

Abstract
Purpose
Following automated variant calling, manual review of aligned read sequences is required to identify a high-quality list of somatic variants. Despite widespread use in analyzing sequence data, methods to standardize manual review have not been described, resulting in high inter- and intralab variability.
Methods
This manual review standard operating procedure (SOP) consists of methods to annotate variants with four different calls and 19 tags. The calls indicate a reviewer’s confidence in each variant and the tags indicate commonly observed sequencing patterns and artifacts that inform the manual review call. Four individuals were asked to classify variants prior to, and after, reading the SOP and accuracy was assessed by comparing reviewer calls with orthogonal validation sequencing.
Results
After reading the SOP, average accuracy in somatic variant identification increased by 16.7% (p value = 0.0298) and average interreviewer agreement increased by 12.7% (p value < 0.001). Manual review conducted after reading the SOP did not significantly increase reviewer time.
Conclusion
This SOP supports and enhances manual somatic variant detection by improving reviewer accuracy while reducing the interreviewer variability for variant calling and annotation.
Keywords: somatic variant refinement, manual review
Introduction
Large genome centers, such as the McDonnell Genome Institute, use a wide variety of sequencing workflows. Typically, extracted nucleic acid is subjected to fragmentation; size selection; KAPA (Wilmington, MA), Swift (Ann Arbor, MI), IDT (San Jose, CA), or Illumina (San Diego, CA) library preparation protocols (end-repair, tailing, ligation, amplification, etc.); NimbleGen (Basel, Switzerland) or IDT custom/exome capture; and subsequent sequencing via Illumina HiSeq 2500/4000 or Novaseq 6000. The sequencing workflow typically follows methods described by Griffith et al.1 Subsequently, the bioinformatics pipeline requires alignment to the reference genome (GRCh37/38) via Burrows–Wheeler Aligner (BWA)2 or BWA-MEM and postprocessing of aligned sequencing reads. Postprocessing requires deduplication of reads via Picard3 and automated somatic variant calling using the intersection or union of Mutect,4 SomaticSniper,5 Strelka,6 VarScan2,7 or others. A multicaller approach is used to identify a preliminary list of high-quality somatic variants from aligned sequence data.8–10 The bioinformatics pipeline can be implemented using the Genome Modeling System.11
Automated pipelines can identify and filter many false variant calls that result from sequencing errors, misalignment of reads, and other factors; however, additional refinement of somatic variants is often required to eliminate variant caller inaccuracies. This additional refinement is critical because inaccurate identification of variants can lead to poor patient management and missed therapeutic opportunities, as outlined in the Association for Molecular Pathology (AMP) guidelines for interpretation and annotation of somatic variation.12,13 Therefore, manual inspection of somatic variants identified by automated variant callers (i.e., manual review) is an important aspect of the sequencing analysis pipeline and is currently the standard for variant refinement. Manual review allows individuals to incorporate information not considered by automated variant callers. For example, a trained eye can discern misclassifications attributable to overlapping errors at the ends of sequence reads, preferential amplification of smaller fragments, or poor alignment in areas of low complexity. Due to computational limitations, automated methods for variant refinement are in early stages of development and manual review remains integral to variant identification workflows.16
Despite extensive use of manual review in clinical diagnostic and molecular pathology settings,17–19 somatic variant refinement strategies are often unstated or only briefly mentioned in studies that report postprocessing of automated variant calls20–25 Lack of formalized procedures for the sequencing pipeline, and specifically for somatic refinement, permits high levels of inter- and intralab variability and can hinder reproducibility of results.26 Thus, development of a procedure to standardize and systematize somatic variant refinement would improve the overall quality of sequencing analysis pipelines.
Here we present a standard operating procedure (SOP) for manual review of paired tumor/normal samples to help standardize somatic variant refinement. We first detail instructions for downloading and using the publicly available Integrative Genomics Viewer (IGV)14,15 and IGVNavigator (IGVNav) software to properly visualize somatic variants during manual review. We also show that adoption of a standardized method for somatic variant refinement through this manual review SOP improves the accuracy of somatic variant calls and reduces overall interreviewer variability.
Materials and methods
Setting up manual review using IGV
The Integrative Genomics Viewer (IGV) is a high-performance genomic data visualization tool. This SOP reviews IGV (v2.4.8) components that can be used to conduct manual review of variants identified by automated somatic variant callers. While we have chosen IGV to develop our SOP, many of the following concepts are applicable to other genomic viewers.27–29 The IGV desktop application is available for all major operating systems.
The IGV interface is composed of three main panels: (1) Genome Ruler, (2) Data Tracks, and (3) Genome Features (Fig. 1). The Genome Ruler provides navigation features to center a genomic locus of interest. A dropdown menu provides reference genome selection, the variant coordinates show the current field of view, the zoom buttons expand/contract the field of view, and other buttons provide additional display and navigation control. Within the Data Tracks section, each horizontal track represents one experiment, sample, or annotation. In Fig. 1, a normal BAM track and a tumor BAM track are loaded. For BAM files, each data track consists of a coverage track and individual read alignments. Reads ideally represent a single originating molecule that was sequenced and aligned to a reference. In default settings, sequenced bases that disagree with the aligned reference sequence are highlighted. The Genome Features section provides reference information that can be used to supplement manual review. The reference DNA and protein sequence tracks are loaded by default. Optionally loaded tracks from the IGV server will typically appear in the Genome Features section.
Fig. 1.
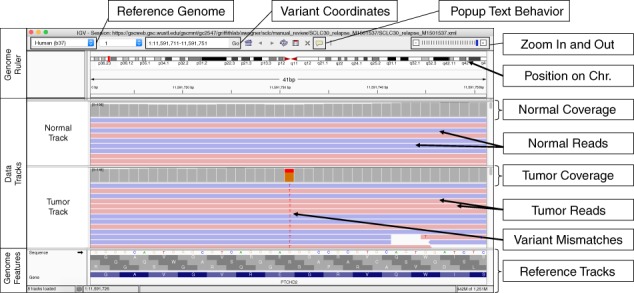
Example of the Integrative Genomics Viewer (IGV) interface with associated features relevant to manual review. The IGV interface is divided into three parts. The Genome Ruler details information about the genome assembly being visualized (Reference Genome), the coordinates currently being visualized (Variant Coordinates), and other navigation/display controls (e.g., Popup Text Behavior, Zoom In and Out, etc.). In this example, a portion of human chromosome 1 (build 37) is shown. The central section of IGV displays Data Tracks. In this case, short read DNA alignment data (e.g., BAM files) are shown for normal and tumor samples and are colored by read strand. Mismatches with the reference genome are highlighted by base: adenine (green), cytosine (blue), guanine (orange), and thymine (red). Coverage tracks summarize the total read depth at each base position. The Genome Features section shows the reference sequence itself, the amino acids for the three possible reading frames, and the gene associated with this locus (PTCHD2 in this example). The default gene track available with IGV is shown (RefSeq). Many other data formats and sources can be loaded as data tracks or genome features.
IGV supports a variety of input files for sequence data visualization. The File dropdown menu details the various supported input files. Indexed BAMs can be efficiently accessed from a local file system. Alternatively, the Load from URL option permits direct URL input from a web service. The Load from Server option downloads tracks from supported data sets (e.g., the Cancer Genome Atlas, Ensembl, etc.).
Setting up manual review using IGVNav
IGVNav software (a Python applet/plugin for IGV), announced here, is available for download under an open access license (GNU) from GitHub (https://github.com/griffithlab/igvnav). When initiated, the user is prompted to open an input file for manual review. The input file is a tab delimited, 0- or 1-based BED-like file with the following columns: chromosome, start coordinate, stop coordinate, reference allele, variant allele, call, tags, and notes. For variants that have not yet been manually reviewed, the call, tags, and notes columns should be blank (Fig. 2b). IGVNav features are shown in Fig. 2a. The navigation bar permits movement through the input variant list. The “S” button sorts alignments by base so that variants appear at the tops of data tracks. Below the navigation bar is the current variant being visualized and the total number of variants in the input file. Editing this section and selecting the Go button will navigate to a specific variant of interest. The three horizontal bars display coordinate information for the current variant. The first bar details the chromosome, start, and stop position; the second bar shows the reference allele; and the third bar shows the variant allele. The Call section allows the manual reviewer to select one of the following: somatic (S) (Fig. S1), germline (G) (Fig. S2), ambiguous (A) (Fig. S3), or fail (F) (Fig. S4). The Tags section allows manual reviewers to annotate variants with commonly observed sequencing patterns. Tags can be used for any call (S, G, A, or F); however, they are especially important for ambiguous and fail calls to indicate the call rationale. Descriptions of calls and tags can be found in Table 1. The IGVNav interface also contains a Notes section, which allows for free text. At any point during a manual review session, the calls, tags, and notes can be saved to the original input file using the Save button (Fig. 2c).
Fig. 2.

Example of the Integrative Genomics Viewer Navigator (IGVNav) interface, associated features, and input/output files. a IGVNav is a simple plugin for IGV that provides a separate application window for recording results of manual review. The 1-Base? button can be selected for 1-base input files (default is 0-base). The “S” button will sort the read sequences in the data tracks so that mismatches appear at the top. The navigation bar displays variant information and allows for movement between variants. The Call, Tags, and Notes sections allow manual reviewers to annotate variants (Table 1), which is reflected in the output file. The Save button is used to update the output file. b An IGVNav input file consists of a header line and data for the first five columns (chromosome [chr], start coordinate [start], stop coordinate [stop], reference allele [ref], and variant allele [var]). Each line represents a variant that will be individually visualized using IGV. c During manual review, the input file is updated by clicking on the Save button. This will print the call, tags, and notes associated with individual variants to the original input file.
Table 1.
List and description of Integrative Genomics Viewer Navigator (IGVNav) calls and tags used to annotate variants in order of appearance on the IGVNav interface with associated supplementary figure number.
| Call Name | Call | Description | Figure |
|---|---|---|---|
| Somatic | S | Variant has sufficient support in the tumor with absence of obvious sequencing artifacts | S1 |
| Germline | G | Variant that has sufficient support in the normal sample beyond what is considered attributable to tumor contamination of the normal | S2 |
| Ambiguous | A | Variant does not meet acceptable criteria for any other label | S3 |
| Fail | F | Variant with low variant support and/or reads that indicate sequencing artifacts | S4 |
| Tag Name | Tag | Description | Figure |
|---|---|---|---|
| Adjacent Indel | AI | Variant is attributable to misalignment caused by a nearby insertion or deletion | S16 |
| Ambiguous Other | AO | Variant is surrounded by inconclusive genomic features that cannot be explained by other tags | S22 |
| Directional | D | Variant is only (or mostly) found on reads in the same direction (positive or negative) | S5 |
| Dinucleotide repeat | DN | Variant is adjacent to a region in the reference genome that has two alternating nucleotides (e.g., TGTGTG…) | S20 |
| End of reads | E | Variant is only seen close to the end (within 30 base pairs) of variant-supporting reads | S18 |
| High Discrepancy Region | HDR | Variant is supported by reads that have other recurrent mismatches across the track and in multiple tracks | S12 |
| Low Count Normal | LCN | Variant has inadequate coverage in the normal track, thus preventing effective comparison with the tumor track | S7 |
| Low Count Tumor | LCT | Variant has inadequate coverage in the tumor track, thus preventing effective comparison with the normal track | S8 |
| Low Mapping quality | LM | Variant is mostly supported by reads that have low mapping quality | S13 |
| Low Variant Frequency | LVF | Variant has low variant allele frequency (VAF) samples | S10 |
| Multiple Mismatches | MM | Variant is supported by reads that have other mismatched base pairs | S11 |
| Mononucleotide repeat | MN | Variant is adjacent to a region in the reference genome that has a single-nucleotide repeat (e.g., AAAAAA…) | S19 |
| Multiple Variants | MV | Variant locus has read support for three or more alleles | S9 |
| No Count Normal | NCN | Variant has no coverage in the normal track, thus preventing effective comparison with the tumor track | S6 |
| Short Inserts | SI | Variant is found mostly on small nucleic acid fragments whereby sequencing from each end results in overlapping reads | S15 |
| Short Inserts Only | SIO | Variant is exclusively found on small nucleic acid fragments such that sequencing from each end results in overlapping reads | S15 |
| Same Start End | SSE | Variant is only observed in reads that start and stop at the same positions | S17 |
| Tumor in Normal | TN | Variant has read support in the normal track | S14 |
| Tandem Repeat | TR | Variant is adjacent to a region in the reference genome that has three or more alternating nucleotides (e.g., GTGGTGGTG…) | S21 |
Step-by-step guide: setting up IGV and IGVNav for manual review
Manual review setup involves six discrete steps (Fig. 3a). First, an IGV session should be opened and the appropriate reference genome should be selected/loaded. The reference genome species and build must match those used for alignment. Second, the IGV session should be populated with data tracks. When tumor DNA, normal DNA, and other DNA or RNA read alignments are available, they can all be loaded within a single IGV session. Step 3, optionally, allows for population of additional tracks that can assist in manual review. Step 4, also optional, recommends that tracks be colored by reads (right click on data track → Color alignments by → read strand) and the centered locus is visualized (View → Preferences → Alignments → Show center line). After initial setup of IGV, step 5 requires opening IGVNav and step 6 requires loading the manual review input file.
Fig. 3.

Step-by-step instructions for setting up and executing somatic variant refinement via manual review. a Method for setting up Integrative Genomics Viewer (IGV) and Integrative Genomics Viewer Navigator (IGVNav) for manual review. b Method for analyzing each variant during manual review.
Step-by-step guide: performing manual review
After initial setup, seven additional steps must be followed to properly review each variant (Fig. 3b). First, the variant must be located by either using the navigation bar in IGVNav or by manually inserting coordinates into the IGV Genome Ruler. Variant-supporting reads can be visualized at the top of each data track by clicking the “S” button in IGVNav, or by using IGV options (right click on data track → Sort alignments by → base).
Step 2 evaluates the quantity of variant support. Selecting the locus of interest within the coverage track will ascertain strand direction, total coverage, and variant allele frequencies (VAFs). Strand direction might indicate a Directional (D) artifact (Fig. S5). Total coverage might indicate No Count Normal (NCN) (Fig. S6), Low Count Normal (LCN) (Fig. S7), or Low Count Tumor (LCT) (Fig. S8). VAFs might indicate Multiple Variants (MV) (Fig. S9) or Low Variant Frequency (LVF) (Fig. S10).
Step 3 evaluates the quality of variant support. Directly visualizing reads identifies Multiple Mismatches (MM) (Fig. S11) or High Discrepancy Regions (HDR) (Fig. S12). Reads that are translucent or transparent indicate Low Mapping (LM) quality (Fig. S13). Mapping quality information can be viewed by clicking on the read in question and viewing the Mapping section (e.g., Mapping = Primary @MAPQ 0). Base quality can also be evaluated in this popup in the Base section (e.g., Base = A @ QV 41). Similar to mapping quality, base quality is reflected by the transparency of the letter. The final part of step 3 is to ensure lack of variant support in normal track(s), (i.e., Tumor in Normal [TN] [Fig. S14]).
Step 4 requires identifying sequencing artifacts. First, toggle between View as pairs (right click each data track → View as pairs) to visualize Short Inserts (SI/SIO) (Fig. S15). Then use the zoom in (“+”) and zoom out (“–”) buttons on the Genome Ruler to identify Adjacent Indels (AI) (Fig. S16), High Discrepancy Regions (HDR) (Fig. S12), exclusive support from reads with Same Start/Ends (SSE) (Fig. S17), and support only at the Ends of reads (E) (Fig. S18). Finally, evaluating the reference sequence elucidates low complexity regions such as Mononucleotide repeats (MN) (Fig. S19), Dinucleotide repeats (DN) (Fig. S20), and Tandem Repeats (TR) (Fig. S21). If reviewer concerns cannot be described with previously defined tags, the reviewer can use the Ambiguous Other (AO) tag and comment in the Notes section (Fig. S22).
Steps 5 through 7 require synthesizing available information to manually review the variant. This involves selecting a call, tag(s), and optionally, providing free text in the Notes section of IGVNav.
Validation of the manual review SOP
We assessed whether the manual review SOP improved accuracy of somatic variant refinement using an acute myeloid leukemia (AML) case with genome sequence data, extensive variant calling, and orthogonal validation (Fig. 4).1 To emulate normal conditions for genome sequencing manual review, we downsampled the unaligned BAM files to 30× and 50× coverage for normal and tumor samples, respectively. Sequencing data was aligned to the reference genome (GRCh38) and variants were detected using the McDonnell Genome Institute’s cancer genomics workflow.30 Using the union of MuTect4 and VarScan,7 143,042 potential variants were identified. A subset of these variants (n = 5,090) had orthogonal validation sequencing at ~1,000× coverage. Coordinates from the platinum variant list, published by Griffith et al., were lifted over to GRCh38 and used to label 1,186 variants as true positives (TPs). The remaining 3,904 variants were labeled as false positives (FPs). A random subset of 300 variants (150 TPs; 150 FPs) were selected for manual review. After receiving basic instruction on how to set up IGV and call variants using the required four classes (S, G, A, F), blinded novice reviewers manually reviewed 200 variants in two batches of 100 using the downsampled genome sequencing BAM files. Subsequently, the reviewers read the SOP and reviewed two more batches of 100 variants. The final batch of 100 variants were among the 200 assessed prior to reading the SOP. Accuracy was assessed by comparing the manual review calls with the orthogonal validation labels. Interreviewer variability was calculated by developing a correlation matrix for all four calls across the four reviewers for each variant. Correlation for identical calls was 1, correlation for conflicting calls (e.g., fail and somatic) was 0, and correlation for semiconflicting calls (e.g., fail and ambiguous) was 0.5 (Table S1). The sum of the matrix was divided by the maximum possible score (i.e., 16 points) to create a relative metric for interreviewer agreement. The average agreement scores from before and after reading the SOP were compared. To determine if reviewers were using tags appropriately, tags assigned to false positives by novice reviewers were compared with gold standard tags created by expert reviewers for false positives reviewed after reading the SOP (Fig. 4a).
Fig. 4.

Validation of the manual review standard operating procedure (SOP). a Sequencing data from an acute myeloid leukemia (AML) case was used to test the impact of the SOP on accurately identifying somatic variants. A total of 300 variants that had genome sequencing and orthogonal sequencing were identified for the experiment. Four novice reviewers assessed 200 variants prior to and after reading the SOP to determine improvement in accuracy, reduction in interreviewer variability, change in reviewer time per variant, and appropriate use of tags. b Reviewer accuracy was assessed before and after reading the SOP. The bar plot shows accuracy stratified by reviewer and the box plot shows the reviewers’ cumulative median accuracy. c Box plot showing the median interreviewer agreement before and after reading the SOP. Agreement for each variant was calculated by assessing the correlation between the four reviewer calls using a correlation matrix as described in the Methods. d Box plot showing the median time required to conduct manual review before and after reading the SOP. e Frequency diagram showing the number of reviewers that correctly annotated false positive variants with gold standard tags, parsed by tag. AI Adjacent Indel, D Directional, DN Dinucleotide repeat, E End of reads, HDR High Discrepancy Region, LM Low Mapping, LVF Low Variant Frequency, MM Multiple Mismatches, MN Mononucleotide repeat, MV Multiple Variants, SSE Same Start End, TN Tumor in Normal, TR Tandem Repeat.
Results
Annotations observed during manual review
Screenshots were created for the 22 annotations used during manual review (Figs. S1–S22). The illustrations and comments emphasize IGV features that highlight sequencing patterns, describe cautions for challenging tumor types, and indicate deviations from standard protocol.
Analysis of four variant calls
This SOP and IGVNav software support four classes of variant calls: somatic (S), germline (G), ambiguous (A), and fail (F) (Table 1). For a call to be labeled as somatic, the variant must have sufficient read data support in the tumor with absence of obvious sequence artifacts (Fig. S1). Conversely, a germline variant is an alteration that has sufficient support in the normal, beyond what can be attributable to tumor contamination (Fig. S2). Barring inadequate sequencing depth and/or impact from copy-number alterations, the VAF for germline variants should be near 100% or 50% in both the normal and tumor tracks, indicative of homozygosity or heterozygosity, respectively. Ambiguous calls should be made when there is insufficient evidence to confidently label a variant with any other call class. The example in Fig. S3 shows no support for the variant in the normal track and 14 reads of support in the tumor. However, most of the reads are on negative strands and some have multiple mismatches. If a reviewer has any residual doubt about failing a variant, then the variant should be labeled ambiguous. To fail a variant, the reviewer must confidently determine that the variant was called because of a sequencing or analysis artifact. For example, Fig. S4 details a variant that was erroneously identified by an automated caller because reads had been aligned to a high discrepancy region.
Analysis of 19 variant tags
It is especially important to annotate fail and ambiguous calls with 1 or more of the 19 tags on the IGVNav interface (Table 1). Each tag represents a sequencing pattern or artifact that is commonly observed during manual review. These patterns can arise during DNA fragmentation, library construction, sequencing, read alignment, or variant calling. Alternatively, some concerns observed during manual review can be caused by simple structural aberrations or more complex issues intrinsic to the tumor being evaluated. Below, we describe how these concerning reads are created within the sequencing pipeline and detail the resulting pattern observed in IGV.
The tumor type and tissue origin can play a role in generating patterns observed during manual review. For example, hematologic tumors or highly metastatic tumors can cause Tumor in Normal (TN) patterns due to the presence of tumor cells in the normal biopsy (Fig. S14). Generally, it is important to characterize the average level of contamination across an individual sample to determine an acceptable threshold for TN. Tumor sample preparation can also impact manual review through sequencing of degraded nucleic acids (e.g., formalin-fixed, paraffin-embedded samples)31 giving rise to Short Inserts (SI) or Short Inserts Only (SIO). When generating paired-end reads, degraded and/or short molecules will produce two sequences that have overlapping alignments. This can exaggerate variant support because most variant callers will consider the overlapping alignments as two independent pieces of evidence, despite representing a single originating DNA fragment (Fig. S15). Short inserts can be visualized in IGV by viewing reads as pairs and looking for horizontal gray bands (representing overlap) in the middle of the paired read alignments.
Additional errors can arise during fragmentation, library construction, and enrichment. DNA quality and quantity, capture reagent balance and efficiency, sample balance in multiplexed preparations, and other factors can impact the uniformity of coverage for a given sample. For example, a selection bias might skew which molecules are amplified/sequenced, resulting in an uneven distribution of sequencing (coverage) across the desired genome space.32 These errors are labeled as No Count Normal (NCN) (Fig. S6), Low Count Normal (LCN) (Fig. S7), and Low Count Tumor (LCT) (Fig. S8). NCN and LCN are defined by no or few reads in the normal tracks and LCT is defined by few reads in the tumor track. Also, given that many real variants have a low VAF, due to tumor heterogeneity or low purity tumors, the combination of Low Variant Frequency (LVF) (Fig. S10) and LCT can prevent a true variant from being confidently called. Our lab has often adopted a minimum VAF threshold of 5% and a coverage threshold of 20 reads for both the tumor and normal tracks. The rationale for the normal track coverage threshold is that if a sequencing artifact is present at a relatively low frequency (<5% occurrence), and if the normal track has <20 reads, it is difficult to confidently rule out the presence of a sequencing artifact. For experiments with higher average coverage, the minimum VAF threshold can be reduced accordingly.
After fragmentation and library preparation, nucleic acids are amplified using polymerase chain reaction (PCR), which can introduce Directional (D) and Same Start/End (SSE) artifacts. Directional artifacts occur when variant support is only apparent on reads in a specific direction (i.e., positive or negative). Typically, this occurs because the sequencing context affects the polymerase in one direction more than the reverse complement (Fig. S5) 33. SSE artifacts occur when a molecule is preferentially amplified and not removed through read deduplication programs.34 This artifact can be confirmed when all variant support reads have the same (or very similar) start and end position after alignment (Fig. S17).
The next step in the pipeline is sequencing. Sequencing errors are defined as nucleotides misread by the sequencing instrument, which can be caused by inefficiencies in sequencing chemistry, technical errors made by the camera system, interference from neighboring clusters, instrument software errors, etc. One type of sequencing error, “dephasing,” occurs when a nucleotide without a proper 3’ -OH blocking group is incorporated or is not properly cleaved. The affected fragment(s) lose synchrony with the cluster, contributing to background noise.35 Ends of reads (E), which occurs when variant support is exclusively found at the end of read sequences (within 30 base pairs), is indicative of a dephasing error (Fig. S18).36 These errors occur with low probability; however, as the read length increases, the summation of errors can pollute the light signal. Because the light signal is used to calculate quality scores, the asynchronous signal should decrease sequence base quality, which may assist in elucidating artifacts caused by dephasing errors.
Many artifacts arise from incorrect alignment of sequence reads to a reference genome. These artifacts include Mononucleotide repeats (MN), Dinucleotide repeats (DN), Tandem Repeats (TR), High Discrepancy Regions (HDR), Low Mapping (LM), Multiple Mismatches (MM), Adjacent Indel (AI), and Multiple Variants (MV). MN (Fig. S19), DN (Fig. S20), and TR (Fig. S21) are attributable to regions of low complexity adjacent to the variant locus. They typically occur when there is a base pair deletion or insertion adjacent to one, two, or greater than two base pair repeats, respectively. HDR, LM, MM, and MV occur when single reads map to multiple and/or incorrect regions. This is typically caused by (1) homologous sequences at multiple loci, (2) highly variable regions between or within individuals (e.g., variable, diversity, and joining (VDJ) regions in immune cells), (3) high error rates in reads, and/or (4) errors in the reference genome. HDRs are apparent when multiple reads contain the same mismatches with the reference genome at various locations (Fig. S12). LM can be determined by looking for translucent reads (Fig. S13). MM is used when variants are supported by reads that disagree with the reference genome at multiple loci across the same read, indicating low sequencing quality or misalignment (Fig. S11). Similarly, MV is defined by read support for three or more different alleles at a given locus, which might indicate poor quality or misaligned reads (Fig. S9). AI is used when a structural variant or a small indel in a repetitive region causes local misalignment and creation of an apparent single-nucleotide variant (SNV)/indel (Fig. S16). Observing these artifacts requires careful scrutiny of the reference genome, base quality, and mapping quality.
In rare instances, if the pre-existing tags cannot adequately annotate a variant, it can be labeled as Ambiguous Other (AO). Given that this tag is nondescriptive, it is recommended to include free text in the Notes section to justify the tag and associated variant call. In the example provided (Fig. S22), the insertion variant shows a low complexity region with increased G/C content that is not contained within a tandem repeat region. This observation can be annotated using the AO tag.
Validation of the manual review SOP
Manual review performed by novice reviewers after reading the SOP improved identification of somatic variants by 16.7% (77.4% vs. 94.1%; p value = 0.0298) (Fig. 4b) and increased the average interreviewer correlation score by 12.7% (80.7 points vs. 93.4 points; p value < 0.0001) (see Methods) (Fig. 4c). The SOP did not significantly impact time required to conduct manual review (Fig. 4d). Additionally, correct use of tags was observed for annotations made after reading the SOP. When evaluating 86 false positives that had 238 tags confirmed by expert reviewers, 143 tags were correctly identified by at least three novice reviewers and only 36 tags were missed by all reviewers (Fig. 4e).
Discussion
Identification and interpretation of variants is crucial for conducting translational research and guiding clinical management of cancer patients.13 In general, implementation of this SOP has improved variant identification consistency, limiting the total number of false positives requiring downstream analysis. Given that variant annotation remains a major bottleneck in translational and clinical research.37,38 reduction in false positives should substantially improve the overall efficiency of lab operations. Therefore, we advocate that others adopt a standardized process for variant refinement such as the SOP presented here.
There are intrinsic limitations associated with manual review that will not be rectified by this SOP. First, manual reviewers have reported reviewer fatigue, especially when evaluating tumors with a high variant burden. Second, despite extensive training, some amount of interreviewer variability will likely remain, especially for ambiguous variants. Third, manual review of variants might change over time as an individual begins to recognize the idiosyncrasies associated with a particular tumor subtype or sequencing platform. Finally, the scope of this SOP is limited to the manual review of somatic SNVs/indels in situations where tumor/normal samples are available; although, many of the aspects of the protocol, including setup and assessment, can be directly applied to other analyses (e.g., structural variant assessment). It is our intent to continuously improve this protocol through subsequent revisions (10.1101/266262). This will include developing an SOP for tumor-only samples, incorporating features that improve somatic variant refinement, and developing machine learning approaches to alleviate manual review burden.
Many of the existing limitations of manual review could be addressed by automating somatic variant refinement. This would further standardize the massively parallel sequencing pipeline and reduce the labor burden required to identify putative somatic variants. Advancements in computational approaches provide an opportunity for the development of such a process.
Electronic supplementary material
Acknowledgements
E.K.B. was supported by the National Cancer Institute (T32GM007200 and U01CA209936). B.J.A. and K.M.C. were supported by the Siteman Cancer Center (T32CA113275). S.J.S. is funded by the National Library of Medicine (NIH NLM R01LM012222 and NIH NLM R01LM012482). A.W. was supported by the National Cancer Institute (NIH NCI F32CA206247). M.G. is funded by the National Human Genome Research Institute (NIH NHGRI R00HG007940). O.L.G. is funded by the National Cancer Institute (NIH NCI K22CA188163 and NIH NCI U01CA209936). Additional funds were provided by the Washington University School of Medicine. This research would not have been possible without exemplary work performed by Michael McLellan, Heather Schmidt, Jennifer Hodges, Yat Tang, Li Ding, and others at the McDonnell Genome Institute to develop the original Medical Genomics/Manual Review Guidelines.
Disclosure
The authors declare no conflicts of interest.
Contributor Information
Malachi Griffith, Email: mgriffit@wustl.edu.
Obi L. Griffith, Email: obigriffith@wustl.edu.
Electronic supplementary material
The online version of this article (10.1038/s41436-018-0278-z) contains supplementary material, which is available to authorized users.
References
- 1.Griffith M, Miller CA, Griffith OL, Krysiak K, Skidmore ZL, Ramu A, et al. Optimizing cancer genome sequencing and analysis. Cell Syst. 2015;1:210–223. doi: 10.1016/j.cels.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broad Institute. Picard tools. http://broadinstitute.github.io/picard/. Accessed 28 June 2018.
- 4.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 7.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krøigård AB, Thomassen M, Lænkholm AV, Kruse TA, Larsen MJ. Evaluation of nine somatic variant callers for detection of somatic mutations in exome and targeted deep sequencing data. PLoS ONE. 2016;11:e0151664. doi: 10.1371/journal.pone.0151664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai L, Yuan W, Zhang Z, He L, Chou KC. In-depth comparison of somatic point mutation callers based on different tumor next-generation sequencing depth data. Sci Rep. 2016;6:36540. doi: 10.1038/srep36540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Callari M, Sammut SJ, De Mattos-Arruda L, Bruna A, Rueda OM, Chin SF, et al. Intersect-then-combine approach: improving the performance of somatic variant calling in whole exome sequencing data using multiple aligners and callers. Genome Med. 2017;9:35. doi: 10.1186/s13073-017-0425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffith M, Griffith OL, Smith SM, Ramu A, Callaway MB, Brummett AM, et al. Genome modeling system: a knowledge management platform for genomics. PLoS Comput Biol. 2015;11:e1004274. doi: 10.1371/journal.pcbi.1004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roy S, Coldren C, Karunamurthy A, Kip NS, Klee EW, Lincoln SE, et al. Standards and guidelines for validating next-generation sequencing bioinformatics pipelines. J Mol Diagn. 2018;20:4–27. doi: 10.1016/j.jmoldx.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23. doi: 10.1016/j.jmoldx.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, Mesirov JP. Variant review with the Integrative Genomics Viewer. Cancer Res. 2017;77:e31–e34. doi: 10.1158/0008-5472.CAN-17-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2012;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mardis ER. The 1,000 genome, the 100,000 analysis? Genome Med. 2010;2:84. doi: 10.1186/gm205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strom SP. Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol Med. 2016;13:3–11. doi: 10.20892/j.issn.2095-3941.2016.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sukhai MA, Craddock KJ, Thomas M, Hansen AR, Zhang T, Siu L, et al. A classification system for clinical relevance of somatic variants identified in molecular profiling of cancer. Genet Med. 2016;18:128–136. doi: 10.1038/gim.2015.47. [DOI] [PubMed] [Google Scholar]
- 19.Kim J, Park WY, Kim NKD, Jang SJ, Chun SM, Sung CO, et al. Good laboratory standards for clinical next-generation sequencing cancer panel tests. J Pathol Transl Med. 2017;51:191–204. doi: 10.4132/jptm.2017.03.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher C, Fulton R, Fulton L, Wallis J, Chen K, Walker J, McDonald S, Bose R, Ornitz D, Xiong D, You M, Dooling DJ, Watson M, Mardis ER, Wilson RK. Genomic landscape of non-small cell lung cancer. Cell. 2012 Sep 14;150:1121‒34. [DOI] [PMC free article] [PubMed]
- 21.Krysiak Kilannin, Gomez Felicia, White Brian S., Matlock Matthew, Miller Christopher A., Trani Lee, Fronick Catrina C., Fulton Robert S., Kreisel Friederike, Cashen Amanda F., Carson Kenneth R., Berrien-Elliott Melissa M., Bartlett Nancy L., Griffith Malachi, Griffith Obi L., Fehniger Todd A. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood. 2016;129(4):473–483. doi: 10.1182/blood-2016-07-729954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C, et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun. 2017;8:268. doi: 10.1038/s41467-017-00296-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rheinbay E, Parasuraman P, Grimsby J, Tiao G, Engreitz JM, Kim J, et al. Recurrent and functional regulatory mutations in breast cancer. Nature. 2017;547:55–60. doi: 10.1038/nature22992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giannakis M, Hodis E, Mu XJ, Yamauchi M, Rosenbluh J, Cibulskis K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014;46:1264–1266. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandmann S, de Graaf AO, Karimi M, van der Reijden BA, Hellström-Lindberg E, Jansen JH, et al. Evaluating variant calling tools for non-matched next-generation sequencing data. Sci Rep. 2017;7:43169. doi: 10.1038/srep43169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiume M, Williams V, Brook A, Brudno M. Savant: genome browser for high-throughput sequencing data. Bioinformatics. 2010;26:1938–1944. doi: 10.1093/bioinformatics/btq332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goecks J, Coraor N, Team Galaxy, Nekrutenko A, Taylor J. NGS analyses by visualization with Trackster. Nat Biotechnol. 2012;30:1036–1039. doi: 10.1038/nbt.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carver T, Harris SR, Otto TD, Berriman M, Parkhill J, McQuillan JA. BamView: visualizing and interpretation of next-generation sequencing read alignments. Brief Bioinform. 2013;14:203–212. doi: 10.1093/bib/bbr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.T Mooney, J Walker, S Siebert, C Miller, M Griffith. cancer-genomics-workflow. McDonnell Genome Institute. https://github.com/genome/cancer-genomics-workflow. Accessed 28 June 2018.
- 31.Yost SE, Smith EN, Schwab RB, Bao L, Jung H, Wang X, et al. Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens. Nucleic Acids Res. 2012;40:e107. doi: 10.1093/nar/gks299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walsh PS, Erlich HA, Higuchi R. Preferential PCR amplification of alleles: mechanisms and solutions. PCR Methods Appl. 1992;1:241–250. doi: 10.1101/gr.1.4.241. [DOI] [PubMed] [Google Scholar]
- 33.Potapov V, Ong JL. Examining sources of error in PCR by single-molecule sequencing. PLoS ONE. 2017;12:e0169774. doi: 10.1371/journal.pone.0169774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aird D, Ross MG, Chen WS, Danielsson M, Fennell T, Russ C, et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011;12:R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakamura K, Oshima T, Morimoto T, Ikeda S, Yoshikawa H, Shiwa Y, et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011;39:e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2009;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 37.Good, Benjamin M., Benjamin J. Ainscough, Josh F. McMichael, Andrew I. Su, and Obi L. Griffith. 2014. “Organizing Knowledge to Enable Personalization of Medicine in Cancer.” Genome Biology 15:438. [DOI] [PMC free article] [PubMed]
- 38.Griffith M, Spies NC, Krysiak K, McMichael JF, Coffman AC, Danos AM, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet. 2017;49:170–174. doi: 10.1038/ng.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.