

Kaposi's sarcoma (KS)-associated herpesvirus is the causative agent of multiple malignancies, predominantly in immunocompromised individuals, including HIV/AIDS patients. Reduced incidence of KS in HIV/AIDS patients receiving antiherpetic drugs to block lytic replication confirms the role of lytic DNA replication and gene products in KSHV-mediated tumorigenesis. Herpesvirus lytic replication results in the production of complex concatemeric DNA, which is cleaved into unit length viral DNA for packaging into the infectious virions. The conserved herpesviral alkaline exonucleases play an important role in viral genome cleavage and packaging. Here, by using the previously described Q129H mutant virus that selectively lacks DNase activity but retains host shutoff activity, we provide experimental evidence confirming that the DNase function of the KSHV SOX protein is essential for viral genome processing and packaging and capsid maturation into the cytoplasm during lytic replication in infected cells. This led to the identification of ORF37’s DNase activity as a potential target for antiviral therapeutics.
KEYWORDS: Kaposi's sarcoma-associated herpesvirus, lytic DNA replication, SOX protein, exonucleases, viral DNA processing
ABSTRACT
The Kaposi's sarcoma-associated herpesvirus (KSHV) alkaline exonuclease SOX, encoded by open reading frame 37 (ORF37), is a bifunctional early-lytic-phase protein that possesses alkaline 5′-to-3′ DNase activity and promotes host shutoff at the mRNA level during productive lytic infection. While the SOX protein is well characterized for drastically impairing cellular gene expression, little is known about the impact of its DNase activity on the KSHV genome and life cycle and the biology of KSHV infections. Here, we introduced a previously described DNase-inactivating Glu129His (Q129H) mutation into the ORF37 gene of the viral genome to generate ORF37-Q129H recombinant virus (the Q129H mutant) and investigated the effects of loss or inactivation of DNase activity on viral genome replication, cleavage, and packaging. For the first time, we provide experimental evidence that the DNase activity of the SOX protein does not affect viral latent/lytic DNA synthesis but is required for cleavage and processing of the KSHV genome during lytic replication. Interestingly, the Q129H mutation severely impaired intranuclear processing of progeny virions compared to the wild-type ORF37, as assessed by pulsed-field and Gardella gel electrophoresis, electron microscopy, and single-molecule analysis of replicating DNA (SMARD) assays. Complementation with ORF37-wt (wild type) or BGLF5 (the KSHV protein homolog in Epstein-Barr virus) in 293L/Q129H cells restored the viral genome encapsidation defects. Together, these results indicated that ORF37’s proposed DNase activity is essential for viral genome processing and encapsidation and, hence, can be targeted for designing antiviral agents to block KSHV virion production.
IMPORTANCE Kaposi's sarcoma (KS)-associated herpesvirus is the causative agent of multiple malignancies, predominantly in immunocompromised individuals, including HIV/AIDS patients. Reduced incidence of KS in HIV/AIDS patients receiving antiherpetic drugs to block lytic replication confirms the role of lytic DNA replication and gene products in KSHV-mediated tumorigenesis. Herpesvirus lytic replication results in the production of complex concatemeric DNA, which is cleaved into unit length viral DNA for packaging into the infectious virions. The conserved herpesviral alkaline exonucleases play an important role in viral genome cleavage and packaging. Here, by using the previously described Q129H mutant virus that selectively lacks DNase activity but retains host shutoff activity, we provide experimental evidence confirming that the DNase function of the KSHV SOX protein is essential for viral genome processing and packaging and capsid maturation into the cytoplasm during lytic replication in infected cells. This led to the identification of ORF37’s DNase activity as a potential target for antiviral therapeutics.
INTRODUCTION
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV), or human herpesvirus 8 (HHV-8), is the most recently discovered γ2-lymphotropic human tumor virus (1, 2) and is associated with KS, primary effusion lymphoma, and a plasmablastic variant of multicentric Castleman's disease in immunocompromised individuals (1, 3–5). KSHV belongs to the genus Rhadinovirus within the family Herpesviridae and is closely related to Epstein-Barr virus (EBV), a B-lymphotropic member of the subfamily Gammaherpesvirus, known to be associated with Hodgkin lymphoma, Burkitt’s lymphoma, and nasopharyngeal cancer (reviewed in reference 6). In common with the gammaherpesviruses, KSHV possesses a large, highly conserved double-stranded DNA genome (approximately 165 kb) with complex protein-coding potential responsible for virus maintenance, replication, virion assembly, and the production of infectious virions in the infected host (7, 8).
Upon de novo infection, KSHV displays two distinct transcriptional programs: a prolonged dormant latency and intermittent productive lytic reactivation (reviewed in reference 9). The process of herpesvirus DNA replication generates branched/longer-than-unit-length concatemeric DNA in the nuclei of infected cells, which is cleaved into linear/unit length monomeric DNA fragments (10). The newly replicated DNA further undergoes a well-coordinated process of encapsidation into immature capsids in the nucleus, resulting in the generation of DNA-filled capsids (C capsids) that mature into the cytoplasm after budding through the nuclear membrane. Mature C capsids are tegumented and enveloped in the cytoplasm by budding into vesicles of the trans-Golgi network and finally released into the extracellular space as mature extracellular virions (reviewed in reference 10). Although the exact mechanism of KSHV lytic DNA replication remains elusive, it appears to initiate from two lytic origins (ori-Lyt-L/R) (11–13) and may follow genome processing and encapsidation similar to those of the other herpesviruses, since multiple alpha- and betaherpesviral proteins are conserved in their gammaherpesvirus homologs.
The onset of productive lytic replication triggers global cellular translational machinery shutoff, defined as a process that involves blocking of host mRNA transcripts to ensure maximum viral gene expression and host immune evasion (14, 15). In KSHV, shutoff activity is initiated by bifunctional KSHV SOX (shutoff alkaline exonuclease) protein, encoded by open reading frame 37 (ORF37) (16). Extensive PCR-based mutagenesis has suggested that these two key functions are distinct and genetically separable and occur within different cellular compartments (17). The SOX-mediated host shutoff activity, which is unique to KSHV/gammaherpesviruses, occurs in the cytoplasm, whereas the residues critical for the DNase activity confined to the nucleus are widely conserved within herpesviruses, notably herpes simplex virus 1 (HSV-1) (UL12) (18, 19), human cytomegalovirus (HCMV) (UL98) (20, 21), EBV (BGLF5) (22, 23), and murine gammaherpesvirus 68 (MHV-68) (muSOX) (14, 24). The 1.46-kb, 55-kDa KSHV SOX is an early lytic gene that has been shown to be expressed 8 to 12 h postreactivation and belongs to the viral alkaline exonuclease family and the restriction endonuclease PD-(D/E)XK superfamily (25). Like other herpesviral alkaline exonucleases, SOX requires Mg2+ for optimal 5′-3′ exonucleolytic activity and processes both single-stranded and double-stranded substrates in vitro for successful viral replication (26). The crystal structure of the SOX protein bound to a DNA duplex indicates that the DNA strand cleavage likely occurs through a bimetal nuclease mechanism that involves the D221 and E244 carboxylate groups (27, 28). The exact mechanism of SOX’s DNase function is still unknown; however, it seems to play a prominent role in processing and packaging of newly synthesized nonlinear DNA into a linear form suitable for encapsidation and nuclear egress.
Although KSHV SOX protein-initiated host shutoff upon productive infection has been extensively studied for many years, much less is known about the conserved DNase activity, which is considered critical for editing of the viral genome during lytic DNA replication (16, 17). Most knowledge about the SOX protein’s intrinsic DNase activity comes from the study of alkaline exonucleases of other herpesviruses, namely, HSV-1 (UL12) and EBV (BGLF5). Recombinant mutant viruses devoid of UL12 (the AN-1 or D340E mutant) and BGLF5 (the ΔBGLF5 mutant) genes have been shown to have effects on infectious virion production, to generate complex viral DNA replication intermediates, and to display impaired nuclear egress and progeny virion production (19, 23, 29, 30). Based on these observations and similarities between herpesvirus replication and packaging machineries, we predict that KSHV SOX most likely possesses DNA-processing activity similar to that of UL12 and BGLF5. However, the exact role of KSHV ORF37 in viral DNA replication and genome editing remains uncharacterized. Interestingly, a point mutation of amino acid residue 129 of ORF37 (Q129H, or Glu129His) has been previously reported to preserve wild-type shutoff activity but to completely abolish the DNase activity of the ORF37 protein (17). Hence, use of ORF37-Q129H recombinant virus with DNase activity abolished will be instrumental in determining the molecular role of ORF37 in viral genome editing and maturation during KSHV lytic replication.
In order to determine the probable role of the ORF37 protein in the context of the KSHV lytic cycle and how the absence of DNase activity may affect virus growth, we constructed an ORF37-DNase-inactivated Q129H recombinant virus (the Q129H mutant) using the bacterial artificial chromosome (BAC) technique (31). Our data suggested that the DNase activity of ORF37 is absolutely essential for the cleavage of newly replicated DNA into a linear form that can be processed and packaged to produce infectious virus, as the Q129H mutant virus was deficient in infectious virus production compared to the BAC16-wt (wild-type) control virus. Moreover, the Q129H mutant was severely impaired in its ability to mediate viral DNA encapsidation and maturation from the nucleus, confirmed by aberrant DNA encapsidation or packaging, migration on a pulsed-field gel, and the presence of more A/B capsids but fewer mature C capsids in the cytoplasm. Importantly, the Q129H mutant virus was rescued upon introduction of ORF37 or BGLF5 (the EBV homolog) protein in trans into the Q129H mutant-harboring cells. These data provide further evidence that ORF37 has a functional role in viral genome editing for efficient progeny virus production and that it can be a promising target for developing therapeutics for preventing KSHV-associated malignancies.
RESULTS
Generation of ORF37-DNase-inactivated (Q129H) recombinant KSHV (the Q129H mutant).
In order to determine the role of the viral alkaline exonuclease ORF37 in lytic DNA replication, we used a KSHV BAC16 clone that allows genetic manipulation of the KSHV genome in Escherichia coli to construct a mutant virus in which the glutamine residue (Q) at position 129 in the ORF37 protein was point mutated to histidine (H) (31). Substitution of H has been previously reported to completely abolish the DNase activity of ORF37 (17). We used a two-step “seamless” homologous-recombineering approach (32) to introduce the desired mutation, and a schematic illustrating the procedure is shown in Fig. 1A. In the first step, the galK-Kanr (kanamycin resistance) cassette was PCR amplified using primers with 50 bp nucleotide sequence flanking amino acid 129 of ORF37. The PCR product containing the target sequence was electroporated into the competent E. coli strain SW106 containing BAC16. The galK-Kanr cassette-containing colonies were screened on chloramphenicol-kanamycin agar plates for the correct insertional mutants by restriction digestion of the DNA with BamHI and hybridization with a GalK probe, which showed the expected signal of 2.6 kb (Fig. 1B). In the second, counterselection step, the galK-Kanr cassette was replaced by recombination with a duplex oligonucleotide containing the desired mutation. The correct colonies were selected on 2-deoxygalactose (DOG) selection plates for the loss of galactokinase and, once again, were confirmed by BamHI restriction digestion of DNA and hybridization with the GalK probe. The correct insertional mutants, the Q129H mutants, were confirmed by the loss of galK-Kanr signal on a Southern blot (Fig. 1B). Introduction of a correct mutation was confirmed by sequencing of the BAC16-Q129H, i.e., ORF37, gene of the mutated BAC16 (Fig. 1C, a). BAC16-Q129H and BAC16-wt were subjected to next-generation sequencing to confirm the integrity of the viral genome, which showed contiguous sequences without rearrangement or deletion. A snapshot of the aligned sequence reads around the mutated region of ORF37 is presented in Fig. 1C, b. The wt and Q129H mutant were transfected into HEK293L cells using metafectene transfection reagent, followed by selection with hygromycin to obtain pure populations of cells latently harboring the KSHV genome, as detected by green fluorescent protein (GFP) signal (Fig. 1D). The hygromycin-resistant and GFP-positive cell clones that survived selection were expanded for further analysis. Expression of a key latent gene encoding latency-associated nuclear antigen (LANA), which is responsible for tethering the viral genome to the host chromosome, was comparably detected in both 293L/wt and 293L/Q129H cell lines, as analyzed by Western blotting, suggesting that the DNase activity of the ORF37 gene does not affect latent persistence of the KSHV genome (Fig. 1E). These stable cells were further induced with sodium butyrate (NaB) and 12-O-tetradecanoyl-phorbol-13-acetate (TPA) to determine whether DNase depletion has an effect on the expression of KSHV’s immediate-early gene encoding replication and transcription activator (RTA), which triggers lytic replication and production of viral particles. At 48 h postinduction (hpi), the levels of RTA and ORF37 expression in 293L/Q129H cells were found to be almost identical to those in 293L/wt cells, indicating that blocking the DNase activity of ORF37 did not affect the RTA-mediated latent-lytic reactivation switch or abrogate ORF37 gene expression (Fig. 1E). To determine if loss of the DNase activity of the Q129H mutant also hampered its host shutoff function, we analyzed the host RNA levels in 293L/Q129H cells using Northern blot analysis. Total RNAs were extracted from uninduced and 48-hpi 293L/wt and 293L/37Q129H cells and resolved on agarose gels, followed by Northern blotting and hybridization with a 32P-labeled GFP DNA probe (GFP was encoded by the BAC16 backbone) (Fig. 1F, a). As seen in the figure, both the wt and the Q129H mutant were able to degrade the GFP mRNA levels, thus retaining the wild-type shutoff activity of SOX. We observed that almost all the 293L/wt and 293L/37Q129H cells entered lytic reactivation at 48 h, as analyzed by immunofluorescence analysis of RTA expression (Fig. 1F, b). In addition, total RNAs extracted from 293L/wt and 293L/37Q129H harvested at 48 hpi were subjected to cDNA synthesis using a high-capacity RNA-to-cDNA kit. Quantification of all KSHV transcripts using a custom real-time quantitative-PCR (qPCR) analysis in a 96-well plate showed almost identical levels of all viral transcripts in 293L/Q129H cells and the 293L/wt control (Fig. 1G).
FIG 1.

Generation of ORF37-DNase-inactivated (Q129H) recombinant KSHV (the Q129H mutant) via homologous recombination. (A) Schematic diagram showing generation of the Q129H mutant, where amino acid residue 129 of ORF37 was point mutated to histidine by homologous recombination and GalK-Kanr counterselection. Insertion of the galK-Kanr cassette was confirmed by BamHI restriction fragment analysis and Southern hybridization with a GalK-specific probe, indicated in red (Probe). (B) EtBr-stained gel and Southern blot of BamHI-digested BAC16-wt (lane 2), an intermediate containing a galK-Kanr cassette (lane 3), and the Q129H mutant (lane 4). The Southern blot with a 32P-labeled GalK probe showed the expected 2.6-kb band in the intermediate (lane 3). (C, a) DNA sequencing of BAC Q129H confirmed the desired mutation in the ORF37 gene. (b) High-throughput sequencing reads of BAC Q129H confirmed mutation in the ORF37 gene. (D) GFP expression levels from HEK293L cells stably transfected with BAC16-wt or the Q129H DNAs post-hygromycin selection were imaged directly from the fixed cells by fluorescence microscopy. The green images show the expression of GFP, confirming the presence of BAC16-wt or the Q129H mutant, and the gray images are the corresponding bright-field images. (E) Comparison of expression levels of LANA, RTA, and ORF37 via Western blot analysis of uninduced and 48-hpi 293L/wt and 293L/Q129H cells confirmed the persistence and reactivation of both viruses. Staining of the same blot with GAPDH-specific antibody served as a loading control. (F, a) Analysis of the host shutoff activity of the Q129H mutant using RNAs extracted from uninduced and 48-hpi 293L/wt and 293L/Q129H cells. RNA was resolved and subjected to Northern blotting and hybridization with a 32P-labeled GFP probe, which confirmed that the Q129H mutant retained its ability to degrade the host mRNA. (b) Immunofluorescence analysis for RTA expression in 293L/wt and 293L/Q129H cells indicated almost all the cells underwent lytic reaction at 48 hpi. DIC, differential interference contrast. (G) Relative quantification of the viral mRNA levels from 48-hpi 293L/Q129H cells compared to the 293L/wt control using a custom real-time qPCR analysis of the KSHV genes in a 96-well format showed almost identical levels of all viral transcripts. The fold changes for the individual KSHV genes were calculated using the ΔΔCT method normalized to GAPDH and relative to BAC16-wt, defined as 1.
Elimination of ORF37’s DNase activity did not affect viral DNA synthesis but exhibited reduced progeny virion production.
The herpesviral genome is replicated upon induction of the lytic cycle to form nonlinear, highly branched concatemeric DNA molecules that are edited to a monomeric linear genome before packaging into virions. Previously, UL12-null HSV-1 (the AN-1 mutant) and a BGLF5-null EBV mutant were shown to exhibit more complex replicative intermediates and to produce significantly reduced (10,000-fold) infectious virus compared to their wt counterparts, suggesting an important role of DNase activity in genome editing for virion production (23, 29, 30). More recently, cells infected with HSV-1 recombinant D340E mutant virus with exonuclease activity abolished were reported to produce ∼60% less replicating viral DNA than wt- and AN-1 mutant-infected cells (19). In order to determine the effect of eliminating the DNase activity of ORF37 on viral DNA replication and the complex genome-editing process, we first compared the levels of viral DNA replication in latent (uninduced) and 48-h lytically induced 293L/wt and 293L/Q129H cells, and the copy number of KSHV genomes was determined by qPCR. Of note, latent cells harboring the wt versus the Q129H mutant contained identical numbers of intracellular KSHV genome copies, reconfirming that ORF37 did affect DNA synthesis (Fig. 2A). Lytically induced 293L/Q129H cells exhibited only a slight reduction in the total amount of intracellular viral DNA compared to 293L/wt cells, suggesting that ORF37 did not significantly affect lytic DNA synthesis (Fig. 2B). Next, we harvested extracellular virions from the supernatants of these cell lines at 5 days postinduction (dpi) and quantified viral DNA production in the supernatants by extracting the KSHV DNA. qPCR analysis revealed that viral supernatants from induced 293L/Q129H cells showed drastic (∼10-fold) reductions in the number of infectious virions with packaged viral DNA (Fig. 2C). At 5 dpi, the recombinant viruses produced in the supernatants of the 293L/wt and 293L/Q129H cell lines were harvested and used to infect equal numbers of permissive 293 cells (known to be stably infected by KSHV), and GFP signals and infection efficiency were measured 2 days postinfection by flow cytometry (Fig. 2D to F). Interestingly, 293L/Q129H cells showed 1.18% GFP-positive cells compared to the 18.6% for 293L/wt cells, confirming that the amount of virus produced from the Q129H mutant was significantly lower than that from the wt control (Fig. 2D to F). Together, these results confirm that the Q129H mutant virus does not affect latent/lytic viral DNA synthesis but is highly deficient in the production of infectious viral particles, possibly due to inefficient viral genome editing and packaging.
FIG 2.

DNase activity of ORF37 did not affect latent/lytic KSHV DNA synthesis but rather virion production. (A and B) Comparison of intracellular KSHV genome copies in latent/uninduced and 48-hpi 293L/wt and 293L/Q129H cells, as analyzed by qPCR. The values were normalized to GAPDH and calculated relative to BAC16-wt, defined as 1. Equal numbers of KSHV genome copies were maintained during the latent phase; however, only a slight decrease in KSHV genome copies was observed during the lytic phase of virus replication. (C) Detection of extracellular KSHV progeny virion production in the supernatants of 5-dpi 293L/wt and 293L/Q129H cells revealed a 10-fold decrease in virus production in 293L/Q129H supernatants compared to wt supernatants. (D and E) HEK293L cells infected with 293L/wt and 293L/Q129H virus supernatants were assayed by flow cytometry 2 days postinfection. The side scatter (SSC) versus forward scatter (FSC) plot was used to identify GFP-positive cells/signals. (F) Mean value of at least three independent experiments from the values determined in panel E. The error bars represent standard deviations of the mean from the results of three different experiments; ***, P < 0.001.
293L/Q129H cells showed inefficient packaging of viral DNA and impaired nuclear egress.
The observation that the Q129H mutant-harboring cells also failed to produce significant amounts of infectious virions prompted us to determine what role the DNase of ORF37 might play in the complex genome-editing and maturation process. Hence, we examined several key steps in the maturation and encapsidation of viral genomes in cells harboring the wt and the Q129H mutant virus. First, in order to look at the efficiency of viral DNA cleavage and packaging into capsids, total and encapsidated (DNase I-resistant) DNAs were extracted from equal numbers of 293L/wt and 293L/Q129H cells, digested with BamHI (to convert concatemeric DNA into monomers), and analyzed by qPCR using primers specific to ORF73 (Fig. 3A). Smaller amounts of DNase I-resistant DNA are indicative of less efficient cleavage of complex, nonlinear replicated viral DNA into monomeric units suitable for packaging into capsids in the nucleus. Although, the wt and the Q129H mutant replicated with similar efficiencies, only ∼20% of the replicated Q129H DNA was recovered in the DNase I-resistant DNA sample compared with the wt genome (Fig. 3A), suggesting improper cleavage of the viral DNA in the absence of DNase activity or a highly compromised encapsidation process due to the cleaved viral DNA being highly unstable and prone to fragmentation. We next analyzed the abilities of the wt and the Q129H mutant virus to package viral DNA into capsids and subsequent nucleocapsid egress, using sucrose gradient centrifugation (Fig. 3B). Data from HSV-1 capsid structure analysis has identified three main types of intracellular capsid forms in the nucleus, namely, A, B, and C capsids, following the expression of late lytic structural proteins (33). A capsids (empty capsids) arising from abortive DNA-packaging events are present in the nucleus and lack both the internal scaffold protein and the viral DNA genome (34, 35). B capsids (scaffold protein-filled capsids) are present in the nucleus and are considered precursors to the mature C capsids (DNA-filled capsids) that result from successful packaging of viral DNA. HSV-1 exonuclease mutants (AN-1 and D340E) have been reported to be defective in the production of viral DNA that can be packaged into infectious virus (19). In addition, a large number of abortive A capsids and fewer DNA-filled C capsids were detected in the cytoplasm, indicating that alkaline exonuclease is required for cleavage of newly replicated viral DNA into a monomer length viral genome for efficient encapsidation and egress of nucleocapsids. The 293L/wt and 293L/Q129H cells were induced for lytic reactivation for 72 h, fractionated into nuclear and cytoplasmic fractions, and subjected to centrifugation on 20 to 50% sucrose gradients. Significant amounts of A, B, and mature C capsids, detected by light-refracting bands, were present in the nuclear fractions of 293L/wt cells; however, fewer C capsids were detected in 293L/Q129H cells harboring the DNase mutant of ORF37 (Fig. 3B; the arrows indicate capsid bands designated A, B, and C). In addition, only B capsids could be observed in the cytoplasmic fractions of 293L/Q129H cells compared to 293L/wt cells. These results imply that in 293L/wt cells, a significant amount of viral DNA is encapsidated in the nucleus, which matures efficiently into the cytoplasm, whereas DNA encapsidation and nuclear egress are not as efficient in 293L/Q129H cells. The smaller numbers of C capsids in the nuclei of the Q129H mutant virus may indicate abortive packaging events that involve unstable encapsidation of improperly resolved viral DNA.
FIG 3.
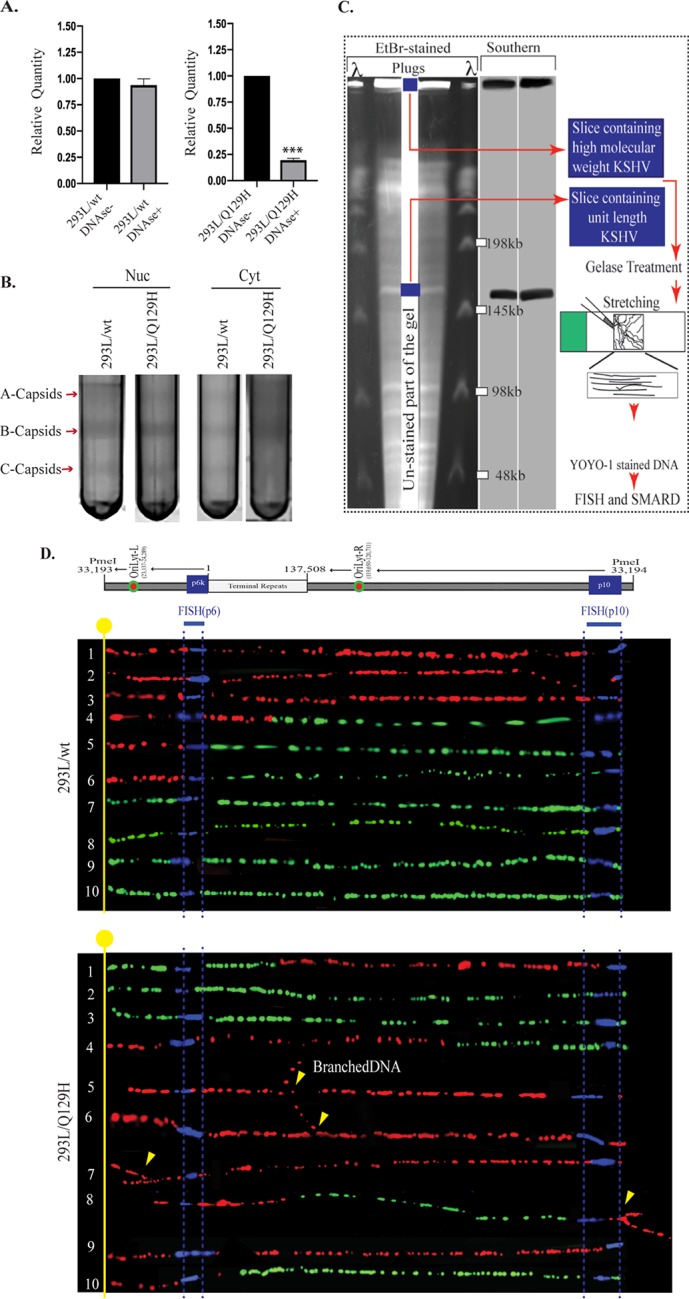
ORF37’s DNase activity is required for efficient packaging of viral DNA and nuclear egress. (A) qPCR analysis of BamHI-digested total and encapsidated (DNase I-resistant) DNAs from 72-hpi 293L/wt and 293L/Q129H cells. When normalized to the total viral DNA, DNA encapsidation in 293L/Q129H cells was significantly reduced compared to the wt control; ***, P < 0.001. (B) Analysis of the KSHV capsids in 72-hpi 293L/wt and 293L/Q129H nuclear and cytoplasmic cell fractions purified by sucrose gradient ultracentrifugation indicated larger amounts of A/B/C capsids for BAC16-wt than for the Q129H mutant (more A/B capsids and fewer C capsids) in the nuclear fraction; however, only B capsids could be seen in the 293L/Q129H cytoplasmic fractions compared to the wt control. The arrows indicate capsid bands designated A, B, and C. (C) Schematic illustrating the SMARD approach, including PFGE of PmeI-digested agarose plugs containing the dually labeled KSHV genome. The middle portion of the gel was stored to extract DNA, and the sides were used for EtBr staining and Southern blotting to localize the KSHV genome. The gel slices containing high-molecular-weight DNA were treated with gelase to isolate the DNA. Stretched DNA was subjected to FISH and SMARD analysis. (D) Microscopy image showing replicating BAC16-wt DNA and Q129H branched DNA. The KSHV genome was detected by FISH probe signals (blue). The first and second labels were detected by immunostaining and are shown in red and green, respectively. Branching of the DNA molecules is indicated by the yellow arrows. The transition site from red to green indicates the location of a replication fork.
The Q129H mutant yielded branched and longer-than-unit-length KSHV DNA genomes following lytic replication.
The proposed model of herpesviral genome replication is through initial bidirectional theta-type replication, followed by a switch to a rolling-circle mechanism in order to generate concatemers of the viral genome (reviewed in reference 36). However, the exact model of replication has not been conclusively determined. Given the defective packaging and maturation of nucleocapsids in the case of the Q129H mutant, we analyzed the nature of newly replicated DNA intermediates generated in the absence of DNase activity, using an innovative single-molecule analysis of replicating DNA (SMARD) assay. The SMARD technique (a schematic is shown in Fig. 3C) consists of several steps starting from sequential labeling of cells with halogenated nucleotides (IdU [5-iodo-2′-deoxyuridine] and CldU [5-chloro-2′-deoxyuridine]), isolation and stretching of DNA on positively charged (silanized) glass slides, and fluorescent in-situ hybridization (FISH) with viral-DNA-specific probes, followed by immunodetection and imaging of the labeled viral DNA molecules with asymmetric probes to determine the unit length molecules. The SMARD assay has been successfully used for identifying replication initiation and termination and replication fork progression using fluorescence microscopy on viral (EBV and KSHV) and cellular DNA molecules (37–41). As SMARD determines the incorporation of nucleotide analogs throughout the replicated DNA, we studied the structure of newly replicated DNA (branched or linear concatemer) generated by both the wt and the Q129H mutant. After labeling the cells with IdU (first label) and CldU (second label), thymidine analogs and their detection on replicated DNA by anti-IdU (red) and anti-CldU (green) antibodies resulted in three types of DNA molecules: (i) molecules that replicated fully during the first labeling period (red signals), (ii) DNA molecules that replicated partially at the end of the first labeling period (red-green signals), and (ii) DNA molecules that replicated/substituted fully during the second labeling period (green signals). The presence of probe signals (blue) after FISH distinguished KSHV DNA from cellular DNA. Analysis of viral DNA molecules with hybridization signals (blue) by FISH probes displayed generation of branched replicated DNA in the Q129H mutant compared to the wt virus (Fig. 3D). Complexity of replication intermediates is indicative of an important role of DNase activity in resolving viral DNA following replication.
Exogenous expression of the ORF37 protein complemented the defective phenotypic traits in the Q129H mutant.
Complementation in trans of the defective mutants using an expression plasmid carrying the previously deleted gene into the mutant indicated that the defects introduced into the mutant were due to the altered gene only and the structural integrity of the remaining viral genome remained intact (42). In order to verify that the impaired phenotype of the Q129H mutant was due specifically to the lack of DNase function of the Q129H mutant, we complemented the DNase-inactive ORF37 gene in 293L/Q129H cells by stably transducing the cells with a lentiviral ORF37-Flag construct (a lentiviral clone expressing the ORF37 gene fused with DsRed-Flag protein), followed by puromycin selection (1 μg/ml), to generate the 293L/Q129H+C37 mutant. When nearly all the cells became positive for GFP and DsRed expression, they were treated with NaB-TPA to induce viral lytic replication and virion production. Whole-cell lysates were prepared from uninduced and 48-hpi 293L/wt, 293L/Q129H, and 293L/Q129H+C37 cells, and lytic genes (RTA and ORF37 genes), as well as Flag expression, was confirmed by Western blot analysis using RTA-, ORF37-, and Flag-specific antibodies (Fig. 4A). The extracellular-virion production in 5-dpi 293L/wt, 293L/Q129H, and 293L/Q129H+C37 cell supernatants was determined using qPCR analysis, and as shown in Fig. 4B, exogenous ORF37 protein in 293L/Q129H+C37 cells could restore virion production compared to the 293L/Q129H supernatants. In order to measure the infectivity of the recombinant virus produced in 293L/Q129H+C37 supernatants, we infected equal numbers of permissive 293 cells (as described previously for wt and Q129H viruses), and GFP signals and infection efficiency were measured 2 days postinfection by flow cytometry (Fig. 4C and D). Interestingly, 293L/Q129H+C37 cell supernatants showed 12.6% GFP-positive cells compared to 1.18% GFP-positive cells in 293L/Q129H cell supernatants (Fig. 2D and E), confirming that the amount of virus produced from the Q129H+C37 mutant was significantly greater than that of the Q129H mutant. To provide an overview of the viral-DNA-packaging ability of the 293L/Q129H+C37 cells, two sets of experiments were performed. In the first set of experiments, mutant viruses produced by 5-dpi 293L/wt, 293L/Q129H, and 293L/Q129H+C37 cells were concentrated, and the capsid forms were separated on 20 to 50% sucrose gradients. The 293L/Q129H+C37 cells were able to readily complement the defect in the production of viral DNA that could be packaged into infectious virus, as indicated by the presence of a more intense C capsid band and thinner A or B capsid bands, similar to those seen for 293L/wt cells (Fig. 4E). In the second set of experiments, thin sections of 3-dpi 293L/wt, 293L/Q129H, and 293L/Q129H+C37 cells were examined by electron microscopy, as described in Materials and Methods. As shown in Fig. 4F, compared to 293L/wt cells, more A or B capsids but very few mature C capsids were identified in the nucleus or extracellular space in 293L/Q129H cells. We could not locate any mature C capsids in the extracellular spaces of several ultrathin sections examined for 293L/Q129H cells, even after multiple attempts. In contrast, all three capsid forms were present in significant amounts in the nuclei, and more mature C capsids and released virions were detectable in the cytoplasm and extracellular spaces of induced 293L/Q129H+C37 cells. This further implies that abolishing the DNase function of ORF37 not only affected the amount and proportion of mature infectious virus produced in the nucleus, but also caused impaired nucleocapsid egress into the cytoplasm.
FIG 4.
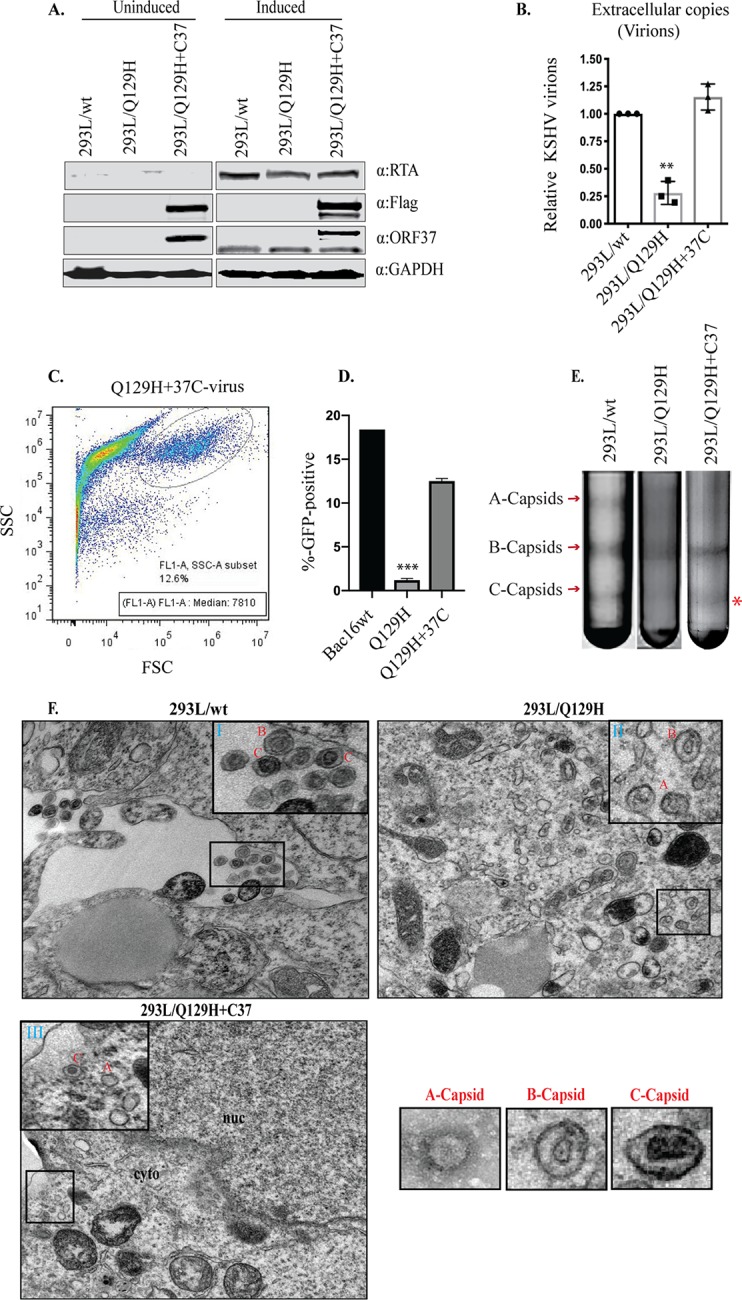
The exogenous expression of ORF37 protein rescued the ORF37-defective phenotype. (A) Western blot analysis of protein extracts from uninduced and 48-hpi 293L/wt, 293L/Q129H, and 293L/Q129H+C37 cells indicated equivalent RTA and ORF37 expression levels in 293L/wt, 293L/Q129H, and 293L/Q129H+C37 cells and confirmed the ORF37-wt trans-complementation as evidenced by Flag expression for 293L/Q129H+C37 cells. (B) Quantitation of extracellular virions in supernatants from 5-dpi 293L/wt, 293L/Q129H, and 293L/Q129H+C37 cells by qPCR assay displayed an increase in virion production in 293L/Q129H+C37 supernatants over 293L/Q129H supernatants, indicating a partial rescue of the defective Q129H phenotype. Mean values and standard deviations from the results of three independent experiments are presented. **, P < 0.01. (C and D) HEK293L cells infected with 293L/Q129H+C37 virus supernatants were assayed by flow cytometry 2 days postinfection (data for 293L/wt and 293L/Q129H are also shown for reference). The side scatter (SSC) versus forward scatter (FSC) plot to identify the GFP-positive cells/signals measured at high fluorescence indicates higher levels of KSHV virions in 293L/Q129H+C37 virus supernatant than in 293L/Q129H supernatants. Mean values of the percentages of infected cells from the results of three independent experiments are plotted. ***, P < 0.001. (E) Analysis of KSHV capsids in 5-dpi 293L/wt, 293L/Q129H, and 293L/Q129H+C37 virus supernatants purified by sucrose gradient ultracentrifugation indicated equivalent amounts of light-refracting A/B/C capsid bands in 293L/wt and 293L/Q129H+C37 supernatants versus 293L/Q129H supernatants. (F) Electron micrographs of 72-hpi 293L/wt, 293L/Q129H, and 293L/Q129H+C37 cells carrying various recombinants displayed more immature nucleocapsids (B capsids) (inset II) in 293L/Q129H cells than in 293L/wt and 293L/Q129H+C37 cells. 293L/Q129H+C37 cells showed DNA-filled nucleocapsids (C capsids) with electron-dense cores (inset III) similar to the 293L/wt cells (inset I). Extracellular virions were not visible. Representative images of individual A, B, and C capsids are shown separately. cyto, cytoplasm; nuc, nucleus; inset bar = 200 nm.
ORF37 complemented the abolished DNase activity of 293L/Q129H cells that showed abnormal linear-genome synthesis.
We next wanted to determine whether complementation of DNase activity could rescue linear-genome synthesis following lytic reactivation. To this end, we used KSHV’s ORF37 and also its homolog in EBV, BGLF5, to determine whether DNase activity among gammaherpesviruses is conserved. Importantly, the mutant of EBV with exonuclease deleted (the BGLF5 mutant) was shown to be defective in genome packaging and generation of branched DNA replication intermediates and had compromised nuclear egress (23). To examine the ability of EBV BGLF5 to complement the impaired DNase activity of the Q129H virus, 293L/Q129H cells were transduced with a lentiviral BGLF5-Flag (Fig. 5A) to generate 293L/Q129H+BG5 stable cells in a manner similar to the generation of 293L/Q129+C37 cells described above. Latent maintenance of the viral genome was determined by the expression of LANA, and the expression of trans-complementing proteins (ORF37 and BGLF5) was confirmed by Flag immunoblotting (Fig. 5B). Lytic reactivation was assessed by the expression of RTA protein in 293L/Q129H+BG5 cells, along with 293L/Q129H+V and 293L/Q129H+C37 cells, by Western blotting (Fig. 5B). Extracellular virion levels obtained from 5-dpi 293L/Q129H+BG5 supernatants were analyzed by qPCR (Fig. 5C) and were found to be ∼5-fold higher than those obtained from cells lacking DNase (293L/Q129H); they were, however, ∼2-fold lower than those obtained from ORF37-complemented (293L/Q129H+C37) cells. These results confirmed that trans-complementation with BGLF5 could only partially rescue the DNase function of the Q129H mutant. To understand the process of defective viral DNA packaging into nucleocapsids by the Q129H mutant, we analyzed the newly replicated viral DNA generated in 72-hpi 293L/Q129H+C37 and 293L/Q129H+BG5 cells and compared it to that of 293L/Q129H+V and 293L/wt cells by pulsed-field gel electrophoresis (PFGE) and Gardella gel electrophoresis and by Southern blot analysis of BamHI-cleaved DNA with a 32P-labeled probe specifically for the KSHV terminal repeat (TR) region. PFGE helps to distinguish between circular and concatemeric DNAs (full-length BamHI-cleaved fragments with all of the TR located on the same 32-kb BamHI-digested fragment, represented by “1” and “2” in the schematic in Fig. 5D) and linear DNA genomes (short BamHI-cleaved fragments with the TR divided into two noncovalent BamHI fragments, represented by “3” in the schematic in Fig. 5D). The Southern blot analysis with a TR-specific probe displayed reduced synthesis of linear genomes extracted from 293L/Q129H+V virus (Fig. 5D, lane 2, compared to 293L/wt (Fig. 5E, lane 1) or trans-complemented 293L/Q129H+C37 and 293L/Q129H+BG5 cells (Fig. 5E, lanes 3 and 4). Additionally, as analyzed by Gardella gel electrophoresis, free linear DNA genomes extracted from 293L/Q129H+V cells (Fig. 5F, lane 2) migrated faster than the viral genomes extracted from 293L/Q129H+C37, 293L/Q129H+BG5, and 293L/wt cells (Fig. 5F, lanes 3, 4, and 1, respectively), despite having similar-size genomes. This abnormal electrophoretic migration behavior of the linear DNase-inactive ORF37 genome further confirms that 293L/Q129H virus is deficient in processing of replicated viral DNA.
FIG 5.

EBV-BGLF5 only partially complements the abolished DNase activity of the Q129H mutant. (A) HEK293T cells transfected with either the pLVXDsRedORF37-Flag or pLVXDsRedBGLF5-Flag lentiviral construct and imaged by fluorescence microscopy for enhanced DsRed expression at 72 h posttransfection. Red indicates the expression of DsRed, confirming the presence of ORF37 and BGLF5, and the gray images are the corresponding bright-field images. (B) 293L/Q129H cells transduced with empty vector (pLVXDsRed-Flag) or with the indicated lentiviruses and selected with puromycin to generate 293L/Q129H cells stably expressing ORF37-wt (293L/Q129H+C37), BGLF5 (293L/Q129H+BG5), or empty vector (293L/Q129H+V) as a control. Western blot analysis of protein extracts from 48-hpi 293L/Q129H-V, 293L/Q129H+C37, and 293L/Q129H+BG5 cells showed equivalent LANA and RTA expression levels, as well as the respective stably expressed proteins with anti-Flag antibody. The GAPDH immunoblot shows equal loading of the cell lysates. (C) Extracellular KSHV progeny virions assessed from 5-dpi 293L/wt, 293L/Q129H+V, 293L/Q129H+C37, and 293L/Q129H+BG5 cells showed substantial but not complete complementation by the BGLF5 protein of EBV. *, P < 0.1; ***, P < 0.001. Horizontal lines represent the means for triplicate samples. (D) Schematic of the KSHV genome concatemer following BamHI digestion. (E) Southern blot analysis of BamHI-cleaved DNA fragments from 72-hpi 293L/wt, 293L/Q129H+V, 293L/Q129H+C37, and 293L/Q129H+BG5 cells, as analyzed by PFGE and hybridization with a 32P-labeled TR probe. BGLF5 complementation of 293L/Q129H cells only partially restored the abolished DNase activity of ORF37. “1” and “2” represent circular and concatemeric DNAs; “3” corresponds to linear DNA segments. (F) Gardella gel electrophoresis to resolve the linear KSHV genome following BamHI digestion, Southern blotting. and hybridization with a 32P-labeled TR probe.
DISCUSSION
Growing interest in the KSHV SOX/ORF37 protein stems from its identification as a bifunctional shutoff alkaline exonuclease protein. The SOX protein has been well characterized as a viral protein responsible for global host shutoff during lytic KSHV infection, as triggered by its intrinsic RNase activity (16, 17). SOX’s homologs in HSV-1 (UL12) and EBV (BGLF5), i.e., alkaline exonucleases encoded by other herpesviruses, have been reported to play important roles in the processing and maturation of complex, newly synthesized double-stranded DNA viral genomes following lytic cycle induction. Also, the Q129H mutation in the highly conserved residues of SOX protein have previously been demonstrated to completely inactivate the intrinsic DNase function while preserving the shutoff activity (17). Despite these research efforts, the precise role of conserved DNase activity in the ORF37 gene in the context of lytic DNA replication and viral growth have never been addressed. In this report, for the first time, we addressed the effect of abolishing DNase activity on KSHV lytic DNA replication and virion production and maturation by constructing the DNase-inactive Q129H recombinant virus. Detailed phenotypic analysis of the Q129H mutant identified a potential role of ORF37 in KSHV genome editing, making it a suitable experimental model to analyze ORF37’s contribution to KSHV lytic reactivation.
We were able to show that both 293L/wt and 293L/Q129H cells maintained nearly equal numbers of latent/lytic KSHV genome copies (Fig. 2A and B); hence, the Q129H mutation is dispensable for viral DNA synthesis. The qPCR analysis and infection experiments with HEK293L cells revealed numbers of infectious virions in 293L/Q129H supernatants significantly lower than those in control wt supernatants (Fig. 2C to F), suggesting that the Q129H mutant is highly deficient in virion production.
These observations prompted us to study key genome maturation steps, i.e., encapsidation of viral DNA into nucleocapsids and primary nuclear egress, in induced 293L/Q129H and 293L/wt cells by DNase I-resistant DNA assay, sucrose gradient centrifugation, and electron microscopy. We concluded that the Q129H mutant with DNase activity abolished was defective in its ability to process viral DNA genomes for encapsidation, as only a small amount of replicated DNA was found to be encapsidated by the Q129H mutant (Fig. 3A) and fewer mature C capsids migrated from the nucleus into the cytoplasm, as represented by an elevated ratio of intranuclear capsids to intracytoplasmic capsids (Fig. 3B). It is currently unknown if ORF37 acts on one or both of these processes. However, the observed impairment in primary egress could itself result from the reduced packaging efficiency, as reported for the HSV-1 alkaline exonuclease UL12 gene (30). There is also a possibility that some of the DNA-filled capsids were packaged with smaller-than-unit-length viral DNA in the nucleus but never matured into the cytoplasm. In line with this, as proposed by Vlazny et al., only capsids that are packaged with a unit length viral genome are able to survive the key conformational change/locking step that permits efficient nuclear egress (43). Therefore, ORF37 could be considered to be involved in this conformation change, leading to abortive packaging and failure of nuclear egress for smaller DNA fragments containing capsids.
As reported in the cases of HSV-1/UL12 and EBV/BGLF5, reduced synthesis of linear viral genomes and a diffused migration pattern of linear viral DNAs in the PFGE assay indicated that elimination of UL12’s nuclease activity leads to a poor resolution of complex replicating DNA structures (29). UL12 was reported to be essential for resolution of branched structures that otherwise would affect the stable packaging of linear genomes into procapsids. In this case, ORF37 has a molecular mechanism similar to that of UL12, and the decreased linear-genome generation in 293L/Q129H cells, along with impaired packaging ability, would be expected to lead to a substantial reduction in the proportion of mature C capsids. However, additional direct effects of ORF37 on virus packaging cannot currently be ruled out. The relationship between the EBV-BGLF5 and the KSHV-ORF37 alkaline exonucleases was analyzed by complementation of the Q129H defective phenotype with ORF37 and BGLF5. Introduction of the intact ORF37 led to a substantial rescue of virion production; however, BGLF5 of EBV could only partially rescue virion production compared to the wild-type ORF37, suggesting that these two proteins may require additional viral factors for their optimal function. Thus, our study validated the previously suggested functional homology of ORF37 and BGLF5 and indicated that, although the DNase activities of the two proteins work on the same processes, the virus-specific factor(s) may regulate the generation of viral DNAs with unique length, genome packaging, and nuclear egress.
In conclusion, the present study used a genetic approach to determine the role of KSHV ORF37/SOX DNase in DNA packaging. The requirement for nuclease activity for efficient KSHV progeny virus production identified this protein as a potential target for antiviral therapeutics. Demonstration of DNase activity by the ORF37 protein now provides a promising target for future studies to develop an additional antiviral therapy that can be used alone or in conjunction with antiviral agents that target KSHV lytic DNA genome maturation to better treat disease caused by KSHV in both immunocompromised and immunocompetent patients. Further insight into the molecular mechanisms that underlie the genome-editing function is required, and the Q129H mutant-harboring cells will be instrumental in this regard.
MATERIALS AND METHODS
Cell lines, plasmids, and chemicals.
Human embryonic kidney (HEK293T and HEK293L) cells (ATCC, Manassas, VA) were routinely maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and penicillin-streptomycin (25 U/ml and 25 μg/ml, respectively). BAC16-wt (the wild-type HHV-8 BAC DNA), which contains the full-length KSHV genome, was kindly provided by Shou-Jiang Gao (University of Southern California). BAC16-ORF37-Q129H DNA was generated by a homologous-recombineering approach (as described below). For lytic reactivation, HEK293L BAC-containing cells were treated with 0.3 M NaB (Sigma-Aldrich, St. Louis, MO) and 20 ng/ml TPA (Sigma-Aldrich, St. Louis, MO) for the indicated times. All the cell lines were grown at 37°C in a humidified environment supplemented with 5% CO2.
Construction of the Q129H mutant.
Mutagenesis of KSHV BAC16 to generate the Q129H mutant was performed by homologous recombination using a two-step “seamless” GalK-positive counterselection scheme (32, 44). In the first step, the galK-Kanr cassette with homologous flanking sequence was PCR amplified using primers with 50-bp homologous sequences (before and after the target sequence) in the sense and antisense primers. The galK-Kanr cassette was amplified by including 20 nucleotides (nt) of homology to the galK-Kanr region. Primers used for galK-Kanr cassette insertion (boldface indicates sequence homologous to residue 129 of the ORF37 locus; lowercase indicates sequence homologous to the galK-Kanr plasmid) were as follows: forward primer: 5′-TGTTCGCAAATGAACGCACATCAGCGGCACCACATCTGCTGTCTAGTGGAGAGGGCCACTAGTAGTCACcctgttgacaattaatcatc-3′; reverse primer, 5′-AGATATAATTCCGTCTCGCAGGGCGTCCCACACGGGGTTCAGACTctcagcaaaagttcgattta-3′.
The PCR product containing the target sequence was subjected to DpnI digestion, followed by agarose gel purification to remove any residual template plasmid. The PCR product was then electroporated into the competent E. coli strain SW102 containing BAC16. The galK-Kanr cassette-containing mutants were selected on chloramphenicol/kanamycin agar plates, and correct insertional mutants were confirmed by restriction digestion with BamHI and Southern blot analysis of fragments containing the galK cassette, using a 32P-labeled GalK probe (45).
The galK-Kanr cassette was replaced by electroporating a double-stranded oligonucleotide (5′-CACATCTGCTGTCTAGTGGAGAGGGCCGGATGAAGATATAATTCCGTCTCGCAG-3′) with homology to the flanking site and plating the bacteria on DOG-containing agar plates for counterselection. Correct colonies were screened and subjected to confirmation by Southern blot analysis using the 32P-labeled GalK probe, PCR amplification of the junctions, and sequence analysis. The integrity of the viral genome and mutation of Q129 to H was confirmed by high-throughput sequencing of the recombinant BAC16 as a standard procedure. Approximately 1 µg of BAC DNA was used for library preparation using a chromatin immunoprecipitation sequencing (ChIP-Seq) DNA library preparation kit (Bioo Scientific) following sonication of BAC DNA to generate small fragments. The ChIP-Seq libraries were validated using a Bioanalyzer 2100 with a high-sensitivity DNA ChIP (Agilent) and quantified by qPCR (Kappa kit). The ChIP-Seq libraries were sequenced using a MiSeq (Illumina) 50-cycle reagent kit on flow cells. The resulting sequencing reads were analyzed using CLC Workbench (CLC Bio).
Stable-clone selection.
BAC16-wt and BAC16-ORF37-Q129H bacmids were transfected into HEK293L cells using Metafectene Pro (Biontex Laboratories GmbH, San Diego, CA) as described previously (46). Two days posttransfection, 50 μg/ml hygromycin B (InvivoGen, San Diego, CA) was added to the culture medium for the selection of stable HEK293L clones carrying the respective genomes. Twelve outgrowing GFP-positive colonies were dislodged, seeded into a new culture flask, and expanded for further investigation. The HEK293L clones containing the BAC16-wt and Q129H viruses used in the present study are referred to as 293L/wt and 293L/Q129H.
trans-complementation of 293L/Q129H using lentiviral vectors.
The pLVXDsRed-Flag lentiviral vectors (Clontech Laboratories, Inc.) carrying ORF37-wt (pLVXDsRed-ORF37-Flag) or BGLF5 (pLVXDsRed-EBV-BGLF5-Flag) were generated by cloning the PCR-amplified ORF37 or EBV-BGLF5 gene into the pLVXDsRed-Flag vector and transfecting them into HEK293T cells along with the lentiviral packaging vectors pCMV-Rev/GP and pCMV-VSVG (Addgene, Inc.) using polyethylenimine (PEI) (Polysciences, Inc.) to produce the respective lentivirus. Supernatants from the transfected HEK293T cells were collected for 5 days, followed by concentration of the virus by ultracentrifugation (25,000 rpm; 2 h; 4°C). The concentrated lentiviral particles were used for transducing 293L/Q129H cells in the presence of 5 μg/ml Polybrene, followed by selection with 1 μg/ml puromycin (Sigma-Aldrich, St. Louis, MO) to obtain a pure population of trans-complemented cells (referred to as 293L/Q129H+V, 293L/Q129H+C37, and 293L/Q129H+BG5). The cells were examined by fluorescence microscopy (DsRed signal encoded by the lentivirus) and Western blot analysis with Flag-specific antibody.
Western blot analysis.
Uninduced and 48-hpi cells were washed with cold PBS (1×), lysed in 100 μl of radioimmunoprecipitation assay (RIPA) lysis buffer (1% NP-40, 50 mM Tris [pH 7.5], 1 mM EDTA [pH 8.0], and 150 mM NaCl) supplemented with protease inhibitors (1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/ml aprotinin, 1 μg/ml pepstatin, 1 μg/ml sodium fluoride, and 1 μg/ml leupeptin), followed by incubation on ice for 30 min. Cell debris was removed by centrifugation (12,000 rpm; 10 min at 4°C), and the lysates were denatured in 50 μl of Laemmli buffer at 95°C for 5 min, resolved by SDS-polyacrylamide gel electrophoresis, and Western blotted onto a 0.45-μm nitrocellulose membrane (Bio-Rad Laboratories) using standard protocols. The proteins of interest were detected by incubating the membrane with specific antibodies overnight at 4°C, followed by incubation with appropriate infrared-dye-tagged secondary antibodies (IR-Dye680/800) and scanning with an Odyssey infrared scanner (Li-Cor Biosciences). The following antibodies were used for this study: mouse anti-LANA (mouse hybridoma), rabbit anti-RTA (custom synthesized at GenScript, Inc.), rabbit anti-ORF37/SOX antisera (kindly provided by Britt Glaunsinger), mouse anti-Flag (M2; Sigma-Aldrich), and mouse anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (G8140) (U.S. Biological, Salem, MA).
Northern blot analysis.
Total RNA was harvested from equal numbers of uninduced and 48-hpi cells using an Illustra RNAspin Mini kit (GE Healthcare) according to the manufacturer’s protocol. Two micrograms of total RNA was resolved on a 1% agarose-formaldehyde gel (80 V for 3 h at 4°C), transferred to a Hybond-N+ membrane (GE Healthcare) using 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) transfer buffer, and hybridized with a 32P-labeled DNA (GFP) probe generated using a random-primer DNA-labeling kit (Clontech Laboratories, Inc.). The autoradiographic signals were detected using a PhosphorImager (Molecular Dynamics, Inc.) according to the manufacturer’s instructions, and the signals were quantified using ImageQuant software (Molecular Dynamics, Inc.).
Immunofluorescence assay.
Forty-eight hours postinduction, 293L/wt and 293L/Q129H cells were grown on UV- and poly-l-lysine-treated coverslips (Thermo Scientific) for 24 h. The cells were washed three times in phosphate-buffered saline (PBS), air dried for 10 min, fixed in 4% paraformaldehyde for 20 min, and permeabilized with 0.2% Triton X-100 in PBS for 10 min. The cells were then blocked with 0.4% fish skin gelatin (FSG)–0.05% Triton X-100 in PBS for 45 min and incubated with specific primary antibodies overnight at 4°C. Subsequently, the cells were washed three times with PBS and stained with Alexa Fluor-conjugated secondary antibodies (Life Technologies Inc.) for 1 h. Nuclear staining was performed using To-Pro-3–PBS for 1 min (Molecular Probes). All incubation steps were performed at room temperature unless otherwise indicated. The cells were visualized and imaged using a confocal laser scanning microscope (Carl Zeiss, Inc.).
Extraction and determination of KSHV DNA.
For quantitation of intracellular KSHV DNA, the total genomic DNA was extracted from uninduced and 48-hpi cells using a modified Hirt lysis method described previously (47, 48). For extracellular viral-DNA extraction, KSHV virions were concentrated (as described below), and 50 µl of virus supernatant diluted with 250 µl of 1× PBS was digested with DNase I (5 U/50 µl of supernatant) at 37°C for 1 h. After DNase I heat inactivation (70°C for 10 min), the supernatants were mixed (1:1 [vol/vol]) with lysis buffer (0.1 mg/ml of proteinase K in water) and incubated at 50°C for 1 h, after which the enzyme was heat inactivated (75°C for 20 min) and subsequently subjected to DNA extraction with phenol-chloroform–isopropanol. The extracted DNA was precipitated and resuspended in 50 µl of sterile water. Amplification reactions were performed in 20-µl volumes containing 10 µl of SYBR Green universal master mix (Bio-Rad Laboratories), 5 µl of forward and reverse ORF57 primers (0.5 µM), and 5 µl of water or intracellular or extracellular KSHV DNA, by real-time qPCR. The primers used were ORF57 (forward primer, TGGACATTATGAAGGGCATCC, and reverse primer, CGGGTTCGGACAATTGCT) and GAPDH (forward primer, GGTCTACATGGCAACTGTGA, and reverse primer, ACGACCACTTTGTCAAGCTC). The KSHV DNA copy numbers were normalized to GAPDH (to account for loading differences) and calculated relative to the respective control samples. All the reactions were run in triplicate.
Quantitation of KSHV RNA.
For transcription analysis, total RNAs were prepared from equal numbers of 48-hpi cells using an Illustra RNAspin Mini kit (GE Healthcare) according to the manufacturer’s protocol. cDNAs were generated using a high-capacity RNA-to-cDNA kit (Applied Biosystems Inc.) according to the manufacturer’s protocol and amplified on an ABI Step One Plus real-time PCR machine (Applied Biosystems Inc.) in a 96-well plate with 20-μl volume per well containing 10 µl of SYBR Green universal master mix (Bio-Rad Laboratories), 5 µl of forward and reverse KSHV ORF-specific primers (0.5 µM), and 5 µl of cDNA diluted in sterile water. Primers for the human β-actin and GAPDH housekeeping genes were also included to normalize the threshold cycle (CT) values. The mRNA levels were calculated by the ΔΔCT method relative to BAC16-wt, defined as 1. As a standard practice, a no-reverse-transcription (no-RT) control reaction was included for all of the prepared RNA samples to ensure their purity. All the reactions were run in triplicate.
KSHV virion purification.
KSHV virions were purified as described previously (30, 49). Briefly, 10 million cells were induced with 0.3 M NaB and 20 ng/ml TPA for 5 days. The culture supernatant was then collected and cleared by centrifugation (4,000 rpm for 30 min at 4°C) and filtered through a 0.45-μm filter to remove cells and cell debris. Virions were pelleted by ultracentrifugation (25,000 rpm for 2 h at 4°C) using a SW28Ti rotor (Beckman Coulter, Brea, CA) and resuspended in 1 ml medium. An aliquot of concentrated virions was used for extracellular viral DNA quantitation using qPCR (as described above). The remaining concentrated virions were layered onto 20 to 50% (wt/vol in PBS) sucrose gradients and centrifuged (35,000 rpm for 2 h at 4°C) using a SW41Ti rotor (Beckman Coulter, Brea, CA). After the ultracentrifugation, KSHV A-, B- and C-type light-refracting capsids were separated at the gradient junction and visualized and photographed by illumination.
Flow cytometry.
Concentrated cell-free virus was used to infect HEK293L cells that were seeded at approximately 1 × 105 cells/well in 24-well plates 24 h prior to infection. The plates were centrifuged with virus at 1,500 × g at room temperature for 30 min in the presence of 8 μg/ml Polybrene. After centrifugation, the medium was replaced with fresh virus-free medium, and the plates were incubated at 37°C for an additional 48 h. Infected cells were trypsinized, washed with PBS, and then fixed in PBS supplemented with 2% paraformaldehyde for 15 min at room temperature. Finally, the cells were washed once and resuspended in PBS. GFP-positive cells were detected using a FACSCalibur flow cytometer equipped with CellQuest Pro software and analyzed using FlowJo software. The titer for each inoculum size, assuming that one infectious particle generates a single GFP-positive cell, was calculated as a percentage of the uninfected cells, according to Poisson's distribution, as described previously (50).
Cell fractionation and analysis of viral capsids.
Nuclear- and cytoplasmic-protein extractions were performed according to standard protocols (51). At 72 hpi, 10 million cells were scraped, collected by low-speed centrifugation (4,000 × g for 5 min at 4°C), and rinsed in 10 ml of ice-cold 1× PBS containing protease inhibitor cocktail. The cells were pelleted by centrifugation (2,000 × g for 5 min at 4°C), washed twice with 1 ml of buffer A (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT], and 0.5 mM PMSF), and centrifuged (4,000 × g for 5 min at 4°C). The supernatant was discarded, and the cellular proteins remaining in the pellet were released by hypotonic lysis in buffer A containing 0.1% NP-40 and centrifugation (10,000 × g for 10 min at 4°C). The supernatant containing the cytosolic fraction was transferred to a new ice-cold Eppendorf tube, while the nuclear proteins remaining in the pellet were obtained by extraction in high-salt buffer B (20 mM HEPES [pH 7.9], 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM Na2 EDTA, pH 8.0, 25% glycerol, 0.5 mM DTT, and 0.5 mM PMSF), followed by centrifugation (10,000 × g for 10 min at 4°C). The supernatant containing nuclear proteins was further diluted with an equal volume of buffer C (20 mM HEPES [pH 7.9], 50 mM KCl, 25% glycerol, 0.5 mM DTT, and 0.5 mM PMSF). The nuclear and cytoplasmic fractions were then layered onto 20 to 50% (wt/vol in PBS) sucrose gradients and centrifuged (35,000 × g for 30 min at 4°C) using an SW41Ti rotor (Beckman Coulter, Brea, CA). The light-refracting capsid bands in each fraction were photographed after centrifugation.
Detection of total and encapsidated DNAs.
The total and encapsidated (DNase-resistant) DNAs were isolated as described previously, with the following modifications (52). Seventy-two-hour-postinduction cells were scraped, collected by low-speed centrifugation, resuspended in Tris-buffered saline (TBS) (137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, 5.5 mM glucose, 25 mM Tris-HCl [pH 7.4]), and divided into two equal samples labeled total cellular and DNase I-resistant DNAs. The cells from both samples were pelleted and resuspended in 184 μl of reticulocyte standard buffer (RSB) (10 mM Tris-HCl [pH 7.5], 10 mM KCl, 1.5 mM MgCl2) containing 0.5% Nonidet P-40 (NP-40). An equal volume of 20 mM Tris-HCl (pH 7.5), 2 mM EDTA,1.2% SDS, and 1 mg/ml proteinase K (Bioline) was either added immediately (for total cellular DNA) or after incubation in the presence of 50 μg of DNase I/ml with occasional mixing for 30 min at 37°C (encapsidated DNA). After the addition of proteinase K, all the samples were incubated for 1 h at 37°C and extracted sequentially with phenol and chloroform, followed by precipitation with 3 M sodium acetate and ethanol. Total and encapsidated DNAs were resuspended in 1× TE (10 mM Tris-HCl, pH 7.5, 1 mM EDTA) containing 5 μg of RNase, digested with BamHI restriction enzyme at 37°C for 3 h, extracted with phenol-chloroform–isoamyl alcohol, and precipitated with ethanol. The extracted total and encapsidated DNAs were resuspended in 50 μl sterile water, and a 5-μl aliquot of the DNA was used to determine viral DNA copy numbers by qPCR using KSHV-ORF73-specific primer (forward primer, CATACGAACTCCAGGTCTGTG, and reverse primer, GGTGGAAGAGCCCATAATCT). Two-fold serial dilutions of the KSHV-ORF73 plasmid were used as the template to produce a standard curve for the quantifications.
Electron microscopy.
Electron microscopy examination of 72-hpi cells was performed in the Shared Instrument Facility at the University of Nevada, Reno, NV, according to the protocols described previously (53, 54). Cells (1 × 105) were grown on poly-l-lysine-treated coverslips and induced for 72 h. The culture medium was removed, and the cells were rinsed with 1× PBS (3 times), fixed in 2% glutaraldehyde in PBS (20 min at 4°C), and postfixed in 2% (wt/vol) osmium tetroxide in 0.2 M sodium-cacodylate buffer, pH 7.4 (1 h at 4°C). The cells were then washed with 0.1 M sodium-cacodylate buffer (twice) and stained with 0.5% uranyl acetate (16 to 20 h at 25°C). Poststaining, the cells were washed twice with water, dehydrated through increasing concentrations of ethanol, and infiltrated with Epon-812 (Electron Microscopy Sciences, Inc.). Embedding was achieved by inverting coverslips onto the Epon-812-filled BEEM capsules (i.e., EMS-embedding capsules). To remove the coverslips from the BEEM capsules with the embedded samples, the samples were transferred between liquid nitrogen and boiling water, causing the glass coverslip to separate from the resin. Ultrathin sections (70 to 80 nm) prepared on a Leica Ultracut UCT ultramicrotome using a Diatome diamond knife, were collected on 200-mesh grids. Images were then recorded using a Philips 400T transmission electron microscope operated at 80,000 eV.
SMARD.
Analysis of the branching in the newly replicated DNA molecules of KSHV was performed by SMARD as described previously (37). Exponentially growing cells were treated with NaB-TPA to induce lytic replication and sequentially labeled with 30 μM IdU (Sigma-Aldrich) at 37°C for 4 h. After 4 h, the cells were collected by centrifugation (2,000 rpm for 5 min at room temperature) and resuspended in PBS (1 × 106 cells per ml) and molten 1% InCert agarose (Lonza Rockland, Inc., Rockland, ME, USA) in PBS (1:1 [vol/vol]). Agarose gel plugs containing DNA were made using a cold plastic mold with 0.5- by 0.2-cm wells with a depth of 0.9 cm, allowed to solidify on ice for 30 min, and lysed in lysis buffer (1% n-lauroylsarcosine [Sigma-Aldrich], 0.5 M EDTA, and 20 mg/ml proteinase K) for 72 h at 50°C. Subsequently, the digested plugs were rinsed with 1× TE and treated with PMSF (Sigma-Aldrich) to remove any residual proteinase K. Next, the plugs were washed with predigestion buffer (10 mM MgCl2 and 10 mM Tris-HCl [pH 8.0]) and digested with 70 units of PmeI (New England Biolabs Inc.) at 37°C overnight to linearize the KSHV genome. The digested gel plugs were rinsed twice with TE, cast into a 0.7% SeaPlaque GTG agarose gel (Lonza Rockland, Inc.), resolved by PFGE, blotted onto a Hybond XL membrane (Amersham, Inc.), and hybridized with a 32P-labeled KSHV TR-specific probe and lambda monocut (molecular weight marker [NEB]). DNA isolation and processing for SMARD were done by melting the gel slices from the appropriate region in the PFGE gel at 72°C for 20 min and digesting the melted DNA with GELase enzyme (Epicentre Biotechnologies, Madison, WI; 1 unit per 50 μl of agarose suspension), followed by incubation of the GELase-DNA-agarose mixture at 45°C for 6 h. The resulting DNA was stretched on 3-aminopropyltriethoxysilane (Sigma-Aldrich)-coated glass slides. After stretching, the DNA was denatured in alkaline denaturing buffer (0.1 N NaOH-70% ethanol and 0.1% β-mercaptoethanol) and fixed with 0.5% glutaraldehyde. The slides containing the DNA were hybridized overnight with biotinylated probes (represented as blue bars on the locus map of the KSHV genome as shown in the top panel of Fig. 3D). Subsequently, the slides were rinsed in 2× SSC-1% SDS, washed in 40% formamide solution containing 2× SSC at 45°C for 5 min, and rinsed in 2× SSC–0.1% IGEPAL CA-630. Following several detergent rinses (4 times in 4× SSC–0.1% IGEPAL CA-630), the slides were blocked with 1% bovine serum albumin (BSA) for at least 20 min and treated with Neutravidin Alexa Fluor 350 (Life Technologies Inc.) for 20 min. The slides were rinsed with PBS containing 0.03% IGEPAL CA-630, treated with biotinylated anti-avidin D (Vector Laboratories, Inc.) for 20 min, and rinsed again. The slides were then treated with Neutravidin Alexa Fluor 350 for 20 min and rinsed again, as described above, followed by incubation with mouse anti-IdU monoclonal antibody and biotinylated anti-avidin D for 1 h. Halogenated nucleotides were detected by incubating the slides with Neutravidin Alexa Fluor 350 and Alexa Fluor 594 goat anti-mouse IgG (Invitrogen Molecular Probes) for 1 h. The coverslips were mounted with ProLong gold antifade reagent (Life Technologies Inc.) after a final PBS and IGEPAL CA-630 wash. Fluorescence microscopy was carried out using a Zeiss microscope to capture the individual DNA molecules.
Gardella and pulsed-field gel electrophoresis.
Gardella gel electrophoresis of 72-hpi cells was performed as described previously with slight modifications (55). Briefly, equal numbers (1 × 106 cells/ml) of cells were harvested, washed with 1× PBS, and resuspended in 1× PBS (molten 1% InCert agarose in PBS; 1:1 [vol/vol]; Lonza Rockland, Inc., Rockland, ME, USA). Agarose gel plugs were made by pipetting the cell-agarose mixture into a cold plastic mold with 0.5- by 0.2-cm wells with a depth of 0.9 cm. The gel plugs were allowed to solidify on ice for 30 min and lysed in lysis buffer (1% n-lauroylsarcosine [Sigma-Aldrich, St. Louis, MO, USA], 0.5 M EDTA, and 20 mg/ml proteinase K) for 48 h at 50°C. The proteinase K-digested plugs were then rinsed in 1× TE at 50°C (5 times; 1 h each time) to remove detergent and proteinase K and stored at 4°C. Restriction enzyme digestion was carried out by incubating the lysed and rinsed plugs in enzyme digestion buffer and BamHI (New England BioLabs Inc.) overnight at 4°C and then for 4 to 6 h at 37°C. The BamHI-digested plugs were rinsed with 1× TE and sealed into a 0.8% agarose gel made in 1× Tris-borate-EDTA (TBE). The gel was run in 0.5× TBE (108 V for 30 h), transferred onto a Hybond N+ membrane (Amersham, Inc.), and hybridized with 32P-labeled DNA fragments specific to the KSHV TR. Lambda monocut DNA was also used as a probe for determining the sizes of the marker bands. Autoradiography was used to determine the location of the appropriate DNA segment. For PFGE, agarose gel plugs from 72-hpi cells were lysed and washed as described above for Gardella gel electrophoresis. The equal-size agarose plugs were digested with BamHI at 37°C overnight and loaded onto a 0.8% agarose gel made in 1× TBE to resolve by PFGE (23, 30). The gel was run in 0.5× TBE buffer at 5 V/cm with a linear pulse increase from 1 s to 6 s at 14°C for 14 h with a Bio-Rad Chef DR II pulsed-field electrophoresis system. Lambda PFGE marker (New England Biolabs, Inc.) was used for size determination. After electrophoresis, the resolved gel was stained with ethidium bromide (EtBr), soaked in 0.25 M HCl and 0.4 M NaOH for 15 min each, blotted onto a Hybond N+ membrane (Amersham, Inc.), and hybridized with a 32P-labeled TR probe (to detect the linear KSHV genome).
Statistical analysis.
Statistical analyses were performed with Prism 7.0 software (GraphPad Inc.), and the P values (significance) were calculated using an unpaired t test.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (AI105000).
We thank Britt A. Glaunsinger (UC, Berkeley) for the generous gift of polyclonal rabbit SOX antisera. We also thank Sally A. DuPré (University of Nevada, Reno) for assistance with the flow cytometry.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Chang Y, Moore P. 2014. Twenty years of KSHV. Viruses 6:4258–4264. doi: 10.3390/v6114258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalt I, Masa S-R, Sarid R. 2009. Linking the Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) to human malignancies, 471:387–407. Mol Microbiol doi: 10.1007/978-1-59745-416-2_19. [DOI] [PubMed] [Google Scholar]
- 4.Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer 10:707. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polizzotto MN, Uldrick TS, Hu D, Yarchoan R. 2012. Clinical manifestations of Kaposi sarcoma herpesvirus lytic activation: multicentric Castleman disease (KSHV-MCD) and the KSHV inflammatory cytokine syndrome. Front Microbiol 3:73. doi: 10.3389/fmicb.2012.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verma SC, Robertson ES. 2003. Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol Lett 222:155–163. doi: 10.1016/S0378-1097(03)00261-1. [DOI] [PubMed] [Google Scholar]
- 7.Renne R, Lagunoff M, Zhong W, Ganem D. 1996. The size and conformation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J Virol 70:8151–8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russo JJ, Bohenzky RA, Chien M-C, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U S A 93:14862. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye F, Lei X, Gao SJ. 2011. Mechanisms of Kaposi's sarcoma-associated herpesvirus latency and reactivation. Adv Virol 2011:1. doi: 10.1155/2011/193860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mettenleiter TC. 2002. Herpesvirus assembly and egress. J Virol 76:1537–1547. doi: 10.1128/JVI.76.4.1537-1547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aneja KK, Yuan Y. 2017. Reactivation and lytic replication of Kaposi’s sarcoma-associated herpesvirus: an update. Front Microbiol 8:613. doi: 10.3389/fmicb.2017.0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.AuCoin DP, Colletti KS, Xu Y, Cei SA, Pari GS. 2002. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) contains two functional lytic origins of DNA replication. J Virol 76:7890–7896. doi: 10.1128/JVI.76.15.7890-7896.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sattler C, Steer B, Adler H. 2016. Multiple lytic origins of replication are required for optimal gammaherpesvirus fitness in vitro and in vivo. PLoS Pathog 12:e1005510. doi: 10.1371/journal.ppat.1005510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richner JM, Clyde K, Pezda AC, Cheng BY, Wang T, Kumar GR, Covarrubias S, Coscoy L, Glaunsinger B. 2011. Global mRNA degradation during lytic gammaherpesvirus infection contributes to establishment of viral latency. PLoS Pathog 7:e1002150. doi: 10.1371/journal.ppat.1002150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abernathy E, Clyde K, Yeasmin R, Krug LT, Burlingame A, Coscoy L, Glaunsinger B. 2014. Gammaherpesviral gene expression and virion composition are broadly controlled by accelerated mRNA degradation. PLoS Pathog 10:e1003882. doi: 10.1371/journal.ppat.1003882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glaunsinger B, Ganem D. 2004. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol Cell 13:713–723. doi: 10.1016/S1097-2765(04)00091-7. [DOI] [PubMed] [Google Scholar]
- 17.Glaunsinger B, Chavez L, Ganem D. 2005. The exonuclease and host shutoff functions of the SOX protein of Kaposi's sarcoma-associated herpesvirus are genetically separable. J Virol 79:7396–7401. doi: 10.1128/JVI.79.12.7396-7401.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henderson JO, Ball-Goodrich LJ, Parris DS. 1998. Structure–function analysis of the herpes simplex virus type 1 UL12 gene: correlation of deoxyribonuclease activity in vitro with replication function. Virology 243:247–259. doi: 10.1006/viro.1998.9054. [DOI] [PubMed] [Google Scholar]
- 19.Grady LM, Szczepaniak R, Murelli RP, Masaoka T, Le Grice SFJ, Wright DL, Weller SK. 2017. The exonuclease activity of HSV-1 UL12 is required for the production of viral DNA that can be packaged to produce infectious virus. J Virol doi: 10.1128/jvi.01380-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuchta AL, Parikh H, Zhu Y, Kellogg GE, Parris DS, McVoy MA. 2012. Structural modelling and mutagenesis of human cytomegalovirus alkaline nuclease UL98. J Gen Virol 93:130–138. doi: 10.1099/vir.0.034876-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheaffer AK, Weinheimer SP, Tenney DJ. 1997. The human cytomegalovirus UL98 gene encodes the conserved herpesvirus alkaline nuclease. J Gen Virol 78:2953–2961. doi: 10.1099/0022-1317-78-11-2953. [DOI] [PubMed] [Google Scholar]
- 22.Stolzenberg MC, Ooka T. 1990. Purification and properties of Epstein-Barr virus DNase expressed in Escherichia coli. J Virol 64:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feederle R, Bannert H, Lips H, Muller-Lantzsch N, Delecluse HJ. 2009. The Epstein-Barr virus alkaline exonuclease BGLF5 serves pleiotropic functions in virus replication. J Virol 83:4952–4962. doi: 10.1128/JVI.00170-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Covarrubias S, Richner JM, Clyde K, Lee YJ, Glaunsinger BA. 2009. Host shutoff is a conserved phenotype of gammaherpesvirus infection and is orchestrated exclusively from the cytoplasm. J Virol 83:9554–9566. doi: 10.1128/JVI.01051-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dahlroth SL, Gurmu D, Schmitzberger F, Engman H, Haas J, Erlandsen H, Nordlund P. 2009. Crystal structure of the shutoff and exonuclease protein from the oncogenic Kaposi's sarcoma-associated herpesvirus. FEBS j 276:6636–6645. doi: 10.1111/j.1742-4658.2009.07374.x. [DOI] [PubMed] [Google Scholar]
- 26.Mendez AS, Vogt C, Bohne J, Glaunsinger BA. 2018. Site specific target binding controls RNA cleavage efficiency by the Kaposi's sarcoma-associated herpesvirus endonuclease SOX. Nucleic Acids Res 46:11968–11979. doi: 10.1093/nar/gky932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee H, Patschull AOM, Bagneris C, Ryan H, Sanderson CM, Ebrahimi B, Nobeli I, Barrett TE. 2017. KSHV SOX mediated host shutoff: the molecular mechanism underlying mRNA transcript processing. Nucleic Acids Res 45:4756–4767. doi: 10.1093/nar/gkw1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bagnéris C, Briggs LC, Savva R, Ebrahimi B, Barrett TE. 2011. Crystal structure of a KSHV–SOX–DNA complex: insights into the molecular mechanisms underlying DNase activity and host shutoff. Nucleic Acids Res 39:5744–5756. doi: 10.1093/nar/gkr111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldstein JN, Weller SK. 1998. In vitro processing of herpes simplex virus type 1 DNA replication intermediates by the viral alkaline nuclease, UL12. J Virol 72:8772–8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez R, Sarisky RT, Weber PC, Weller SK. 1996. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J Virol 70:2075–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36–e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tandon R, Mocarski ES, Conway JF. 2015. The A, B, Cs of herpesvirus capsids. Viruses 7:899–914. doi: 10.3390/v7030899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown JC, Newcomb WW. 2011. Herpesvirus capsid assembly: insights from structural analysis. Curr Opin Virol 1:142–149. doi: 10.1016/j.coviro.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henaff D, Rémillard-Labrosse G, Loret S, Lippé R. 2013. Analysis of the early steps of herpes simplex virus 1 capsid tegumentation. J Virol 87:4895–4906. doi: 10.1128/JVI.03292-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muylaert I, Tang KW, Elias P. 2011. Replication and recombination of herpes simplex virus DNA. J Biol Chem 286:15619–15624. doi: 10.1074/jbc.R111.233981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verma SC, Lu J, Cai Q, Kosiyatrakul S, McDowell ME, Schildkraut CL, Robertson ES. 2011. Single molecule analysis of replicated DNA reveals the usage of multiple KSHV genome regions for latent replication. PLoS Pathog 7:e1002365. doi: 10.1371/journal.ppat.1002365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norio P, Schildkraut CL. 2001. Visualization of DNA replication on individual Epstein-Barr virus episomes. Science 294:2361–2364. doi: 10.1126/science.1064603. [DOI] [PubMed] [Google Scholar]
- 39.Norio P, Schildkraut CL. 2004. Plasticity of DNA replication initiation in Epstein-Barr virus episomes. PLoS Biol 2:e152. doi: 10.1371/journal.pbio.0020152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. 2009. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guan Z, Hughes CM, Kosiyatrakul S, Norio P, Sen R, Fiering S, Allis CD, Bouhassira EE, Schildkraut CL. 2009. Decreased replication origin activity in temporal transition regions. J Cell Biol 187:623–635. doi: 10.1083/jcb.200905144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feederle R, Mehl-Lautscham AM, Bannert H, Delecluse H-J. 2009. The Epstein-Barr virus protein kinase BGLF4 and the exonuclease BGLF5 have opposite effects on the regulation of viral protein production. J Virol 83:10877–10891. doi: 10.1128/JVI.00525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vlazny DA, Kwong A, Frenkel N. 1982. Site-specific cleavage/packaging of herpes simplex virus DNA and the selective maturation of nucleocapsids containing full-length viral DNA. Proc Natl Acad Sci U S A 79:1423–1427. doi: 10.1073/pnas.79.5.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strahan RC, McDowell-Sargent M, Uppal T, Purushothaman P, Verma SC. 2017. KSHV encoded ORF59 modulates histone arginine methylation of the viral genome to promote viral reactivation. PLoS Pathog 13:e1006482. doi: 10.1371/journal.ppat.1006482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rossetto CC, Pari G. 2012. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog 8:e1002680. doi: 10.1371/journal.ppat.1002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McDowell ME, Purushothaman P, Rossetto CC, Pari GS, Verma SC. 2013. Phosphorylation of Kaposi's sarcoma-associated herpesvirus processivity factor ORF59 by a viral kinase modulates its ability to associate with RTA and oriLyt. J Virol 87:8038–8052. doi: 10.1128/JVI.03460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 48.Verma SC, Lan K, Choudhuri T, Cotter MA, Robertson ES. 2007. An autonomous replicating element within the KSHV genome. Cell Host Microbe 2:106–118. doi: 10.1016/j.chom.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purushothaman P, Thakker S, Verma SC. 2015. Transcriptome analysis of Kaposi's sarcoma-associated herpesvirus during de novo primary infection of human B and endothelial cells. J Virol 89:3093–3111. doi: 10.1128/JVI.02507-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Full F, Jungnickl D, Reuter N, Bogner E, Brulois K, Scholz B, Sturzl M, Myoung J, Jung JU, Stamminger T, Ensser A. 2014. Kaposi's sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLoS Pathog 10:e1003863. doi: 10.1371/journal.ppat.1003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosner M, Hengstschlager M. 2008. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum Mol Genet 17:2934–2948. doi: 10.1093/hmg/ddn192. [DOI] [PubMed] [Google Scholar]
- 52.Hodge PD, Stow ND. 2001. Effects of mutations within the herpes simplex virus type 1 DNA encapsidation signal on packaging efficiency. J Virol 75:8977–8986. doi: 10.1128/JVI.75.19.8977-8986.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mascorro JA, Bozzola JJ. 2007. Processing biological tissues for ultrastructural study. Methods Mol Biol 369:19–34. doi: 10.1007/978-1-59745-294-6_2. [DOI] [PubMed] [Google Scholar]
- 54.Mingo RM, Han J, Newcomb WW, Brown JC. 2012. Replication of herpes simplex virus: egress of progeny virus at specialized cell membrane sites. J Virol 86:7084–7097. doi: 10.1128/JVI.00463-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gardella T, Medveczky P, Sairenji T, Mulder C. 1984. Detection of circular and linear herpesvirus DNA molecules in mammalian cells by gel electrophoresis. J Virol 50:248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]