

Abstract
Cervical cancer is driven by persistent infection of human papillomavirus (HPV), which is influenced by HPV type and intratypic variants, yet the impact of HPV type and intratypic variants on patient outcomes is far less understood. Here, we examined the association of cervical cancer stage and survival with HPV type, clade, lineage, and intratypic variants within the HPV E6 locus. Of 1,028 HPV-positive cases recruited through the CerGE study, 301 were in-situ and 727 were invasive cervical cancer (ICC), with an average post-diagnosis follow-up of 4.8 years. HPV sequencing was performed using tumor-isolated DNA to assign HPV type, HPV 16 lineage, clade, and intratypic variants within the HPV 16 E6 locus, of which nonsynonomous variants were functionally annotated by molecular modeling. HPV 18-related types were more prevalent in ICC compared to in-situ disease and associated with significantly worse recurrence-free survival (RFS) compared to HPV 16-related types. The HPV 16 Asian American lineage D3 and Asian lineage A4 associated more frequently with ICC than with in situ disease and women with an intratypic HPV 16 lineage B exhibited a trend toward worse RFS than those with A, C, or D lineages. Participants with intratypic E6 variants predicted to stabilize the E6–E6AP–p53 complex had worse RFS. Variants within the highly immunogenic HPV 16 E6 region (E14–I34) were enriched in ICC compared to in-situ lesions but were not associated with survival. Collectively, our results suggest that cervical cancer outcome is associated with HPV variants that affect virus-host interactions.
Keywords: cervical cancer, human papillomavirus (HPV), HPV variants, cancer risk, HPV types, E6 oncoprotein, patient outcome
Introduction
Most cervical cancer is caused by persistent infection with human papillomavirus (HPV), with viral sequences detected in more than 95% of cases.1 While HPV infections are common, persistent viral infection leading to ICC is less common. Of the >200 different HPV types to date, 20 have been linked to development of invasive cancer (ICC).1–4 Other factors that may increase an individual’s likelihood of developing ICC include smoking,5,6 use of hormonal contraception,7 and immunosuppression.8 Specific viral types and intratypic variants have also been linked to increased risk of disease.9–11 However, the impact of HPV type, intratypic variants, and functional variants within the viral DNA sequence on cervical cancer outcome remains poorly understood.
HPV type and intratypic variants are determined by analyzing the HPV genome sequence, which encodes genes for two capsid proteins (L1 and L2) and six nonstructural regulatory proteins (E1, E2, E4, E5, E6, and E7).11 HPV types are designated by the variations in the nucleotide sequence of the L1 capsid,1,2,11 with the most common oncogenic HPV types 16 and 18 being detected in 50% and 20% of cervical cancer cases, respectively.11 Although HPV 16 is the most prevalent type in cervical cancer, HPV 18 typically associates with worse survival.5,12–16 HPV types are further classified into phylogenic clades. High-risk HPV clades have been identified as alpha-9 (A9; comprised of HPV 16, 31, 33, 35, 52, 58, and 67) and alpha-7 (A7; comprised of HPV 18, 39, 45, 59, 68, 70, 85, and 97).2,9 We previously demonstrated that the most common high-risk HPV types can also be defined as HPV 16-related (HPV 16, 31, and 52) or HPV 18-related (HPV 18 and 45).5,17–21 Furthermore, HPV 16 can be classified into four major lineages (A, B, C, D) and several alphanumerical sublineages based on the broader genetic profile of the viral genome.2
HPV typing is based on variations in the conserved region of the L1 capsid protein. However, the mechanism(s) by which certain HPV types drive cervical cancer involves other gene products in the HPV genome, such as the viral oncogenes E6 and E7.4 The viral E6 oncoprotein facilitates ubiquitin-mediated proteasomal degradation of the tumor suppressor protein, p53, and works in conjunction with the viral E7 protein to promote the initiation and progression of cervical cancer in cells infected with a high risk HPV.22 Some intratypic HPV 16 variants have also been associated with viral persistence and progression to CIN3.10,23 However, how these intratypic HPV 16 variants may correlate with cervical cancer survival has yet to be studied in detail.
Here, we examine the association of HPV lineage and sequence variants with survival and clinical parameters in a population of 1,028 women with ICC and in situ disease. We used previously identified viral type associations and a novel molecular modeling approach to classify HPV E6 variants into functional mechanisms that affect the interface between the virus and host systems. Our study revealed that differences in viral type, HPV 16 lineage, and nucleotide sequence variants within the E6 binding motif of HPV 16 significantly associate with survival from cervical cancer.
Methods
Study population
We investigated 1,028 HPV-positive cervical tumors from participants enrolled in the Cervical Cancer Genetic Epidemiology (CerGE) study.12 Subjects in the CerGE study were recruited to a trio study developed to investigate inherited genetic polymorphisms and HPV subtypes and variants that contribute to cervical cancer. Patients had ICC or CIN3 (CIN3 and/or adenocarcinoma in situ). The participants completed a questionnaire that included Ethnic group and the average follow-up was 4.8 years after diagnosis. All participants with available tumor had HPV typing completed on their tumor DNA. All HPV16 and HPV18-postive cases were considered for analysis in our study. Characteristics of the participants, their tumors, and terms for analysis exclusion are provided in Table 1 and accompanying CONSORT diagram (Supporting Information Fig. 1). Of note, most of the analyses conducted in our study were confined to ICC cases. Comparison between the prevalence of ICC and in situ disease was only tested in the examination of HPV types and lineages. All data presented in Figures 1–4 represents analysis of ICC cases only. Our study protocol was approved by the Institutional Review Board of the Medical College of Wisconsin.
Table 1.
Cohort characteristics
| White | Black | Other | p-value | |
| Total participants | 853 (83.0%) | 165 (16.0%) | 10 (1.0%) | 0.997 |
| Mean age at diagnosis (± SD) | 38.6 ± 12.1 | 41.0 ± 13.2 | 41.3 ± 9.9 | 0.065 |
| Stage | ||||
| 0 | 250 (29.3%) | 48 (29.1%) | 3 (30.0%) | 0.042 |
| I–II | 544 (63.8%) | 91 (55.2%) | 7 (70.0%) | |
| III–IV | 53 (6.2%) | 25 (15.2%) | 0 (0.0%) | |
| Unstaged | 6 (0.7%) | 1 (0.6%) | 0 (0.0%) | |
| Histology - ICC | <0.001 | |||
| Squamous cell carcinoma | 412 (68.3%) | 102 (87.2%) | 2 (28.6%) | |
| Adenocarcinoma | 139 (23.1%) | 6 (5.1%) | 3 (42.9%) | |
| Adenosquamous | 31 (5.1%) | 5 (4.3%) | 2 (28.6%) | |
| Other | 21 (3.5%) | 4 (3.4%) | 0 (0.0%) | |
| Genotypes and lineages of HPVs compared to cervical cancer stage | ||||
| Stage 0 | Stage I-II | Stage III-IV | p-value | |
| HPV type(s)1 | ||||
| HPV16-related | 199 (85.8%) | 377 (72.2%) | 43 (82.7%) | <0.001 |
| HPV18-related | 33 (14.2%) | 145 (27.8%) | 9 (17.3%) | |
| A7 clade1 | 34 (13.6%) | 142 (28.0%) | 13 (20.6%) | <0.001 |
| A9 clade | 216 (86.4%) | 365 (72.0%) | 50 (79.4%) | |
| HPV 16 lineages1 | ||||
| A1–A3; European | 140 (84.8%) | 217 (73.6%) | 26 (76.5%) | 0.037 |
| A4; Asian | 2 (1.2%) | 16 (5.4%) | 1 (2.9%) | |
| B; African (Af1) | 5 (3.0%) | 11 (3.7%) | 2 (5.9%) | |
| C; African (Af2) | 2 (1.2%) | 4 (1.4%) | 1 (2.9%) | |
| D1; North American | 7 (4.2%) | 5 (1.7%) | 2 (5.9%) | |
| D3; Asian American | 9 (5.5%) | 42 (14.2%) | 2 (5.9%) | |
| HPV 16 E6 variants | ||||
| Destabilize protein structure | 10 (6.2%) | 29 (9.4%) | 4 (10.8%) | 0.472 |
| Destabilize E6–E6AP–p53 complex | 27 (16.7%) | 72 (23.3%) | 7 (18.9%) | 0.436 |
| Stabilize E6–E6AP–p53 complex | 4 (2.5%) | 12 (3.9%) | 3 (8.1%) | 0.344 |
| Immunogenic region 1 (E14–I34) | 22 (13.6%) | 81 (26.2%) | 8 (21.6%) | 0.030 |
| Immunogenic region 1 (L45–A68) | 22 (13.6%) | 51 (16.5%) | 5 (13.5%) | 0.649 |
Some tumors had more than one HPV type, HPV Clade, and HPV 16 lineage and were excluded from the analysis. Stage 0 samples were not used in survival analyses.
Figure 1.
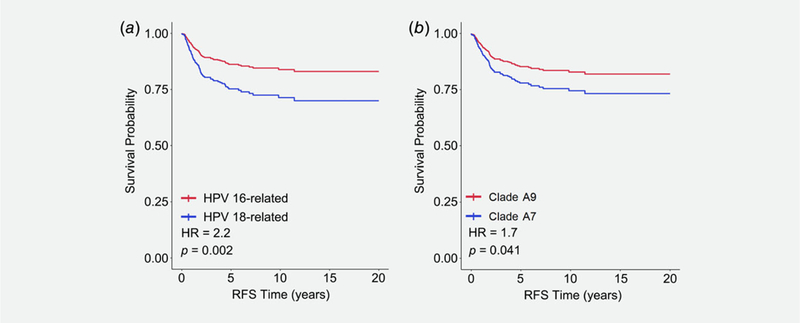
Kaplan–Meier recurrence-free survival in ICC patients that is associated with infection by (a) HPV16-related (red line) and HPV18-related (blue line) strains and (b) A9 (red line) and A7 clades (blue line).
Figure 4.
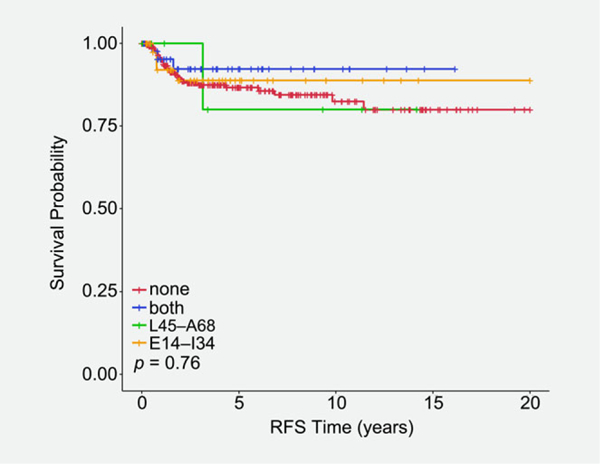
Kaplan–Meier recurrence-free survival in ICC patients that is associated with nonsynonymous variants that fall within the highly immunogenic regions of the E6 protein: E14–I34 (orange line), L45–A68 (green line), or both (blue line). ICC cases with no E6 variants in these regions are represented by the red line.
Sequencing methods
All samples were obtained at the time of diagnosis and cervical tissue was either snap frozen in Tissue-Tek optimum cutting temperature compound (Sakura Finetek USA, Inc.) or formalin-fixed and embedded in paraffin. DNA was then extracted and subjected to HPV typing as previously reported.17,18 The E6 gene of HPV 16-positive cases was amplified using a three- or four- primer set system. The PCR products were purified using the Agencourt AMPure PCR purification system (Beckman Coulter Genomics, Danvers, MA), and sequenced by Sanger sequencing at the Washington University Genome Sequencing Center in St. Louis or the Human and Molecular Genetics Center’s Sequencing Service Core Facility at the Medical College of Wisconsin. The resulting HPV 16 E6 sequences were aligned with those of the E6 reference sequence (Genbank, K02718), using the BLAST 2.0 software server (http://www.ncbi.nih.gov/BLAST/). Cases were then assigned E6 lineages, and all the variants were listed according to the E6 reference sequence. The E6 locus could not be completely sequenced in 74 cases due to failed PCR amplification or insufficient sample. There were no significant differences in overall survival (p = 0.98), recurrence-free survival (p = 0.52), age (p = 0.45), Ethnic group (p = 0.52), stage (p = 0.32), or histology (p = 0.68) between patients with complete or incomplete E6 sequence data. Out of 350 HPV 16- positive ICC patients with a fully sequenced E6 locus, we observed 130 patients with zero nonsynonymous E6 variants, 103 patients with one nonsynonymous E6 variant, 43 patients with two nonsynonymous E6 variants, and 74 patients with three or more nonsynonymous E6 variants. Participants with phylogenetically related HPV types were grouped according to our previous work as HPV 16-related or HPV 18-related strains5,17–21 and by variant A7 or A9 clades.2,9
Structural bioinformatics
An experimental structure of the HPV E6 protein bound to the human E6AP and P53 proteins,24 referred to as the E6–E6AP–p53 complex, was used to model the effects of intratypic variants in the HPV 16 E6 locus. Protein annotations were gathered from UniProt25 (E6, P03126; E6AP, Q05086; P53, P04637) and protein structures for the complex and several individual E6 domains solved by NMR spectroscopy26–28 from the Protein Data Bank (https://www.rcsb.org/).29 Using FoldX version 4 (http://foldxsuite.crg.eu/), we calculated changes in folding stability (ΔΔGfold) for each variant, both for E6 alone and in the E6–p53–E6AP complex. Intratypic HPV 16 variants leading to amino acid changes were categorized according to their likelihood to impact three key aspects E6 protein function: 1) direct destabilization of E6 protein structure, 2) destabilization of the E6–E6AP–p53 complex, or 3) stabilization of the E6–E6AP–p53 complex (Table 2). Protein structure images were generated using PyMol version 1.9.0 (Schrödinger, Cambridge, MA).
Table 2.
HPV 16 E6 variants
| Structural/functional characteristic |
Polymorphisms |
|---|---|
| Destabilize protein structure | R17K, R17T, R17G, R17I, K41E, I59V |
| Destabilize E6-E6AP-p53 complex | R17G, Q21H, T29S, K41E, I59V, H85Y, Q98R |
| Stabilize E6-E6AP-p53 complex | R17I and Q21D |
| Immunogenic region 1 | E14–I34 |
| Immunogenic region 2 | L45–A68 |
Statistical analysis
One-way ANOVA was used to compare patient age across Ethnic group in all cervical cancer patients (Table 1). Chi-square tests were used to compare Ethnic group with histological classification of cervical cancer and stage of all cervical cancer patients (Table 1). Chi-square tests were also used for comparison of cervical cancer stage (0, I-II, and III/IV) with HPV type, clade, lineage, and intratypic E6 locus variants in cervical cancer patients in which the variables could be assigned (i.e., patients with missing variables were excluded from the analysis; Table 1). Kaplan–Meier curves were generated for survival, and the log-rank statistic was used for testing the association of HPV type, clade, lineage, and intratypic E6 locus variants with outcome in patients with ICC (in situ cases were not included in these analyses). RFS was defined as the time elapsed since the cancer was diagnosed until death and/or first recurrence of the tumor within a 20-year window. Cox regressions were used to measure the effects of HPV type and clade on RFS, with Ethnic group and histopathological type considered as covariates. To avoid over stratification of the data, only univariate analysis with the log-rank statistic was used to test whether lineage and intratypic nonsynonomous variants in the HPV16 E6 locus had a significant impact on RFS. For all analyses, a p-value of equal or less than 0.05 was considered as statistically significant. All statistical analyses were performed using R version 3.5.0 statistical software.
Results
Ethnic group
Self-reported Ethnic group within the CerGE study identified 853 white patients (83%) and 164 black patients (16%), which were evenly distributed between in-situ disease and ICC (Table 1). Age at diagnosis was slightly younger in white patients (38.6 ± 12.1) compared to black patients (41.0 ± 13.2), but that difference was not statistically significant. Ethnic group correlated significantly with disease stage at diagnosis (p = 0.042) and histological type (p < 0.001), with black patients being more likely to be diagnosed with more advanced disease (Table 1). Neither HPV type nor clade associated significantly with Ethnic group (data not shown).
HPV type and clade
HPV types were assessed for 301 patients with stage 0, in-situ disease and 720 patients with ICC stages I–IV (Table 1). As demonstrated previously,1 HPV 16-related types were more prevalent in cervical cancer (~77% of HPV-typed cases) compared to HPV 18-related types (~23% of HPV-typed cases). However, the prevalence of HPV 18-related types was significantly higher in ICC compared to in-situ disease (p < 0.001; Table 1). Participants with HPV 18-related types also had significantly lower recurrence-free survival (RFS; HR = 2.2; p = 0.002) than those with HPV 16-related types (Fig. 1a). Also, patients with HPV clade A7 were far more likely to present with ICC (p < 0.001) compared to patients infected with HPV clade A9 (Table 1), corresponding to a worse RFS in ICC patients with clade A7 (HR = 1.7; p = 0.041; Fig. 1b).
HPV 16 lineage
DNA sequences from L1 and the entire E6 locus were used to assign HPV 16 lineages to 495 in situ and ICC stages I-IV cases. The European lineage group A1–A3 was the most prevalent in the cohort (n = 383 cases), followed by the Asian American lineage D3 (n = 53 cases), Asian lineage A4 (n = 19), African lineage B (n = 18), North American lineage D1 (n = 14), and African lineage C (n = 7; Table 1). The six HPV lineage groups showed a more significant correlation with cervical cancer stage (p = 0.037). The HPV lineage groups also showed a moderate correlation with outcomes in patients with ICC (p = 0.069), with the African lineage group B exhibiting a trend toward worse RFS than the other lineage groups (Fig. 2).
Figure 2.
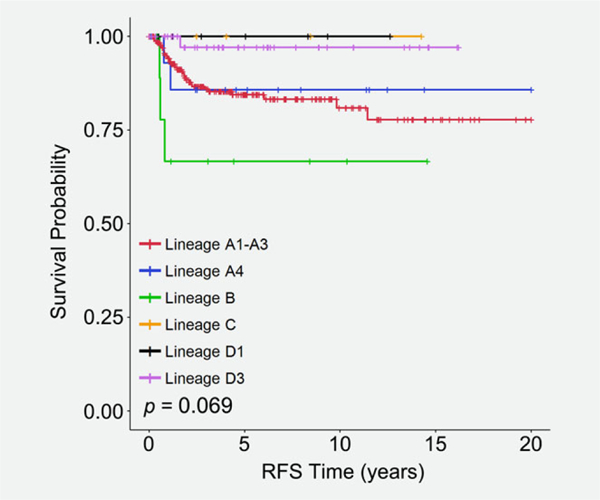
Kaplan–Meier recurrence-free survival in ICC patients that is associated with HPV16 lineages: A1–A3 (red line), A4 (blue line), B (green line), C (orange line), D1 (black line), and D3 (pink line).
HPV 16 variants in the E6 oncoprotein
Based on differences in protein structure, we classified the E6 variants as potentially impacting the stability of the E6 protein or affecting the binding interface of E6 with either E6AP or p53. We hypothesized that inactivation of the p53 tumor suppressor by E6 would be attenuated by intratypic HPV 16 variants that destabilized either the E6 protein or the interaction of E6 with the E6AP–p53 complex. Thus, destabilization would predict less aggressive disease and better outcomes. Alternatively, we hypothesized that intratypic HPV 16 variants that stabilized the interaction of E6 with E6AP–p53 would ultimately enhance the ability of E6 to sequester the p53 tumor suppressor, resulting in more aggressive disease and worse outcomes. To test these hypotheses, we identified DNA sequence variants across the E6 locus from 500 cases of in situ disease and ICC and, using a protein structure-based approach, evaluated which of them likely alter stability of the E6 protein or its interaction within the E6–E6AP–p53 complex (Figs. 3a and 3b). No significant correlation between E6 variant classes and disease stage was detected (Table 1).
Figure 3.
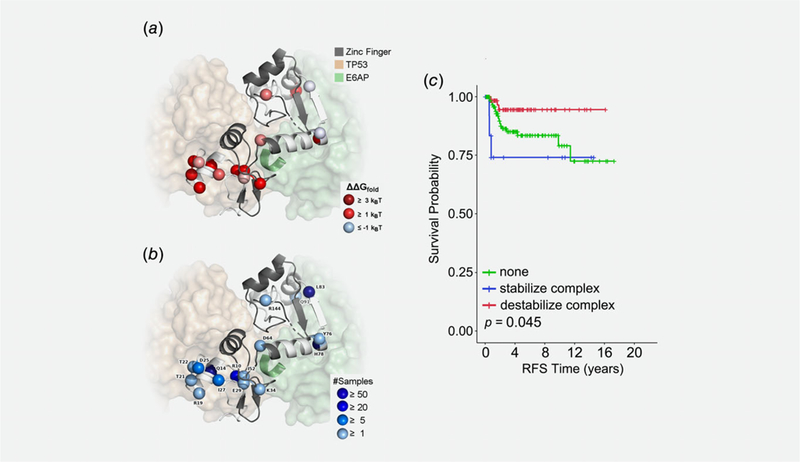
Structural and functional annotation of E6 variants and their association with outcome. We used existing experimental protein structures of E6 to calculate changes in stability for E6 alone and E6 in complex with TP53 (p53) and E6AP. (a) Cartoon of E6 with the residues in the two zinc finger domains colored gray and the other residues colored white. Spheres mark the variant sites, which are colored by their effects on folding stability of the protein complex. Proteins bound to E6 are shown in transparent surfaces, and the LXXLL motif of E6AP is shown in the cartoon. (b) The same sites are shown in spheres, now labeled by their WT residues and colored according to the number of samples they were observed. (c) Kaplan–Meier recurrence-free survival in ICC patients that is associated with HPV 16 nonsynonymous variants that were predicted to stabilize the E6–E6AP–p53 complex (blue line) or destabilize the E6–E6AP–p53 complex (red line) compared to ICC patients with no E6 variants (green line).
Protein structure modeling revealed 130 ICC cases with no E6 variants that were predicted to alter function, 33 ICC cases with only E6 variants that were predicted to destabilize the E6 protein structure (R17K, R17T, R17G, R17I, K41E, and I59V), 70 ICC cases with only E6 variants that were predicted to destabilize the E6–E6AP–p53 complex (R17G, Q21H, T29S, K41E, I59V, H85Y, and Q98R), and 15 cases with E6 variants that were predicted to stabilize the E6–E6AP–p53 complex (R17I and Q21D). Of note, 11 cases within the E6–E6AP–p53 complex stabilizing group also had at least one variant from the other two groups. No association with outcome was observed for the protein destabilizing group (data not shown). In contrast, nonsynonymous E6 variants in the complex stabilizing and complex destabilizing groups were associated with RFS in ICC patients, with worse outcomes observed in the E6–E6AP–p53 complex stabilizing group compared to the complex destabilizing group (Fig. 3c; HR = 6.4, p = 0.023).
Villada et al.30 identified two regions of the E6 protein (E7–I27 and L38–A61) that were predicted to be highly immunogenic for HPV 16-specific T cell responses in vulvar intra-epithelial neoplasia. We postulated that variants within these two regions might affect clinical stage at diagnosis and/or the survival of patients with cervical cancer. Patients with ICC had significantly more variants (p = 0.006) in the E14–I34 immunogenic region of E6 compared to patients with in-situ lesions (Table 1). In contrast, there was no significant difference (p = 0.671) in the number of variants detected in the L45–A68 immunogenic region between patients with in- situ lesions versus patients with ICC (Table 1). The RFS for ICC patients with variants in either immunogenic region trended toward better survival but did not reach statistical significance (Fig. 4).
Discussion
In a large cohort of cervical cancer patients, we explored the association of HPV types, lineages, clades, and intratypic HPV 16 E6 variants with clinical outcome. We found that the incidence of HPV 18-related types was significantly higher in ICC patients and correlated with worse outcome compared to HPV 16-related types, as has been reported previously.5,12–16 Likewise, we found that patients with HPV clade A7 were more likely to be diagnosed with ICC than those with HPV clade A9, which coincided with HPV clade A7 also being associated with worse RFS within the ICC group. Due to the high prevalence of HPV 16 in cervical cancer patients, we explored whether outcome could be linked with intratypic variants that define HPV 16 lineages or alter function and immunogenicity of the HPV 16 E6 protein. Our study revealed for the first time that multiple classes of nonsynonymous variants within the HPV 16 E6 locus are correlated with ICC outcome. Women with intratypic HPV 16 E6 variants that were predicted to stabilize the E6–E6AP–p53 complex had worse RFS, in agreement with the expected mechanism that increased binding would enhance p53 degradation and the silencing of cellular defense mechanisms. We also detected a strong association of HPV 16 E6 intratypic variants within a highly immunogenic region (E14–I34)30 with a higher likelihood of ICC compared to in situ disease, suggesting the mutation of highly antigenic residues within the E6 protein is a potential mechanism for HPV to suppress immune surveillance. Collectively, our study provides novel evidence that intratypic HPV variants play a potentially important role in the risk of ICC and disease outcome.
Interpreting the association of HPV type and clade with ICC and outcome
In our study, we confirmed that HPV 18-related types were more frequent in ICC patients and correlated with worse outcome of ICC compared to HPV 16-related types, as has been reported previously.5,12–16 In addition to reduced survival, HPV 18 associates with tumors showing deeper cervical stromal invasion, more nodal metastases, and higher rates of recurrent disease for women receiving radiation for stage 1 disease.31 Although all HPV-positive cancers have detectable expression of HPV E6- and E7-oncogene mRNAs,1 cancer progression might depend on the functional consequences of specific viral characteristics. For example, HPV 18 more frequently integrates into the host genome, producing more chromosomal aberrations; and has a higher ratio of unspliced to spliced E6 transcripts than HPV 16.1 Tang et al.32 showed differential host gene mRNA expression from cervical cancers containing different levels of viral oncogene expression and found that cancers with full-length E6 expression associated with a “dedifferentiated” host signature.32 HPV 18-positive cancers have lower levels of miR-375 (tumor suppressive) and higher levels of miR-944 (oncogenic) than cancers containing HPV 16.1,33,34 Thus, infection with HPV 18-related types may result in tumors that are more aggressive or more difficult to detect through Pap tests than those infected with other HPV types. HPV 18 frequently associates with glandular cancers (adeno in situ and adenocarcinoma) that are above the cervical transformation zone, high in the endocervical canal, and away from the mucosal surface, making sampling more difficult.35 This is highlighted by the increased adeno in situ detection when concurrent high grade squamous lesions are present in the cervix because they are identified during further work-up of the squamous lesion.36
Interpreting the association of intratypic HPV 16 variants with ICC and outcome
Individual HPV types can be assembled into phylogenetic trees grouped by DNA nucleotide variants, whose branches often correlate with major ethnic groups and cluster in regions of the world.2 We observed moderate differences in the RFS of ICC patients with different intratypic HPV 16 lineage groups (p = 0.069), with the HPV 16 African B lineage group correlating with the worst RFS compared to the remaining lineages. This contrasts with the findings of Zuna et al., who reported a statistically significant increased risk of death in patients harboring European variants compared to patients with non-European variants.37 Their cohort consisted of 155 women with HPV 16-positive ICC from the central United States, with a median follow up of 26.7 months.37 However, as the authors pointed out, the women with European variants had a higher stage than those with non-European variants (43% vs. 18%, stage II–IV), and stage and lymph node status are the most significant prognostic factors in cervical cancer.38 In our study, the six HPV lineage groups also showed a significant correlation with the incidence of in situ disease compared to ICC (p = 0.037). The most prevalent association with ICC was seen with the Asian American lineage D3 (83%) and the Asian lineage A4 (89%). Further research into differential outcomes between HPV 16 lineages in racially diverse populations should provide additional insights into the contributions of viral lineages to biological outcomes in cervical cancer.
We also examined variants in the E6 oncoprotein that might affect interactions with host proteins, because binding of the HPV 16 E6 oncoprotein to E6AP and p53 is vital to the E6 protein’s function. E6 induces the degradation of p53 and other cellular proteins. E6AP is a cellular E3 ubiquitin-protein ligase that is extensively involved in the ubiquitin-mediated degradation of E6-dependent substrates, including proteasomal degradation of p53 and the proinflammatory cytokine IL-1β in malignant cervical cancer cells.39 In a transgene mouse model, E6AP was required for HPV16 E6 to synergize with estrogen to promote cervical cancer,40 further supporting the role of the E6–E6AP–p53 complex in cervical cancer. E6AP was the first protein shown to interact with E6.41,42 Early studies identified E6 residues important for E6AP binding,43–45 but more recently mutations in these residues have been more specifically tied to global E6 stability.26
Here, we describe for the first time that genetic variants within the HPV 16 E6 locus were predicted to stabilize the E6–p53–E6AP complex (R17I and Q21D) and are associated with worse RFS compared to the complex destabilizing group (HR = 6.4, p = 0.023). Moreover, in some cases the dominant effects of the E6–E6AP–p53 complex stabilizing variants were observed in the presence of E6 variants from the other two functional groups. This observation agrees with the expected mechanism that the increased binding of E6 to the E6AP–p53 complex likely enhances the degradation of p53 and silencing of cellular defense mechanisms. E6 binds to E6AP at the LXXLL motif in E6AP, but previous studies have postulated that this would be impossible in the presence of p53 because p53 also binds to LXXLL, thus occluding the binding site for E6. However, the N-terminal zinc finger domain of E6 also has an LXXLL motif.28 Therefore, we believe that the previous observations highlight the importance of the LXXLL motif and the potential for a concentration-dependent switch in E6AP binding: at a high concentration of E6, self-association could compete with the binding of E6 to E6AP. These results suggest that strengthening the formation and function of this complex is more important to clinical outcomes in cancer than any factors that may interrupt this process. By examining nonsynonymous/synonymous rate ratios, Chen et al. identified R17 and Q21 as two of three codons in E6 that evolve under the influence of strong diversifying selection.46 Our data suggest that the selective pressure of these codons might be due to the functional consequences of these variants on interaction with the E6AP–p53 protein complex.
In addition to p53, the E6-E6-AP complex has also been shown to bind to human proteins Bak,47 NFX1,44 c-myc,48 SIPA1L1 (E6TP149), and MCM7.50 Thus, it is possible that HPV 16 E6 variants that affect the stabilization of the E6-E6AP-p53 complex may also affect the interaction of E6 with these proteins. On the other hand, although it is well established that HPV 16 E6 activates telomerase in epithelial cells,51,52 it has more recently been demonstrated that the regulation of hTERT by E6 does not require E6-E6AP binding.53 We would therefore not expect the E6-E6AP-p53 destabilizing variants to affect the ability of E6 to regulate hTERT. Future in vitro studies are needed to functionally validate our modeling data to specifically determine if these variants can indeed stabilize/destabilize the E6 protein alone or as a complex with E6AP and other host proteins.
Another possible implication of codon selection within the HPV 16 E6 locus is evasion of host immunity and to date two highly immunogenic regions of the HPV 16 E6 protein (E14–I34 and L45–A68) have been identified.30 The E6 protein is strongly immunogenic because it contains multiple epitopes that are presented to CD8+ T-cells on class I and II MHC complexes. Here, we investigated for the first time whether variants within the immunogenic regions of the HPV 16 E6 protein correlated with the risk of ICC and disease outcome. We found that patients with ICC had a significantly higher number of variants (p = 0.006) in the E14–I34 strongly immunogenic region30 compared to patients with in-situ lesions, but these differences did not impact disease outcome within the ICC group. Although we focused on these two well-described immunogenic regions of HPV 16 E6, it is important to note that we cannot rule out the possibility that there are additional, yet to be identified regions of E6 with similar immunogenic properties that may or may not associate with ICC. One interpretation of our immunogenic association data is that the mutation of highly antigenic residues within the E6 protein is a potential mechanism for HPV to suppress host immune surveillance. Supporting this hypothesis is evidence that the E6 sequence variant at R17 alters an HLA–B7 peptide-binding epitope that likely influences immune recognition.54 This region also contains two codons that were predicted to stabilize the E6–E6AP–p53 complex (R17I and Q21D), potentially confounding the interpretation of both results. Interestingly, while variants in the immunogenic region (E14–I34) were more prevalent in ICC compared to in situ disease and did not correlate with ICC survival, the variants that were predicted to stabilize the E6–E6AP–p53 complex (R17I and Q21D) correlated with worse ICC survival, yet were not enriched in ICC. Collectively, these data suggest that distinct mechanisms within these functional groupings of variants likely influence the development of ICC and disease outcome. An interesting avenue for future research would be to gather and analyze HLA typing data from these patients to explore how they may correlate with HPV 16 E6 variants in these highly immunogenic regions.
Despite the relatively large size of the current study, the lower frequency of individual variants within the HPV 16 E6 locus precluded the testing of individual variants in cervical cancer outcome. Larger studies are needed to better understand how viral variants independently affect survival in patients with invasive cervical cancer and how they may impact treatment. Further, the current research was limited to investigation of HPV 16 E6 variants and how they may influence E6 function. To date, there is very little information regarding how HPV 18 E6 variants may affect disease progression, outcome, and/or E6 function. In the few studies that have examined HPV 18 E6 variants in cervical disease, there were no significant associations with disease risk55 or disease progression.56 However, certain HPV 18 E6 intratypic variants have been reported to differentially affect gene expression and cellular pathways involved in driving cervical carcinogenesis.57 Future work is needed to elucidate the functional effects of HPV18 E6 variants in cervical disease.
Conclusions
The strengths of our study include large sample size, complete E6 sequence data, and well- annotated clinical outcomes with longer-term follow up, which enabled the determination of HPV types and intratypic variants that confer different biological and pathological consequences in cervical cancer. HPV intratypic variants may have functional differences in either their oncogenic potential or ability to circumvent host immune surveillance. We provide evidence that genetic variants that alter binding between the E6 oncoprotein and its critical host partners associate with cancer survival. Our mechanistic and structure-based model leads to more granular interpretation of the data – it is not region-based groupings of variants in E6, but function-based groupings that elucidate outcome associations. Further experimental work on the interaction/relationship between host and viral sequences will provide new insights into the contributions of viral variants to the development of invasive and metastatic cervical cancer.
Supplementary Material
What’s new?
Cervical cancer is driven by persistent infection of human papillomavirus (HPV), which is influenced by HPV type and intratypic variants. The impact of HPV type and intratypic variants on cervical cancer outcome remains poorly understood, however. The current study provides novel evidence that intratypic HPV variants play an important role in cervical cancer risk and disease outcome. Specifically, differences in viral type, HPV 16 lineage, and sequence variants within the E6 binding motif of HPV 16 significantly associate with survival of cervical cancer patients. These findings may drive the development of better prognostic markers and novel therapeutics for cervical cancer.
Acknowledgements
This work was supported by the NCI R01CA193343 (M. J. F), NCI R01CA095713 (JSR), the Mary Kay Foundation Grant No. 024–16 (M.J.F), the METAvivor Foundation (M.J.F), the Advancing a Healthier Wisconsin Endowment (M.J.F.), and the Women’s Health Research Program in the Department of Obstetrics and Gynecology, Medical College of Wisconsin.
Grant sponsor: Department of Obstetrics and Gynecology, Medical College of Wisconsin
Grant sponsor: Advancing a Healthier Wisconsin Endowment
Grant sponsor: METAvivor Foundation
Grant sponsor: Mary Kay Foundation
Grant number: 024–16
Grant sponsor: NCI
Grant numbers: R01CA095713, R01CA193343 DOI: 10.1002/ijc.32038
Footnotes
We have no conflicts of interest or financial disclosures to report.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Cancer Genome Atlas Research Netwotk, Albert Einstein College of Medicine, Analytical Biological Services et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017; 543:378–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burk RD, Harari A, Chen Z. Human papillomavirus genome variants. Virology 2013;445:232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu P, Iden M, Fye S, et al. Targeted, deep sequencing reveals full methylation profiles of multiple HPV types and potential biomarkers for cervical cancer progression. Cancer Epidemiol Biomarkers Prev 2017;26:642–50. [DOI] [PubMed] [Google Scholar]
- 4.Galloway DA, Laimins LA. Human papillomaviruses: shared and distinct pathways for pathogenesis. Curr Opin Virol 2015;14:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright JD, Li J, Gerhard DS, et al. Human papillomavirus type and tobacco use as predictors of survival in early stage cervical carcinoma. Gynecol Oncol 2005;98:84–91. [DOI] [PubMed] [Google Scholar]
- 6.International Collaboration of Epidemiological Studies of Cervical C, Appleby P, Beral V, et al. Carcinoma of the cervix and tobacco smoking: collaborative reanalysis of individual data on 13,541 women with carcinoma of the cervix and 23,017 women without carcinoma of the cervix from 23 epidemiological studies. Int J Cancer 2006;118:1481–95. [DOI] [PubMed] [Google Scholar]
- 7.International Collaboration of Epidemiological Studies of Cervical C, Appleby P, Beral V, et al. Cervical cancer and hormonal contraceptives: collaborative reanalysis of individual data for 16,573 women with cervical cancer and 35,509 women without cervical cancer from 24 epidemiological studies. Lancet 2007;370:1609–21. [DOI] [PubMed] [Google Scholar]
- 8.Madeleine MM, Finch JL, Lynch CF, et al. HPV-related cancers after solid organ transplantation in the United States. Am J Transplant2013;13:3202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burk RD, Chen Z, Van Doorslaer K. Human papillomaviruses: genetic basis of carcinogenicity. Public Health Genomics 2009;12:281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mirabello L, Yeager M, Cullen M, et al. HPV16 sublineage associations with histology-specific cancer risk using HPV whole-genome sequences in 3200 women. J Natl Cancer Inst 2016;108: djw100 10.1093/jnci/djw100. Print 2016 Sep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tommasino M The human papillomavirus family and its role in carcinogenesis. Semin Cancer Biol 2014;26:13–21. [DOI] [PubMed] [Google Scholar]
- 12.Yang SH, Kong SK, Lee SH, et al. Human papillomavirus 18 as a poor prognostic factor in stage I-IIA cervical cancer following primary surgical treatment. Obstet Gynecol Sci 2014;57:492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burger RA, Monk BJ, Kurosaki T, et al. Human papillomavirus type 18: association with poor prognosis in early stage cervical cancer. J Natl Cancer Inst 1996;88:1361–8. [DOI] [PubMed] [Google Scholar]
- 14.Lombard I, Vincent-Salomon A, Validire P, et al. Human papillomavirus genotype as a major determinant of the course of cervical cancer. J Clin Oncol 1998;16:2613–9. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz SM, Daling JR, Shera KA, et al. Human papillomavirus and prognosis of invasive cervical cancer: a population-based study. J Clin Oncol 2001;19:1906–15. [DOI] [PubMed] [Google Scholar]
- 16.Lai C-H, Chang C-J, Huang H-J, et al. Role of human papillomavirus genotype in prognosis of early-stage cervical cancer undergoing primary surgery. J Clin Oncol 2007;25:3628–34. [DOI] [PubMed] [Google Scholar]
- 17.Hu X, Zhang Z, Ma D, et al. TP53, MDM2, NQO1, and susceptibility to cervical cancer. Cancer Epidemiol Biomarkers Prev 2010;19:755–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z, Borecki I, Nguyen L, et al. CD83 gene polymorphisms increase susceptibility to human invasive cervical cancer. Cancer Res 2007;67:11202–8. [DOI] [PubMed] [Google Scholar]
- 19.Ma D, Hovey RL, Zhang Z, et al. Genetic variations in EGFR and ERBB4 increase susceptibility to cervical cancer. Gynecol Oncol 2013;131: 445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Fye S, Borecki IB, et al. Polymorphisms in immune mediators associate with risk of cervical cancer. Gynecol Oncol 2014;135:69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin MP, Borecki IB, Zhang Z, et al. HLA-Cw group 1 ligands for KIR increase susceptibility to invasive cervical cancer. Immunogenetics 2010;62 (11–12):761–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vande Pol SB, Klingelhutz AJ. Papillomavirus E6 oncoproteins. Virology 2013;445:115–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mirabello L, Yeager M, Yu K, et al. HPV16 E7 genetic conservation is critical to carcinogenesis. Cell 2017;170:1164–74.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Zapien D, Ruiz FX, Poirson J, et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016;529:541–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The UniProt C. UniProt: the universal protein knowledgebase. Nucleic Acids Res 2017;45: D158–D69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nomine Y, Masson M, Charbonnier S, et al. Structural and functional analysis of E6 oncoprotein: insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol Cell 2006;21:665–78. [DOI] [PubMed] [Google Scholar]
- 27.Zanier K, Charbonnier S, Sidi AO, et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science (New York, NY) 2013;339:694–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zanier K, Ould M’Hamed Ould Sidi A, Boulade-Ladame C, et al. Solution structure analysis of the HPV16 E6 oncoprotein reveals a self-association mechanism required for E6-mediated degradation ofp53. Structure 2012;20:604–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000;28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bourgault Villada I, Moyal Barracco M, Berville S, et al. Human papillomavirus 16-specific T cell responses in classic HPV-related vulvar intraepithelial neoplasia. Determination of strongly immunogenic regions from E6 and E7 proteins. Clin Exp Immunol 2010;159:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Im SS, Wilczynski SP, Burger RA, et al. Early stage cervical cancers containing human papillomavirus type 18 DNA have more nodal metastasis and deeper stromal invasion. Clin Cancer Res 2003;9:4145. [PubMed] [Google Scholar]
- 32.Tang KW, Alaei-Mahabadi B, Samuelsson T, et al. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun 2013;4:2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palatnik A, Ye S, Kendziorski C, et al. Identification of a serum-induced transcriptional signature associated with metastatic cervical cancer. PLoS One 2017;12:e0181242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bierkens M, Krijgsman O, Wilting SM, et al. Focal aberrations indicate EYA2 and hsa-miR-375 as oncogene and tumor suppressor in cervical carcinogenesis. Genes Chromosomes Cancer 2013;52:56–68. [DOI] [PubMed] [Google Scholar]
- 35.Kalir T, Simsir A, Demopoulos HB, et al. Obstacles to the early detection of endocervical adenocarcinoma. Int J Gynecol Pathol 2005;24: 399–403. [DOI] [PubMed] [Google Scholar]
- 36.Chaump M, Pirog EC, Panico VJ, et al. Detection of in situ and invasive endocervical adenocarcinoma on ThinPrep pap test: morphologic analysis of false negative cases. Cytojournal 2016;13:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuna RE, Tuller E, Wentzensen N, et al. HPV16 variant lineage, clinical stage, and survival in women with invasive cervical cancer. Infect Agent Cancer 2011;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kidd EA, Siegel BA, Dehdashti F, et al. Lymph node staging by positron emission tomography in cervical cancer: relationship to prognosis. J Clin Oncol 2010;28:2108–13. [DOI] [PubMed] [Google Scholar]
- 39.Niebler M, Qian X, Hofler D, et al. Posttranslational control ofIL-1beta via the human papillomavirus type 16 E6 oncoprotein: a novel mechanism of innate immune escape mediated by the E3-ubiquitin ligase E6-AP and p53. PLoS Pathog 2013;9:e1003536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shai A, Pitot HC, Lambert PF. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res 2010;70:5064–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheffner M, Huibregtse JM, Vierstra RD, et al. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination ofp53. Cell 1993;75:495–505. [DOI] [PubMed] [Google Scholar]
- 42.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J 1991;10:4129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crook T, Tidy JA, Vousden KH. Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and transactivation. Cell 1991;67:547–56. [DOI] [PubMed] [Google Scholar]
- 44.Gewin L, Myers H, Kiyono T, et al. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev 2004;18:2269–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Chen JJ, Gao Q, et al. Multiple functions of human papillomavirus type 16 E6 contribute to the immortalization of mammary epithelial cells. J Virol 1999;73:7297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Z, Terai M, Fu L, et al. Diversifying selection in human papillomavirus type 16 lineages based on complete genome analyses. J Virol 2005; 79:7014–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas M, Banks L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J Gen Virol 1999;80(Pt 6):1513–7. [DOI] [PubMed] [Google Scholar]
- 48.Kinoshita T, Shirasawa H, Shino Y, et al. Transactivation of prothymosin alpha and c-myc promoters by human papillomavirus type 16 E6 protein. Virology 1997;232:53–61. [DOI] [PubMed] [Google Scholar]
- 49.Gao Q, Kumar A, Singh L, et al. Human papillomavirus E6-induced degradation of E6TP1 is mediated by E6AP ubiquitin ligase. Cancer Res 2002;62:3315–21. [PubMed] [Google Scholar]
- 50.Kukimoto I, Aihara S, Yoshiike K, et al. Human papillomavirus oncoprotein E6 binds to the C-terminal region of human minichromosome maintenance 7 protein. Biochem Biophys Res Commun 1998;249:258–62. [DOI] [PubMed] [Google Scholar]
- 51.Stoppler H, Hartmann DP, Sherman L, et al. The human papillomavirus type 16 E6 and E7 oncoproteins dissociate cellular telomerase activity from the maintenance of telomere length. J Biol Chem 1997;272:13332–7. [DOI] [PubMed] [Google Scholar]
- 52.Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996;380: 79–82. [DOI] [PubMed] [Google Scholar]
- 53.Sekaric P, Cherry JJ, Androphy EJ. Binding of human papillomavirus type 16 E6 to E6AP is not required for activation of hTERT. J Virol 2008; 82:71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ellis JR, Keating PJ, Baird J, et al. The association of an HPV16 oncogene variant with HLA-B7 has implications for vaccine design in cervical cancer. NatMed 1995;1:464–70. [DOI] [PubMed] [Google Scholar]
- 55.Chen AA, Gheit T, Franceschi S, et al. Human papillomavirus 18 genetic variation and cervical cancer risk worldwide. J Virol 2015;89:10680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu HH, Zheng LZ, Lin AF, et al. Human papillomavirus (HPV) 18 genetic variants and cervical cancer risk in Taizhou area, China. Gene 2018; 647:192–7. [DOI] [PubMed] [Google Scholar]
- 57.Fragoso-Ontiveros V, Maria Alvarez-Garcia R, Contreras-Paredes A, et al. Gene expression profiles induced by E6 from non-European HPV18 variants reveals a differential activation on cellular processes driving to carcinogenesis. Virology 2012; 432:81–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.