

Summary
Inflammatory responses are known to facilitate tissue recovery following injury. However, the precise mechanisms that enhance lung alveolar regeneration remain unclear. Here, using an organoid-based screening assay, we find that interleukin-1 (IL-1) and tumor necrosis factor α (TNFα) enhance the proliferation of AEC2s while maintaining their differentiation capacity. Furthermore, we find that expression of IL-1β and TNFα are induced in the AEC2 niche following influenza-induced injury in vivo, and lineage tracing analysis revealed that surviving AEC2s around the damaged area contribute to alveolar regeneration. Through genetic and pharmacological modulation of multiple components of the IL-1-nuclear factor κB (NF-κB) signaling axis, we show that cell-intrinsic as well as stromal mediated IL-1 signaling are essential for AEC2 mediated lung regeneration. Taken together, we propose that the IL-1/TNFα-NF-κB signaling axis functions as a component of an inflammation-associated niche to regulate proliferation of surviving AEC2s and promote lung regeneration.
Keywords: lung regeneration, inflammatory niche, alveolar epithelium, influenza virus, alveolar organoids, alveolar damage, repair, IL-1, TNFα, NF-κB
Graphical Abstract
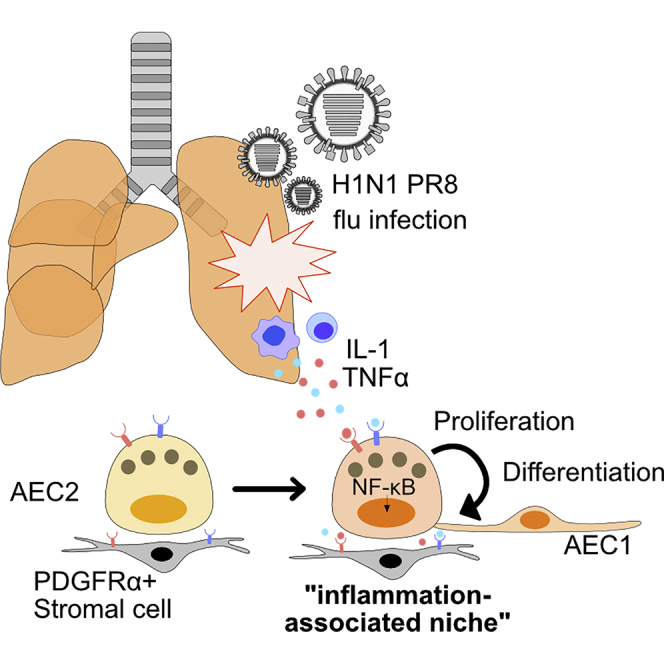
Highlights
-
•
IL-1/TNFα enhance the growth of lung alveolar stem cells (AEC2s) in organoid culture
-
•
AEC2s treated with IL-1 or TNFα maintain differentiation ability
-
•
AEC2s proliferate and contribute to lung repair after influenza virus infection
-
•
NF-κB pathway is activated in AEC2s treated with IL-1 or TNFα
The role of inflammatory responses in tissue repair and stem cell behavior is largely unexplored in the lung. Katsura et al. demonstrate that the cytokines interleukin-1 and tumor necrosis factor α, upregulated after influenza infection, enhance the proliferation of alveolar type 2 epithelial cells (AEC2s), endogenous lung stem cells. Moreover, these AEC2s generate AEC1s and contribute to alveolar regeneration.
Introduction
Rapid and efficient repair is essential after injury to restore organ function. In the alveolar region of the adult lung multiple stem/progenitor cells, including SFTPC+ type 2 alveolar epithelial cells (AEC2s), AXIN2+ SFTPC+ alveolar progenitor cells, and putative bronchioalveolar stem cells contribute to regeneration (Barkauskas et al., 2013, Desai et al., 2014, Kim et al., 2005, Zacharias et al., 2018). Some injuries, for example influenza virus infection or exposure to bleomycin, may mobilize a population of KRT5+ TRP63+ cells derived from airways to damaged regions (Kanegai et al., 2016, Kumar et al., 2011, Vaughan et al., 2015, Zuo et al., 2015). Moreover, recent studies have identified a critical role for a subset of immune cells, identified as CCR2+ monocytes and M2-like macrophages, in pneumonectomy-induced compensatory lung regrowth (Lechner et al., 2017). In many of the alveolar injury models studied, it has been shown that the critical cellular interactions required for repair involve conserved intercellular signaling pathways such as WNT, BMP, FGF, and SHH (Chung et al., 2018, Nabhan et al., 2018, Peng et al., 2015, Zacharias et al., 2018). However, it is unclear whether inflammatory cytokines can directly act on AEC2s to promote repair.
Here, we utilize an easily tractable alveolar organoid-based culture platform (Barkauskas et al., 2013) to screen cytokines that are upregulated after influenza virus infection and test their effect on AEC2 behavior. Further characterization of candidates in ex vivo organoid cultures and in vivo genetic models leads to the conclusion that the IL-1/TNFα-nuclear factor κB (NF-κB) signaling axis plays a critical role in alveolar regeneration by enhancing repair mediated by surviving AEC2s.
Results
Organoid-Based Screening Reveals IL-1 and TNFα as Potent Inducers of AEC2 Proliferation
We treated organoid cultures with 11 different cytokines known to be transiently upregulated after influenza virus infection (Guo and Thomas, 2017, Pociask et al., 2013, Shoemaker et al., 2015, Watanabe et al., 2013). After 15 days, type I interferon (α and β) was the only cytokine that gave a dramatic change in colony-forming efficiency (CFE) (Figure 1A). However, other cytokines appeared to promote larger organoids than controls. Indeed, classification of organoids based on their perimeters as small (150–450 μm), medium (450–1,500 μm), and large (>1,500 μm) (Figures S1A and S1B) revealed a significant increase in larger organoids in response to IL-1α/β, TNFα, and IL-17A/F (Figure 1B). Since IL-1α/β and TNFα gave the biggest effect we focused on them for this study. The effect of both cytokines was dose dependent, with a maximum at 10 ng/mL (Figure S1C). To distinguish between an increase in cell number versus size due to hypertrophy, we performed flow-cytometric (fluorescence-activated cell sorting [FACS]) analysis of cells isolated from organoids and found a 7-fold increase in TOMATO+ cells with IL-1β and TNFα (Figure 1C). Analysis for Ki67, a marker for proliferating cells, corroborated our FACS data (Figure 1D). Taken together, these results indicate that IL-1β and TNFα can enhance the proliferation of AEC2.
Figure 1.

IL-1α/β and TNFα Enhance Growth of AEC2s in Organoid Culture
(A–D) The CFE (A) and size (B) of organoids treated with indicated cytokines was quantified at day 15. Organoids at day 10 were analyzed for fold increase of TOMATO+ cells after FACS (C) and cell proliferation as judged by Ki67 staining (D).
(E) Representative differential interference contrast (DIC) and fluorescence microscopy images of organoids at day 10 (top) and day 15 (bottom).
(F and G) Representative immunofluorescence images of sections stained for RAGE and SFTPC (top) or HOPX and T1α cells (bottom) at day 10 (F) and day 15 (G). Insets show higher-magnification images of RAGE+ and SFTPC+ cells. Although organoids treated with IL-1β or TNFα are larger than controls, the results are consistent regardless of organoid size. Scale bars, 50 μm.
All bar graphs show mean ± SEM of three independent experiments. ∗p < 0.05; n.s, not significant.
IL-1β- and TNFα-Treated AEC2s Maintain Their Ability to Differentiate
To test whether AEC2s treated with IL-1β and TNFα maintain their phenotype and ability to differentiate into AEC1s, we examined organoids for expression of markers of AEC2s (SFTPC) and AEC1s (RAGE [AGER], T1α [PODOPLANIN], and HOPX). After 10 days, control organoids mostly consist of a monolayer of AEC2s and AEC1s (Figures 1E and 1F) but by day 15 they were organized into a multilayered epithelium, with AEC1s preferentially localized within the interior, as described previously (Figures 1E and 1G) (Barkauskas et al., 2013). Significantly, treated organoids do not differ from controls in their cellular composition despite their increase in size, suggesting that IL-1β or TNFα enhance AEC2 proliferation while maintaining their ability to differentiate.
Influenza Injury Induces IL-1β, TNFα, and Target Gene Expression in the AEC2 Niche
To study the in vivo relevance of the organoid studies, we examined the spatial expression of IL-1β and TNFα in influenza virus-infected mouse lungs. Section in situ hybridization showed a few cells expressing transcripts in uninfected lungs (Figure S2A). However, at 7 days post infection (7 dpi), Il1b and Tnfa transcripts were clearly elevated, both in the damaged areas where there were few SFTPC+ AEC2s and immediately outside (Figure 2A). To examine whether surviving AEC2s are responding to IL-1β and TNFα, we performed in situ hybridization for Neurl3, a downstream target gene (Figures 2B, 2C, and 4B). This revealed expression of Neurl3 in AEC2s close to the damaged areas, with the levels falling off further away, as expected if the cells are responding directly to the inflammatory cytokines (Figure S2B).
Figure 2.

IL-1β and TNFα Are Expressed in Damaged Area after Influenza Virus Infection and AEC2s Express Target Gene
(A) Il1b (top panels) and Tnfa (bottom panels) transcripts (green) were detected by PLISH in lungs 7 days after influenza virus infection.
(B) Neurl3 transcripts (green) were detected by PLISH in lungs 7 days after infection. H&E staining shows whole structure of the region. AEC2s (red) were visualized by SFTPC staining. Yellow dashed lines indicate the edge of the damaged area.
(C) High-magnification images of the inset in (B) Arrowheads indicate SFTPC+ AEC2s expressing Neurl3 transcripts.
In all experiments, n = 3 animals/group. Scale bars, 50 μm.
Figure 4.

NF-κB Pathway Is Involved in the Enhanced Proliferation of AEC2s after IL-1 or TNFα Stimulation
(A) Schematic of sample preparation for RNA-seq experiment.
(B) Heatmap of 165 genes whose expression was significantly different in IL-1β- or TNFα-treated AEC2s compared with controls.
(C and D) Top 10 pathway (C) and GO terms (D) enriched in the 165 differentially expressed genes shown in (B).
(E) Schematic diagram of IL-1 downstream signaling. Myd88 KO cells and IRAK4 inhibitor AS2444697 were used to disrupt the pathway.
(F) Representative images of organoids using Myd88 KO cells at day 15.
(G) Representative images for sections of lungs from wild-type (right panel) and Myd88 KO mice (left panel) at 10 dpi. White arrowheads indicate EdU+ proliferating AEC2s. Scale bar, 50 μm.
(H) Representative images of organoids treated with 0.1 ng/mL IL-1β or 10 μM IRAK4 inhibitor AS2444697 together with 0.1 ng/mL of IL-1β. All experiments were independently performed three times.
(I) Schematic model for the role of IL-1 and TNFα in inflammation-associated alveolar stem cell niche. We suggest that inflammatory cytokines IL-1 and TNFα released from inflamed area serve as a facultative “inflammation-associated niche,” resulting in the enhanced proliferation of AEC2s. Once AEC2s replicate, these cells give rise to AEC1s and contribute to lung repair.
Surviving AEC2s Proliferate and Contribute to Alveolar Regeneration after Influenza Injury
Previous molecular studies on alveolar repair after influenza virus infection have focused on the KRT5+ cells that mobilize to damaged alveoli and have paid less attention to the behavior of surviving AEC2s in relation to parenchymal damage. In damaged areas, extensive proliferation of infiltrating CD45+ cells is observed at 10 dpi (Figure 3A). Significantly, at this time many of the SFTPC+ AEC2s immediately outside these regions are also proliferating, particularly within 200 μm of the lesions (Figures 3A, 3B, and 3D). To follow the temporal and spatial dynamics of the proliferation and differentiation of AEC2s near damaged area, we performed lineage tracing using Sftpc-CreERT2 and Rosa26-tdTomato (Figure 3E). At 10 dpi we found many lineage-labeled AEC2s near damaged areas that were labeled by 5-ethynyl-2′-deoxyuridine (EdU), and by 15 dpi these cells appeared in clusters, indicating active clonal expansion. By 20 dpi proliferation had largely declined around damaged areas but was now seen toward the periphery of the lung (Figure S3A). At 20 dpi, many lineage-labeled cells express RAGE, an AEC1 marker, and have morphologies indistinguishable from AEC1s (Figure 3F). To determine the relationship between the proliferation and differentiation of lineage-labeled AEC2s and KRT5+ epithelial “pods,” we localized the two populations in adjacent sections at different times after infection (Figure S3). Significantly, the lineage-labeled AEC2s were never contiguous with KRT5+ clusters, which were always confined to areas of maximal damage, defined by the absence of AEC2s.
Figure 3.

IL-1 Signaling Is Important for Alveolar Regeneration Mediated by Surviving AEC2s
(A–C) Influenza virus-infected lungs at 10 dpi stained for CD45 (green), SFTPC (red), and EdU (white) (A). Yellow dashed lines show damaged area lacking SFTPC+ cells. Inset (1) shows that some CD45+ immune cells in the damaged area are replicating; inset (2) shows proliferating AEC2s (EdU+ SFTPC+) around damaged area. Representative images of SFTPC (red) and EdU (white) staining in virus-infected lungs of wild-type (B) and Il1r1 KO mice (C) at 10 dpi. Green indicates EdU+ SFTPC+ proliferating AEC2s visualized by Fiji software. Yellow dashed lines show damaged areas lacking SFTPC+ cells. To estimate the overall degree of damage, we estimated the area in which AEC2s are absent in 5–7 individual sections from three different wild-type or Il1r1 KO mice at 10 dpi. There was no significant difference in the values, suggesting there was no difference in viral infectivity between two genotypes (data not shown). Scale bars, 1 mm.
(D) Quantification of proliferating SFTPC+ cells in influenza virus-infected lungs of wild-type (gray) and Il1r1 KO mice (red). EdU+ SFTPC+ cells were counted and the numbers were divided by the corresponding areas shown on the x axis.
(E) Schematic diagram of lineage-tracing analysis of SFTPC+ cells.
(F) Representative images of lineage-tracing analysis 10 (left), 15 (middle), and 20 days (right) after influenza virus infection. Lineage-labeled cells (red) were visualized by using RFP antibody and AEC1s (white) were stained by RAGE antibody. Cell proliferation was assessed by EdU (cyan) detection. Arrowheads indicate EdU+ TOMATO+ proliferating lineage-labeled cells. Scale bars, 50 μm.
(G) Representative images of organoids derived from IL-1 receptor KO cells with IL-1β at day15.
(H) Quantification of organoid size in each combination. These experiments were performed independently three times.
For all experiments, n = 3 animals/group. ∗p < 0.05.
IL-1 Signaling Is Essential for AEC2 Replication in the Lung after Influenza Injury
Previous studies have implicated IL-1β and TNFα in lung development (Lappalainen et al., 2005, Stouch et al., 2016) but little is known about their role in lung regeneration. We therefore used a genetic approach to investigate the function of these cytokines after influenza virus infection. We found a drastic decrease in the number of proliferating AEC2s around damaged areas in mice lacking IL-1 receptor (which binds both IL-1α and IL-1β) versus wild-type controls even though the extent of viral damage appeared to be the same as in wild-type at 10 dpi (Figures 3C and 3D). While this result is consistent with a positive effect of IL-1β on AEC2 proliferation, we cannot rule out that the recruitment of T cells and neutrophils is also impaired (Schmitz et al., 2005) leading to a reduction in the level of cytokine production and an indirect effect on AEC2 proliferation. To investigate a direct role of IL-1 signaling, we used sorted AEC2s (EPCAM+ Lysotracker+) (Van der Velden et al., 2013) and platelet-derived growth factor receptor α-positive (PDGFRα+) stromal cells that were either wild-type or null mutant for IL-1 receptor and performed organoid assays. As shown in Figures 3G and 3H, after 15 days organoids derived from AEC2s lacking IL-1 receptor are significantly smaller than organoids in which both cell populations are wild-type (Figures 3G and 3H). Deleting the receptors from stromal cells also reduced organoid size, but to a lesser extent. Organoids derived from both mutant AEC2s and stromal cells are the smallest. Together, we conclude that IL-1 signaling is critical in alveolar epithelial repair and has a direct effect on AEC2 proliferation.
Our previous data showed that TNFα also enhances AEC2 proliferation. We therefore established organoid cultures with cells from TNFα receptor null mutant mice (Figures S4A and S4B), and found a significant decrease in organoid size in cultures derived from EPCAM+ Lysotracker+ AEC2s lacking TNFα receptor. Deleting the receptors from stromal cells also reduced organoid size, but to lesser extent. Organoids were smallest when both AEC2s and stromal cells were derived from TNFα receptor null mutant mice. These data indicate that TNFα enhances AEC2 proliferation by AEC2-intrinsic mechanisms, as well as through stromal cell-mediated effects. However, there was no reduction in AEC2 proliferation in TNFα receptor knockout (KO) mice at 10 dpi in influenza virus-infected lungs (Figures S4C and S4D). It is therefore likely that other mechanisms can compensate for the absence of TNFα receptor in vivo.
RNA-Seq Analysis Reveals that IL-1 and TNFα Treatment Activates NF-κB Pathway in AEC2s
To examine the downstream mechanisms that enhance AEC2 proliferation after IL-1β and TNFα treatments, we performed RNA sequencing (RNA-seq) analysis of AEC2s isolated from organoids 6 h after exposure to either cytokine (Figure 4A). We identified 165 differentially expressed genes that were common in both IL-1β- and TNFα-treated AEC2s compared with saline-treated controls (Figure 4B and Table S2). Pathway and gene ontology (GO) analysis revealed enrichment for many genes related to NF-κB signaling, immune responses, and cytokine signaling (Figures 4C and 4D). Some of the prominent genes include Neurl3 and Tnfa, two known target genes of NF-κB signaling. We also found a significant upregulation of NF-κB1, NF-κB2, and RELB, key effector transcription factors involved in the NF-κB pathway. These data implicate a role for the IL-1-TNFα/NF-κB signaling axis in AEC2 replication.
Genetic and Pharmacologic Modulation Reveals an Essential Role for the IL-1/NF-κB Signaling Axis in AEC2 Proliferation
To assess the role of NF-κB signaling in AEC2s replication, we genetically or pharmacologically disrupted several downstream components. In the IL-1 signaling pathway, MYD88 is an essential adaptor protein for activation of the NF-κB pathway (Figure 4E). Organoids generated from EPCAM+ epithelial cells and PDGFRα+ stromal cells lacking Myd88 are significantly smaller than controls (Figure 4F). In addition, the frequency of EdU+ proliferating AEC2s was dramatically decreased in the lungs of Myd88 KO mice after influenza virus infection (Figure 4G), indicating that MYD88 is critical for AEC2-mediated alveolar regeneration. In addition, IRAK4 is a major kinase in the IL-1 signaling pathway (Figure 4E). When we disrupted IRAK4 by AS2444697 in culture, the enhanced organoid growth seen with IL-1β was inhibited (Figure 4H). Taken together, these results demonstrate that the NF-κB pathway is important for enhanced AEC2 proliferation after IL-1β and TNFα stimulation.
Discussion
In this study, we have established several new findings in relation to lung repair by endogenous stem cells after injury, specifically after influenza virus infection. First, we provide evidence from organoid assays that several cytokines, including IL-1 and TNFα, can robustly promote the proliferation of AEC2s. We further exploit the organoid assays by combining mutant epithelial and stromal cells, and specific inhibitors, to show that the effect of the cytokines is, at least in part, through direct action on AEC2s via surface receptors and the NF-κB signaling pathway. A second important finding relates to the source of the epithelial cells that repair the alveolar region after influenza infection. As previously reported by others, we found that KRT5+ epithelial “pods” slowly appear in areas of maximal alveolar damage (Figure S3A). However, evidence now suggests that these TRP63+ KRT5+ epithelial cells generated in response to infection are likely very inefficient at differentiating into alveolar cells in vivo (Kanegai et al., 2016, Vaughan et al., 2015). By contrast, our lineage-tracing studies clearly show that in the areas around maximal damage surviving AEC2s, that are not contiguous with the KRT5+ pods, robustly proliferate at 10 dpi, and differentiate into AEC1s. These findings, together with in situ hybridization studies on tissue sections, suggest a model (Figure 4I) in which inflammatory responses promote repair through the production within and around the damaged areas of cytokines such as IL-1 and TNFα that locally promote the expansion of surviving endogenous alveolar stem cells. Final confirmation of this model will require the deletion of genes encoding these receptors or NF-κB pathway components specifically in AEC2s and the generation of mice lacking multiple cytokines in the lung. Intriguingly, AEC2s distant from damaged areas eventually proliferate and give rise to AEC1s at 25 dpi (Figures S3A and S3B). This raises the possibility that there are two parts to the overall response of the lung to damage by influenza virus. A more local response occurs around the damaged areas, which is regulated directly by inflammatory cytokines such as IL-1 and TNFα, and another component occurs distant from damaged areas that is analogous to the compensatory regrowth observed in uninfected lungs after reduction in the gas exchange capacity by left lobe pneumonectomy (Ding et al., 2011, Lechner et al., 2017, Liu et al., 2016).
It has been reported that IL-1 and TNFα signaling are involved in regeneration processes after tissue disruption caused by various injuries in other tissues (Kulkarni et al., 2017, Naik et al., 2017, Wang et al., 2017). Here we have uncovered a new mechanism of alveolar stem cell proliferation controlled by an inflammation-associated niche in which IL-1 and TNFα are involved. We believe that this is a key finding that connects inflammatory responses and tissue repair in lung.
Experimental Procedures
Organoid Culture
Organoid culture was as described by Barkauskas et al. (2013) with modifications. In brief, FACS-sorted cells were resuspended in Basic medium (Chen et al., 2012) containing 10% fetal bovine serum (FBS) and insulin-transferrin-selenium and mixed with equal amount of growth factor-reduced Matrigel (BD Biosciences #356230). Basic medium/Matrigel (100 μL) containing 2–5 × 103 AEC2s (SFTPC-tdTomato or EPCAM+ Lysotracker+ cells) and 5 × 104 stromal cells was seeded in 24-well 0.4-μm Transwell inserts (Falcon). Medium was changed every other day. ROCK inhibitor (10 μM, Y-27632; Sigma-Aldrich) was added for the first 48 h, and transforming growth factor-β inhibitor (10 μM, SB431542; Abcam) was added for 7 days. Mouse recombinant proteins were used at 10 ng/mL except where stated: IFN-α (5 ng/mL; BioLegend), IFN-β1 (5 ng/mL; BioLegend), TNF-α (BioLegend), IL-1α (BioLegend), IL-1β (BioLegend), IL-6 (R&D Systems), IL-15 (BioLegend), IL-17A (5 ng/mL; R&D Systems), IL-17F (5 ng/mL; R&D Systems), IL-18 (Medical & Biological Laboratories), IL-22 (R&D systems), IL-23 (R&D Systems), and IL-33 (R&D Systems). IRAK4 inhibitor (10 μM, AS2444697) was from Tocris Bioscience. Organoids >150 μm in perimeter were counted on day 15 post culture.
Immunofluorescence Analysis
Immunofluorescence of paraffin sections was exactly as described by Chung et al. (2018). EdU (50 mg/kg, E10187, Life Technologies) was injected intraperitoneally 4 h before fixation and Click-iT EdU Alexa Fluor 647 Imaging Kit (Life Technologies) was used to detect positive cells. Primary antibodies were as follows: surfactant protein C (Millipore, ab3786, 1:500), RAGE/AGER (R&D Systems, MAB1179, 1:200), HOPX (Santa Cruz Biotechnology, sc-30216, 1:250), T1α (DSHB, clone 8.1.1, 1:1,000), RFP (Rockland, 600401379, 1:250), Ki67 (Abcam, 15580, 1:500), and KRT5 (BioLegend, Poly19055, 1:500). Images were obtained by using confocal microscopes (Zeiss LSM 780, Olympus FV3000) or a wide-field fluorescence microscope (Zeiss Axio Imager).
Sample Preparation for RNA Sequencing
Organoids were dissociated from Matrigel with Dispase followed by trypsin-EDTA digestion. Cells were washed with 10% FBS in DMEM/F12 and Sftpc-CreER; tdTomato+ cells were sorted by FACS. Total RNA was extracted using Direct-zol RNA Miniprep Kit (Zymo Research) and mRNA was enriched from ∼26 ng of each total RNA using NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs). Libraries were prepared using NEBNext Ultra II RNA Library Prep Kit for Illumina (New England BioLabs). Paired-end sequencing (150 bp for each read) was performed using HiSeq X with the depth of 24 million reads per sample.
In Situ Hybridization
The PLISH protocol was kindly provided by Dr. Tushar Desai (Stanford University, CA, USA) (Nagendran et al., 2018). Frozen sections were fixed with 3.7% formaldehyde for 20 min, treated with 20 μg/mL proteinase K for 8 min at 37°C, and dehydrated with ethanol. The sections were incubated with gene-specific probes (Table S1) in hybridization buffer (1 M sodium trichloroacetate, 50 mM Tris [pH 7.4], 5 mM EDTA, 0.2 mg/mL heparin in diethyl pyrocarbonate (DEPC)-treated H2O) for 2 h at 37°C followed by washing with hybridization buffer. Common bridge and circle probes were added to the section and incubated for 1 h followed by T4 ligase reaction for 2 h. Rolling circle amplification was performed by using phi29 polymerase (Lucigen) overnight. The section was washed with label probe hybridization buffer (2× saline-sodium citrate/20% formamide in DEPC-H2O) and fluorophore-conjugated detection probe was applied and incubated for 1 h at 37°C. After washing with PBS containing 0.05% Tween 20, conventional immunofluorescence assay was performed.
Statistics
All bar graphs show mean ± SEM of three independent experiments. Statistical significance was calculated by using ANOVA followed by the Tukey-HSD method. p < 0.05 was considered statistically significant.
Author Contributions
Conceptualization, H.K. and B.L.M.H.; Methodology, H.K. and Y.K.; Investigation, H.K. and Y.K.; Writing, H.K., Y.K., P.R.T., and B.L.M.H.; Funding acquisition, P.R.T. and B.L.M.H.
Acknowledgments
We thank members of the Hogan and Tata laboratories for helpful discussion, Christina Barkauskas for critical comments on the manuscript, Yoshihiro Kawaoka and Nicholas Heaton for providing influenza virus, Tushar Desai for providing the PLISH protocol, Robin Yeh for quantification analysis, and Gregory Sempowski, Charles McGee, Kristina Riebe, and Chris Sample of the Duke Human Vaccine Institute for support. This work was funded by the National Institutes of Health (U01-HL111018 to B.L.M.H. and R00-HL127181 to P.R.T.), with additional support from United Therapeutics Corporation. H.K. is supported by the Naito Foundation.
Published: March 28, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.02.013.
Contributor Information
Purushothama Rao Tata, Email: purushothamarao.tata@duke.edu.
Brigid L.M. Hogan, Email: brigid.hogan@duke.edu.
Accession Numbers
The accession number for the RNA-seq data reported in this paper is GEO: GSE125662.
Supplemental Information
References
- Barkauskas C.E., Cronce M.J., Rackley C.R., Bowie E.J., Keene D.R., Stripp B.R., Randell S.H., Noble P.W., Hogan B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Matsumoto K., Brockway B.L., Rackley C.R., Liang J., Lee J.H., Jiang D., Noble P.W., Randell S.H., Kim C.F. Airway epithelial progenitors are region specific and show differential responses to bleomycin-induced lung injury. Stem Cells. 2012;30:1948–1960. doi: 10.1002/stem.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung M.I., Bujnis M., Barkauskas C.E., Kobayashi Y., Hogan B.L.M. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development. 2018;145 doi: 10.1242/dev.163014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai T.J., Brownfield D.G., Krasnow M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. 2014;507:190–194. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B.S., Nolan D.J., Guo P., Babazadeh A.O., Cao Z., Rosenwaks Z., Crystal R.G., Simons M., Sato T.N., Worgall S. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell. 2011;147:539–553. doi: 10.1016/j.cell.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X.J., Thomas P.G. New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 2017;39:541–550. doi: 10.1007/s00281-017-0636-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanegai C.M., Xi Y., Donne M.L., Gotts J.E., Driver I.H., Amidzic G., Lechner A.J., Jones K.D., Vaughan A.E., Chapman H.A. Persistent pathology in influenza-infected mouse lungs. Am. J. Respir. Cell Mol. Biol. 2016;55:613–615. doi: 10.1165/rcmb.2015-0387LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.F., Jackson E.L., Woolfenden A.E., Lawrence S., Babar I., Vogel S., Crowley D., Bronson R.T., Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Kulkarni N.N., Adase C.A., Zhang L.J., Borkowski A.W., Li F., Sanford J.A., Coleman D.J., Aguilera C., Indra A.K., Gallo R.L. IL-1 receptor-knockout mice develop epidermal cysts and show an altered innate immune response after exposure to UVB radiation. J. Invest. Dermatol. 2017;137:2417–2426. doi: 10.1016/j.jid.2017.07.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.A., Hu Y., Yamamoto Y., Hoe N.B., Wei T.S., Mu D., Sun Y., Joo L.S., Dagher R., Zielonka E.M. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell. 2011;147:525–538. doi: 10.1016/j.cell.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen U., Whitsett J.A., Wert S.E., Tichelaar J.W., Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am. J. Respir. Cell Mol. Biol. 2005;32:311–318. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- Lechner A.J., Driver I.H., Lee J., Conroy C.M., Nagle A., Locksley R.M., Rock J.R. Recruited monocytes and type 2 immunity promote lung regeneration following pneumonectomy. Cell Stem Cell. 2017;21:120–134.e7. doi: 10.1016/j.stem.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Wu H., Jiang K., Wang Y., Zhang W., Chu Q., Li J., Huang H., Cai T., Ji H. MAPK-mediated YAP activation controls mechanical-tension-induced pulmonary alveolar regeneration. Cell Rep. 2016;16:1810–1819. doi: 10.1016/j.celrep.2016.07.020. [DOI] [PubMed] [Google Scholar]
- Nabhan A.N., Brownfield D.G., Harbury P.B., Krasnow M.A., Desai T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. 2018;359:1118–1123. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagendran M., Riordan D.P., Harbury P.B., Desai T.J. Automated cell-type classification in intact tissues by single-cell molecular profiling. Elife. 2018;7 doi: 10.7554/eLife.30510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S., Larsen S.B., Gomez N.C., Alaverdyan K., Sendoel A., Yuan S., Polak L., Kulukian A., Chai S., Fuchs E. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature. 2017;550:475–480. doi: 10.1038/nature24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T., Frank D.B., Kadzik R.S., Morley M.P., Rathi K.S., Wang T., Zhou S., Cheng L., Lu M.M., Morrisey E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature. 2015;526:578–582. doi: 10.1038/nature14984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pociask D.A., Scheller E.V., Mandalapu S., McHugh K.J., Enelow R.I., Fattman C.L., Kolls J.K., Alcorn J.F. IL-22 is essential for lung epithelial repair following influenza infection. Am. J. Pathol. 2013;182:1286–1296. doi: 10.1016/j.ajpath.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz N., Kurrer M., Bachmann M.F., Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J. Virol. 2005;79:6441–6448. doi: 10.1128/JVI.79.10.6441-6448.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker J.E., Fukuyama S., Eisfeld A.J., Zhao D., Kawakami E., Sakabe S., Maemura T., Gorai T., Katsura H., Muramoto Y. An ultrasensitive mechanism regulates influenza virus-induced inflammation. PLoS Pathog. 2015;11:e1004856. doi: 10.1371/journal.ppat.1004856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouch A.N., McCoy A.M., Greer R.M., Lakhdari O., Yull F.E., Blackwell T.S., Hoffman H.M., Prince L.S. IL-1beta and inflammasome activity link inflammation to abnormal fetal airway development. J. Immunol. 2016;196:3411–3420. doi: 10.4049/jimmunol.1500906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Velden J.L., Bertoncello I., McQualter J.L. LysoTracker is a marker of differentiated alveolar type II cells. Respir. Res. 2013;14:123. doi: 10.1186/1465-9921-14-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan A.E., Brumwell A.N., Xi Y., Gotts J.E., Brownfield D.G., Treutlein B., Tan K., Tan V., Liu F.C., Looney M.R. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Chen H., Tian R., Zhang Y., Drutskaya M.S., Wang C., Ge J., Fan Z., Kong D., Wang X. Macrophages induce AKT/beta-catenin-dependent Lgr5(+) stem cell activation and hair follicle regeneration through TNF. Nat. Commun. 2017;8:14091. doi: 10.1038/ncomms14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T., Kiso M., Fukuyama S., Nakajima N., Imai M., Yamada S., Murakami S., Yamayoshi S., Iwatsuki-Horimoto K., Sakoda Y. Characterization of H7N9 influenza A viruses isolated from humans. Nature. 2013;501:551–555. doi: 10.1038/nature12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias W.J., Frank D.B., Zepp J.A., Morley M.P., Alkhaleel F.A., Kong J., Zhou S., Cantu E., Morrisey E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. 2018;555:251–255. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo W., Zhang T., Wu D.Z., Guan S.P., Liew A.A., Yamamoto Y., Wang X., Lim S.J., Vincent M., Lessard M. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature. 2015;517:616–620. doi: 10.1038/nature13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.