

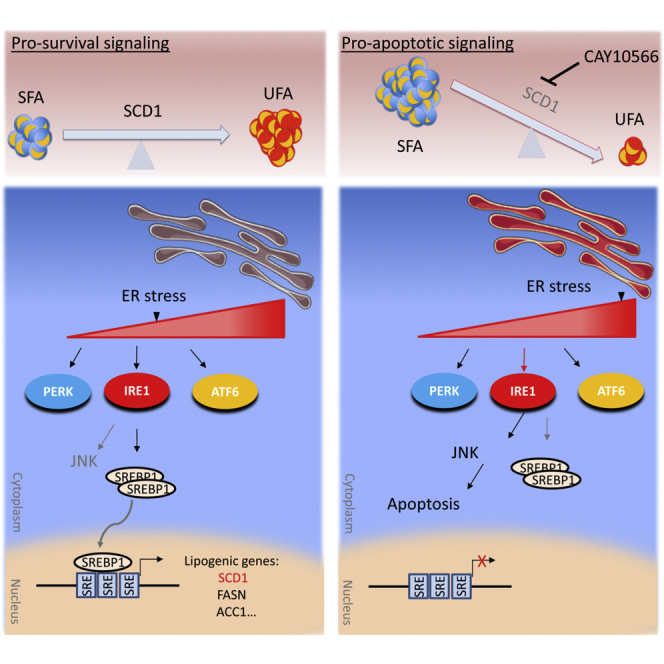
Summary
Inherent plasticity and various survival cues allow glioblastoma stem-like cells (GSCs) to survive and proliferate under intrinsic and extrinsic stress conditions. Here, we report that GSCs depend on the adaptive activation of ER stress and subsequent activation of lipogenesis and particularly stearoyl CoA desaturase (SCD1), which promotes ER homeostasis, cytoprotection, and tumor initiation. Pharmacological targeting of SCD1 is particularly toxic due to the accumulation of saturated fatty acids, which exacerbates ER stress, triggers apoptosis, impairs RAD51-mediated DNA repair, and achieves a remarkable therapeutic outcome with 25%–100% cure rate in xenograft mouse models. Mechanistically, divergent cell fates under varying levels of ER stress are primarily controlled by the ER sensor IRE1, which either promotes SCD1 transcriptional activation or converts to apoptotic signaling when SCD1 activity is impaired. Taken together, the dependence of GSCs on fatty acid desaturation presents an exploitable vulnerability to target glioblastoma.
Keywords: glioblastoma, glioma stem cells, stearoyl CoA desaturase, ER stress, unfolded protein response, inositol-requiring enzyme 1
Graphical Abstract
Highlights
-
•
SCD1 is essential for GSC maintenance and confers tumor growth advantage in vivo
-
•
Therapeutic targeting of SCD1 is highly effective in GBM mouse models
-
•
SCD1 exerts a cytoprotective function in GSCs by regulating ER homeostasis
-
•
ER stress promotes lipogenesis through the activation of IRE1-SREBP1-SCD1 axis
In this article, Pinkham and colleagues demonstrate that the fatty acid desaturase stearoyl CoA desaturase (SCD1) is essential for the maintenance of glioblastoma cancer stem cells. SCD1 is activated by ER stress and exerts a cytoprotective function by regulating ER homeostasis, thus favoring survival and tumor growth. Pharmacological targeting of SCD1 exhibits potent therapeutic efficacy in brain tumor mouse models.
Introduction
Nearly half of the patients diagnosed with glioblastoma (GBM), the most common malignant primary brain tumor, will die within a year (Ostrom et al., 2016), underlining the urgency for effective GBM therapies. Contributing to the genetic and phenotypic variability of GBM is a highly tumorigenic subpopulation of glioma stem-like cells (GSCs), which drives tumor initiation, growth, and recurrence (Lathia et al., 2015). Contingent on their inherent plasticity and survival cues that are not fully elucidated, these cells can survive and proliferate in a relatively hostile environment whereby hypoxia, acidic pH, and low nutrient levels prevail (Lathia et al., 2015). Such conditions induce cellular stress, in particular ER stress, which is known to promote pathogenesis in many diseases including cancer. ER stress, which is aberrantly high in tumors (Wang and Kaufman, 2014) and supports various hallmarks of cancer (Urra et al., 2016), is counteracted by the activation of the unfolded protein response (UPR), an adaptation signaling mechanism that enables tumor cells to survive under severe stress conditions (Urra et al., 2016). The UPR engages three signaling sensors, inositol-requiring enzyme 1 (IRE1), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Ron and Walter, 2007), that act jointly to alleviate and resolve ER stress. UPR signaling also promotes tumor growth through various mechanisms, including the activation of lipid synthesis pathways (Cubillos-Ruiz et al., 2017). GBM presents an upregulated lipid metabolism characterized by increased activation of sterol regulatory element-binding protein 1 (SREBP1), a master transcriptional regulator of lipid synthesis (Guo et al., 2009), and an abundance of unsaturated fatty acids (UFAs) (Srivastava et al., 2010). One of the target genes regulated by SREBP1 is stearoyl coenzyme A (CoA) desaturase 1 (SCD1), a key enzyme responsible for the conversion of saturated fatty acids (SFAs) to UFAs, necessary for tumor proliferation in several malignancies (Igal, 2010), and the maintenance of lung and ovarian cancer stem cells (Li et al., 2017, Noto et al., 2013). A defining role of SCD1 in GBM tumor initiation and growth, and the regulation mechanisms governing its activity, remain largely unexplored.
In this study, we demonstrate that persistent activation of ER stress promotes SCD1 expression through the activation of IRE1 and SREBP1. We propose that, through its unique role of controlling intracellular levels of SFAs, SCD1 is an essential regulator of ER stress that confers a selective advantage and promotes survival of GSCs in their tumor microenvironment.
Results
SCD1 Is a Therapeutic Target in Highly Proliferative GSC Populations
We have recently described a subpopulation of GSCs that escapes differentiation conditions and has enhanced stem cells properties, tumorigenic potential, and superior therapeutic resistance compared with the parental GSCs (Teng et al., 2017). To identify targeted inhibitors with increased toxicity toward this subpopulation that we termed floating cells (FCs), we have previously performed a small-scale inhibitors screen whereby GSCs and corresponding FCs were treated with target-selective inhibitors (Teng et al., 2017). Out of 141 compounds tested, only two inhibitors for SCD1 and nicotinamide phosphoribosyltransferase (NAMPT), a recently identified therapeutic target for GBM (Tateishi et al., 2016), showed increased sensitivity to the FCs compared with the parental GSCs (Figure S1A). Intriguingly, while SCD1 is a prominent target gene for SREBP1, NAMPT has also been reported to be positively regulated by this transcription factor (Rome et al., 2008). We primarily focused on SCD1, and first validated an increased sensitivity of FCs derived from two additional GSC lines to SCD1 inhibitors (MK-8245 and CAY10566 [CAY]) compared with the parental GSCs (Figures S1B and S1C). Gene expression analysis showed increased expression of SCD1 and other SREBP1 targets genes such as acyl-CoA synthetase short chain family member 2 (ACSS2), acetyl-CoA carboxylase α (ACC1), and fatty acid synthase (FASN) in the FC subpopulations (Figures S1D and S1E). An upregulated mRNA and protein expression of SCD1 (Figures S1D and S1F), along with vulnerability to SCD1 inhibitors, suggests an increased dependency of the FCs on SCD1 activity. We have previously reported that FCs are characterized by aberrant activation of nuclear factor κB (NF-κB) and increased mesenchymal properties. Treatment of previously characterized proneural and mesenchymal GSCs (Mao et al., 2013) showed a clear sensitivity to the SCD1 inhibitor in the latter subtype (Figure S1G). Overall, these findings suggest an increased expression and dependence on SCD1 in highly tumorigenic GBM subpopulations.
SCD1 Is Essential for GSC Maintenance and In Vivo Tumor Initiation
Palmitic Acid (PA; C16:0) is the most abundant SFA in human serum and the direct substrate of SCD1 (Carta et al., 2017). To functionally assess SCD1 activity in vivo, we tested whether PA supplementation could increase neutral lipid accumulation in GBM, detected using BODIPY, a fluorescent dye that stains neutral lipids. Indeed, tumor sections from mice receiving a daily dose of PA displayed a strong lipid staining (Figure 1A), suggesting that fractions of free PA are readily taken up by the tumor, desaturated by SCD1 before downstream processing into neutral lipids. Genetic silencing of SCD1 with short hairpin RNA (shRNA) decreased the expression of the pluripotency markers SOX2, NESTIN, OLIG2, OCT4, and NANOG (Figures 1B–1D), as well as stem cell frequency (Figure 1E). Silencing of SCD1 also decreased cell viability in five patient-derived GSCs (Figures 1F and 1G) but not in normal human astrocytes (NHAs) or immortalized neural stem cells (NSCs) maintained under serum-free conditions (Figure 1F). Supplying GSCs with oleic acid (OA; C18:1), the main product of SCD1, rescued from shSCD1-mediated cell death (Figure 1G). The role of SCD1 in tumor initiation was further evaluated by implanting GSCs expressing shSCR or shSCD1 into the striatum of nude mice. Silencing of SCD1 completely prevented tumor growth as assessed by Firefly luciferase (Fluc) bioluminescence imaging as well as overall survival up to 150 days after implantation when the experiment was terminated (Figure 1H). Ectopic expression of SCD1 using a lentivirus system (SCD1-OE; Figure S2A) resulted in a greater than 5-fold increase in stem cell frequency in two GSCs (Figure S2B). We repeated these experiments in one GSC specimen that was propagated long-term as neurospheres (>50 passages) and presented a low stem cell frequency. Forced expression of SCD1 in these cells increased stem cell frequency (Figure S2C), cell proliferation (Figure S2D), and secondary neurosphere formation (Figure S2E). Following intracranial implantation of these high-passage GSCs in mice, four of five mice in the SCD1-OE group developed tumors that gradually grew over time (Figure 1I), and three of five succumbed to tumor burden by day 135 post implantation when this experiment was terminated (Figure S2F). None of the mice in the control arm developed any tumor detectable by Fluc imaging or H&E staining (Figures 1I, 1J, and S2F). In sum, SCD1 is essential for GSC maintenance and confers a tumor growth advantage in vivo.
Figure 1.

SCD1 Is Essential for GSC Maintenance and Tumor Initiation
(A) Mice bearing 83-Fluc GSC tumors received daily injections over 7 days of BSA or PA (3 mg/kg of body weight). Representative brain sections were stained with BODIPY (red) and DAPI (blue). Asterisks depict the tumor injection site.
(B) Immunostaining for NESTIN, SOX2, and nuclei (DAPI) in 19-GSCs expressing shSCR or shSCD1 at day 7 after shRNA transduction. Scale bar, 100 μm.
(C) Immunoblot analysis of SCD1, SOX2, and NESTIN in GSCs expressing shSCR or shSCD1.
(D) Relative expression of stem cell markers determined by qPCR in GSCs transduced with shSCR or shSCD1 for 7 days.
(E) Stem cell frequency in 157 GSCs expressing shSCR or shSCD1 determined using the limited dilution analysis algorithm.
(F) Cell viability in three GSCs, NHA, and NSC expressing shSCR or shSCD1, at day 7 after transduction. Data is expressed as percentage of shSCR.
(G) GSCs (326 and 1123) expressing shSCR or shSCD1 were cultured in the presence of BSA or OA (50 μM). Cell viability was determined 7 days post transduction.
(H) 83-Fluc GSCs transduced with shCtrl or shSCD1 and intracranially implanted in mice (shCtrl, n = 4; shSCD1, n = 6) after 24 h. Survival analysis is shown using Kaplan-Meier curves. p = 0.0025 (two-sided log-rank test). Representative Fluc imaging of brain tumors at day 10 post implantation is also shown.
(I) High-passage 157-Fluc GSCs (5 × 104) expressing control (Ctrl) or SCD1 (SCD1-OE) were implanted in the brain of nude mice (n = 5/group). Longitudinal Fluc imaging shown for individual mice in each group.
(J) H&E staining of brain sections of representative mice from both groups at day 135 post implantation.
∗p < 0.05, ∗∗p < 0.001, Student’s t test. See also Figure S2.
Perturbation of SCD1 Activity Depletes GSCs Regardless of Their Genetic Background
We tested three pharmacological inhibitors of SCD1: PluriSIn, CAY, and the ACC1 inhibitor TOFA, which reportedly also inhibits SCD1 (Mason et al., 2012), and identified CAY as the most cytotoxic inhibitor with an average IC50 < 100 nM in different GSC specimens (Figure S3A and data not shown). GSCs treated with CAY showed increased caspase-3 and caspase-7 activities indicative of apoptotic cell death (Figure 2A), along with increased histone H2AX phosphorylation (γ-H2AX), a marker of DNA damage (Figure 2B). NHAs were indifferent to CAY treatment under serum-free conditions (Figure S3B). To confirm that CAY inhibits SCD1 activity, we analyzed fatty acid composition in GSCs treated with a control vehicle or CAY. We confirmed a decrease in UFAs (Figures 2C and S3C) and observed an overall increase of SFAs, which was more pronounced around C20:0–C26:0, thereby suggesting an accumulation of SFAs after CAY treatment (Figure 2C). Next, we performed rescue experiments after treatment with this inhibitor in the presence of SFAs (16:0; 18:0), monounsaturated FAs (18:1; 16:1; 18:2), and polyunsaturated FAs (20:4 n-6; 22:6 n-3). All of the UFAs tested almost completely protected against CAY-induced cell death (Figure 2D). We treated ten patient-derived GSCs with CAY and assessed short-term (4 days) and long-term (9 days) survival. We observed two different patterns of short-term response whereby four GSCs were highly sensitive to CAY-induced cell death with an IC50 < 60 nM, while six GSCs showed poor or no response (Figure 2E). However, long-term (9 days) treatment with CAY in this more resistant GSC subset (MGG8 and 157) resulted in >80% decrease in cell viability and neurosphere formation (Figure 2F). These results establish that inhibitors of SCD1 can effectively target patient-derived GBM cells in culture regardless of subtype.
Figure 2.

Pharmacological Targeting of SCD1 Depletes GSCs
(A) Fold change in caspase-3/7 activation in 326 GSCs treated with CAY (200 nM).
(B) Immunostaining for γ-H2AX in CAY-treated GSCs. Scale bar, 200 μm. Representative images of single nuclei depicting γ-H2AX foci are also shown (inset).
(C) Heatmap representing the quantitative ratio of SFAs and UFAs in 83 CAY-treated GSCs relative to the untreated control. Values below or above 1 are indicative of decreased or increased fatty acids ratios, respectively.
(D) Cell viability in 83-GSCs treated with CAY (100 nM) in the presence of the indicated fatty acids.
(E) Cell viability at day 4 in ten GSC specimens treated with CAY at the indicated doses.
(F) Cell viability and representative bright-field micrographs of secondary spheres at day 9 in 157-GSCs treated with CAY (100 nM).
∗p < 0.05, ∗∗p < 0.001, Student’s t test. See also Figure S3.
Pharmacological Targeting of SCD1 Achieves a Strong Therapeutic Effect in GBM Mouse Models
To evaluate the therapeutic efficacy of SCD1 inhibition in GSCs xenograft mouse models, we elected to deliver CAY via the intranasal route, which allows bypassing of the blood-brain barrier (Chauhan and Chauhan, 2015). Mice were implanted with mesenchymal 83-Fluc GSCs and received a daily intranasal dose of 5 mg/kg of CAY (Figure 3A). Treatment with CAY did not cause any weight loss (Figure S3D) and resulted in an average decrease of 56% in Fluc signal at day 12 post-implantation (Figure 3B), along with decreased tumor proliferation assessed by Ki67 staining (Figure 3C). This translated into a significantly extended survival of 35 days in the treated group compared with 18 days for the control group (Figure 3D). Additionally, two of eight mice in the treated group were still alive at day 156 post implantation with no detectable tumors (Figure S3E). Under a similar experimental setup, mice receiving a lower dose of CAY (1.5 mg/kg) also showed a significantly extended survival of 22 days in the treated group compared with 16 days for the control group (p = 0.0009; Figure S3F). These experiments were repeated in mice bearing proneural 157-GSC brain tumors. All mice (8/8) in the control group showed growing tumors (Figure 3E) and eventually died from tumor burden (Figure 3F). Remarkably, none of the CAY-treated mice (0/8) had any detectable tumor and survived up to 158 days when the experiment was terminated (Figures 3E and 3F). We did not observe a significant change in liver weight following CAY treatment, suggesting an absence of hepatomegaly (Figure 3G). Therefore, intranasal delivery of CAY in patient-derived xenograft mouse models is a highly effective therapeutic strategy.
Figure 3.

Therapeutic Targeting of SCD1 in Preclinical GBM Mouse Models
(A) Overview of experimental setup.
(B–D) Mice implanted with 83-Fluc GSCs (2 × 104; n = 8/group) received a daily intranasal dose of DMSO (Control) or CAY (5 mg/kg) for 10 days. (B) Overtime monitoring of tumor growth with Fluc imaging in individual mice from Ctrl and CAY-treated groups. Hash depicts the time of death due to tumor burden. (C) Ki67 immunostaining in one Ctrl and one CAY-treated mouse. Scale bar, 100 μm. (D) Kaplan-Meier curves showing median survival in both groups (∗p = 0.008, two-sided log-rank test).
(E–G) Mice implanted with 157-Fluc GSCs (1 × 105; n = 8/group) were treated with vehicle or CAY (5 mg/kg). (E) Overtime Fluc imaging demonstrates the absence of tumor growth in all eight CAY-treated mice. (F) Survival curves in both groups (p = 0.0002; two-sided log-rank test). (G) The ratio of liver weight to the body weight in both experimental groups is shown. ns, non-significant by Student’s t test.
See also Figure S3.
SCD1 Inhibition Exacerbates ER Stress through the Accumulation of SFAs
SFAs are known to induce ER stress (Volmer et al., 2013), hence the increased expression of the UPR transcripts CHOP and sXBP1 in GSCs treated with PA (Figure S4A). Similarly, GSCs treated with CAY displayed a strong increase in the ER stress markers BiP (also known as GRP78), CHOP, sXBP1, and GADD34 (Figure 4A), which was further accentuated over time (Figure 4A), in line with an increased accumulation of SFAs following SCD1 inhibition. Treatment with CAY also increased BiP (in two of the four GSCs tested) and CHOP proteins levels along with increased phosphorylation of eIF2α and γ-H2AX (Figure 4B). We hypothesized that SFA accumulation after CAY treatment primarily induces apoptotic cell death through an overwhelming ER stress response. The combination of CAY and a low dose of PA resulted in increased expression of CHOP and sXBP1 transcripts (Figure S4B), as well as protein levels of BiP, CHOP, and γ-H2AX (Figure S4C). This combination also increased caspase-3/7 activities (Figure 4C) and cell death in GSCs but not in NHAs (Figure 4D). Similarly, silencing of SCD1 or treatment with a different SCD1 inhibitor increased PA-induced cytotoxicity (Figures S4D and S4E). The SFA stearic acid (SA; C18:0) also increased cell death when combined with CAY (Figure S4F). Alleviating ER stress with the chemical chaperone phenylbutyrate (PBA) significantly repressed caspase-3/7 activation (Figure 4E) and protected against SCD1 inhibition (Figure 4F). Further, azoramide, which protects against chemically induced ER stress (Fu et al., 2015), also significantly rescued mice from CAY-induced cytotoxicity (Figure S4G). Overall, these data demonstrate that targeting SCD1 results in chronic ER stress and renders GSCs susceptible to SFA-induced lipotoxicity.
Figure 4.

SCD1 Inhibition Promotes ER Stress and Triggers UPR-Mediated Apoptotic Signaling
(A) Relative mRNA expression of ER stress markers in GSCs treated with CAY (200 nM) for 24 and 48 h.
(B) Immunoblot analysis of four GSCs treated with CAY for 48 h.
(C) Fold change in caspase-3/7 activation after treatment with CAY (50 nM) in the presence or absence of PA (50 μM) or OA (50 μM).
(D) Cell viability in five GSC specimens and NHA, treated with CAY (50 nM) or the combination of CAY and PA (50 μM) for 4 days.
(E) Fold change in caspase-3/7 activation after treatment with CAY (50 nM) in the presence or absence of PBA (5 mM).
(F) Cell viability in GSCs pretreated with CAY for 24 h followed by PBA treatment for 4 days.
(G) Relative mRNA expression of sXBP1 and CHOP in GSCs treated with CAY and/or OA (50 μM).
(H) Immunoblot analysis of phosphorylated c-Jun in GSCs treated with CAY.
(I) Cell viability of GSCs treated with CAY (50 nM) or the combination of CAY with: JNK inhibitor (SP 600125: 20 μM), IRE1 inhibitors (4μ8C: 25 μM and KIRA6: 5 μM), PERK inhibitor (GSK2656157: 10 μM), and ATF6 inhibitor (Ceapin A7: 20 μM).
See also Figures S4 and S5.
SCD1-Mediated UFA Synthesis Mitigates ER Stress
We postulated that transcriptional activation of SCD1 and subsequent increase in UFAs mediates a cytoprotective function in GSCs. In line with the protective effect of OA following SCD1 targeting, treatment with OA reversed PA-induced cytotoxicity (Figure S5A) and prevented caspase-3/7 activation after treatment with CAY (Figure 4C). Importantly, OA suppressed CHOP and sXBP1 upregulation following CAY treatment (Figure 4G), thus suggesting that the protective role of UFAs such as OA after SCD1 inhibition is largely due to their potential role in alleviating ER stress. Ectopic expression of SCD1 reduced ER stress induced by PA or thapsigargin (Tg), as assessed by BiP and sXBP1 mRNA expression (Figure S5B), and protected against PA-induced cytotoxicity (Figure S5C). Collectively, these results support that SCD1 acts as an essential regulator of ER stress to prevent prolonged UPR, thus favoring survival in GSCs.
Cell Death Caused by SCD1 Targeting Is Contingent on UPR Signaling
Persistent activation of UPR signals the inability of cells to adapt to ER stress, thus activating an apoptotic switch executed by CHOP or JNK (Szegezdi et al., 2006). Silencing of CHOP failed to protect against CAY-mediated cell death (Figures S5D and S5E). IRE1 kinase mediates proapoptotic signaling though the activation of JNK, which directly phosphorylates and activates the transcription factor c-Jun (Hibi et al., 1993). The phosphorylation of c-Jun was transiently increased in response to CAY treatment (Figure 4H). On the other hand, treatment with JNK inhibitor SP600125 prevented caspase-3/7 activation (Figure S5F) and reverted CAY-mediated cytotoxicity (Figure 4I). Additionally, the overexpression of a dominant-negative IRE1 (Tirasophon et al., 1998) (K599A) but not wild-type IRE1 produced significant rescue from cell death mediated by CAY or PA treatment (Figure S5G). Furthermore, treatment with two IRE1 inhibitors, 4μ8C (Cross et al., 2012) and KIRA6 (Ghosh et al., 2014), prevented caspase-3/7 activation (Figure S5F) and almost completely abrogated CAY-mediated cytotoxicity (Figures 4I, S5H, and S5I). The ATF6 inhibitor Ceapin A7 (Gallagher et al., 2016) failed to prevent cell death, while the PERK inhibitor GSK2656157 produced significant rescue from cell death in two of the three GSCs tested (Figures 4I, S5H, and S5I). It should be noted that both PERK and IRE1 have redundant functions and are able to switch from ER stress regulators to apoptosis effectors when ER homeostasis is unattainable (Han et al., 2009). Overall, SCD1 inhibition induces terminal UPR signaling and triggers apoptotic cell death via IRE1 and JNK.
Adaptive ER Stress Response Promotes SCD1 Expression through SREBP1 Activation
Given its cytoprotective properties, we asked whether SCD1 is transcriptionally activated under ER stress in order to support GSC survival. Tg-induced ER stress resulted in a dose-dependent increase of SREBP1 transcriptional targets involved in de novo lipid synthesis including SCD1, FASN, ACC1, acyl-CoA synthetase short chain family member 2 (ACSS2), and ELOVL fatty acid elongase 6 (ELOVL6) (Figures 5A and 5B). Silencing of SREBP1 effectively decreased the expression of these genes (Figure S6A) and, concurrently with SCD1 downregulation, resulted in a strong decrease (>60%) in mRNA expression of stem cell markers (Figure S6B). Furthermore, the upregulation of SREBP1 target genes by Tg, confirmed in a second GSC specimen (Figure S6C), was reversed after SREBP1 silencing (data not shown) or concomitant treatment with 25-hydroxycholesterol (25-HC), an inhibitor of SREBP processing (Adams et al., 2004) (Figure S6C). ER stress-mediated increase of SCD1 protein expression was further confirmed after treatment with PA or SA (Figure 5C) or with the synthetic compound HA15, which triggers ER stress by specifically targeting BiP (Cerezo et al., 2016) (Figure S6D). Conversely, alleviating ER stress with PBA, azoramide, or tauroursodeoxycholic acid (TUCDA) downregulated SCD1 protein levels (Figures 5D and 5E). Thus, endogenous ER stress also contributes to SCD1 transcriptional regulation.
Figure 5.

ER Stress Promotes a Lipogenic Signature through SREBP1 and IRE1 Signaling
(A) De novo lipogenesis pathway.
(B) Relative mRNA expression of SREBP1 target genes in GSCs treated with Tg (200 nM).
(C–E) Protein expression of SCD1 in GSCs treated with PA and SA (C) or the ER stress inhibitors PBA (2.5, 5, and 10 mM), TUCDA (0.5 mM), and azoramide (50 μM) (D and E).
(F) Immunoblot analysis showing an increased SCD1 expression in four GSCs treated with CAY (200 nM).
(G) Cell viability in GSCs treated with the indicated doses of CAY in combination with T0901317 (25 μM) or SR9243 (10 μM) for 3 days.
(H) Immunoblot analysis of SCD1 expression following IRE1 knockdown.
(I) Relative mRNA expression of SREBP1 target genes in GSCs treated with Tg (300 nM) in the presence or absence of IRE1 inhibitor 4μ8C.
∗p < 0.05, ∗∗p < 0.001, Student’s t test. See also Figures S6 and S7.
Intriguingly, due to the accumulation of SFAs, we observed an increased SCD1 expression following CAY treatment (Figure 5F). Blocking SCD1 transcriptional activation would prevent de novo synthesis of this desaturase and likely increase cytotoxicity of SCD1 inhibitors. Since the nuclear receptor liver-X-receptor (LXR) directly activates the expression of SREBP1, we used a selective LXR agonist T0901317, and an inverse agonist SR9243, to forcibly activate or repress LXR, respectively. The LXR inverse agonist effectively decreased SCD1 expression, which was increased after treatment with the LXR agonist (Figure S6E). Concomitant treatment with SR9243 and CAY further increased ER stress markers compared with the SCD1 inhibitor alone (Figure S6F). Unexpectedly, despite a strong downregulation of SCD1 by SR9243, the latter did not cause any significant increase in ER stress (Figure S6F) or cytotoxicity (Figure S6G). This is likely justified by the lack of SFA biosynthesis and accumulation after LXR inhibition. On the other hand, the combined treatment of SR9243 with low doses of CAY (<25 nM) decreased GSC viability (Figures 5G and S6H), while the LXR agonist T0901317 protected from CAY-mediated cell death (Figure 5G). Collectively, our data suggest that cytotoxicity caused by SCD1 inhibition is primarily due to toxic accumulation of SFAs. Therefore, targeting SCD1 and not upstream lipogenesis regulators is likely to achieve the best therapeutic outcome.
IRE1 Signaling Is Essential for Transcriptional Activation of De Novo Lipid Synthesis Genes
Given that sustained IRE1 activity promotes cell survival under ER stress, we asked whether IRE1 is an upstream regulator of SREBP1 transcriptional activity. The overexpression of IRE1 that is sufficient to activate IRE1 signaling increased XBP1 splicing (Figure S7A) and upregulated SREBP1 target genes (Figure S7B). On the other hand, silencing of IRE1 resulted in a decreased expression of SREBP1 targets including SCD1 (Figures 5H and S7C) and prevented their upregulation following Tg treatment (Figure S7C). Finally, concurrent treatment with Tg and the IRE1 inhibitor 4μ8C prevented XBP1 splicing (Figure S7D) and essentially suppressed the upregulation of lipogenesis target genes (Figure 5I). Taken together, these results indicate that IRE1 controls the transcriptional activation of SREBP1 targets including SCD1.
The Expression of BiP Predicts Response to SCD1 Inhibition
Elevated expression of BiP, the putative marker of ER stress, is commonly observed in various malignancies including GBM (Pyrko et al., 2007). Analysis of The Cancer Genome Atlas (TCGA) database confirmed that BiP expression is significantly increased in GBM compared with non-tumor tissue (Figure 6A), and elevated levels of BiP correlated with poor survival (Figure 6B). Furthermore, concordant with an increased SCD1 expression by ER stress, we observed a positive correlation between the transcriptional levels of BiP and SCD1 in GBM patients using the TCGA datasets (Pearson's r = 0.2686; p < 0.0001; Figure S7E). We then compared SCD1 and BiP protein expression across different GSCs. Interestingly, we observed a strong correlation between high BiP expression and sensitivity to SCD1 inhibitors; all four GSCs (L0, L1, 83, and 326) that were highly responsive to CAY treatment (Figure 2E) presented the highest BiP protein expression (Figure 6C), suggesting that GSCs with elevated endogenous ER stress are more dependent on SCD1. This was confirmed in the FC population which, in addition to increased expression of SREBP1 targets (Figure S1D), showed increased levels of ER stress markers compared with their parental neurospheres (Figures S7F and S7G). Additionally we initially reported that GSCs with mesenchymal properties are highly sensitive to short-term cytotoxicity of SCD1 inhibitors (Figure S1G). TCGA analysis of BiP and CHOP, across the three main GBM subtypes, shows that mesenchymal GBM expresses high levels of BiP and CHOP compared with proneural and classical subtypes (Figure S7H). Overall, GBM tumors with increased ER stress, which is often linked to an aggressive tumor behavior and poor prognosis, are increasingly vulnerable to SCD1 inhibition.
Figure 6.

The Expression of BiP Is Increased in GBM and Predicts Sensitivity to SCD1 Inhibition
(A) TCGA analysis of BiP mRNA expression in normal non-tumor and GBM.
(B) Survival analysis in 525 GBM patients from the TCGA dataset based on high versus low BiP expression levels. p = 0.0005, log-rank test.
(C) Immunoblot analysis of SCD1 and BiP expression in NHA and eight GSC specimens.
See also Figure S7.
SCD1 Inhibition Downregulates DNA-Repair Mechanisms and Enhances Temozolomide Cytotoxicity
ER stress-inducing compounds can impair DNA-repair mechanisms and therefore sensitize to the conventional GBM therapeutic temozolomide (TMZ) (Weatherbee et al., 2016, Xipell et al., 2016), which in itself induces ER stress in GBM (Pyrko et al., 2007). Treatment of GSCs with TMZ resulted in a modest increase in mRNA expression of CHOP and sXBP1 (<3-fold) (Figure 7A). The combination of CAY and TMZ displayed an overall increase in ER stress markers compared with CAY alone (Figure 7A). To assess whether this increase is caused by SFAs, we treated GSCs with TMZ and a relatively low dose of PA. Remarkably, the combination of TMZ with PA led to a prominent upregulation of all four markers (Figure 7A). Treatment with CAY in GSCs specimens consistently decreased the expression of the DNA-repair protein RAD51, which contributes to GSC resistance to radiation therapy and TMZ (Short et al., 2011) (Figure 7B). As such, treatment with CAY and TMZ resulted in increased levels of γ-H2AX (Figures 7B and 7C) and caspase-3/7 activation even in MGG23, a TMZ-resistant GSC with unmethylated MGMT promoter (Wakimoto et al., 2012) (Figure 7D). Importantly, treatment with CAY (10–100 nM) increased TMZ cytotoxicity in TMZ-responsive GSCs and sensitized to TMZ in TMZ-resistant GSCs (Figure 7E). For instance, in MGG23 this combination decreased cell viability by ∼60% (Figure S7I) and depleted secondary sphere formation (Figures 7F and 7G). Treatment with PA (but not OA) also strongly sensitized to TMZ (Figures S7J and S7K). Finally, CAY-mediated sensitization to TMZ, which was confirmed in one additional GSC specimen (Figure 7H), could be completely reversed by alleviating ER stress or by inhibiting PERK or IRE1 (Figure 7H). Overall, these data indicate that ER stress induced by the accumulation of SFAs impairs RAD51-mediated DNA-repair mechanisms, thus sensitizing to TMZ.
Figure 7.

SCD1 Inhibition Compromises DNA Damage Repair and Increases Temozolomide Cytotoxicity
(A) Relative mRNA expression of ER stress markers in 83-GSCs treated with CAY (200 nM), PA (200 μM), TMZ (100 μM), or their respective combination as indicated.
(B) Immunoblot analysis of Rad51 and γ-H2AX in GSCs treated with CAY and TMZ.
(C) Immunostaining for γ-H2AX in 83-GSCs treated with CAY (50 nM) and TMZ (100 μM). Scale bar, 200 μm. Representative images of single nuclei depicting γ-H2AX foci are also shown (inset).
(D) Fold change in caspase-3/7 activation in GSCs pretreated with CAY (200 nM) followed by TMZ treatment (100 μM).
(E) GSCs were pretreated with CAY for 24 h prior to TMZ treatment. Cell viability was measured after 5 days. MGG8 were treated with CAY (100 nM) and TMZ (0–10 μM). GSCs (83 and L0) were treated with CAY (10 nM) and TMZ (0–100 μM).
(F and G) MGG23 were pretreated with CAY (100 nM) for 24 h followed by TMZ (100 μM) for 7 days. Secondary spheres were counted 15 days after treatment (F). Micrographs of neurospheres are shown in (G). Scale bar, 100 μm.
(H) Cell viability in 326-GSCs after 7 days of treatment with CAY (10 nM), TMZ (50 μM), or their combination in the presence of OA, PBA, azoramide, PERK, or IRE1 inhibitor.
See also Figure S7.
Discussion
Aberrant activation of UPR signaling is intimately associated with several hallmarks of cancer and is reported in different tumor types including GBM (Obacz et al., 2017). This high dependence on UPR signaling creates a vulnerability that could be exploited to target these tumors. In this study, we have identified an increased activation and dependence on SCD1 activity that is necessary for GSC maintenance and proliferation. IRE1 signaling typically promotes survival under low or moderate ER stress and contributes to GBM progression (Lhomond et al., 2018). However, under irremediable ER stress, IRE1 converts to a proapoptotic signaling (Han et al., 2009). Following a similar paradigm, we show that in GBM, IRE1 activation of SREBP1 transcriptional targets, which include SCD1, promotes survival under ER stress. While we have not addressed the mechanism by which IRE1 activates SREBP1, it was previously reported that sXBP1, a key transcription factor downstream of IRE1, can directly interact with SREBP1 promoter (Ning et al., 2011). Disruption of SCD1 activity in GSCs prevents synthesis of UFAs required for membrane synthesis and cell proliferation and causes a toxic accumulation of SFAs, thus leading to terminal UPR signaling mediated by JNK. In line with our findings, SREBP1 and its target SCD1 were shown to be upregulated under hypoxic conditions in GBM (Lewis et al., 2015). This is possibly caused by IRE1 activation since hypoxia is known to activate the UPR, including IRE1-XBP1 signaling (Romero-Ramirez et al., 2004). Additionally, IRE1 activates downstream oncogenic signaling such as NF-κB (Cubillos-Ruiz et al., 2017), a regulator of SCD1 in ovarian cancer stem cells (Li et al., 2017). Therefore, it is likely that IRE1-mediated activation of SREBP1 targets is not limited to GBM. Our work also extends our previous findings whereby we have characterized a subpopulation of GBM stem cells with increased tumorigenic potential and aberrant activation of NF-κB (Teng et al., 2017). Interestingly, we have previously determined that these cells are enriched under acidic pH or hypoxia. Here, we provide mechanistic insights detailing an activated ER stress and SREBP1 signature as well as an increased vulnerability to SCD1 inhibition in these cells. Therefore, it is likely that IRE1-XBP1-NF-κB signaling is a key driver of aggressive GBM subpopulations.
An upregulation of SREBP1-mediated lipid synthesis is both a prerequisite and a consequence of the increased proliferation of tumor cells, potentially fueled by the biochemical environment of GBM, or endogenously by oncogenic stress, genomic instability, or misfolded proteins, all of which promote ER stress and UPR signaling. ER stress is further compounded by an avid uptake of free SFAs such as PA, the most abundant SFA in human serum and in most diets. Based on our mechanistic findings, and given its cytoprotective function, we propose that SCD1 activation is a metabolic adaptation by tumor cells to mitigate cellular damage under prolonged ER stress. This, in addition to its critical role in UFA synthesis essential for tumor cell proliferation and signaling, creates a “non-oncogene addiction” to SCD1 activity in GBM.
Prominent ER stress characterized by elevated expression of BiP renders GBM cells highly susceptible to SCD1 inhibition. An increased expression of BiP is commonly observed in cancer and correlates with chemoresistance (Luo and Lee, 2013, Roller and Maddalo, 2013). Since the upregulation of SCD1 is part of the cellular adaptation to ER stress, and SCD1 expression promotes self-renewal and tumor growth, increased SCD1 expression is de facto linked to therapeutic resistance. Indeed, SCD1 was found to be upregulated in TMZ-resistant GBM cell lines, and this upregulation reportedly promotes resistance to TMZ through the activation of AKT signaling (Dai et al., 2017). ER stress can affect genomic stability and DNA-repair mechanisms (Chevet et al., 2015). SCD1 inhibition resulted in increased DNA damage. Furthermore, the UPR triggers aberrant proteasomal degradation of proteins, a process referred to as ER-associated degradation (Travers et al., 2000). Accordingly, we propose an alternative mechanism of TMZ sensitization in GSCs with impaired SCD1 activity, primarily driven by overwhelming ER stress and the ensuing UPR-mediated degradation of RAD51.
Our study provides a proof of concept that effective pharmacological targeting of SCD1 achieves a strong therapeutic outcome, since intranasal delivery of CAY at relatively low doses significantly improved overall survival in one GSCs xenograft mouse model and completely prevented tumor growth with a 100% cure rate in a second model. This strong in vivo therapeutic outcome is likely attributed to a heightened ER stress and therefore increased vulnerability to SCD1 inhibition in the tumor environment. While this outcome is very promising, there are significant challenges that remain pertinent. One key question is the ideal delivery route of such inhibitors in humans in a way to maximize brain tumor penetrance and minimize systemic toxicity. We propose that delivery of therapeutics such as SCD1 inhibitors through nasal instillation warrants careful consideration and should be evaluated. This technique has several advantages, chief among which is its ability to bypass the blood-brain barrier (Chauhan and Chauhan, 2015) and minimize systemic toxicity due to rapid delivery of molecules directly to the brain. For clinical evaluation of such therapy, target engagement and effectiveness of SCD1 inhibitors could be evaluated in patients through in vivo proton magnetic resonance spectroscopy, which can readily detect lipid peaks and predominantly those corresponding to UFAs (Griffin et al., 2003).
Our in vivo results strongly suggest that free UFAs cannot circumvent or protect from SCD1 inhibition. It should be noted that all experimental animals were fed ad libitum with a chow diet, which contains a significant amount of fatty acids, in particular OA. Furthermore, we argue that the overwhelming cytotoxicity caused by SCD1 inhibition is due to toxic accumulation of SFAs in the tumor and ensuing ER stress. Based on our results, targeting of SCD1, but not general regulators of lipogenesis (SREBP1, LXR) or other de novo lipogenesis enzymes upstream of SCD1, is likely to be most effective because of the resulting accumulation of SFAs.
Taken together, our findings reveal that SCD1 activity provides a survival advantage for GBM cancer stem cells, which presents a metabolic vulnerability to SCD1 inhibition. We provide preclinical evidence that effective targeting of this enzyme can achieve a robust therapeutic outcome for this incurable brain cancer.
Experimental Procedures
Cell-Based Assays
CellTiter-Glo (Promega) was used to measure cell viability. CellTiter 96 Aqueous One Solution (Promega) was used to measure cell proliferation. Caspase-3/7 activity was detected using Caspase-Glo 3/7 (Promega). All reagents were used as recommended by the manufacturer. For data analysis, each data point in the treated samples was normalized to its respective vehicle or pretreatment control. Detailed methods for different experiments can be found in the Supplemental Information.
Immunoblot Analysis
Cells were lysed in RIPA buffer (Boston Bio Products) supplemented with protease and phosphatase inhibitors. Proteins were quantified using the Bradford protein determination assay (Bio-Rad), and 20–30 μg of protein were loaded and resolved on 10% NuPAGE Bis-Tris gels (Life Technologies) then transferred to nitrocellulose membranes (Bio-Rad) before incubation with the indicated antibodies. Proteins were detected using SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Immunocytochemistry
Cells grown as neurospheres were fixed with ice-cold acetone for 20 min. Cells were mounted on slides, air dried, permeabilized with 0.1% Triton X-100, and simultaneously blocked with 5% BSA for 1 h at room temperature. Cells were incubated overnight at 4°C with γ-H2AX antibody (1:400) or SOX2 and NESTIN antibodies (1:100). Fluorophore-conjugated secondary antibodies (Life Technologies, 1:100) were then added and incubated for 1 h. Cell nuclei were counterstained with DAPI (Life Technologies), mounted on a microscope slide, and analyzed by fluorescence microscopy.
Mouse Orthotopic Brain Tumor Models
All animal studies were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care and complied with guidelines set forth by the NIH Guide for the Case and Use of Laboratory Animals. GSCs (at the indicated dose) expressing Firefly luciferase (Fluc) were stereotactically implanted into the left forebrain of nude mice (2.5 mm lateral and 0.5 mm anterior to bregma, at a 2.5-mm depth from the skull surface). Tumor initiation and growth were monitored by imaging Fluc bioluminescence activity using a Xenogen IVIS 200 Imaging System (PerkinElmer), after intraperitoneal (i.p.) injections of D-luciferin (150 mg/kg body weight) (Gold Biotechnology). Image intensity was quantitated using the Living Image software 4.3.1 (PerkinElmer). CAY was delivered by topical instillation in the nose; this compound was initially dissolved in DMSO (100 mM). CAY was freshly resuspended in a 20% solution of (2-hydroxypropyl)-β-cyclodextrin (Sigma) and injected dropwise intranasally (final volume 20 μL). PA was delivered by i.p. injections.
Histological Analysis
Brains were collected following euthanasia, fixed in 4% paraformaldehyde, and cryoprotected in sucrose solution. Brain sections were prepared from fresh-frozen brain and subjected to H&E, BODIPY 493/503 (Thermo Fisher), or Ki67 staining according to standard protocols.
Statistical Analysis
All cell culture experiments (with the exception of fatty acid analyses, which were performed in one replicate) consisted of a minimum of three independent replicates and were repeated at least three times. Statistical significance was calculated using a two-tailed Student's t test, and p values of 0.05 or less were considered significant. The results are presented as the mean ± SD. Experiments involving animal survival were analyzed using a log-rank (Mantel-Cox) test and plotted as Kaplan-Meier survival curves using GraphPad Prism. Group size was solely determined based on preliminary tests, and no statistical method was used to determine sample size.
Author Contributions
Conceptualization, C.E.B.; Methodology, A.K.I and C.E.B.; Investigation, K.P., D.J.P., A.H., A.B.K., I.A., K.R., C.C.d.H., J.T., P.S.C., L.C., G.G.-I., A.K., and C.E.B.; Writing – Original Draft, K.P., D.J.P., and C.E.B. Writing – Review & Editing, K.P., D.J.P., and C.E.B.; Visualization: C.E.B.; Funding Acquisition, C.E.B.; Resources, C.E.B.; Supervision, C.E.B.
Acknowledgments
We are very grateful to Drs. Hiroaki Wakimoto (MGH), Ichiro Nakano (University of Alabama Birmingham) and Brent Reynolds (University of Florida) for providing primary GBM cells; Dr. Peter Walter (USCF/HHMI) for providing Ceapin A7; Dr. Bakhos Tannous for critical reading of the manuscript; and Dr. Anders Naar for helpful discussion and input. We also thank Dr. Luzia Sampaio and Mohamed El-Abtah for technical assistance. We acknowledge the MGH Vector Core supported by NIH/NINDS P30NS04776 and 1S10RR025504 Shared Instrumentation grant for the IVIS imaging system. This work was supported by the NIH, NCI grant K22CA197053 (to C.E.B.)
Published: March 28, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.02.012.
Supplemental Information
References
- Adams C.M., Reitz J., De Brabander J.K., Feramisco J.D., Li L., Brown M.S., Goldstein J.L. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J. Biol. Chem. 2004;279:52772–52780. doi: 10.1074/jbc.M410302200. [DOI] [PubMed] [Google Scholar]
- Carta G., Murru E., Banni S., Manca C. Palmitic acid: physiological role, metabolism and nutritional implications. Front. Physiol. 2017;8:902. doi: 10.3389/fphys.2017.00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerezo M., Lehraiki A., Millet A., Rouaud F., Plaisant M., Jaune E., Botton T., Ronco C., Abbe P., Amdouni H. Compounds triggering ER stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell. 2016;29:805–819. doi: 10.1016/j.ccell.2016.04.013. [DOI] [PubMed] [Google Scholar]
- Chauhan M.B., Chauhan N.B. Brain uptake of neurotherapeutics after intranasal versus intraperitoneal delivery in mice. J. Neurol. Neurosurg. 2015;2 [PMC free article] [PubMed] [Google Scholar]
- Chevet E., Hetz C., Samali A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015;5:586–597. doi: 10.1158/2159-8290.CD-14-1490. [DOI] [PubMed] [Google Scholar]
- Cross B.C., Bond P.J., Sadowski P.G., Jha B.K., Zak J., Goodman J.M., Silverman R.H., Neubert T.A., Baxendale I.R., Ron D., Harding H.P. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. U S A. 2012;109:E869–E878. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos-Ruiz J.R., Bettigole S.E., Glimcher L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168:692–706. doi: 10.1016/j.cell.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S., Yan Y., Xu Z., Zeng S., Qian L., Huo L., Li X., Sun L., Gong Z. SCD1 confers temozolomide resistance to human glioma cells via the Akt/GSK3beta/beta-catenin signaling axis. Front. Pharmacol. 2017;8:960. doi: 10.3389/fphar.2017.00960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S., Yalcin A., Lee G.Y., Li P., Fan J., Arruda A.P., Pers B.M., Yilmaz M., Eguchi K., Hotamisligil G.S. Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci. Transl. Med. 2015;7:292ra298. doi: 10.1126/scitranslmed.aaa9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher C.M., Garri C., Cain E.L., Ang K.K., Wilson C.G., Chen S., Hearn B.R., Jaishankar P., Aranda-Diaz A., Arkin M.R. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6alpha branch. Elife. 2016;5 doi: 10.7554/eLife.11878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R., Wang L., Wang E.S., Perera B.G., Igbaria A., Morita S., Prado K., Thamsen M., Caswell D., Macias H. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158:534–548. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J.L., Lehtimaki K.K., Valonen P.K., Grohn O.H., Kettunen M.I., Yla-Herttuala S., Pitkanen A., Nicholson J.K., Kauppinen R.A. Assignment of 1H nuclear magnetic resonance visible polyunsaturated fatty acids in BT4C gliomas undergoing ganciclovir-thymidine kinase gene therapy-induced programmed cell death. Cancer Res. 2003;63:3195–3201. [PubMed] [Google Scholar]
- Guo D., Prins R.M., Dang J., Kuga D., Iwanami A., Soto H., Lin K.Y., Huang T.T., Akhavan D., Hock M.B. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009;2:ra82. doi: 10.1126/scisignal.2000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D., Lerner A.G., Vande Walle L., Upton J.P., Xu W., Hagen A., Backes B.J., Oakes S.A., Papa F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi M., Lin A., Smeal T., Minden A., Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Igal R.A. Stearoyl-CoA desaturase-1: a novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis. 2010;31:1509–1515. doi: 10.1093/carcin/bgq131. [DOI] [PubMed] [Google Scholar]
- Lathia J.D., Mack S.C., Mulkearns-Hubert E.E., Valentim C.L., Rich J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis C.A., Brault C., Peck B., Bensaad K., Griffiths B., Mitter R., Chakravarty P., East P., Dankworth B., Alibhai D. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene. 2015;34:5128–5140. doi: 10.1038/onc.2014.439. [DOI] [PubMed] [Google Scholar]
- Lhomond S., Avril T., Dejeans N., Voutetakis K., Doultsinos D., McMahon M., Pineau R., Obacz J., Papadodima O., Jouan F. Dual IRE1 RNase functions dictate glioblastoma development. EMBO Mol. Med. 2018;10 doi: 10.15252/emmm.201707929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Condello S., Thomes-Pepin J., Ma X., Xia Y., Hurley T.D., Matei D., Cheng J.X. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell. 2017;20:303–314.e5. doi: 10.1016/j.stem.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B., Lee A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–818. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P., Joshi K., Li J., Kim S.H., Li P., Santana-Santos L., Luthra S., Chandran U.R., Benos P.V., Smith L. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. U S A. 2013;110:8644–8649. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason P., Liang B., Li L., Fremgen T., Murphy E., Quinn A., Madden S.L., Biemann H.P., Wang B., Cohen A. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS One. 2012;7:e33823. doi: 10.1371/journal.pone.0033823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning J., Hong T., Ward A., Pi J., Liu Z., Liu H.Y., Cao W. Constitutive role for IRE1alpha-XBP1 signaling pathway in the insulin-mediated hepatic lipogenic program. Endocrinology. 2011;152:2247–2255. doi: 10.1210/en.2010-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noto A., Raffa S., De Vitis C., Roscilli G., Malpicci D., Coluccia P., Di Napoli A., Ricci A., Giovagnoli M.R., Aurisicchio L. Stearoyl-CoA desaturase-1 is a key factor for lung cancer-initiating cells. Cell Death Dis. 2013;4:e947. doi: 10.1038/cddis.2013.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obacz J., Avril T., Le Reste P.J., Urra H., Quillien V., Hetz C., Chevet E. Endoplasmic reticulum proteostasis in glioblastoma—from molecular mechanisms to therapeutic perspectives. Sci. Signal. 2017;10 doi: 10.1126/scisignal.aal2323. [DOI] [PubMed] [Google Scholar]
- Ostrom Q.T., Gittleman H., de Blank P.M., Finlay J.L., Gurney J.G., McKean-Cowdin R., Stearns D.S., Wolff J.E., Liu M. American brain tumor association adolescent and young adult primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 2016;18(Suppl 1):i1–i50. doi: 10.1093/neuonc/nov297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrko P., Schonthal A.H., Hofman F.M., Chen T.C., Lee A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–9816. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- Roller C., Maddalo D. The molecular chaperone GRP78/BiP in the development of chemoresistance: mechanism and possible treatment. Front. Pharmacol. 2013;4:10. doi: 10.3389/fphar.2013.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rome S., Lecomte V., Meugnier E., Rieusset J., Debard C., Euthine V., Vidal H., Lefai E. Microarray analyses of SREBP-1a and SREBP-1c target genes identify new regulatory pathways in muscle. Physiol. Genomics. 2008;34:327–337. doi: 10.1152/physiolgenomics.90211.2008. [DOI] [PubMed] [Google Scholar]
- Romero-Ramirez L., Cao H., Nelson D., Hammond E., Lee A.H., Yoshida H., Mori K., Glimcher L.H., Denko N.C., Giaccia A.J. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–5947. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Short S.C., Giampieri S., Worku M., Alcaide-German M., Sioftanos G., Bourne S., Lio K.I., Shaked-Rabi M., Martindale C. Rad51 inhibition is an effective means of targeting DNA repair in glioma models and CD133+ tumor-derived cells. Neuro Oncol. 2011;13:487–499. doi: 10.1093/neuonc/nor010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava N.K., Pradhan S., Gowda G.A., Kumar R. In vitro, high-resolution 1H and 31P NMR based analysis of the lipid components in the tissue, serum, and CSF of the patients with primary brain tumors: one possible diagnostic view. NMR Biomed. 2010;23:113–122. doi: 10.1002/nbm.1427. [DOI] [PubMed] [Google Scholar]
- Szegezdi E., Logue S.E., Gorman A.M., Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K., Iafrate A.J., Ho Q., Curry W.T., Batchelor T.T., Flaherty K.T., Onozato M.L., Lelic N., Sundaram S., Cahill D.P. Myc-driven glycolysis is a therapeutic target in glioblastoma. Clin. Cancer Res. 2016;22:4452–4465. doi: 10.1158/1078-0432.CCR-15-2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng J., Carla da Hora C., Kantar R.S., Nakano I., Wakimoto H., Batchelor T.T., Chiocca E.A., Badr C.E., Tannous B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro Oncol. 2017;19:820–832. doi: 10.1093/neuonc/now253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W., Welihinda A.A., Kaufman R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers K.J., Patil C.K., Wodicka L., Lockhart D.J., Weissman J.S., Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Urra H., Dufey E., Avril T., Chevet E., Hetz C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer. 2016;2:252–262. doi: 10.1016/j.trecan.2016.03.007. [DOI] [PubMed] [Google Scholar]
- Volmer R., van der Ploeg K., Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U S A. 2013;110:4628–4633. doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto H., Mohapatra G., Kanai R., Curry W.T., Jr., Yip S., Nitta M., Patel A.P., Barnard Z.R., Stemmer-Rachamimov A.O., Louis D.N. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol. 2012;14:132–144. doi: 10.1093/neuonc/nor195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Kaufman R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- Weatherbee J.L., Kraus J.L., Ross A.H. ER stress in temozolomide-treated glioblastomas interferes with DNA repair and induces apoptosis. Oncotarget. 2016;7:43820–43834. doi: 10.18632/oncotarget.9907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xipell E., Aragon T., Martinez-Velez N., Vera B., Idoate M.A., Martinez-Irujo J.J., Garzon A.G., Gonzalez-Huarriz M., Acanda A.M., Jones C. Endoplasmic reticulum stress-inducing drugs sensitize glioma cells to temozolomide through downregulation of MGMT, MPG, and Rad51. Neuro Oncol. 2016;18:1109–1119. doi: 10.1093/neuonc/now022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.