

The pathogenesis of Ebola virus disease (EVD) in humans is complex, and the mechanisms contributing to immunity are poorly understood. In particular, it appears that the quality and magnitude of the human B cell response early after recovery from EVD may be reduced compared to most viral infections. Here, we isolated human monoclonal antibodies from B cells of four survivors of EVD at 1 or 3 months after hospital discharge. Ebola-specific memory B cells early in convalescence were low in frequency, and the antibodies they encoded demonstrated poor neutralizing potencies. One neutralizing antibody that protected mice from lethal infection, EBOV237, was identified in the panel of 25 human antibodies isolated. Recognition of the glycan cap epitope recognized by EBOV237 suggests that this antigenic site should be considered in vaccine design and treatment strategies for EVD.
KEYWORDS: antibodies, human, neutralizing, antiviral, Ebola virus, Filoviridae infections, hemorrhagic fever, Ebola, protection
ABSTRACT
The human B cell response to natural filovirus infections early after recovery is poorly understood. Previous serologic studies suggest that some Ebola virus survivors exhibit delayed antibody responses with low magnitude and quality. Here, we sought to study the population of individual memory B cells induced early in convalescence. We isolated monoclonal antibodies (MAbs) from memory B cells from four survivors treated for Ebola virus disease (EVD) 1 or 3 months after discharge from the hospital. At the early time points postrecovery, the frequency of Ebola-specific B cells was low and dominated by clones that were cross-reactive with both Ebola glycoprotein (GP) and with the secreted GP (sGP) form. Of 25 MAbs isolated from four donors, only one exhibited neutralization activity. This neutralizing MAb, designated MAb EBOV237, recognizes an epitope in the glycan cap of the surface glycoprotein. In vivo murine lethal challenge studies showed that EBOV237 conferred protection when given prophylactically at a level similar to that of the ZMapp component MAb 13C6. The results suggest that the human B cell response to EVD 1 to 3 months postdischarge is characterized by a paucity of broad or potent neutralizing clones. However, the neutralizing epitope in the glycan cap recognized by EBOV237 may play a role in the early human antibody response to EVD and should be considered in rational design strategies for new Ebola virus vaccine candidates.
IMPORTANCE The pathogenesis of Ebola virus disease (EVD) in humans is complex, and the mechanisms contributing to immunity are poorly understood. In particular, it appears that the quality and magnitude of the human B cell response early after recovery from EVD may be reduced compared to most viral infections. Here, we isolated human monoclonal antibodies from B cells of four survivors of EVD at 1 or 3 months after hospital discharge. Ebola-specific memory B cells early in convalescence were low in frequency, and the antibodies they encoded demonstrated poor neutralizing potencies. One neutralizing antibody that protected mice from lethal infection, EBOV237, was identified in the panel of 25 human antibodies isolated. Recognition of the glycan cap epitope recognized by EBOV237 suggests that this antigenic site should be considered in vaccine design and treatment strategies for EVD.
INTRODUCTION
Ebola virus is a member of the Filoviridae family and causes intermittent outbreaks of severe human Ebola virus disease (EVD). Currently, there is an outbreak in the North Kivu province of the Democratic Republic of Congo (DRC), declared by the DRC Ministry of Health on 1 August 2018. As of 15 August 2018, there are confirmed 51 cases of Ebola, including 17 deaths (https://wwwnc.cdc.gov/travel/notices/watch/ebola-democratic-republic-of-the-congo). In 2014, a large outbreak occurred in West Africa, with a total of 28,616 cases of EVD and 11,310 deaths that were reported in Guinea, Liberia, and Sierra Leone, and an additional 36 cases and 15 deaths that occurred when the outbreak spread outside these three countries (https://www.cdc.gov/vhf/ebola/history/2014-2016-outbreak/index.html). Experimental vaccines, antibodies, small molecules, and RNA treatments for Ebola virus infection are in development, which are intended to prevent or treat infection using immunologic principles. Indeed, we and others have shown that at late time points after recovery, survivors possess memory B cells in circulation specifying broad and potent protective antibodies (1–6). Monoclonal antibodies (MAbs) isolated from human B cells of survivors can exert potent therapeutic effects in experimental infection models in nonhuman primates using cocktails of MAbs or even monotherapy (7–9).
Ebola virus is an enveloped, nonsegmented, negative-sense strand RNA virus (http://www.who.int/en/news-room/fact-sheets/detail/ebola-virus-disease). Together with the Marburg and Cuevaviruses, these members constitute the Filoviridae family. There are six species of Ebola virus: Zaire ebolavirus (EBOV), Bundibugyo ebolavirus (BDBV), Sudan ebolavirus (SUDV), Taï Forest ebolavirus (TAFV), Reston ebolavirus (RESV), and Bombali ebolavirus (BOMV). Of these species, EBOV, BDBV, and SUDV are the primary species associated with lethal disease in humans (1). The surface glycoprotein (GP) of Ebola virus is the major target of neutralizing antibodies. The envelope glycoprotein gene of Ebola virus encodes two GPs through transcriptional editing. The major product of the gene is a 364-residue soluble dimeric form of the GP (sGP). The precise role of sGP in Ebola virus infection is not clear. However, it is thought that sGP may serve as a decoy to distract the immune response for virus evasion or contribute to pathogenesis (10). The minor product of the gene is a 676-residue viral surface GP that is anchored to the viral envelope. The GP consists of two subunits, GP1 and GP2, which form a trimeric structure on the virus surface. The GP1 subunit contains the heavily glycosylated mucin-like domain and a glycan cap, which are cleaved proteolytically by cathepsins within the endosome during virus entry into the host cell (10, 11). This exposes the receptor-binding site on the GP1 subunit that interacts with the endosomal receptor, domain C of the Niemann-Pick C1 protein (NPC1) (12). Upon receptor binding, the fusion loop of the GP2 subunit becomes exposed, enabling fusion of the virus with the endosomal membrane (10, 11). The involvement of GP in virus attachment and entry into host cells, receptor binding within the endosome, and membrane fusion with the endosomal membrane result in the GP being the major target of neutralizing MAbs (5).
Cocktails of murine-human chimeric (ZMapp, ZMab, and MB-003) and human MAbs administered as a monotherapy have shown potent neutralizing and nonneutralizing activities for the prevention and treatment of Ebola virus (EBOV) in experimental infection animal models, including ZMapp protection of nonhuman primates (7–9). The MAbs characterized target diverse antigenic sites on GP, including the glycan cap, GP1 receptor-binding site, GP1 head, GP1/GP2 interface, GP2 fusion loop, GP2 stalk, and HR2/MPER region (1–6, 12–18). Several MAbs that recognize antigenic sites present on the glycan cap and GP1 head are cross-reactive with sGP (1, 5, 10, 13, 18). The ZMapp cocktail (2G4, 4G7, and 13C6) consists of three murine-human chimeric MAbs that recognize EBOV GP. 2G4 and 4G7 recognize the base of the GP at the GP1/GP2 interface and exhibit potent neutralization activity in vitro (16–18). The other component of ZMapp is a weakly neutralizing MAb, 13C6, which recognizes the glycan cap and cross-reacts with EBOV sGP.
The characterization of MAbs isolated at late time points after recovery has been described previously; however, the early human B cell response to EBOV is still poorly understood, especially at the clonal level. To investigate the early response to EBOV infection, we took advantage of an unprecedented opportunity to study the acute response to infection of four U.S. heath care workers, infected in West Africa, who had received intensive care treatment and monitoring at Emory University Hospital. The acute response to infection was characterized by an unusually long period of immune activation, as evidenced by the persistence of activated CD8 and CD4 T cells and plasmablasts even 1 month after the patients’ discharge from the hospital and other persistent signs of immune activation (19). Studies also have revealed very prolonged maintenance of infectious virus in immune-privileged sites, including the eye (20, 21) and semen (22), that can last over 2 years. Both polyclonal and monoclonal antibodies have been shown to exert potent antiviral effects against filovirus infection and disease in preclinical models. Indeed, we and others have shown that at late time points after recovery, survivors possess memory B cells in circulation specifying broad and potent protective antibodies (1–6). However, the early human B cell response to EBOV is poorly defined. Here, we investigated the B cell response in survivors 1 and 3 months postdischarge, at a time when immune activation persisted and virus may have persisted in immune-privileged sites. The results show that soon after the resolution of viremia, the memory B cell response in blood is characterized by a low frequency of EBOV-specific B cells, with the antibodies encoded by those cells displaying low neutralizing activity.
RESULTS
Isolation of human GP-reactive MAbs from Ebola survivors.
Peripheral blood mononuclear cells (PBMCs) were isolated from patients who survived EVD (designated subjects EVD2, EVD5, EVD9, and EVD15) 1 or 3 months after their discharge from Emory University Hospital. PBMCs were transformed with EBV, and after expansion, cell supernatants were screened to determine the relative magnitude of B cell responses to soluble forms of the Ebola virus (EBOV), Bundibugyo virus (BDBV), Sudan virus (SUDV), and Marburg virus (MARV) glycoprotein (GP) ectodomains. From the corresponding enzyme-linked immunosorbent assay (ELISA) data, Circos plots were generated to illustrate the cross-reactivity of the B cell responses for each donor to the various glycoproteins (Fig. 1A and B). The relative height of each bar in Fig. 1 indicates the optical density (OD) values at 405 nm as determined by ELISA. Subjects EVD2 and EVD5 had a greater and broader B cell response to these GPs 1 month after hospital discharge than subjects EVD9 and EVD15. However, an increase in B cell response to these GPs was observed for subject EVD15 at 3 months after hospital discharge compared to 1 month. The reactivities of EBOV GP and secreted GP (sGP)-specific B cell lines were primarily EBOV GP/sGP cross-reactive (Fig. 1C) for all subjects at both time points, except for EVD15 at the 1-month time point (EVD15_1). Additionally, a majority of the B cell reactivity was toward EBOV GP and sGP. There was an increase in cross-reactivity to BDBV GP of subject samples at 3 months compared to 1 month postdischarge, indicating that development of these antibodies occurred over the period of recovery. A minor percentage of reactive B cells were cross-reactive to EBOV, BDBV, and SUDV GP and again increased at the 3-months compared to the 1-month postdischarge time point.
FIG 1.

Circos plot representation of the four EVD survivors’ cross-reactive B cell responses. (A and B) PBMC samples isolated from subjects EVD2, EVD5, EVD9, and EVD15 1 month (A) or 3 months (B) postdischarge were transformed with EBV and subsequently expanded. The supernatants from the transformed B cell lines were screened initially for binding to the soluble form of full-length extracellular domain of MARV, SUDV, EBOV, or BDBV GP. The height of the lines indicates the relative intensity of the optical density (OD) values at 405 nm as determined by ELISA using the indicated GP. Black lines indicated the antibody bound only to GP from one species. Orange, blue, and red lines represent cross-reactive B cell responses to GP from two, three, and four virus species, respectively. For PBMC samples isolated from donors EVD9 and EVD15 at 3 months postdischarge, supernatant reactivity from transformed B cell lines was not tested against MARV GP. (C) The reactivity of EBOV, BDBV, SUDV GP, and EBOV B cell lines are represented as the percentage of total GP and/or sGP-specific B cell lines. Positive reactivity was determined by >0.8 optical density values at 405 nm. The values are heat mapped according to percent reactivity with >60% in dark green, 30% to 60% in lighter green, and <30% in light green.
We isolated human MAbs specific to EBOV, BDBV, and SUDV GPs or EBOV sGP from transformed B cells. A panel of 25 human MAbs was isolated. The half-maximal effective concentration (EC50) of binding values for the EBOV GP-reactive human MAbs as determined via ELISA are indicated in Fig. 2A. The MAbs exhibited values for EC50 in diverse ranges, including 0 to 100 ng/ml, 100 to 1,000 ng/ml, 1,000 to 10,000 ng/ml, or greater than 10,000 ng/ml, as indicated in the heat map by dark red, orange, or yellow color, or a greater than sign (>), respectively. The EBOV GP-reactive human MAbs are separated into four groups, with two subgroups (A and B) for groups 1 and 2 designated based on pattern of reactivity to the various GPs. The A subgroup indicates MAbs that bound GP and EBOV sGP, suggesting the GP1 glycan cap as the GP binding site for these MAbs, whereas MAbs from the B subgroup bound GP only. Increasing in cross-reactivity, group 1 bound EBOV, group 2 bound EBOV and BDBV or SUDV, and group 3 bound EBOV, BDBV, and SUDV. Group sGP MAbs bound to EBOV sGP only. None of these MAbs bound to MARV GP. The isolation of MAbs, primarily from subjects EVD2 and EVD5 at 1 and 3 months postdischarge, reflects the intensity and broad B cell response measured initially with EBV-transformed B cell line supernatants, as indicated by the Circos plots (Fig. 1A and B).
FIG 2.

Binding and neutralization characterization of MAb EBOV237. (A) Twenty-five human monoclonal antibodies (MAbs) were isolated from the four EVD survivors, with respective subject number and month of PBMC isolation postdischarge indicated. These MAbs are organized into groups and subgroups based on their breadth of binding for diverse GPs. Of the 25 MAbs, all but two bound to EBOV GP. These MAbs bound only to full-length EBOV sGP and are referred to as “sGP.” Subgroup A includes MAbs that are able to bind to full-length EBOV GP and EBOV sGP. Subgroup B includes the MAbs that bound only to full-length EBOV GP. Groups 1, 2, and 3 indicate the breadth of the MAb to EBOV, EBOV and BDBV, and EBOV, BDBV, and SUDV GP, respectively. The EC50 values <100 ng/ml are in red, <1,000 ng/ml in orange, and <10,000 ng/ml in yellow. EC50 values >10,000 ng/ml are in white and indicated with the symbol >. The EC50 values shown are an average of technical triplicates and are representative of two duplicate ELISAs, with similar results. Neutralization data for each MAb are shown as either positive or negative, with EBOV237 shown in red. (B) Binding curve for EBOV237 to full-length EBOV, BDBV, SUDV, MARV GP, or EBOV sGP, as measured by OD at 405 nm with increasing MAb concentration. (C) Neutralization data of EBOV237 as determined by plaque reduction neutralization tests at 50% (PRNT50) or 80% (PRNT80) reduction, corresponding with the indicated IC50 values of 0.75 μg/ml and 1.56 μg/ml, respectively. ND, not tested.
EBOV237, a neutralizing MAb.
To determine the antiviral activity of the EBOV GP reactive human MAbs, neutralization assays were performed against Ebola Zaire virus (EBOV/Kik-9510621, CDC no. 807224). Plaque reduction neutralization test (PRNT) assays were performed to determine the endpoint titer for 50% (PRNT50) or 80% (PRNT80) reduction in plaques compared to a virus-only control. From the panel of 25 human EBOV MAbs, one MAb (designated EBOV237) was neutralizing with a PRNT50 of 0.78 μg/ml and a PRNT80 of 1.56 μg/ml (Fig. 2C).
A representative curve for binding of EBOV237 to full-length EBOV, BDBV, SUDV, or MARV GP, or EBOV sGP is shown in Fig. 2B. EBOV237 bound to EBOV GP, BDBV GP, and EBOV sGP with EC50 values of 25, 3,367, and 9 ng/ml, respectively. EBOV237 did not bind SUDV and MARV GPs. Thus, EBOV237 primarily binds to EBOV GP, EBOV sGP, and BDBV GP, albeit with lower affinity. Stronger binding of EBOV237 to EBOV GP in comparison to the other EBOV human MAbs correlated with its neutralization activity toward EBOV.
Epitope mapping of EBOV237 to the glycan cap region of EBOV GP1.
To determine the epitopes recognized by the panel of EBOV human MAbs, competition-binding assays were performed with MAbs that bound to EBOV GP or sGP as determined by a >0.19-nm shift in the interference pattern of light through biolayer interferometry. Four epitope groups were identified on EBOV GP from the panel of EBOV human MAbs. Recombinant forms of two MAbs that are components of the ZMapp therapeutic cocktail (2G4 and 13C6) were used as controls for binding to EBOV GP. The epitopes for 2G4 and 13C6 are known, as they bind to the base of the GP or to the GP1 glycan cap, respectively (17, 18). None of the MAbs tested competed with 2G4, indicating that these MAbs do not bind to the base of EBOV GP. EBOV237 did not compete for binding with either 2G4 or 13C6 (Fig. 3A), indicating that EBOV237 binds to a distinct site on EBOV GP. EBOV237 competed for binding with EBOV173 and EBOV236, suggesting that these human EBOV MAbs bind similar sites on EBOV GP. Several unidirectional pairs were identified as well, such as EBOV173 and 13C6. This may be due to difference in angle of binding of the first MAb relative to the second MAb. This could allow for binding of the MAb in one direction but not in the other. Additionally, the relative kinetics of binding for the first MAb may also account for the unidirectional competition of MAb binding. EBOV237 and a previously isolated neutralizing BDBV MAb, BDBV43 (1), were competed with the human EBOV MAbs on EBOV sGP (Fig. 3B). In addition to competition with BDBV43, EBOV237 competed partially with EBOV29 or EBOV102 on EBOV sGP. EBOV237 did not compete for binding with the previously isolated nonneutralizing MAb BDBV91, which is known to bind to the hydrophobic pocket at the EBOV sGP dimer interface (10).
FIG 3.

Epitope mapping of EBOV237 indicates binding to the glycan cap. (A and B) Competition-binding analysis of MAbs to full-length EBOV GP (A) and EBOV sGP (B) are indicated. The numbers indicate residual percent binding of the second antibody in the presence of the first antibody, in comparison to the absence of the first antibody. A reduction in percent binding of the second antibody to <30% due to the presence of the first antibody (black boxes with white numbers) indicates full competition for binding. A reduction in percent binding of the second antibody from 30% to 70% due to the presence of the first antibody (gray boxes with black numbers) indicates intermediate competition. A reduction in percent binding of the second antibody to <30% due to the presence of the first antibody (white boxes with red numbers) indicates a lack of competition. (C to E) EBOV237 was epitope mapped by screening on an EBOV Δmucin GP alanine scan mutation library expressed in HEK-293T cells, with binding by EBOV237 assayed by flow cytometry. This identified N278A and P279A mutants as critical clones that showed specifically reduced binding for EBOV237 Fab (<20% and <30% of binding to WT EBOV GP, respectively; red bar) but a high level of binding to control MAbs EBOV296, EBOV442, or EBOV520 (gray/white bars) (C). Error bars represent the mean and range (half of the maximum minus minimum values) of at least two replicate data points. (D and E) The monomer (D; side view) or trimer (E; top view) form of EBOV Δmucin GP, with GP1 in yellow and GP2 in red (PDB 5JQ3) is shown (31). The critical binding residues for EBOV237 (N278, P279, and I260) to EBOV Δmucin GP are indicated by the green spheres on either structure. S322 is not shown, as this residue corresponds to the mucin-like domain of EBOV GP. (F) Four antibody neutralization escape mutant viruses were isolated, each with the mutations I260R and S322G. A PRNT was done using 10, 1, 0.1, 0.01, or 0.001 μg of EBOV237 incubated with either wild-type (WT) virus or escape mutant 4. The numbers for the assays, performed in triplicate, indicate the PFU per well.
To identify key residues recognized by EBOV237, mutations were introduced into GP through alanine-scanning shotgun mutagenesis of EBOV GPΔmucin (Ebola virus H.sapiens-tc/COD/1976/Yambuku-Mayinga [23], Δ311–461). Identification of the residues N278 and P279 as critical residues for binding confirmed that the EBOV237 epitope lies within the glycan cap region of GP1 (Fig. 3C to E). The critical residues for EBOV237 are conserved in BDBV, supporting the cross-reactivity profile of EBOV237 to BDBV GP. However, the reduction in binding of EBOV237 to BDBV GP (EC50 value, 3,367 ng/ml) suggests that other factors may be involved. This is also consistent with the cross-reactivity profile of EBOV237 in SUDV (not recognized by EBOV237) residues 278 and 279 are D278 and A279, respectively. The glycan cap region of GP1 also is bound by the ZMapp cocktail MAb 13C6. Residues G264 to W275, identified for binding of this MAb to EBOV GP, lie at the tip of the glycan cap (10, 18). The difference in residues bound by EBOV237 and 13C6 is consistent with the differences observed in the competition-binding assays done with EBOV GP (Fig. 3A). Thus, even though these two MAbs bind to the glycan cap of GP1, EBOV237 and 13C6 recognize distinct residues or epitopes in this domain.
To identify key residues selectively pressured to mutate in the presence of EBOV237, we isolated EbolaΔVP30-GFP neutralization escape mutant viruses. Sequence analysis of EBOV GP for the mutant viruses identified the mutated residues I260R and S322G. These two mutations were identified in four different independently selected neutralization escape mutant viruses (Fig. 3F). PRNTs confirmed the ablation of neutralization for escape mutant 4 in the presence of different concentrations of EBOV237 (Fig. 3F). Plaque titers for escape mutant 4 were similar to those of the control MAb VP35 for wild-type or mutant viruses. Thus, EBOV237 selectively pressures the virus to mutate at these key residues as it binds the glycan cap of GP1, leading to escape of the virus from the neutralization capabilities of EBOV237. The escape mutation at I260 is consistent with glycan cap location and EBOV237 epitope (critical residues N278 and P279). However, the escape mutation S322G presumably exerts an allosteric effect, since it is not located within the glycan cap but instead within the mucin-like domain. The EBOV237 cross-reactivity with sGP, which lacks residue S322, is also inconsistent with inclusion of S322 as part of the epitope.
In vivo studies revealed a correlate of protection.
Mouse challenge studies were performed to determine the prophylactic efficacy of EBOV237. Initially, groups of 10 female BALB/c mice, age 6 to 8 weeks, were treated intraperitoneally (i.p.) 24 h prior to virus inoculation (day −1) with a single dose of 100 μg of EBOV237. For the positive-control group, mice were treated i.p. with 100 μg of MAb 13C6. For the negative-control group, mice were treated i.p. with an irrelevant IgG antibody. On day 0 (d0), mice were inoculated i.p. with 100 PFU of mouse-adapted Ebola Zaire virus (Mayinga) (ma-ZEBOV). Mice were monitored daily for 28 days after virus inoculation. EBOV237 provided a similar level of protection against ma-ZEBOV infection as the positive-control MAb 13C6 (Fig. 4A). An irrelevant negative-control IgG antibody did not protect mice from lethal infection. To observe clinical signs of disease, mice were monitored twice daily for 28 days after virus inoculation (Fig. 4B). Individual disease score parameters included normal (0), reduced grooming/ruffled fur (1), subdued but normal when stimulated (2), lethargic, hunched posture, subdued even when stimulated (3), and nasal discharge/bleeding/unresponsive when stimulated/weak/paralysis (4). Animals treated with the irrelevant negative-control IgG antibody exhibited several signs of disease. Animals treated with MAb 13C6 or EBOV237 also showed some signs of disease. However, there was a reduction in signs of clinical disease for both of these groups, with a slight improvement shown for EBOV237. Group body weights were taken daily for 28 days after virus inoculation (Fig. 4C). Groups treated with EBOV237 or 13C6 showed little variation in average weight of mice over the 28 days. The group treated with irrelevant negative-control IgG antibody showed variation in average weight, corresponding with survival of the mice (Fig. 4A). These data confirm the prophylactic efficacy of EBOV237 against ma-ZEBOV infection.
FIG 4.
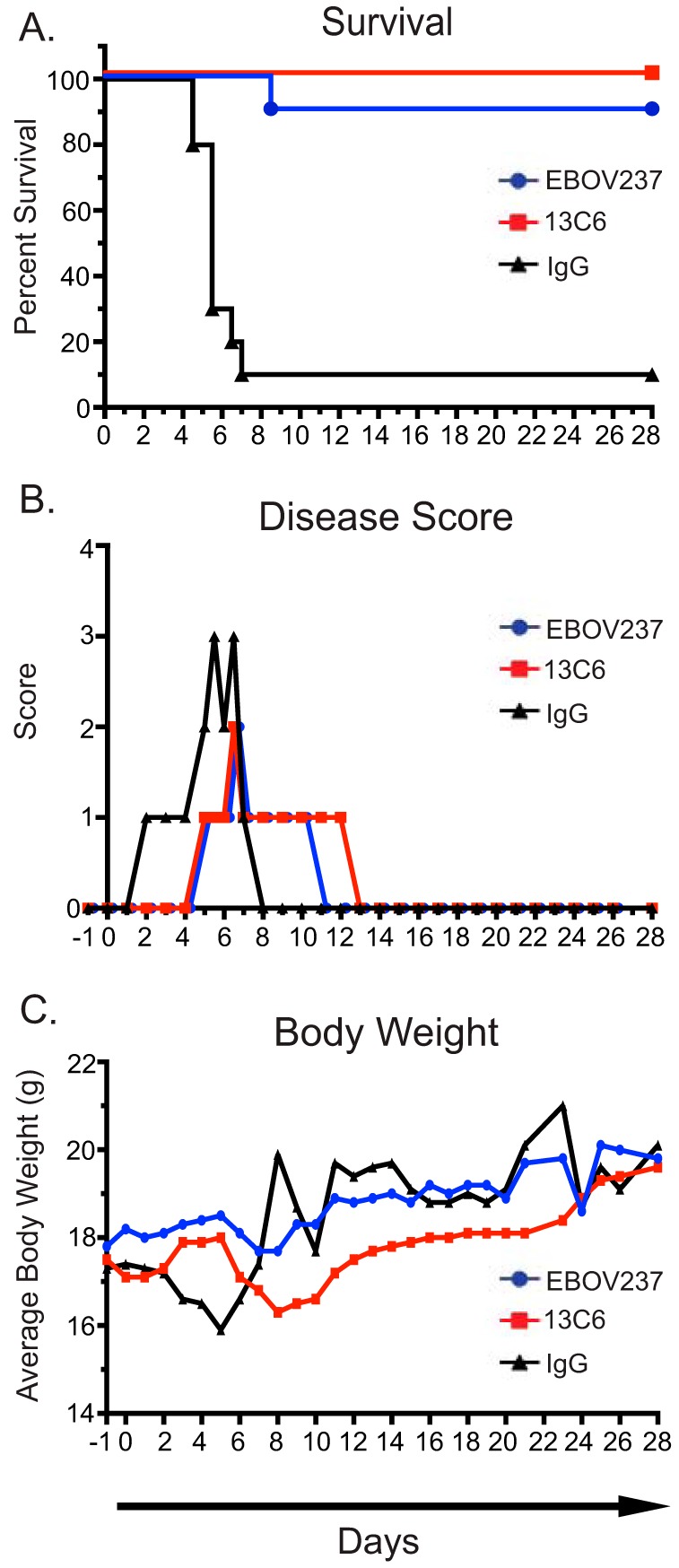
EBOV237 provided prophylactic protection from mouse-adapted Ebola Zaire virus (Mayinga) infection. BALB/c mice in groups of 10 were each given 100 μg of EBOV237 intraperitoneally 24 h prior to challenge with 100 PFU of Mayinga virus intraperitoneally. (A) Kaplan-Meier survival curve indicates that EBOV237 provides similar protection against lethal disease as MAb 13C6, in comparison to the irrelevant negative-control IgG. (B) Disease scores were recorded based on the sickest animal in each treatment group over the course of 28 days (normal [0], reduced grooming/ruffled fur [1], subdued but normal when stimulated [2], lethargic, hunched posture, subdued even when stimulated [3], nasal discharge/bleeding/unresponsive when stimulated/weak/paralysis [4]). Animals treated with MAb 13C6 or the irrelevant negative-control IgG exhibited several signs of disease, with an improvement shown for EBOV237. (C) Average weight of mice (calculated from group weight divided by number of mice weighed) taken over a course of 28 days. Mice treated with EBOV237 or 13C6 showed little variation in average weight of mice over the course of study. Mice treated with the irrelevant negative-control IgG antibody showed variation in average weight.
A dose-response curve also was performed to determine the efficacy of protection for EBOV237 against mouse-adapted Ebola Zaire virus (Mayinga) (ma-ZEBOV) infection. At 24 h prior to virus inoculation, groups of 10 female BALB/c mice, age 6 to 8 weeks, were treated i.p. with a single dose of 200, 100, 50, or 25 μg of EBOV237. The positive- and negative-control groups were the same as described for the initial study. On day 0, mice were inoculated i.p. with 100 PFU of ma-ZEBOV. Mice were monitored daily for 21 days after virus inoculation. Survival depended on the dose of EBOV237 administered, with a dose of 200 μg providing similar protection against ma-ZEBOV infection to the positive-control MAb 13C6 administered at 100 μg (Fig. 5A). Animals treated with EBOV237 showed a reduction in signs of disease in a dose-dependent manner. Similar improvement of disease was observed for mice treated with 200 μg of EBOV237 and with the positive-control MAb 13C6 (Fig. 5B). Little variation in the average weight of mice over the course of 21 days was observed for animals treated with any dose of EBOV237 compared to animals treated with the irrelevant negative-control antibody.
FIG 5.
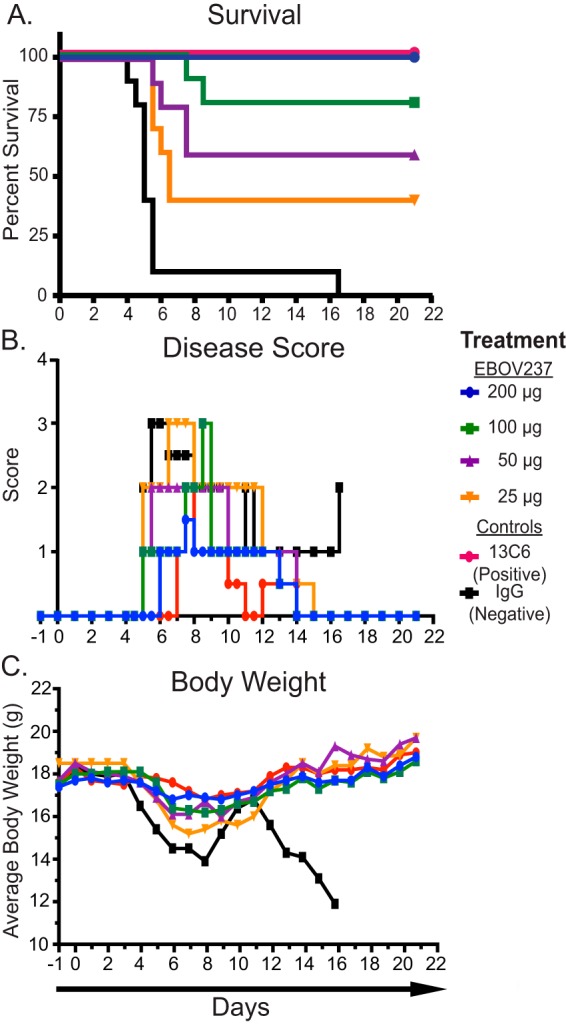
EBOV237 provides dose-dependent efficacy. Groups of 10 mice were given either 200, 100, 50, or 25 μg EBOV237, 100 μg of a positive-control MAb 13C6, or 100 μg of an irrelevant negative-control IgG intraperitoneally 24 h prior to challenge with 100 PFU of Mayinga virus intraperitoneally. (A) Kaplan-Meier survival curve over the course of 21 days demonstrates the extent of survival depended on the dose of MAb. EBOV237 given at 200 μg provided a similar level of protection against lethal disease caused by Mayinga virus as MAb 13C6, in comparison to the irrelevant negative-control IgG. (B) Individual disease scores were determined over the course of 21 days (normal [0], reduced grooming/ruffled fur [1], subdued but normal when stimulated [2], lethargic, hunched posture, subdued even when stimulated [3], nasal discharge/bleeding/unresponsive when stimulated/weak/paralysis [4]). Reduction of disease score was observed in a dose-dependent manner for animals treated with EBOV237. (C) Average weight of mice (calculated from group weight divided by number of mice weighed) taken over a course of 21 days. Mice treated with any dose of EBOV237 or 13C6 showed little variation in the average weight of mice over the course of the study compared to mice treated with the irrelevant negative-control IgG.
DISCUSSION
In this study, we found that the overall human B cell response to Zaire ebolavirus (EBOV) infection early in convalescence is low in EBOV-specific B cell frequency and the antibodies encoded are generally of limited neutralizing potency. Most of the previous work on the human B cell response to EBOV infection has focused on cells obtained long after infection. At later time points, we and others have observed high frequency of circulating B cells that include clones encoding broad and potent protective antibodies (1–6). Previous serological studies suggest that some human EBOV survivors do not possess high-titer neutralizing antibody responses until later after recovery from infection. It was uncertain if this observation was due to just a delayed or low level of secretion of antibodies by plasma cells or if the response was fundamentally deficient in the frequency of virus-specific B cells secreting high-quality antibodies. Using B cell clones, here we show that the early response is characterized by both low magnitude of B cell response and poor functional quality of the antibodies specified by those B cells. Variation in antibody response was observed among these subjects, however, with EVD2 and EVD5 exhibiting a higher and broader response to diverse EBOV species. It is unclear why there are differences in B cell reactivity from these subjects. It is possible that differences in the treatments administered potentially contribute to the specific immune response for each subject, as subjects EVD2 and EVD5 both received ZMapp, whereas EVD9 and EVD15 received a small interfering RNA (siRNA) product and a DNA polymerase inhibitor, respectively, in addition to convalescent-phase serum (19). Additionally, severity of disease may also account for these differences. Further studies are necessary to address the influence on the induction of a patient’s B cells following a specific treatment regimen for EVD.
In total, we isolated 25 MAbs from four U.S. donors who survived EVD. Blood samples were collected at 1 to 3 months postdischarge to represent early convalescence time points. Out of these MAbs, only one, EBOV237, possessed detectable neutralization activity, which was isolated from subject EVD5 3 months postdischarge. It was interesting that the early B cell response was dominated by clones that recognized the secreted form of EBOV glycoprotein (sGP). This finding may indicate that the initial B cell response to EBOV infection is driven principally by sGP, which might serve as a decoy to allow for virus evasion of the immune response. The low frequency of neutralization activity from the isolated MAb panel suggests that the B cell response is of low quality and quantity early during infection and further implicates the importance of aggressive treatment of disease early during infection and during early convalescence.
Identification of the neutralizing epitope recognized by EBOV237 through several laboratory methods revealed that EBOV237 recognizes a unique site within the glycan cap of GP1. In particular, residues N278 and P279 and residues I260 and S322 were identified through alanine-scanning mutagenesis and neutralization escape mutant viruses, respectively. This epitope is in a location near the top of the glycan cap, similar to that of chimerized mouse antibodies included in the ZMapp cocktail, MAb 13C6, and the ZMab cocktail, 1H3, that also recognize the glycan cap of GP1. However, MAbs 1H3 and 13C6 are weakly neutralizing, and the differences in epitope recognition by EBOV237 suggest that this new MAb binds to a slightly different antigenic determinant within the glycan cap of GP1 that enables it to have stronger neutralization activity against EBOV. In addition to in vitro virus neutralization activity, EBOV237 also exhibited prophylactic activity, similar in comparison to the ZMapp cocktail MAb 13C6, in a dose-dependent manner against infection and disease caused by the mouse-adapted Ebola Zaire virus (Mayinga) (ma-ZEBOV) strain.
The results presented here suggest that the human B cell response to EVD early in recovery is characterized by targeting the EBOV GP glycan cap or sGP, with a paucity of broad or potent neutralizing B cell clones. However, the neutralizing epitope in the glycan cap recognized by EBOV237 may play a role in the early human antibody response to EVD and should be considered in rational design strategies for new EBOV vaccine candidates.
MATERIALS AND METHODS
Research subjects.
The subjects, designated EVD2, EVD5, EVD9, and EVD15, were patients who survived EVD and received treatment at Emory University Hospital in 2014. The clinical course and immune activation profiles for these patients were previously described (19, 24). Peripheral blood from the subjects was collected 1 month and 3 months postdischarge from the hospital. The sample that yielded the MAb EBOV237 is designated EVD5_3, indicating the subject sample identification (EVD5) and time point of peripheral blood collection (3 months postdischarge). Multiple PCR tests were performed to detect the presence of virus, which were negative, indicating the absence of virus at these time points. The institutional review boards (IRBs) at Emory University and Vanderbilt University Medical Center approved the protocol for the recruitment and collection of blood samples used in this study. Specifically, the subjects were enrolled under an Emory protocol, IRB00076700, The Longitudinal Characterization of Immune Responses to Ebola Virus Infections. The samples were obtained after the subjects gave written informed consent. PBMCs were isolated and cryopreserved in liquid nitrogen until use.
Generation of human hybridomas.
Previously cryopreserved PBMCs were thawed rapidly in a 37°C water bath. The cells were washed and resuspended in prewarmed medium A (catalog no. 03801; ClonaCell-HY) prior to Epstein-Barr virus (EBV) transformation. In 16 ml of B cell growth medium (medium A, 50 μl CpG [2.5 mg/ml; Invitrogen oligonucleotide ZOEZOEZZZZZOEEZOEZZZT], 15 μl Chk2 inhibitor [10 mM in dimethyl sulfoxide {DMSO}; catalog no. C3742; Sigma], and 20 μl cyclosporine [1 mg/ml in ethanol; catalog no. C1832; Sigma]), 4.5 ml of filtrate containing EBV from the B95.8 cell line (ATCC CRL-1612) was added. The PBMCs then were added and plated at 50 μl/well for 8 to 10 million cells per 384-well plate (catalog no. 164688; Thermo Scientific). After 7 to 10 days at 37°C, cells were expanded into four 96-well plates (catalog no. 353072; Falcon) at 200 μl/well in 20 ml of B cell expansion medium (medium A, 50 μl CpG, 15 μl Chk2 inhibitor, and irradiated [9,000 rads] heterologous human PBMCs [Nashville Red Cross] at 10 million cells/ml) per 96-well plate. The plates were incubated for an additional 4 days at 37°C, prior to screening by ELISA with purified glycoproteins (described below and as previously described [1]). Cells from positive wells containing supernatant reactive for an EBOV-specific ELISA were added to 1 ml of prewarmed BTX cytofusion medium (300 mM sorbitol [catalog no. 36021; Sigma], 0.1 mM calcium acetate [catalog no. AC21105-2500; Fisher], 0.5 mM magnesium acetate [catalog no. AC42387-0050; Fisher], and 1 mg/ml bovine serum albumin [BSA; catalog no. A9418; Sigma]) in microcentrifuge tubes. The microcentrifuge tubes were centrifuged at 3,000 rpm for 4 min, and the supernatant was decanted. This procedure was repeated for a total of three times. HMMA 2.5 cells were washed 3× in BTX cytofusion medium and resuspended in BTX cytofusion medium for a total of 10 million cells/ml. HMMA 2.5 nonsecreting myeloma cells then were added to the microcentrifuge tubes containing the positive EBV-transformed B cell pellet, for a total of 1.15 million cells per fusion. These cells were resuspended and transferred to a cytofusion cuvette (catalog no. 450125; BTX). Electrofusion of the cells was performed as described previously (25). The cuvettes were incubated at 37°C for 30 min. The cells then were added to 21 ml of HAT medium (400 ml of medium A, 100 ml of medium E [catalog no. 03805; ClonaCell-HY], 50× HAT medium supplement [100 μM hypoxanthine, 0.4 μM aminopterin, 16 μM thymidine; catalog no. H0262; Sigma], 150 μl ouabain octahydrate [1 mg/ml; catalog no. O3125; Sigma]) and plated at 50 μl/well in 384-well plates. The plates were incubated for a total of 18 days at 37°C in 7% CO2 prior to screening by ELISA.
Human MAb production and purification.
Positive wells from hybridoma fusions containing supernatant reactive for an EBOV-specific ELISA were cloned biologically by single-cell fluorescence-activated cell sorting. These clones then were expanded serially in 1, 2, and then 30 ml of medium E into 48-well plates (catalog no. 3548; Corning), 12-well plates (catalog no. 3513; Corning), and T-75-cm2 flasks (catalog no. 430641; Corning), respectively. For each T-75-cm2 flask, the cells were scraped and expanded further into four T-225-cm2 flasks (catalog no. 431082; Corning) in 250 ml of serum-free medium (Hybridoma-SFM, catalog no. 12045-076; Gibco). After 21 days at 37°C, the supernatants were filtered using a 0.2-μm-pore-size filter. Antibodies were purified from the filtrate using HiTrap protein G or HiTrap MabSelectSure columns (catalog no. 17040501 and 11003494, respectively; GE Healthcare Life Sciences).
Screening ELISA.
Soluble forms of the full-length extracellular domain of Ebola Zaire virus (EBOV), Bundibugyo virus (BDBV), Sudan virus (SUDV), and Marburg virus (MARV) glycoproteins (GPs) (1 μg/ml) or EBOV secreted GP (sGP) (1 μg/ml) were diluted in 1× Dulbecco’s phosphate-buffered saline without calcium and magnesium (D-PBS) to coat 384-well ELISA plates (catalog no. 265203; Thermo Scientific) at 25 μl/well and incubated at 4°C overnight. The plates were washed 3× with D-PBS-T (1× D-PBS plus 0.05% Tween 20) and blocked for 1 h at room temperature with blocking solution (1% nonfat dry milk [blotting-grade blocker, catalog no. 170-6404; Bio-Rad], 1% goat serum [catalog no. 16210-072; Gibco] in D-PBS-T). After blocking, the plates then were washed 3× with D-PBS-T, and 10 μl/well of supernatant from wells containing EBV-transformed B cells was added. Plates were incubated for 2 h at room temperature and then washed 3× with D-PBS-T. A solution of secondary antibodies (goat anti-human IgG Fc gamma fragment-specific alkaline phosphatase [AP] conjugated, catalog no. W99008A; Meridian Life Science) at a 1:4,000 dilution in blocking solution was then added at 25 μl/well for 1 h at room temperature. Alkaline phosphatase substrate solution (phosphatase substrate tablets [catalog no. S0942; Sigma] in AP substrate buffer (1 M Tris aminomethane [catalog no. BP152-5; Fisher], 30 mM MgCl2 [catalog no. M1028; Sigma]) was added at 25 μl/well following plate washing 6× with 1× D-PBS-T. Plates were incubated at room temperature in the dark for 2 h then read at an optical density of 405 nm with a Biotek plate reader. Reactive wells were chosen based of an optical density of >0.5 nm. For data visualization, the Circos software package was used (26).
Recombinant glycoprotein.
For the production of recombinant EBOV, BDBV, SUDV, and MARV GP ectodomains (amino acids 1 to 636) and EBOV sGP (amino acids 1 to 316), stable Drosophila S2 cell lines were generated. Briefly, Effectene transfection reagent (Qiagen) was used to transfect S2 cells with modified pMTpuro vector plasmids containing the GP gene of interest, followed by stable selection of transfected cells with 6 μg/ml puromycin in complete Schneider’s medium. Stable cells were transitioned to Insect Xpress medium (Lonza) in shaker culture, and GP ectodomain expression was induced with 0.5 mM CuSO4. Supernatants were harvested after 4 days. All proteins were engineered with a double strep tag at the C terminus to facilitate purification using Strep-Tactin resin (Qiagen), and the GPs were further purified by Superdex 200 size-exclusion chromatography in Tris-buffered saline (TBS) or PBS buffer.
Viruses and cells.
Neutralization assays were performed using a virus stock containing Ebola virus Zaire (EBOV/Kik-9510621, CDC no. 807224, Vero E6p2), which was passed additionally in Vero cell monolayer cultures once and Vero E6 cell monolayer cultures twice (Vero E6p2, Vero p1, E6p2). Mouse-adapted Ebola Zaire (ma-ZEBOV) was prepared from the original Bray stock (1976 strain, Mayinga) (27) with an additional passage on Vero E6 cell monolayer cultures (Mp3, Vp2, Mp9, ppGH, Vp1). For plaque reduction neutralization tests (PRNTs), a biologically contained Ebola virus, EbolaΔVP30 virus, was generated as previously described possessing the green fluorescent protein (GFP) reporter gene instead of the VP30 open reading frame in the viral genome (28). VeroVP30 cells were maintained in complete growth medium (10% fetal bovine serum [FBS], 1× minimal essential medium [MEM] with antibiotics and supplements), and EbolaΔVP30-GFP virus was propagated in a reduced medium (2% FBS, 1× MEM with antibiotics and supplements) as previously described (28). EbolaΔVP30 viruses were approved for use under biosafety level 2 containment at the University of Wisconsin—Madison by the NIH and CDC in February 2016.
Half-maximal effective concentration (EC50) binding ELISA analysis.
EBOV, BDBV, SUDV, or MARV GP, or EBOV sGP (1 μg/ml) was prepared in 1× D-PBS to coat 384-well ELISA plates at 25 μl/well and incubated at 4°C overnight. The plates were washed 3× with D-PBS-T and blocked for 1 h at room temperature with blocking solution (1% nonfat dry milk, 1% goat serum, D-PBS-T). After blocking, the plates were washed 3× with D-PBS-T, and 25 μl/well of 3-fold serially diluted purified EBOV human MAb (30 μg/ml to 170 ng/ml) in blocking solution was added. A solution of secondary antibodies (goat anti-human IgG Fc gamma fragment-specific alkaline phosphatase conjugated, catalog no. W99008A; Meridian Life Science) at a 1:4,000 dilution in blocking solution was added at 25 μl/well for 1 h at room temperature. Alkaline phosphatase substrate solution was added at 25 μl/well following plate washing 6× with D-PBS-T. Plates were incubated at room temperature in the dark for 2 h and then read at an optical density of 405 nm with a Biotek plate reader. EC50 values were calculated using a nonlinear regression analysis of the curves generated in Prism version 5 (GraphPad Software).
Neutralization assay.
Virus-specific neutralizing antibody responses were titrated in a plaque reduction neutralization test (PRNT), as previously described (2). Antibodies were diluted serially in minimal essential medium (Corning Cellgro, Manassas, VA) containing 5% heat-inactivated fetal bovine serum (Gibco-Invitrogen, Gaithersburg, MD), 1× antibiotic-antimycotic (Gibco-Invitrogen) (MEM complete), and incubated 1 h at 37°C with virus. After incubation, the antibody-virus mixture was added in duplicate to 6-well plates containing 90 to 95% confluent monolayers of Vero E6 cells. Plates were incubated for 1 h at 37°C with gentle rocking every 15 min. Following the incubation, wells were overlaid with 0.5% agarose in supplemented Eagle’s basal minimum essential (EBME) medium, 10% heat-inactivated fetal bovine serum (Gibco-Invitrogen), and 2× antibiotic-antimycotic (Gibco-Invitrogen), and plates were incubated at 37°C in 5% CO2 for 7 days. On day 7, cells were stained by the addition of a second overlay prepared as described above containing 4 to 5% neutral red. Plates were incubated for 18 to 24 h at 37°C in 5% CO2. The endpoint titer was determined to be the highest dilution with ≥50% or ≥80% reduction (PRNT50 or PRNT80, respectively) in the number of plaques observed to virus-only control wells.
Plaque reduction neutralization test (PRNT).
To determine the neutralizing activity of EBOV237, a standard PRNT was performed with EbolaΔVP30-GFP virus (28). Approximately 200 PFU of virus was incubated with EBOV237 (serial 10-fold dilutions of antibody in reduced medium) or a VP35 MAb as a control for 60 min at 37°C. After incubation, standard plaque assays were performed (in triplicate) in which the virus-antibody mixture was inoculated on VeroVP30 cell monolayers for 60 min. After washing off unbound virus, cells were overlaid with 1.25% methylcellulose medium and incubated for 7 days. Cells were fixed, an immunostaining assay using an antibody against VP40 was performed as previously described (28), and the number of plaques was counted in each well.
Generation of antibody escape mutant viruses.
To generate mutant viruses that no longer were neutralized, approximately 10,000 PFU of EbolaΔVP30-GFP virus was incubated with 5 μg/ml of EBOV237 at 37°C for 60 min. After incubation, the virus-antibody mixture was inoculated onto VeroVP30 cells and incubated for 60 min at 37°C, and after incubation, cells were washed three times with reduced medium. Any escape mutant viruses were propagated for 9 days in reduced medium containing 5 μg/ml of MAb EBOV237. Four individual escape mutant viruses were isolated by selection of resistant plaques (28), and virus stocks were generated for each escape mutant virus in reduced medium in the presence of EBOV237 (5 μg/ml). The sequence of the virus GP was determined using viral RNA isolated from the cell culture supernatant of the stock virus (RNeasy minikit; Qiagen). A reverse transcription-PCR (RT-PCR; Verso 1-Step RT-PCR kit; Thermo Fisher Scientific) was performed to amplify the viral GP gene using forward and reverse primers at positions 6000 and 8110 of the Zaire Ebola virus (strain Mayinga 1976) genome that flank the 5′ and 3′ open reading frames of the GP gene, respectively. The resulting RT-PCR product was blunt end cloned into the TOPO pCR-XL vector (Thermo Fisher Scientific), and the sequence of the GP was determined by Sanger DNA sequencing method for plasmid DNA from 10 to 20 bacterial clones for each virus escape mutant.
Competition-binding analysis using biolayer interferometry.
For competition-binding experiments, EBOV GP or EBOV sGP was biotinylated using an EZ-link Micro NHS-PEG4-biotinylation kit (catalog no. 21955; Thermo Scientific). The biosensors were soaked for 10 min in 200 μl of 1× kinetics buffer (catalog no. 18-1092; ForteBio), followed by a baseline signal measurement for 60 s. The biotinylated proteins (5 μg/ml) then were immobilized onto streptavidin-coated biosensor tips (catalog no. 18-5019; ForteBio) for 120 s. After washing the biosensor tips by immersion into 1× kinetics buffer for 60 s, the biosensors were immersed into 1× kinetics buffer containing the first antibody (100 μg/ml) for 600 s. The tips then were immersed into kinetics buffer containing the second antibody (100 μg/ml) for 300 s. Comparison between the maximal signal of the second antibody in the absence or presence of the first antibody was used to determine the percent binding of the second antibody. If the percent binding of the second antibody was reduced to <30% of the maximal signal that occurred in the absence the first antibody, the antibodies were considered to display full competition for binding. If the percent binding was reduced from 30% to 70% of the maximal signal that occurred in the absence of the first antibody, the antibodies were considered to display intermediate competition for binding. If the percent binding was >70% of the maximal signal that occurred in the absence of the first antibody, the antibodies were considered noncompeting.
Fab production.
To generate the Fab fragment of EBOV237, purified MAb IgG protein was digested with papain (0.1 mg/ml stock solution, catalog no. P3125; Sigma) at a ratio of 20:1 (wt:wt) MAb-papain. Digestion was performed at 37°C for 1 h before termination by the addition of a 1/10 volume of 0.3 M iodoacetamide in PBS, followed by immediate 10-fold dilution in 10% normal goat serum (NGS) and storage at 4°C.
Alanine-scanning shotgun mutagenesis for epitope mapping.
We created an alanine-scanning mutagenesis library for EBOV GP (18) using an expression construct of EBOV GPΔmucin (Ebola virus H.sapiens-tc/COD/1976/Yambuku-Mayinga [23], Δ311–461). Each of the amino acid residues 33 to 310 and 462 to 676 of EBOV GPΔmucin were substituted in individual GP constructs one at a time to alanine (or alanine residues to serine). The signal peptide sequence, residues 1 to 32, was not altered. The alanine-scanning mutagenesis library clones constituted 99.9% of the target residues (492 of 493), and the presence of the mutations was confirmed by DNA sequencing. Clones were arrayed at one mutant per well per into 384-well plates. HEK-293T cells were transfected with the mutant library. After 22 h of incubation to allow for expression of the mutant GPs, the cells were either fixed with 4% paraformaldehyde in PBS with calcium and magnesium or unfixed. The cells then were incubated with the Fab fragment of EBOV237 in 10% normal goat serum (NGS) (Sigma-Aldrich, St. Louis, MO) for 1 h at room temperature. An Alexa Fluor 488-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) in 10% NGS was added, and the cells were incubated for 30 min at room temperature. The cells then were washed twice in PBS without calcium and magnesium and resuspended in Cellstripper (Cellgro, Manassas, VA) with 0.1% BSA (Sigma-Aldrich). A multiwell automated flow cytometer (HTFC; Intellicyt, Albuquerque, NM) then was used to detect cellular fluorescence. The relative EBOV237 Fab fragment reactivity to the mutants in comparison to the wild-type EBOV GPΔmucin was determined by subtracting the background signal given by mock vector-transfected control cells and normalizing the signal to the wild-type EBOV GPΔmucin-transfected control. Critical clones were determined if they did not bind the EBOV237 Fab fragment but bound other control EBOV MAbs (EBOV296, EBOV442, and EBOV520). This approach excludes mutants with misfolding or expression defects (29). The mutagenesis data were analyzed by detailed algorithms, as described previously (30).
In vivo protection studies.
All animal research was conducted under an IACUC-approved protocol in compliance with the Animal Welfare Act, PHS Policy, and other federal statutes and regulations relating to animals and experiments involving animals. The facility where this research was conducted is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 2011. Female BALB/c mice, age 6 to 8 weeks, were purchased from Charles River Laboratories. Upon arrival, mice were housed in microisolator cages in an animal biosafety level 4 containment area and provided chow and water ad libitum. An initial study evaluated the efficacy of EBOV237 at a single concentration. Twenty-four hours before infection (day −1), groups of mice (10 mice per group) were treated intraperitoneally (i.p.) with a single dose (100 μg) of antibody. Negative-control mice received an irrelevant, IgG antibody. Positive-control mice received 100 μg of the previously described MAb 13C6. On day 0, mice were inoculated by the i.p. route with 100 PFU of ma-ZEBOV. Mice were monitored daily (twice daily if clinical signs of disease were noted) for 28 days after virus inoculation. Group weights were recorded daily after virus inoculation. Body weights were calculated by dividing the group weight by the number of mice weighed. In a second study, efficacy was evaluated at various decreasing doses. For this study, mice (n = 10/group) were administered a single dose of 200, 100, 50, or 25 μg of EBOV237 at 24 h before virus exposure. Negative-control mice received an irrelevant, IgG antibody. Positive-control mice received a single dose of 100 μg of MAb 13C6. On d0, mice were inoculated by the i.p. route with 100 PFU of ma-ZEBOV. Mice were monitored daily (twice daily if there were clinical signs of disease) for 21 days after virus inoculation. Group weights were recorded daily after virus inoculation. Body weights were calculated by dividing the group weight by the number of mice weighed.
ACKNOWLEDGMENTS
We thank the Emory Serious Communicable Diseases team for their efforts to collect samples from these patients and the members of the Rafi Ahmed laboratory at Emory who separated and cryopreserved the survivor PBMCs. We thank Hannah King, Rebecca Lampley, and Gopal Sapparapu for technical support with antibody protein preparation.
This work was supported by U.S. NIH grants U19 AI109711 (to J.E.C.) and U19 AI109762 (to E.O.S.), Defense Threat Reduction Agency grant HDTRA1-13-1-0034 (to J.E.C.), HHS contract HHSN272201400058C (to J.E.C. and B.J.D.), and Defense Advanced Research Project Agency grant W31P4Q-14-1-0010 (to J.E.C. and Y.K.). Flow cytometry experiments were performed in the VUMC Flow Cytometry Shared Resource, supported by NIH grants P30 CA68485 and DK058404.
L.E.W., A.I.F., P.J.G. Y.K., and J.E.C. planned the studies. L.E.W., A.I.F., N.K., R.B., S.R., E.D., M.L.F., E.O.S, P.J.H., A.E.P., and P.J.G. conducted the experiments. A.B. generated the Circos plots. L.E.W., E.D., B.J.D., P.J.H, Y.K, P.J.G., and J.E.C. interpreted the studies. L.E.W. and J.E.C. wrote the first draft of the paper. E.O.S., B.J.D., Y.K., P.J.G., and J.E.C. obtained funding.
S.R., E.D., and B.D. are employees of Integral Molecular. B.J.D. is a shareholder of Integral Molecular. J.E.C. is a consultant for Sanofi, is on the Scientific Advisory Boards of PaxVax, CompuVax, GigaGen, and Meissa Vaccines, is a recipient of previous unrelated research grants from Moderna and Sanofi, and is founder of IDBiologics. All other authors declare no competing interests. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the DOD.
REFERENCES
- 1.Flyak AI, Shen X, Murin CD, Turner HL, David JA, Fusco ML, Lampley R, Kose N, Ilinykh PA, Kuzmina N, Branchizio A, King H, Brown L, Bryan C, Davidson E, Doranz BJ, Slaughter JC, Sapparapu G, Klages C, Ksiazek TG, Saphire EO, Ward AB, Bukreyev A, Crowe JE Jr. 2016. Cross-reactive and potent neutralizing antibody responses in human survivors of natural ebolavirus infection. Cell 164:392–405. doi: 10.1016/j.cell.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, Piper AE, Goff A, Shamblin JD, Wollen SE, Sprague TR, Fusco ML, Pommert KB, Cavacini LA, Smith HL, Klempner M, Reimann KA, Krauland E, Gerngross TU, Wittrup KD, Saphire EO, Burton DR, Glass PJ, Ward AB, Walker LM. 2016. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science 351:1078–1083. doi: 10.1126/science.aad5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wec AZ, Herbert AS, Murin CD, Nyakatura EK, Abelson DM, Fels JM, He S, James RM, La Vega M-AA, de Zhu W, Bakken RR, Goodwin E, Turner HL, Jangra RK, Zeitlin L, Qiu X, Lai JR, Walker LM, Ward AB, Dye JM, Chandran K, Bornholdt ZA. 2017. Antibodies from a human survivor define sites of vulnerability for broad protection against ebolaviruses. Cell 169:878–890.e15. doi: 10.1016/j.cell.2017.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flyak AI, Kuzmina N, Murin CD, Bryan C, Davidson E, Gilchuk P, Gulka CP, Ilinykh PA, Shen X, Huang K, Ramanathan P, Turner H, Fusco ML, Lampley R, Kose N, King H, Sapparapu G, Doranz BJ, Ksiazek TG, Wright DW, Saphire EO, Ward AB, Bukreyev A, Crowe JE Jr. 2018. Broadly neutralizing antibodies from human survivors target a conserved site in the Ebola virus glycoprotein HR2-MPER region. Nat Microbiol 3:670–677. doi: 10.1038/s41564-018-0157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saphire EO, Schendel SL, Fusco ML, Gangavarapu K, Gunn BM, Wec AZ, Halfmann PJ, Brannan JM, Herbert AS, Qiu X, Wagh K, He S, Giorgi EE, Theiler J, Pommert KBJB, Krause TB, Turner HL, Murin CD, Pallesen J, Davidson E, Ahmed R, Aman MJ, Bukreyev A, Burton DR, Crowe JE Jr, Davis CW, Georgiou G, Krammer F, Kyratsous CA, Lai JR, Nykiforuk C, Pauly MH, Rijal P, Takada A, Townsend AR, Volchkov V, Walker LM, Wang C-I, Zeitlin L, Doranz BJ, Ward AB, Korber B, Kobinger GP, Andersen KG, Kawaoka Y, Alter G, Chandran K, Dye JM, Viral Hemorrhagic Fever Immunotherapeutic Consortium. 2018. Systematic analysis of monoclonal antibodies against ebola virus GP defines features that contribute to protection. Cell 174:938–952.e13. doi: 10.1016/j.cell.2018.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilchuk P, Kuzmina N, Ilinykh PA, Huang K, Gunn BM, Bryan A, Davidson E, Doranz BJ, Turner HL, Fusco ML, Bramble MS, Hoff NA, Binshtein E, Kose N, Flyak AI, Flinko R, Orlandi C, Carnahan R, Parrish EH, Sevy AM, Bombardi RG, Singh PK, Mukadi P, Muyembe-Tamfum JJ, Ohi MD, Saphire EO, Lewis GK, Alter G, Ward AB, Rimoin AW, Bukreyev A, Crowe JE Jr. 2018. Multifunctional pan-ebolavirus antibody recognizes a site of broad vulnerability on the Ebolavirus glycoprotein. Immunity 49:363–374.e10. doi: 10.1016/j.immuni.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, Fausther-Bovendo H, Wei H, Aviles J, Hiatt E, Johnson A, Morton J, Swope K, Bohorov O, Bohorova N, Goodman C, Kim D, Pauly MH, Velasco J, Pettitt J, Olinger GG, Whaley K, Xu B, Strong JE, Zeitlin L, Kobinger GP. 2014. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 514:47–53. doi: 10.1038/nature13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, Ploquin A, Doria-Rose NA, Staupe RP, Bailey M, Shi W, Choe M, Marcus H, Thompson EA, Cagigi A, Silacci C, Fernandez-Rodriguez B, Perez L, Sallusto F, Vanzetta F, Agatic G, Cameroni E, Kisalu N, Gordon I, Ledgerwood JE, Mascola JR, Graham BS, Muyembe-Tamfun J-JJ, Trefry JC, Lanzavecchia A, Sullivan NJ. 2016. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 351:1339–1342. doi: 10.1126/science.aad5224. [DOI] [PubMed] [Google Scholar]
- 9.Gilchuk P, Mire CE, Geisbert JB, Agans KN, Deer DJ, Cross RW, Slaughter JC, Flyak AI, Mani J, Pauly MH, Velasco J, Whaley KJ, Zeitlin L, Geisbert TW, Crowe JE Jr. 2018. Efficacy of human monoclonal antibody monotherapy against bundibugyo virus infection in nonhuman primates. J Infect Dis 218:S565–S573. doi: 10.1093/infdis/jiy295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pallesen J, Murin CD, de Val N, Cottrell CA, Hastie KM, Turner HL, Fusco ML, Flyak AI, Zeitlin L, Crowe JE Jr, Andersen KG, Saphire EO, Ward AB. 2016. Structures of Ebola virus GP and sGP in complex with therapeutic antibodies. Nat Microbiol 1:16128. doi: 10.1038/nmicrobiol.2016.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. 2008. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454:177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bornholdt ZA, Ndungo E, Fusco ML, Bale S, Flyak AI, Crowe JE Jr, Chandran K, Saphire EO. 2016. Host-primed Ebola virus GP exposes a hydrophobic NPC1 receptor-binding pocket, revealing a target for broadly neutralizing antibodies. mBio 7:e02154-15. doi: 10.1128/mBio.02154-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashiguchi T, Fusco ML, Bornholdt ZA, Lee JE, Flyak AI, Matsuoka R, Kohda D, Yanagi Y, Hammel M, Crowe JE Jr, Saphire EO. 2015. Structural basis for Marburg virus neutralization by a cross-reactive human antibody. Cell 160:904–912. doi: 10.1016/j.cell.2015.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Misasi J, Gilman MS, Kanekiyo M, Gui M, Cagigi A, Mulangu S, Corti D, Ledgerwood JE, Lanzavecchia A, Cunningham J, Muyembe-Tamfun JJ, Baxa U, Graham BS, Xiang Y, Sullivan NJ, McLellan JS. 2016. Structural and molecular basis for Ebola virus neutralization by protective human antibodies. Science 351:1343–1346. doi: 10.1126/science.aad6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wec AZ, Nyakatura EK, Herbert AS, Howell KA, Holtsberg FW, Bakken RR, Mittler E, Christin JR, Shulenin S, Jangra RK, Bharrhan S, Kuehne AI, Bornholdt ZA, Flyak AI, Saphire EO, Crowe JE Jr, Aman MJ, Dye JM, Lai JR, Chandran K. 2016. A “Trojan horse” bispecific-antibody strategy for broad protection against ebolaviruses. Science 354:350–354. doi: 10.1126/science.aag3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Audet J, Wong G, Wang H, Lu G, Gao GF, Kobinger G, Qiu X. 2014. Molecular characterization of the monoclonal antibodies composing ZMAb: a protective cocktail against Ebola virus. Sci Rep 4:6881. doi: 10.1038/srep06881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murin CD, Fusco ML, Bornholdt ZA, Qiu X, Olinger GG, Zeitlin L, Kobinger GP, Ward AB, Saphire EO. 2014. Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc Natl Acad Sci U S A 111:17182–17187. doi: 10.1073/pnas.1414164111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davidson E, Bryan C, Fong RH, Barnes T, Pfaff JM, Mabila M, Rucker JB, Doranz BJ. 2015. Mechanism of binding to Ebola virus glycoprotein by the ZMapp, ZMAb, and MB-003 cocktail antibodies. J Virol 89:10982–10992. doi: 10.1128/JVI.01490-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McElroy AK, Akondy RS, Davis CW, Ellebedy AH, Mehta AK, Kraft CS, Lyon GM, Ribner BS, Varkey J, Sidney J, Sette A, Campbell S, Ströher U, Damon I, Nichol ST, Spiropoulou CF, Ahmed R. 2015. Human Ebola virus infection results in substantial immune activation. Proc Natl Acad Sci U S A 112:4719–4724. doi: 10.1073/pnas.1502619112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shantha JG, Mattia JG, Goba A, Barnes KG, Ebrahim FK, Kraft CS, Hayek BR, Hartnett JN, Shaffer JG, Schieffelin JS, Sandi JD, Momoh M, Jalloh S, Grant DS, Dierberg K, Chang J, Mishra S, Chan AK, Fowler R, O’Dempsey T, Kaluma E, Hendricks T, Reiners R, Reiners M, Gess LA, ONeill K, Kamara S, Wurie A, Mansaray M, Acharya NR, Liu WJ, Bavari S, Palacios G, Teshome M, Crozier I, Farmer PE, Uyeki TM, Bausch DG, Garry RF, Vandy MJ, Yeh S. 2018. Ebola Virus Persistence in Ocular Tissues and Fluids (EVICT) study: reverse transcription-polymerase chain reaction and cataract surgery outcomes of Ebola survivors in Sierra Leone. EBioMedicine 30:217–224. doi: 10.1016/j.ebiom.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varkey J, Shantha J, Crozier I, Kraft C, Lyon M, Mehta A, Kumar G, Smith J, Kainulainen M, Whitmer S, Ströher U, Uyeki T, Ribner B, Yeh S. 2015. Persistence of Ebola virus in ocular fluid during convalescence. N Engl J Med 372:2423–2427. doi: 10.1056/NEJMoa1500306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer WA, Brown J, Wohl DA, Loftis AJ, Tozay S, Reeves E, Pewu K, Gorvego G, Quellie S, Cunningham CK, Merenbloom C, Napravnik S, Dube K, Adjasoo D, Jones E, Bonarwolo K, Hoover D. 2017. Ebola virus ribonucleic acid detection in semen more than two years after resolution of acute Ebola virus infection. Open Forum Infect Dis 4:ofx155. doi: 10.1093/ofid/ofx155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuhn JH, Andersen KG, Bào Y, Bavari S, Becker S, Bennett RS, Bergman NH, Blinkova O, Bradfute S, Brister JR, Bukreyev A, Chandran K, Chepurnov AA, Davey RA, Dietzgen RG, Doggett NA, Dolnik O, Dye JM, Enterlein S, Fenimore PW, Formenty P, Freiberg AN, Garry RF, Garza NL, Gire SK, Gonzalez J-PP, Griffiths A, Happi CT, Hensley LE, Herbert AS, Hevey MC, Hoenen T, Honko AN, Ignatyev GM, Jahrling PB, Johnson JC, Johnson KM, Kindrachuk J, Klenk H-DD, Kobinger G, Kochel TJ, Lackemeyer MG, Lackner DF, Leroy EM, Lever MS, Mühlberger E, Netesov SV, Olinger GG, Omilabu SA, Palacios G, et al. 2014. Filovirus RefSeq entries: evaluation and selection of filovirus type variants, type sequences, and names. Viruses 6:3663–3682. doi: 10.3390/v6093663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyon MG, Mehta AK, Varkey JB, Brantly K, Plyler L, McElroy AK, Kraft CS, Towner JS, Spiropoulou C, Ströher U, Uyeki TM, Ribner BS, Emory Serious Communicable Diseases Unit. 2014. Clinical care of two patients with Ebola virus disease in the United States. N Engl J Med 371:2402–2409. doi: 10.1056/NEJMoa1409838. [DOI] [PubMed] [Google Scholar]
- 25.Yu X, McGraw PA, House FS, Crowe JE Jr. 2008. An optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J Immunol Methods 336:142–151. doi: 10.1016/j.jim.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. 1999. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis 179:S248–S258. doi: 10.1086/514292. [DOI] [PubMed] [Google Scholar]
- 28.Halfmann P, Kim JH, Ebihara H, Noda T, Neumann G, Feldmann H, Kawaoka Y. 2008. Generation of biologically contained Ebola viruses. Proc Natl Acad Sci U S A 105:1129–1133. doi: 10.1073/pnas.0708057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paes C, Ingalls J, Kampani K, Sulli C, Kakkar E, Murray M, Kotelnikov V, Greene TA, Rucker JB, Doranz BJ. 2009. Atomic-level mapping of antibody epitopes on a GPCR. J Am Chem Soc 131:6952–6954. doi: 10.1021/ja900186n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davidson E, Doranz BJ. 2014. A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology 143:13–20. doi: 10.1111/imm.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, Bowden TA, Padilla-Parra S, Fry EE, Stuart DI. 2016. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 535:169–172. doi: 10.1038/nature18615. [DOI] [PMC free article] [PubMed] [Google Scholar]