). In addition, individual vPIC components are rapidly degraded when expressed alone but are more stable when all six are coexpressed (27). The EBV protein kinase BGLF4 stimulates the expression of L genes independently and in concert with the vPIC (26), suggesting that posttranslational modifications modulate the action of the vPIC to stimulate the expression of L genes. However, the regulation of the vPIC has not been extensively studied.
In this study, we found that a series of CDK inhibitors suppressed EBV late gene expression and progeny production without affecting viral replication. BDLF4 underwent Cyc A/CDK2 and Cyc E/CDK2 complex-mediated phosphorylation. Mechanistically, BDLF4 was phosphorylated at threonine 91 (T91), preventing its proteasomal degradation. We conclude that phosphorylation of BDLF4 controls its stability and contributes to temporally regulated viral gene expression during EBV lytic replication.
RESULTS
The vPIC component BDLF4 is phosphorylated in the absence of EBV.
During our investigation of EBV lytic genes, we found that a BDLF4 knockout virus exhibits a specific defect in late gene expression (25). Since the kinase activity of BGLF4, the only protein kinase encoded by EBV, is required for BGLF4 stimulation of L gene expression (26), we first investigated the phosphorylation status of the vPIC component BDLF4. FLAG-tagged BDLF4 protein was expressed in EBV-negative HEK293 cells or EBV-positive HEK293/EBV(WT) cells, and then the migration of each protein in SDS-PAGE was compared (Fig. 1A). We found that the phosphorylated form of BDLF4 migrated more slowly on a phosphate affinity (Phos-tag) gel, and we confirmed this finding by phosphatase treatment (Fig. 1A). Phosphorylation was observed both in the presence and in the absence of EBV (Fig. 1A). Although BGLF4, which phosphorylates several proteins at CDK1 target sites (32), modulates the cellular conditions for viral lytic replication (33), BGLF4 did not affect the phosphorylation of BDLF4 (Fig. 1B), suggesting the involvement of cellular kinases in BDLF4 phosphorylation.
FIG 1.
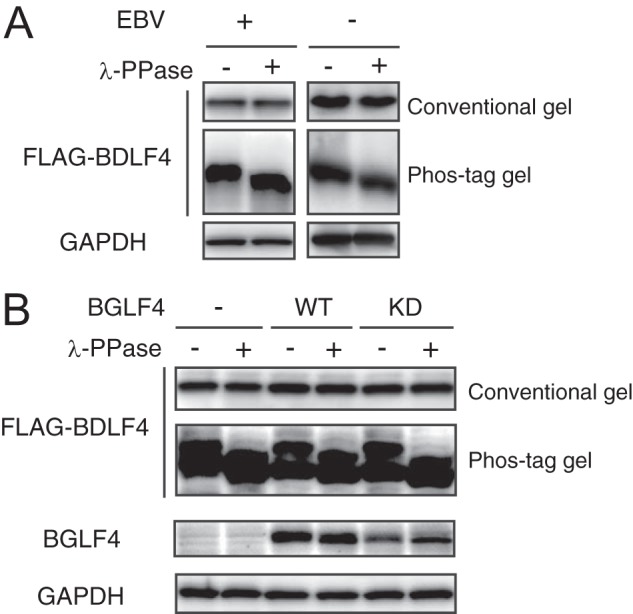
EBV BDLF4 protein is phosphorylated in a viral kinase-independent manner. (A) HEK293/EBV(WT) cells were transfected with FLAG-BDLF4 and BZLF1 expression plasmids (left). HEK293 cells were transfected with a FLAG-BDLF4-expressing plasmid (right). Lysates were either left untreated (–) or treated (+) with λ-protein phosphatase (λ-PPase), separated by conventional or phosphate affinity (Phos-tag) gel electrophoresis, and analyzed by immunoblotting using anti-FLAG and anti-GAPDH antibodies. (B) Lysates from HEK293/EBV(dBGLF4) cells coexpressing BZLF1 protein with WT BGLF4 or a kinase-dead (KD) mutant were analyzed as described for panel A.
CDK inhibitors regulate the stability of the BDLF4 protein.
To identify the kinase responsible for BDLF4 phosphorylation, we performed an inhibitor screen (Fig. 2). The finding that vPIC components are unstable when expressed alone (27) suggests that phosphorylation contributes to the stability of the BDLF4 protein. Cells expressing wild-type (WT) BDLF4 were first treated with kinase inhibitors and then subjected to immunoblot analysis in order to evaluate BDLF4 protein levels. We identified a series of CDK inhibitors that reduced the expression of BDLF4 protein (Fig. 2C). Treatment with the CDK2/CDK9 inhibitor (CDK2/9i) or alsterpaullone, 2-cyanoethyl (A2CE) decreased BDLF4 protein levels, although levels of enhanced green fluorescent protein (EGFP), which was exogenously expressed from the CMV IE promoter, were not reduced upon addition of a CDK inhibitor (Fig. 3A). In addition, CDK2/9i and A2CE did not alter BDLF4 mRNA expression from that with dimethyl sulfoxide (DMSO) treatment (Fig. 3B). These findings suggest that CDK inhibitors target BDLF4 protein but not mRNA. To investigate the mechanisms responsible for the CDK inhibitor-induced reduction in BDLF4 protein levels, we assessed the effects of the proteasome inhibitors MG132 and bortezomib (BTZMB). Treatment with MG132 or BTZMB inhibited the reduction in BDLF4 protein levels (Fig. 3C), indicating that CDK inhibitors enhance the degradation of the BDLF4 protein. Proteasome-mediated protein degradation follows protein ubiquitination. Therefore, we next examined polyubiquitin (Ub) chain conjugation on BDLF4 after CDK inhibitor treatment. As shown in Fig. 3D, hemagglutinin (HA)-Ub immunoprecipitation (IP) showed that CDK2/9i increased the level of ubiquitinated BDLF4. In agreement with these findings, CDK2/9i and A2CE inhibited the expression of BDLF4 protein during EBV lytic replication (Fig. 3E). Thus, CDK inhibitors suppressed the expression of BDLF4 protein by enhancing its proteasomal degradation. Also, the addition of a CDK inhibitor reduced the intensity of the lower migrating band (BDLF4) in the Phos-tag gel (Fig. 3F), indicating that the inhibitors suppressed BDLF4 phosphorylation. However, we cannot rule out the possibility that a kinase that is not a CDK also phosphorylates BDLF4; the lower BDLF4 band was not completely eliminated.
FIG 2.

Screen for kinase inhibitors downregulating BDLF4 expression. (A) Workflow of the screen. (B) Summary of screen data. IB, immunoblotting. (C) Heat map showing the levels of BDLF4 protein in cells treated with the indicated inhibitors. Signals higher than the baseline level are shown in red; signals lower than baseline are shown in blue. The numbers on the heat map key indicate log2-fold changes relative to the DMSO-treated control. (D) Immunoblot data derived using anti-FLAG and anti-GAPDH antibodies. Relative (Rel.) signal intensities are ratios of the FLAG band intensity to the GAPDH band intensity.
FIG 3.

CDK inhibitors suppress BDLF4 phosphorylation, destabilizing the protein. (A) (Left) HEK293 cells were transfected with a BDLF4 expression plasmid together with an EGFP expression plasmid (as a control) and were then treated with CDK inhibitors. Lysates were analyzed by immunoblotting using anti-FLAG, anti-GFP, and anti-GAPDH antibodies. (Right) The FLAG-BDLF4 band intensities were quantified and normalized to those of GAPDH. The results shown are means ± SDs from at least three independent experiments. Double asterisks indicate P values of <0.01. (B) qPCR measurements of BDLF4 transcript levels in HEK293/FLAG-BDLF4 cells treated with CDK inhibitors. Results shown are means ± SDs from three independent experiments. n.s., not significant. (C) Lysates from HEK293/FLAG-BDLF4 cells treated with CDK inhibitors were analyzed by immunoblotting using the indicated antibodies. (D) HA-Ub immunoprecipitation showed that a CDK inhibitor enhanced Ub conjugation of BDLF4 protein. HA-Ub was transiently transfected into HEK293/FLAG-BDLF4 cells, which were subsequently treated with a CDK inhibitor and BTZMB. (E) Lysates from HEK293/EBV(WT) cells in which lytic replication had been induced and which had been treated with CDK inhibitors were analyzed by immunoblotting using the indicated antibodies. (F) HEK293/FLAG-BDLF4 cells were treated with CDK inhibitors and BTZMB. Lysates either were not (–) or were incubated (+) with λ-protein phosphatase (λ-PPase), subjected to conventional or phosphate affinity (Phos-tag) gel electrophoresis, and immunoblotted using anti-FLAG and anti-GAPDH antibodies.
CDK inhibitors regulate the expression of EBV L genes.
To validate the results of the screen described above in terms of EBV lytic infection, we assessed the impact of CDK inhibitors on L gene expression using EBV-positive HEK293/EBV(WT) cells. Lytic replication was induced by transfection with a BZLF1 protein expression plasmid, and then CDK inhibitors were added to the medium 24 h posttransfection (p.t.) (Fig. 4A, left). Total RNA and genomic DNA were prepared at 48 h p.t. and were used for quantitative PCR (qPCR) analysis. Treatment with CDK2/9i or A2CE at 24 h p.t. decreased the expression of gp350 (encoded by an L gene) without any effect on the expression of BALF5 (encoded by an E gene) (Fig. 4A, center) or viral replication (Fig. 4A, right). In contrast, CDK inhibitor treatment at 6 h p.t. decreased the expression of this E gene (Fig. 4B), consistent with previous work on S-like-phase CDKs using alternative CDK inhibitors such as purvalanol A and roscovitine (34). Therefore, blocking CDK activity at various times after the induction of lytic replication enables evaluation of the effects of CDK inhibitors on L gene expression. Moreover, a CDK inhibitor-mediated reduction in L gene expression was also observed by immunoblotting (Fig. 4C) and reporter assays (Fig. 4D). In agreement with these findings, progeny virus production was significantly reduced by CDK inhibitor treatment (Fig. 4E), indicating that CDK may be a therapeutic target for EBV.
FIG 4.

CDK inhibitors regulate the expression of late genes during EBV lytic replication. (A) qPCR of viral RNAs (center) and viral replication levels (right) in HEK293/EBV(WT) cells treated with CDK inhibitors at 48 h after the induction of lytic replication. Cells were transfected with the BZLF1 expression plasmid and were treated with CDK 2/9 inhibitor (CDK2/9i; 500 nM) or alsterpaullone, 2-cyanoethyl (A2CE; 1 μM) as indicated on the schedule (left). Results shown are means ± SDs from three independent experiments. Double asterisks, P < 0.01; n.s., not significant. (B) qPCR analyses of viral RNA levels (center) and viral replication levels (right) in HEK293/EBV(WT) cells treated with CDK inhibitors 48 h after the induction of lytic replication. Cells were transfected with a BZLF1-expressing plasmid and were treated with CDK2/9i (500 nM) or A2CE (1 μM) as indicated on the schedule (left). Results shown are means ± SDs from three independent experiments. Double asterisks, P < 0.01. (C and D) HEK293/EBV(WT) cells were transfected with a TATT-oriLyt-Luc reporter plasmid together with pSV40-Rluc as an internal control and a BZLF1 expression plasmid as indicated. DMSO or a CDK inhibitor was added to the medium at 24 h p.t. (C) Lysates harvested from the cells at 48 h p.t. were subjected to immunoblot analysis with the indicated antibodies. BZLF1 is an IE gene; BMRF1 and BALF2 are E genes; and BRRF2, BALF4 (gB), and BKRF4 are L genes (11, 48, 55, 56). (D) Lysates were also analyzed using luciferase reporter assays. The results are presented as means ± SDs from three independent transfections after normalization to the internal control (Renilla luciferase activity). Double asterisks, P < 0.01. (E) HEK293/EBV(WT) cells were first transfected with BZLF1 expression vectors and then treated with CDK inhibitors. The virus yields were determined by counting GFP-positive Akata(–) cells. The results are shown as means ± SDs from three independent experiments and are relative to the virus yield of BZLF1 with DMSO treatment (infectivity value, 1). Double asterisks, P < 0.01.
CDK2 complexes phosphorylate BDLF4 to protect it from proteasomal degradation.
Our next goal was to determine which CDK was involved in the phosphorylation of BDLF4. Because CDKs that interact with multiple cyclins primarily regulate the cell cycle, we assessed whether the stability of BDLF4 was impacted by the cell cycle. A double thymidine block and release assay in HEK293 cells stably expressing FLAG-BDLF4 demonstrated that the levels of BDLF4 changed in a cell cycle-dependent manner (Fig. 5A), suggesting that cell cycle-related CDKs phosphorylated BDLF4. HEK293 cells have been used extensively in studies of the cell cycle; these cells exhibit sequential CDK activation, although they express the adenovirus E1A protein, which inhibits pRb activity (35–37).
FIG 5.

CDK2 is responsible for the phosphorylation-mediated stabilization of BDLF4. (A) The cell cycle of HEK293/FLAG-BDLF4 cells was synchronized using a double thymidine block (61). Cells were harvested at the indicated times after release of the block and were analyzed by immunoblotting using the indicated antibodies. AS, asynchronized; h.p.r., hours postrelease. (B and C) HEK293/FLAG-BDLF4 cells were transfected with plasmids expressing shRNA targeting CDK1 or CDK2 mRNA. Cells were lysed at 48 h p.t. for immunoblotting using the indicated antibodies. (D) HEK293/FLAG-BDLF4 cells were transfected with FLAG-BZLF1 and/or shRNA expression plasmids as indicated, followed by CHX treatment (50 mg/ml). The protein levels of FLAG-BDLF4 and FLAG-BZLF1 (control) were detected using anti-FLAG antibodies. Results are presented as means ± SDs from three independent experiments and are shown relative to the protein levels in the absence of CHX treatment. Asterisks, P < 0.05; n.s., not significant. (E) HEK293/FLAG-BDLF4 cells transfected with shCDK2 were treated with MG132 for 24 h before harvesting. Lysates were analyzed by immunoblotting using the indicated antibodies. (F) (Left) Lytic replication was induced by transfection of 293/EBV(WT) cells with a BZLF1-expressing plasmid, followed by transfection with plasmids carrying shCDK2 at 8 h p.t.; the cells were harvested at 48 h p.t. (Right) Lysates were subjected to immunoblotting using the indicated antibodies.
Next, we examined whether CDK2, which is dispensable for S-phase entry and progression (38), regulated the levels of BDLF4. Knockdown of CDK2 by short hairpin RNA (shRNA) significantly reduced the levels of BDLF4 protein from those with shCDK1 (Fig. 5B and C). To compare the stability of BDLF4 protein under the CDK2 knockdown conditions, cells were cotransfected with FLAG-BDLF4 and FLAG-BZLF1 expression plasmids and were then treated with cycloheximide (CHX), an inhibitor of protein synthesis, followed by immunoblotting. As shown in Fig. 5D, knockdown of CDK2 by shRNA reduced the half-life of BDLF4 protein but not that of BZLF1 protein. Further experiments confirmed that the reduction of BDLF4 by shCDK2 was prevented by MG132 treatment (Fig. 5E). To further evaluate the role played by CDK2 in lytic replication, we used shRNA to knock down CDK2 in the middle-to-late phase of lytic replication in HEK293/EBV(WT) cells (Fig. 5F, left). CDK2 knockdown reduced the expression of BALF4 (glycoprotein B [gB], encoded by an L gene) but had no effect on the expression of BMRF1 (encoded by an E gene) (Fig. 5F, right). These findings suggest that CDK2 is responsible for stabilizing the BDLF4 protein.
The other screen that we used to identify inhibitors of L gene expression in Tet-BZLF1/B95-8 cells (20) also identified four CDK inhibitors that regulated L gene expression (Fig. 6).
FIG 6.

Screening for kinase inhibitors that specifically inhibit EBV late gene expression but do not inhibit viral replication. (A) Workflow. (B) Summary results of the screen. (C) Heat map showing the viral early/late gene expression ratios and the extents of viral replication in B95-8 cells in which lytic replication was induced, followed by the addition of the indicated inhibitors. Signals higher than the baseline values are shown in red; signals lower than the baseline are shown in blue. The numbers on the heat map are log2-fold changes from the levels with DMSO alone. (D) qPCR analyses of viral mRNA (left) and DNA (right) levels. Bars represent the log fold changes in E/L ratios (BMRF1/MCP expression levels) (left) and relative replication levels (normalized to those for the DMSO controls) (right). Rel., relative.
Phosphorylation of BDLF4 protein by Cyc A/CDK2 and Cyc E/CDK2 is required for L gene expression.
To validate these observations, we phosphorylated BDLF4 in vitro using recombinant Cyc A/CDK2 and Cyc E/CDK2 complexes. Escherichia coli-derived WT BDLF4 was phosphorylated by both the Cyc A/CDK2 and Cyc E/CDK2 complexes, but not by the Cyc B/CDK1 complex (Fig. 7A). Elevations in Cyc E- and Cyc A-associated CDK activities modulate the cellular conditions obtaining during the S-like phase of EBV lytic replication (19). We next identified the residues of BDLF4 protein that were phosphorylated. BDLF4 phosphorylated in vitro by Cyc A/CDK2 and Cyc E/CDK2 complexes was digested with chymotrypsin and analyzed by liquid chromatography–tandem mass spectrometry (LC–MS-MS). Eight phosphopeptides consistent with phosphorylation at serine 8 (S8), T16, S26, T91, T97, T220, T222, and T223 were identified (Fig. 7B and Table 1). To investigate whether the phosphorylation of BDLF4 by CDK2 complexes influences the function of BDLF4, we expressed WT BDLF4, a 6×A mutant (in which six S/T sites phosphorylated by Cyc A/CDK2 were mutated to A), or a 4×A mutant (in which four S/T sites phosphorylated by Cyc E/CDK2 were mutated to A) (Fig. 7C) together with a BZLF1 expression plasmid and a TATT-oriLyt reporter in HEK293/EBV(dBDLF4) cells. As shown in Fig. 7D, the 6×A and 4×A mutants exhibited significantly lower activity than WT BDLF4. In addition to performing the reporter assays, we confirmed that the BDLF4 6×A and 4×A mutants did not complement progeny virus production in HEK293/EBV(dBDLF4) cells (Fig. 7E). These findings indicate that phosphorylation of BDLF4 by CDK2 complexes stimulates BDLF4-mediated L gene expression.
FIG 7.

Phosphorylation of BDLF4 by CDK2 complexes enhances L gene expression. (A) In vitro phosphorylation assay showing that BDLF4 protein was phosphorylated by recombinant Cyc A1/CDK2 and Cyc E1/CDK2, but not by Cyc B1/CDK1. Histone H1 served as the control. The phosphorylation assay used ATP-γ-S as the phosphodonor and an anti-thiophosphate ester (anti-ThioP) antibody to detect substrate phosphorylation. Immunoblotting (IB) using an anti-BDLF4 or anti-histone H1 antibody shows the levels of the recombinant protein used for the assay. (B) List of the serine/threonine residues in BDLF4 phosphorylated by Cyc A1/CDK2 and Cyc E1/CDK2. These residues were identified by LC–MS-MS analysis. (C) Schematic representation of the S/T sites mutated in the A×6 and A×4 mutants. (D) HEK293/EBV(dBDLF4) cells were transfected with the pTATT-oriLyt-Luc reporter plasmid together with pSV40-Rluc as an internal control and BZLF1 and BDLF4 (WT or mutant) expression plasmids as indicated. Luciferase activities were measured at 48 h posttransfection. The results are presented as means ± SDs from three independent transfections after normalization against the internal control (Renilla luciferase activity). Lysates were subjected to immunoblotting using the indicated antibodies. Asterisks, P < 0.05. (E) HEK293/EBV(dBDLF4) cells were transfected with BZLF1 and BDLF4 (WT or mutant) expression plasmids as indicated. The viral yields were determined by counting GFP-positive Akata(–) cells. The results are shown as means ± SDs from three independent experiments relative to the viral yield of BZLF1 plus WT BDLF4 (infectivity value, 1). Double asterisks, P < 0.01.
TABLE 1.
Full list of the class 1 phosphorylation sites in the BDLF4 protein identified by LC–MS-MS
| Position | Amino acid | Localization probability with: |
|||
|---|---|---|---|---|---|
| CycA Rep1a | CycA Rep2 | CycE Rep1 | CycE Rep2 | ||
| 8 | S | 1.000 | 1.000 | 1.000 | 0.997 |
| 16 | T | 1.000 | 1.000 | 1.000 | |
| 26 | S | 0.994 | 0.955 | 0.998 | |
| 91 | T | 0.936 | 0.979 | 0.954 | 0.962 |
| 97 | T | 0.994 | 0.988 | 0.860 | |
| 220 | T | 0.997 | 0.984 | 0.968 | |
| 222 | T | 0.861 | 0.932 | 0.855 | |
| 223 | T | 0.794 | 0.797 | 0.815 | |
Rep, replicate.
Phosphorylation at T91 controls the stability of BDLF4 protein.
To investigate which phosphorylated residue is responsible for BDLF4 stability, phosphoinactive mutants (in which S or T was replaced with A) were generated and transiently expressed in HEK293 cells. The T91A mutant was expressed at protein levels much lower than those of the WT and other mutant proteins (Fig. 8A). The reductions in BDLF4 T91A levels were rescued by treatment with MG132 or BTZMB (Fig. 8B). Furthermore, an acidic amino acid substitution (T91E) partially rescued the protein level of the T91A mutant but did not rescue transcriptional activity (Fig. 8C). However, we cannot rule out the possibility that a glutamic acid mutant may not reliably mimic the phosphorylated form of BDLF4, because glutamic acid is singly charged whereas phosphorylated threonine is nominally doubly charged at physiological pH (39). On the other hand, the inhibitory effects of CDK inhibitors on BDLF4-mediated reporter expression were reduced upon mutation of T91 (Fig. 8D), suggesting that T91 of BDLF4 is a target site for a cellular kinase(s) such as CDK2. Indeed, the T91A mutant did not complement the viral progeny production of a BDLF4 deletion mutant of EBV as well as WT BDLF4 did (Fig. 8E). Hence, these results suggest that although phosphorylation at T91 stabilizes BDLF4, phosphorylation at multiple S/T residues is required for control of BDLF4 transcriptional activity.
FIG 8.

Phosphorylation of BDLA4 at T91 plays an important role in the stabilization of BDLF4 protein. (A) Lysates from HEK293 cells transfected with WT BDLF4 or BDLF4 A mutants were analyzed by immunoblotting using anti-FLAG and anti-GAPDH antibodies. (B) HEK293 cells were first transfected with a BDLF4 WT or T91A mutant expression plasmid and then treated with MG132 or BTZMB. Lysates were analyzed by immunoblotting using the indicated antibodies. (C) HEK293/EBV(dBDLF4) cells were transfected with the pTATT-oriLyt-Luc reporter plasmid, pSV40-Rluc (internal control), and plasmids expressing BZLF1 and BDLF4 (WT, T91A mutant, or T91E mutant) as indicated. Luciferase activities were measured at 48 h posttransfection. The results are presented as means ± SDs from three independent transfections after normalization to the internal control (Renilla luciferase activity) levels. Lysates were subjected to immunoblotting using the indicated antibodies. Asterisks, P < 0.05. (D) HEK293/EBV(dBDLF4) cells were transfected with the pTATT-oriLyt-Luc reporter plasmid, pSV40-Rluc (internal control), and plasmids expressing BZLF1 and BDLF4 (WT or T91A mutant). Luciferase activities in cells treated with CDK inhibitors (CDK2/9i or A2CE) were assayed. The effects of CDK inhibitors on reporter expression are expressed as percentages of decrease from the level of activity for samples treated with DMSO. The results are means ± SDs from three independent experiments. Asterisks, P < 0.05; double asterisks, P < 0.01, respectively. (E) HEK293/EBV(dBDLF4) cells were transfected with BZLF1 and BDLF4 expression vectors as indicated. The viral yields were determined by counting GFP-positive Akata(–) cells. The results are shown as means ± SDs from three independent experiments relative to the viral yield of BZLF1 plus WT BDLF4 (infectivity value, 1). Double asterisks, P < 0.01.
DISCUSSION
In this study, we identified S-like-phase CDKs (Cyc A/CDK2 and Cyc E/CDK2) as host cell factors in the regulation of the EBV vPIC, which plays a central role in viral L gene expression. Upon CDK2 knockdown, the level of BDLF4 protein was reduced. This effect was also seen in phosphoinactive BDLF4 mutants, suggesting that phosphorylation of BDLF4 impacted its stability. HA-Ub IP and proteasome inhibitor treatment showed the Ub-mediated degradation of BDLF4 when it was dephosphorylated. Therefore, our data suggest that the cellular environment affects the temporal control of L gene expression via posttranslational modification of the BDLF4 protein (Fig. 9).
FIG 9.
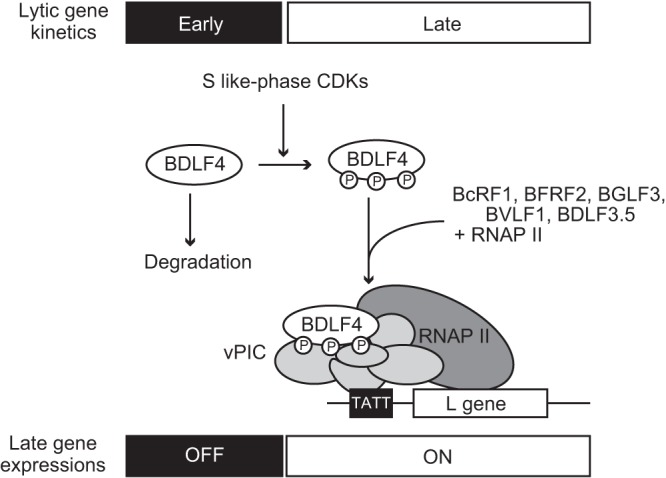
Model illustrating how S-like-phase CDKs temporally control late gene transcription during the EBV lytic phase. BDLF4 (encoded by an EBV E gene) (17, 25) is expressed during early lytic replication, at which time L gene expression is suppressed, because the unphosphorylated form of BDLF4 is rapidly degraded. As the lytic cycle progresses, the activities of cyclin E- and cyclin A-associated CDK proteins (S-phase CDKs) increase (19). Activated S-phase CDKs phosphorylate BDLF4, stabilizing the protein. Phosphorylated BDLF4 engages in vPIC formation with five other viral proteins and transactivates L gene expression in the middle-to-late phase of lytic replication. P, phosphorylation; RNAP II, RNA polymerase II complex.
Switch-like regulation of phosphorylation through kinase cascades is able to affect coordinated and well-organized changes and is fundamental to the proper progression of biological reactions. For instance, the decision to enter into a new phase during cell cycle progression is not taken lightly and is controlled by multistep and positive-feedback mechanisms. The Ub-mediated disruption of the cell cycle regulator is controlled by its phosphorylation (40). SKP2 associates with the Ub E3 ligase SCF complex and targets p27 for degradation only when it is phosphorylated on T187 (41–43). In contrast to phosphorylation-driven ubiquitination, several reports have demonstrated phosphorylation-induced stabilization due to protection from Ub-mediated degradation (44, 45). In this study, we showed that S-like-phase CDKs phosphorylated BDLF4 protein at multiple S/T sites and inhibited the Ub-mediated degradation of BDLF4, which contributed to the temporal regulation of L gene expression. These findings suggest that phosphorylation at these S/T residues is essential for BDLF4 to serve as the L gene regulator. However, we cannot rule out the possibility that phosphorylation at other, unknown or uncharacterized sites plays additional regulatory roles; phosphorylation both positively and negatively influenced BDLF4 stability (Fig. 8A). Further detailed analysis may shed light on additional regulation of the L gene regulator.
BDLF4 protein (25) and the other vPIC components are members of the EBV E-gene group (17). Although individual vPIC components are rapidly degraded when expressed in isolation (27), such proteins expressed during early lytic replication may form the vPIC. This implies that additional regulation of the vPIC is required to control L gene expression. Similarly, effective EBV lytic replication requires the specific cell cycle-associated actions involved in progression from G1 to S phase (S-like phase) (19). In this study, we demonstrated that S-like-phase CDKs phosphorylated and stabilized BDLF4 protein, indicating a direct link between S-like-phase CDK activity and L gene expression during EBV lytic replication. Thus, two-step regulation of L gene transcription by expression of the vPIC components and stabilization of the BDLF4 protein generates a robust viral gene cascade system (Fig. 9).
Three-dimensional surface reconstruction imaging reveals spatially different distributions between viral E and L gene mRNAs within replication compartments (13). While mRNAs of the E gene are located outside the BMRF1 cores, the subdomain structures of replication compartments, L gene mRNAs are identified inside the BMRF1 cores, where BcRF1 is recruited at the late stage of lytic replication (13). This is consistent with the recent finding that ongoing viral DNA replication maintains the integrity of a replication/transcription complex that is required for the recruitment and retention of factors essential for L gene transcription (16). Linkage between viral replication and L gene transcription, from bacteriophages to herpesviruses, is a widely accepted concept. Bacteriophage T4 recognizes a late transcriptional promoter via a small protein that links RNA polymerase to the DNA processivity factor by functioning as a sliding clamp to provide access to promoters as viral DNA synthesis proceeds (46). The interaction between the DNA polymerase processivity factor KSHV ORF59 and RNA polymerase II (47) also supports this spatial regulation of viral L gene expression via vPIC recruitment, in addition to the temporal regulation presented here. To clarify the entire regulatory mechanism, further studies, including proteomic and chromosomal conformation capture analyses, are required.
Expression of L genes is required for completion of the herpesvirus productive cycle. A drug that specifically inhibits this process could be useful for the treatment of herpesvirus infection/disease. Accumulating evidence from beta- and gammaherpesviruses shows that a distinct set of conserved genes, which are not present in alphaherpesviruses, constitute the vPIC and are involved in the regulation of L gene expression (reviewed in reference 23). Taken together, these data suggest that CDK inhibitors have inhibitory effects not only on EBV, but also on KSHV and HCMV. The combination of CDK inhibitors with existing inhibitors of herpesvirus replication, such as ganciclovir, may synergistically reduce the growth of beta- and gammaherpesviruses.
In summary, our findings explain the link between S-like-phase CDKs and viral L gene expression through phosphorylation of the vPIC subunit BDLF4.
MATERIALS AND METHODS
Cells and reagents.
HEK293, HEK293T, HEK293/FLAG-BDLF4, HEK293/EBV(WT), HEK293/EBV(dBDLF4) (25), and HEK293/EBV(dBGLF4) (48) cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). HEK293/FLAG-BDLF4 cells were established by transfecting HEK293 cells with linearized pcDNA3-FLAG-BDLF4 (WT) (25) and selecting for transfected cells using G418. The generation of Akata(–) (49) and Tet-BZLF1/B95-8 (20) cells has been described previously. CDK2/9i (250 to 500 nM; Merck Millipore, Billerica, MA, USA) and A2CE (1 μM; Merck Millipore) were used to inhibit CDK activity. For the MG132 (Sigma-Aldrich, St. Louis, MO, USA) and BTZMB (AdooQ BioScience, Irvine, CA, USA) experiments, cells were treated with MG132 (10 μM) or BTZMB (1 μM) for 4 to 24 h before harvesting. CHX was purchased from Sigma-Aldrich.
Plasmids.
Mammalian expression vectors pcDNA4-BZLF1, pcDNA3-FLAG-BDLF4 (WT), pGEX-BDLF4 (WT), pcDNA3-BGLF4 (WT and kinase dead [KD]), pcDNA3-HA-Ub, and pcDNA4-EGFP have been described previously (25, 48, 50–52). FLAG-tagged BDLF4 mutant (S8A, T16A, S26A, T91A, T97A, T220A, T222A, T223A, 6×A, 4×A, and T91E in pcDNA3) expression plasmids were generated by site-directed mutagenesis. The inserted DNA sequence of each vector was confirmed by direct DNA sequencing. The reporter plasmid pTATT-oriLyt-Luc (15) was kindly provided by Eric Johannsen (University of Wisconsin School of Medicine and Public Health, Madison, WI, USA). The control reporter plasmid pSV40-Rluc and shRNA expression plasmids against CDK1 (shCDK1#1 [TRCN0000582; numbers beginning with TRCN were developed by the RNA1 Consortium to identify the TRC library] and shCDK1#2 [TRCN0196603]), CDK2 (shCDK2#1 [TRCN0039961] and shCDK2#2 [TRCN0010470]), and a nonmammalian control (shScramble [SHC202]), were purchased from Promega (Madison, WI, USA) and Sigma-Aldrich, respectively.
Kinase inhibitor screen.
For the original screen, 5 × 104 HEK293/EBV(dBDLF4) cells, which were cotransfected with BZLF1 and FLAG-BDLF4 expression plasmids using the Neon transfection system, were first seeded in 100 μl DMEM supplemented with 10% FBS in 96-well plates and then treated with 3 μM kinase inhibitors. At 48 h after incubation, lysates from the cells were prepared and subjected to immunoblot analysis using antibodies against FLAG and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Nine kinase inhibitors exhibited relative signal intensities (normalized to that with DMSO) of <0.5 and thus were considered to reduce BDLF4 protein levels.
For an additional screen, 3 × 104 Tet-BZLF1/B95-8 cells were seeded in 100 μl RPMI medium supplemented with 10% FBS and 2 μg/ml doxycycline (Dox) in 96-well plates. Test compounds (kinase inhibitors), which were provided by the Screening Committee of Anticancer Drugs (Tokyo, Japan), were added to the medium to a concentration of 3 μM. After 72 h, total RNA was prepared and subjected to qPCR analysis using specific primers for BMRF1 and major capsid protein (MCP) mRNAs (53). The ratio between BMRF1 and MCP expression (E/L ratio) was calculated. The sum of the average E/L ratio (DMSO) and two standard deviations (E/L ratio, 1.6) was used as an initial cutoff to identify hits. The effects of these 32 compounds on viral replication were subsequently analyzed. Genomic DNA was prepared at 72 h after treatment and was then subjected to qPCR analysis using specific primers and probes for BALF2 and 18S rRNA (54). Ten kinase inhibitors that exhibited relative BALF2/18S rRNA values (normalized to the DMSO control) of >0.95 were considered to specifically inhibit EBV L gene expression without impacting viral replication.
qPCR analysis.
Genomic DNA was prepared using the DNeasy Blood & Tissue kit (Qiagen, Gaithersburg, MD, USA). Total RNA was prepared and was subsequently reverse transcribed to cDNA using the Cell Lysis & RT kit for qPCR (Toyobo, Osaka, Japan). Viral DNA and mRNA levels were analyzed by qPCR using the 7500 Fast DX real-time PCR system (Applied Biosystems, Foster City, CA, USA) as described previously (25).
Immunoblotting and antibodies.
Immunoblotting and signal detection were performed as described previously (50). For the separation and detection of phosphoprotein, SuperSep Phos-tag gel (Wako Pure Chemical Industries, Osaka, Japan) was used according to the manufacturer’s instructions. Treatment with λ protein phosphatase was performed as described previously (55).
Anti-cyclin A (B-8), anti-cyclin B1 (GNS1), and anti-cyclin E (E-4) mouse monoclonal antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-CDK1 and anti-CDK2 rabbit polyclonal antibodies were obtained from Bethyl Laboratories (Montgomery, TX, USA). Rabbit anti-GAPDH, rabbit anti-α/β-tubulin, and horseradish peroxidase-conjugated secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit anti-FLAG (F7425; Sigma-Aldrich), mouse anti-HA (12CA5; Roche, Basel, Switzerland), rabbit anti-histone H1 (Active Motif, Carlsbad, CA, USA), and rabbit anti–thiophosphate ester (Abcam, Cambridge, UK) antibodies were also used. Affinity-purified anti-BZLF1, anti-BALF2, anti-BMRF1, anti-BALF4, anti-BDLF4, anti-BRRF2, and anti-BKRF4 antibodies were prepared as described previously (11, 25, 48, 55, 56). Mouse and rabbit TrueBlot (eBioscience, San Diego, CA, USA) were used as secondary antibodies for immunoblotting of immunoprecipitation samples.
Determination of progeny virus titers.
For titration of virus yields, HEK293/EBV(dBDLF4) cells were transfected with BZLF1 and BDLF4 (WT or mutant) expression plasmids using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Cells and media were harvested and freeze-thawed, after which cell debris was removed. The supernatant from the centrifugation was used as a virus stock. EBV-negative Akata(–) cells were infected with the virus, and EGFP-positive cells were counted by fluorescence-activated cell sorter (FACS) to measure the viral titer.
Luciferase reporter assay.
Cells (5 × 104 cells/well in 96-well plates) were transfected with expression plasmids for BZLF1 (100 ng) and BDLF4 (WT or A mutants) (25 ng) together with pTATT-oriLyt-Luc (100 ng) and pSV40-Rluc (30 ng) using Lipofectamine 2000 (Thermo Fisher Scientific). Luciferase assays were performed as described previously (57).
HA-Ub immunoprecipitation.
HEK293T cells were transfected with pcDNA3-BDLF4 (WT) together with pcDNA3-HA-Ub using the Neon transfection system (Thermo Fisher Scientific). CDK2/9 inhibitor (500 nM) or DMSO was added to the medium at 24 h p.t. These cells were treated with 1 μM BTZMB for 4 h before harvesting. At 48 h p.t., cells were harvested and subjected to immunoprecipitation with anti-HA magnetic beads (Thermo Fisher Scientific) as described previously (50, 57).
In vitro phosphorylation assay.
Glutathione S-transferase-tagged WT BDLF4 [GST-BDLF4 (WT)] protein was purified from E. coli using standard techniques. A total of 200 ng of each cyclin/CDK complex (CDK2/cyclin A1, CDK2/cyclin E1. and CDK1/cyclin B1 active recombinant proteins purchased from SignalChem [Richmond, BC, Canada]) was incubated with 500 ng GST-BDLF4 (WT), 50 mM Tris-HCl (pH 8), 2 mM dithiothreitol (DTT), 1 mM ATP-γ-S, 5 mM beta-glycerophosphate, and 10 mM MgCl2 at 37°C for 30 min. Native histone H1 protein (1 μg; SignalChem) served as a control. Following the kinase reaction, the mixtures were further reacted with 2.5 mM p-nitrobenzyl mesylate for 2 h at room temperature, stopped with SDS-PAGE loading buffer, subsequently boiled for 5 min, and then resolved by SDS-PAGE. Phosphorylation was detected by immunoblotting with an anti–thiophosphate ester antibody.
Preparation of digested peptides for LC–MS-MS.
Peptides from in vitro kinase assay samples were prepared for LC–MS-MS analysis. Samples were first diluted twice with 8 M urea and then alkylated with iodoacetamide (final concentration, 82.5 mM) for 30 min at 37°C. The samples were further diluted 5-fold with ultrapure water and then mixed with chymotrypsin (protein/enzyme ratio, 25:1). The digestion step was conducted overnight at 37°C. Digestion was stopped by adding trifluoroacetic acid (final concentration, 1%). The samples were subjected to a desalting and washing step using C18/SCX-StageTips (58). Digested peptides were eluted with elution buffer (0.5 M ammonium acetate, 30% acetonitrile) and evaporated.
Mass spectrometric analysis.
Digested peptides were analyzed by a Q Exactive mass spectrometer (Thermo Scientific, Bremen, Germany) coupled to the UltiMate 3000 Nano LC system (Thermo Scientific) and an HTC-PAL autosampler (CTC Analytics, Zwingen, Switzerland). The Q Exactive mass spectrometer was operated in the data-dependent mode. Analytical columns (75 μm by 30 cm) were packed in-house with ReproSil-Pur C18-AQ 1.9-μm resin (Dr. Maisch, Ammerbuch, Germany). Peptides were separated using a gradient from 5% to 35% solvent B (solvent A, 0.1% formic acid and 2% acetonitrile; solvent B, 0.1% formic acid and 90% acetonitrile) at a flow rate of 280 nl/min for 85 min. Dynamic exclusion was set to 20 s. Other typical mass spectrometric conditions were as described previously (58).
Phosphopeptide identification.
The data measured were analyzed using MaxQuant, version 1.5.1.2, for peptide identification (59). Searches of MS-MS spectra were conducted against a database of amino acid sequences obtained from UniProt (60). The Uniprot identification numbers (IDs) for the amino acid sequences were P0CK56, P24941, P78396, and P24864. Enzyme specificity was set to chymotrypsin. Missed cleavages were tolerated at ≤2 sites. Carbamidomethylation of cysteine was set as a fixed modification. Methionine oxidation, N-terminal protein acetylation, and phosphorylation of serine, threonine, and tyrosine were set as variable modifications. False discovery rates at the peptide and protein levels were set to <1%, which was calculated based on the number of accepted hits from the reverse database. The identified phosphopeptides were filtered for an Andromeda score of 40 and a localization probability of >0.75.
Data analysis and statistics.
Results are shown as means ± standard deviations (SDs) from >3 independent experiments. Statistical analyses were performed using Microsoft Excel. Differences between the two groups were determined by Welch’s t test and were considered statistically significant at a P value of <0.05.
ACKNOWLEDGMENTS
We thank Eric Johannsen, Makoto Ohashi, and the Screening Committee of Anticancer Drugs (SCADS), Japan, for providing invaluable materials; Tomoko Kunogi, Kenta Nakagiri, and the Division for Medical Research Engineering, Nagoya University Graduate School of Medicine, for technical support; and all members of the Kimura laboratory for helpful discussions.
This work was supported in part by grants from the JSPS KAKENHI (grants JP16H06231, JP16H06867, and JP17H04081 to Y.S., T.W., and H.K., respectively) and from the Japan Agency for Medical Research and Development (AMED) to T.M. (grant JP17fm0208016) and H.K. (grant JP15ek0109098), by the GSK Japan Research Grant to Y.S. and T.W., by a Takeda Science Foundation grant to Y.S., and by grants from the 24th General Assembly of the Japanese Association of Medical Sciences and the Kitamura Memorial Foundation for Research of Blood Diseases (both to Y.S.). T.I. and T.S. are supported by Takeda Science Foundation scholarships.
REFERENCES
- 1.Cohen JI, Fauci AS, Varmus H, Nabel GJ. 2011. Epstein-Barr virus: an important vaccine target for cancer prevention. Sci Transl Med 3:107fs107. doi: 10.1126/scitranslmed.3002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chien YC, Chen JY, Liu MY, Yang HI, Hsu MM, Chen CJ, Yang CS. 2001. Serologic markers of Epstein-Barr virus infection and nasopharyngeal carcinoma in Taiwanese men. N Engl J Med 345:1877–1882. doi: 10.1056/NEJMoa011610. [DOI] [PubMed] [Google Scholar]
- 3.Tsai MH, Raykova A, Klinke O, Bernhardt K, Gartner K, Leung CS, Geletneky K, Sertel S, Munz C, Feederle R, Delecluse HJ. 2013. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep 5:458–470. doi: 10.1016/j.celrep.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. 2012. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 12:233–245. doi: 10.1016/j.chom.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, Jankowska-Gan E, Burlingham WJ, Sun X, Gulley ML, Tang W, Gumperz JE, Kenney SC. 2011. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol 85:165–177. doi: 10.1128/JVI.01512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu ZG, Iwatsuki K, Oyama N, Ohtsuka M, Satoh M, Kikuchi S, Akiba H, Kaneko F. 2001. The latency pattern of Epstein-Barr virus infection and viral IL-10 expression in cutaneous natural killer/T-cell lymphomas. Br J Cancer 84:920–925. doi: 10.1054/bjoc.2000.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zuo J, Thomas WA, Haigh TA, Fitzsimmons L, Long HM, Hislop AD, Taylor GS, Rowe M. 2011. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathog 7:e1002455. doi: 10.1371/journal.ppat.1002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu CC, Liu MT, Chang YT, Fang CY, Chou SP, Liao HW, Kuo KL, Hsu SL, Chen YR, Wang PW, Chen YL, Chuang HY, Lee CH, Chen M, Wayne Chang WS, Chen JY. 2010. Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res 38:1932–1949. doi: 10.1093/nar/gkp1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu SH, Wu CC, Fang CY, Yu SL, Hsu HY, Chow YH, Chen JY. 2014. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget 5:8583–8601. doi: 10.18632/oncotarget.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furnari FB, Adams MD, Pagano JS. 1992. Regulation of the Epstein-Barr virus DNA polymerase gene. J Virol 66:2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daikoku T, Kudoh A, Fujita M, Sugaya Y, Isomura H, Shirata N, Tsurumi T. 2005. Architecture of replication compartments formed during Epstein-Barr virus lytic replication. J Virol 79:3409–3418. doi: 10.1128/JVI.79.6.3409-3418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hau PM, Deng W, Jia L, Yang J, Tsurumi T, Chiang AK, Huen MS, Tsao SW. 2015. Role of ATM in the formation of the replication compartment during lytic replication of Epstein-Barr virus in nasopharyngeal epithelial cells. J Virol 89:652–668. doi: 10.1128/JVI.01437-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugimoto A, Sato Y, Kanda T, Murata T, Narita Y, Kawashima D, Kimura H, Tsurumi T. 2013. Different distributions of Epstein-Barr virus early and late gene transcripts within viral replication compartments. J Virol 87:6693–6699. doi: 10.1128/JVI.00219-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenzie JL, Dhillon RS, Schulte PM. 2015. Evidence for a bimodal distribution of hybrid indices in a hybrid zone with high admixture. R Soc Open Sci 2:150285. doi: 10.1098/rsos.150285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Djavadian R, Chiu YF, Johannsen E. 2016. An Epstein-Barr virus-encoded protein complex requires an origin of lytic replication in cis to mediate late gene transcription. PLoS Pathog 12:e1005718. doi: 10.1371/journal.ppat.1005718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li D, Fu W, Swaminathan S. 2018. Continuous DNA replication is required for late gene transcription and maintenance of replication compartments in gammaherpesviruses. PLoS Pathog 14:e1007070. doi: 10.1371/journal.ppat.1007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Djavadian R, Hayes M, Johannsen E. 2018. CAGE-seq analysis of Epstein-Barr virus lytic gene transcription: 3 kinetic classes from 2 mechanisms. PLoS Pathog 14:e1007114. doi: 10.1371/journal.ppat.1007114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato Y, Shirata N, Murata T, Nakasu S, Kudoh A, Iwahori S, Nakayama S, Chiba S, Isomura H, Kanda T, Tsurumi T. 2010. Transient increases in p53-responsible gene expression at early stages of Epstein-Barr virus productive replication. Cell Cycle 9:807–814. doi: 10.4161/cc.9.4.10675. [DOI] [PubMed] [Google Scholar]
- 19.Kudoh A, Fujita M, Zhang L, Shirata N, Daikoku T, Sugaya Y, Isomura H, Nishiyama Y, Tsurumi T. 2005. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J Biol Chem 280:8156–8163. doi: 10.1074/jbc.M411405200. [DOI] [PubMed] [Google Scholar]
- 20.Kudoh A, Fujita M, Kiyono T, Kuzushima K, Sugaya Y, Izuta S, Nishiyama Y, Tsurumi T. 2003. Reactivation of lytic replication from B cells latently infected with Epstein-Barr virus occurs with high S-phase cyclin-dependent kinase activity while inhibiting cellular DNA replication. J Virol 77:851–861. doi: 10.1128/JVI.77.2.851-861.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsurumi T, Fujita M, Kudoh A. 2005. Latent and lytic Epstein-Barr virus replication strategies. Rev Med Virol 15:3–15. doi: 10.1002/rmv.441. [DOI] [PubMed] [Google Scholar]
- 22.Serio TR, Cahill N, Prout ME, Miller G. 1998. A functionally distinct TATA box required for late progression through the Epstein-Barr virus life cycle. J Virol 72:8338–8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gruffat H, Marchione R, Manet E. 2016. Herpesvirus late gene expression: a viral-specific pre-initiation complex is key. Front Microbiol 7:869. doi: 10.3389/fmicb.2016.00869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gruffat H, Kadjouf F, Mariame B, Manet E. 2012. The Epstein-Barr virus BcRF1 gene product is a TBP-like protein with an essential role in late gene expression. J Virol 86:6023–6032. doi: 10.1128/JVI.00159-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe T, Narita Y, Yoshida M, Sato Y, Goshima F, Kimura H, Murata T. 2015. The Epstein-Barr virus BDLF4 gene is required for efficient expression of viral late lytic genes. J Virol 89:10120–10124. doi: 10.1128/JVI.01604-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Guindy A, Lopez-Giraldez F, Delecluse HJ, McKenzie J, Miller G. 2014. A locus encompassing the Epstein-Barr virus bglf4 kinase regulates expression of genes encoding viral structural proteins. PLoS Pathog 10:e1004307. doi: 10.1371/journal.ppat.1004307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aubry V, Mure F, Mariame B, Deschamps T, Wyrwicz LS, Manet E, Gruffat H. 2014. Epstein-Barr virus late gene transcription depends on the assembly of a virus-specific preinitiation complex. J Virol 88:12825–12838. doi: 10.1128/JVI.02139-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKenzie J, Lopez-Giraldez F, Delecluse HJ, Walsh A, El-Guindy A. 2016. The Epstein-Barr virus immunoevasins BCRF1 and BPLF1 are expressed by a mechanism independent of the canonical late pre-initiation complex. PLoS Pathog 12:e1006008. doi: 10.1371/journal.ppat.1006008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brulois K, Wong LY, Lee HR, Sivadas P, Ensser A, Feng P, Gao SJ, Toth Z, Jung JU. 2015. Association of Kaposi's sarcoma-associated herpesvirus ORF31 with ORF34 and ORF24 is critical for late gene expression. J Virol 89:6148–6154. doi: 10.1128/JVI.00272-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chapa TJ, Perng YC, French AR, Yu D. 2014. Murine cytomegalovirus protein pM92 is a conserved regulator of viral late gene expression. J Virol 88:131–142. doi: 10.1128/JVI.02684-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Omoto S, Mocarski ES. 2014. Transcription of true late (γ2) cytomegalovirus genes requires UL92 function that is conserved among beta- and gammaherpesviruses. J Virol 88:120–130. doi: 10.1128/JVI.02983-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu J, Liao G, Shan L, Zhang J, Chen MR, Hayward GS, Hayward SD, Desai P, Zhu H. 2009. Protein array identification of substrates of the Epstein-Barr virus protein kinase BGLF4. J Virol 83:5219–5231. doi: 10.1128/JVI.02378-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato Y, Tsurumi T. 2010. Noise cancellation: viral fine tuning of the cellular environment for its own genome replication. PLoS Pathog 6:e1001158. doi: 10.1371/journal.ppat.1001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kudoh A, Daikoku T, Sugaya Y, Isomura H, Fujita M, Kiyono T, Nishiyama Y, Tsurumi T. 2004. Inhibition of S-phase cyclin-dependent kinase activity blocks expression of Epstein-Barr virus immediate-early and early genes, preventing viral lytic replication. J Virol 78:104–115. doi: 10.1128/JVI.78.1.104-115.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. 2002. PKB/Akt mediates cell-cycle progression by phosphorylation of p27Kip1 at threonine 157 and modulation of its cellular localization. Nat Med 8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 36.Maddika S, Ande SR, Wiechec E, Hansen LL, Wesselborg S, Los M. 2008. Akt-mediated phosphorylation of CDK2 regulates its dual role in cell cycle progression and apoptosis. J Cell Sci 121:979–988. doi: 10.1242/jcs.009530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pozner A, Terooatea TW, Buck-Koehntop BA. 2016. Cell-specific Kaiso (ZBTB33) regulation of cell cycle through cyclin D1 and cyclin E1. J Biol Chem 291:24538–24550. doi: 10.1074/jbc.M116.746370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hinds PW. 2003. Cdk2 dethroned as master of S phase entry. Cancer Cell 3:305–307. doi: 10.1016/S1535-6108(03)00084-9. [DOI] [PubMed] [Google Scholar]
- 39.Pearlman SM, Serber Z, Ferrell JE Jr. 2011. A mechanism for the evolution of phosphorylation sites. Cell 147:934–946. doi: 10.1016/j.cell.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakayama KI, Nakayama K. 2006. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 41.Carrano AC, Eytan E, Hershko A, Pagano M. 1999. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 42.Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U, Krek W. 1999. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol 1:207–214. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- 43.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. 1999. p27Kip1 ubiquitination and degradation is regulated by the SCFSkp2 complex through phosphorylated Thr187 in p27. Curr Biol 9:661–664. doi: 10.1016/S0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 44.Wang VY, Li Y, Kim D, Zhong X, Du Q, Ghassemian M, Ghosh G. 2017. Bcl3 phosphorylation by Akt, Erk2, and IKK is required for its transcriptional activity. Mol Cell 67:484–497. doi: 10.1016/j.molcel.2017.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ulery PG, Rudenko G, Nestler EJ. 2006. Regulation of ΔFosB stability by phosphorylation. J Neurosci 26:5131–5142. doi: 10.1523/JNEUROSCI.4970-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geiduschek EP, Kassavetis GA. 2010. Transcription of the T4 late genes. Virol J 7:288. doi: 10.1186/1743-422X-7-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, Jager S, Shales M, Horner J, Hernandez RD, Krogan NJ, Glaunsinger BA. 2015. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 57:349–360. doi: 10.1016/j.molcel.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murata T, Isomura H, Yamashita Y, Toyama S, Sato Y, Nakayama S, Kudoh A, Iwahori S, Kanda T, Tsurumi T. 2009. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 389:75–81. doi: 10.1016/j.virol.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 49.Shimizu N, Tanabe-Tochikura A, Kuroiwa Y, Takada K. 1994. Isolation of Epstein-Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt's lymphoma (BL) line Akata: malignant phenotypes of BL cells are dependent on EBV. J Virol 68:6069–6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sato Y, Kamura T, Shirata N, Murata T, Kudoh A, Iwahori S, Nakayama S, Isomura H, Nishiyama Y, Tsurumi T. 2009. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog 5:e1000530. doi: 10.1371/journal.ppat.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murata T, Shimotohno K. 2006. Ubiquitination and proteasome-dependent degradation of human eukaryotic translation initiation factor 4E. J Biol Chem 281:20788–20800. doi: 10.1074/jbc.M600563200. [DOI] [PubMed] [Google Scholar]
- 52.Sato Y, Ochiai S, Murata T, Kanda T, Goshima F, Kimura H. 2017. Elimination of LMP1-expressing cells from a monolayer of gastric cancer AGS cells. Oncotarget 8:39345–39355. doi: 10.18632/oncotarget.16996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshida M, Watanabe T, Narita Y, Sato Y, Goshima F, Kimura H, Murata T. 2017. The Epstein-Barr virus BRRF1 gene is dispensable for viral replication in HEK293 cells and transformation. Sci Rep 7:6044. doi: 10.1038/s41598-017-06413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Narita Y, Murata T, Ryo A, Kawashima D, Sugimoto A, Kanda T, Kimura H, Tsurumi T. 2013. Pin1 interacts with the Epstein-Barr virus DNA polymerase catalytic subunit and regulates viral DNA replication. J Virol 87:2120–2127. doi: 10.1128/JVI.02634-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe T, Sakaida K, Yoshida M, Masud HM, Sato Y, Goshima F, Kimura H, Murata T. 2017. The C-terminus of Epstein-Barr virus BRRF2 is required for its proper localization and efficient virus production. Front Microbiol 8:125. doi: 10.3389/fmicb.2017.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masud H, Watanabe T, Yoshida M, Sato Y, Goshima F, Kimura H, Murata T. 2017. Epstein-Barr virus BKRF4 gene product is required for efficient progeny production. J Virol 91:e00975-17. doi: 10.1128/JVI.00975-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sato Y, Shirata N, Kudoh A, Iwahori S, Nakayama S, Murata T, Isomura H, Nishiyama Y, Tsurumi T. 2009. Expression of Epstein-Barr virus BZLF1 immediate-early protein induces p53 degradation independent of MDM2, leading to repression of p53-mediated transcription. Virology 388:204–211. doi: 10.1016/j.virol.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 58.Adachi J, Hashiguchi K, Nagano M, Sato M, Sato A, Fukamizu K, Ishihama Y, Tomonaga T. 2016. Improved proteome and phosphoproteome analysis on a cation exchanger by a combined acid and salt gradient. Anal Chem 88:7899–7903. doi: 10.1021/acs.analchem.6b01232. [DOI] [PubMed] [Google Scholar]
- 59.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 60.The UniProt Consortium. 2018. UniProt: the universal protein knowledgebase. Nucleic Acids Res 46:2699. doi: 10.1093/nar/gky092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bostock CJ, Prescott DM, Kirkpatrick JB. 1971. An evaluation of the double thymidine block for synchronizing mammalian cells at the G1-S border. Exp Cell Res 68:163–168. doi: 10.1016/0014-4827(71)90599-4. [DOI] [PubMed] [Google Scholar]