

Abstract
As an NNN-tridentate ligand, the 2,2’:6’,2”-terpyridine plays an important role in coordination chemistry. With three coordination sites and low LUMO, terpyridine and its derivatives are one of the typical Pincer ligand and/or non-innocent ligands in transition metal catalysis. Interesting catalytic reactivities have been obtained with these tpy-metal complexes targeting some challenging transformations, such as C-C bond formation and hydrofunctionalization. On the other hand, terpyridine ligands can form “closed-shell” octahedral <tpy-M2+-tpy> complexes, which provide a linear and stable linkage in supramolecular chemistry. Numerous supramolecular architectures have been achieved using modified terpyridine ligands including Sierpiński triangles, hexagonal gasket and supramolecular rosettes. This review presents a summary of recent progress regarding transition metal-terpyridine complexes with the focus on their applications in catalysis and supramolecular structure construction. Facile synthesis of terpyridine derivatives is also described. We hope this article can serve to provide some general perspectives of the terpyridine ligand and their applications in coordination chemistry.
Keywords: Terpyridine, Pincer ligand, Non-innocent ligand, Supermolecule, Iteration
Organometallic chemistry is the study of interaction between metal cations and ligands. Ligands often play a crucial role in tuning metal cation reactivity [1]. Often, some “prestigious” ligand frameworks could be crucial and provide a strong impact that can lead to the paradigm shift for coordination chemistry research [2–5]. With inherent lone pair electrons, nitrogen can coordinate with various transition metal cations. Thus, N-containing ligands are a crucial component in coordination chemistry [6–8]. Based on the binding pattern, these N-containing compounds can be categorized into mono-dentate and multi-dentate ligands, based on the number of available coordination sites. Among those reported nitrogen ligand systems, the 2,2’:6’,2”-terpyridine is one fascinating ligand, which has caught great attentions over the past two decades [9,10]. In this review, we would like to explore some investigations reported recently regarding transition metal complexes, based on this unique ligand system. Due to the fast pace of research growth in this topic, we will focus on two areas: chemical catalysis and supramolecular structure construction. It is our hope that this article can provide some general perspectives of the terpyridine ligands and their applications in coordination chemistry.
1. Introduction
The 2,2’:6’,2”-terpyridine here was simplified to terpyridine and abbreviated as tpy. With three nitrogen coordination sites, terpyridine is an NNN-type Pincer ligand, which can provide tight chelation with various metal cations in a nearly planar geometry. Depending on the number of coordination terpyridine applied, there are two possible coordination modes, mono-tpy Pincer complexes and bis-tpy {M(tpy)2} complexes (Scheme 1).
Scheme 1.

Basic binding mode of terpyridine ligands.
Before the metal coordination, nitrogens on 2-pyridine present a trans–trans geometry to avoid lone-pair electron repulsion. Upon coordination, a cis–cis geometry will be formed through metal chelation [12,13]. As a result, three pyridines adopt perfect coplanar conformation, allowing a good conjugation between the aromatic rings and metal cation. This feature makes tpy a potential “non-innocent” ligand, with the capability to stabilize the low valency metal, including nickel [14–23], iron [24,25], cobalt [26– 29] and manganese [30,31] etc. Interesting catalytic reactivity has been obtained with these tpy-metal complexes targeting some challenging transformations, such as C–C unsaturated bond hydrofunctionalization and C–C bond formation. Discussions on recent examples of tpy-complexes catalytic reactivity is provided in Section 3.
Another important binding mode of terpyridine ligand is the formation of octahedral complexes {M(tpy)2} containing two tpy ligands in a perpendicular geometry (Scheme 1). Comparing to tri-bipyridine (bpy) metal complexes {M(bpy)3}, another class of “closed-shell” metal complexes [32], {M(tpy)2} offers a good binding ability and good stability with the tridentate association model. Meanwhile, unlike {M(bpy)3} which can be chiral complexes (formation of fac and mer isomers), {M(tpy)2} are achiral complexesin most cases. This makes terpyridine a ligand system in supramolecular structure construction by avoiding unnecessary stereoselectivity issues. Similar to many other ligand systems, {M (tpy)2} complex stability is strongly associated with the coordinating metal cations. According to literature [33], the binding affinity of terpyridine towards transition metals has the following order: Ru2+ > Os2+ > Fe2+ > Zn2+ > Cd2+. Both ruthenium and osmium form stable tpy complexes with a very slow ligand exchange rate at room temperature. In contrast, Zn2+ and Cd2+ exhibit weaker binding with tpy even under ambient conditions, making their {M (tpy)2} complexes more dynamic. This feature allows the design and application of terpyridine in the formation of complexed supramolecular networks.
As a “closed-shell” organometallic complex, {M(tpy)2} can theoretically offer effective photo-electron interactions through metal-ligand charge transfer process, similar to {M(bpy)3} complexes [34,35]. However, unlike bpy ligand, tpy has a rather short excited state life time [36]. Although some progress has been made in improving the excited state life time using tpy derivatives, practical photo-electronic systems developed using {M(tpy)2} core structures are rare. Currently, one of the most popular applications of these type of tpy complexes is in the formation of metallo- supramolecular structures by effectively using {M(tpy)2} as an dynamic linker for linear geometry [13,37–40]. Over the past two decades, interesting 2D and 3D supramolecular structures have been reported based on this {M(tpy)2} linkage strategy [41–51]. Compared to other linear supramolecular linker, such as organoplatinium, the “closed-shell” {M(tpy)2} offers an improved structural stability and coordination reliability, which makes it a popular motif in metallosupramolecular research. Notably, New- kome and coworkers have recently published a comprehensive review regarding the previously reported examples from several well-established terpyridine modules and their application in supramolecular construction [52]. As a very active research area, new examples emerge rapidly. In this review, we summarized some recent examples in the last 5 years with the focus on the supramolecular shape iteration, hoping to provide a complementary prespective of terpyridine ligand in ongoing coordination chemistry research.
2. Synthesis of terpyridine
The first synthesis of terpyridine was reported by Gilbert Morgan in 1932 [11]. It was obtained as the byproduct from the oxidative coupling between pyridines using FeCl3 catalyst. Treating 8 kg pyridine at elevated temperature (340 °C) and in a sealed steel autoclave, around 60 g of terpyridine was isolated along with bipyridine (major product) and other poly-pyridines. With the interest in obtaining this compound and its derivatives, new synthetic routes have been developed. Currently, two general synthetic routes are used in the preparation of terpyridine and its derivatives [53,54]. The first and more prevalent method for terpyridine synthesis is the condensation between dione and ammonium, as shown in Scheme 2A.
Scheme 2.

Functionalized terpyridine synthesis through a condensation reaction [12,53,55–59].
As shown in Scheme 2A [12,53,55,57,58,60], reaction between ethyl picolinate and acetone gave a triketone intermediate. Subsequent condensation with ammonium acetate followed by chlorination with PCl5 or POCl3 gave the 4’-chloro(terpyridine). Notably, as a versatile intermediate, 4’-chloro(terpyridine) can be easily converted into other terpyridine derivatives simply through a Williamson ether synthesis. This approach allows the incorporation of various functional groups into the C-4’ position of the central pyridine ring, including hydroxy, thiol, carboxylic acid and amino groups. Alternatively, switching pyridine ester to pyridine ketone (acetopyridine) to react with a substituted aryl aldehyde can give similar condensation products (Scheme 2B) [56,59]. Using this method, various substituted aryl groups can be easily incorporated at the tpy 4’-position. This method has been widely applied for the preparation of functional terpyridine for self-assembly studies.
Although this condensation strategy is easy to handle, it suffered from limited reaction scope, especially for different substitutions on the two side of pyridines. To overcome this problem, a metal mediated coupling has been developed as shown in Scheme 3 [61]. Taking advantage of the well-developed palladium-catalyzed, cross-coupling reactions, researchers developed various revised conditions of effective Suzuki, Negishi and Stille couplings for the construction of C-C bond. The main challenge for this coupling strategy resulted in relatively low reactivity due to the competing binding between terpyridine and palladium catalyst. Thus, Stille coupling often gave optimal results (higher yields and good functional group tolerability) with the faster transmetalation step using aryl tin coupling partners. The reactions can be conducted by coupling either 2,6-dihalopyridine with 2-stannylpyridines or 2,6- distannylpyridine with 2-bromopyridine. The overall reaction is clean with good functional group tolerability. According to literature, various functional groups have been successfully introduced into the pyridine at 2,4,6-positions on the ring [54]. Representative examples of symmetrical and unsymmetrical terpyridines are shown in Scheme 3B with reported yields. Notably, this coupling strategy has been successfully used for gram-scale synthesis, assuring the accessibility of the desired substituted terpyridines. The main drawback of this approach is the low atom economy and requirement of toxic tin reagents.
Scheme 3.

Terpyridine synthesis by Stille-coupling [61].
3. Catalysis
3.1. Introduction
Based on the three nitrogen-based coordination sites and free- rotatable σ-bonds between pyridine rings, the 2,2’:6’,2’’-terpyri dine can serve as tridentate Pincer ligand to stabilize various transition metal cations. Terpyridine contains three electron-deficient pyridine heterocycles, which makes terpyridine not only a strong σ-donor but also a very good π-receptor [10]. This nature makes tpy as a “non-innocent ligand”, due to delocalization of electron from metal center to low-lying LUMO’s of pyridine rings under redox chemistry conditions. On the other hand, the nucleophilicity of the N-moiety (σ-donor ability) is not simply due to the presence of an electron pair on this atom, but also the geometrical/steric advantages of the pyridine moiety. Therefore, Metal center at lower oxidation state (with high electron density on the overall complexes) benefit from these features with enhanced stability and reactivity towards challenging transformations. For example, diverse C-C bond formation was achieved by terpyridine-Ni complexes catalyzed cross-coupling reactions [14–23].
Another important feature of terpyridine ligand is its ability to assist single electron processes, allowing metal-promoted radical processes in catalytic processes. This reactivity was observed with tpy-Cu [62–64] and tpy-Pd [65–68] complexes. Similarly, the nature of Pincer complexes containing electron-deficient ligands will exhibit interesting metal-hydride M-H reactivity. Some promising examples were reported in the metal hydride chemistry including by tpy-Ru [69–73] and tpy-Rh [74–76] complexes/catalysts. More recently, tpy-Mn, tpy-Fe and tpy-Co complexes have been successfully identified for C-C multiple bond hydrofunctionalization and cross-coupling [24–31]. These recent successes triggered great excitement and enthusiasm in further exploring potential new reactivity of this unique terpyridine. Some representative catalytic reactivity recently developed with terpyridine complexes will be discussed and summarized in the following section.
3.2. Tpy-nickel catalysis
Low valence nickel (Ni0 and NiI) complexes have good reactivity toward oxidative addition of carbon-halide bond [77]. A practical concern was how to stabilize the highly reactive Ni center to facilitate effective catalytic turnover. With the ability to form Pincer complexes with electron deficient π-receptor, terpyridine was found to be effective in promoting these Ni catalyzed transformation. In 2010, Weix and co-workers used tert-butyl modified tpy to assist the nickel catalyzed alkyl-halide reductive coupling [21]. In their study, direct dimerization of primary and secondary alkyl halides, alkyl pseudohalides, and allylic acetates were achieved with high yield under mild conditions. Mn powder were used as the reductant. Notably, this challenging C(sp3)-C(sp3) coupling was efficiently achieved using terpyridine while other ligands gave low yields. Some transformations have been also performed under ambient conditions. Some challenging cross-couplings using less active substrates, such as alkyl chloride, alkyl mesylates and alkyl trifluoroacetates, was initially problematic. However, upon addition of catalytic sodium iodide (up to 50%), this problem was overcome, highlighting the high efficiency of this new catalytic system (Scheme 4).
Scheme 4.

Reductive dimerization of alkyl bromides. Figure was reproduced from Ref. [21] with permission of the copyright holders.
When monitoring the reaction kinetics, researchers observed that the secondary, tertiary and hindered primary haloalkanes gave very slow reaction rate under these nickel catalyzed reductive coupling conditions. Based on this result, the authors developed an effective cross coupling method by tuning the substrate reaction kinetics. Recently, Weix’s group further extended the reaction scope to alkene substrates with the utilization of metal-alkane migratory insertion. As shown in Scheme 5A [23], under previous conditions (Scheme 4), the reaction between an alkyl halide and cyclic enone resulted in a formal Michael addition with the assistance of silanechloride, giving the desired silyl enol ethers in good yields. Notably, in this case, sterically hindered alkane halides are needed to prevent substrate dimerization.
Scheme 5.

Very recently, the same group further extended the reaction scope to simple primary alkanes as shown in Scheme 5B [17]. To prevent alkyl bromide dimerization, authors proposed the application of sterically hindered 6,6”-dibromo-2,2’:6’,2”-terpyridine. With this new ligand, successful addition using primary alkyl bromide was achieved under mild conditions. Impressively, the C-Br bond in tpy remained unreactive in this case. This result revealed the possibility of applying this strategy for other challenging transformations while the undesired dimerization was effectively prevented.
When coordinating to nickel center with low oxidation state, Ni (0) for example, terpyridine stabilized the metal cation as well enriched the electron density, therefore, this Ni(0) complex can be feasible towards oxidative insertion into challenging C-N bond. Recently, the Grag group disclosed the nickel-catalyzed esterification of aliphatic amides (Scheme 6) [16]. This reaction was initiated with oxidative addition of nickel(0) to amide C-N bond. Based on computational results, oxidative addition is the rate- determine step with a higher oxidative addition barrier of aliphatic amides over the aromatic ones. Authors suggested that tuning the electronic nature of terpyridine plays a crucial role in the oxidative addition of aliphatic amides. Only terpyridine gave satisfying yields while all other tested monodentate or bidentate ligands resulted in either no reaction or very low yields of the desired products.
Scheme 6.

Esterification of aliphatic amides [16].
Similarly, the Han group later reported Ni-catalyzed amide C-N bond activation, followed by the reductive cross-coupling with aryl iodide. In this reaction, terpyridine was used as ligand with a catalytic amount NiI2 (10%), and zinc powder as the reductant (2.0 equiv, Scheme 7) [19]. This work presented the first example using an amide as the electrophilic reagent to couple with another electrophile with broad reaction scope under mild conditions. Based on the control experiments, no transmetallation between organozinc reagent and nickel intermediates was observed. The radical oxidation to intermediate A followed by reductive elimination process was proposed. Interestingly, this reaction demonstrated a good oxidative addition selectivity toward amide C-N bond over an aryl C-I bond, revealing the orthogonal reactivity compared to the Pd catalyzed processes.
Scheme 7.

Reductive cross-coupling of amides with electrophiles [20].
Inspired by Weix’s work, Biscoe and co-workers later developed a Ni-catalyzed Negishi cross-coupling by reacting alkylzinc with aryl iodides (Scheme 8) [18]. The desired coupling products were obtained in good to modest yield under mild conditions. Impressively, while Pd-catalyzed Negishi reaction often suffers from undesired “chain-walking” isomerization products due to the competing β-hydride elimination, this nickel catalyzed process showed excellent selectivity with retention and isomerization products in a ratio over 100:1. This work again highlighted that terpyridine was crucial for both the reactivity and chemoselectivity.
Scheme 8.
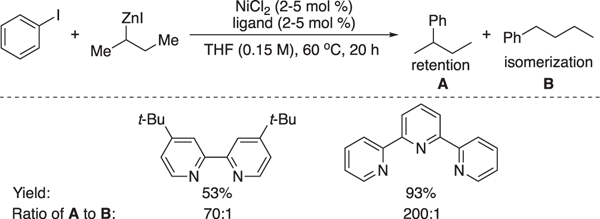
Negishi coupling of alkylzinc and aryl iodides [18].
Recently, the Giri group disclosed the combination of tpy- nickel-catalyzed alkylhalide-olefin cyclization and Negishi cross-coupling for the preparation of some lactone-based natural products [22]. The reaction tolerates a wide range of functional groups, giving the desired products with excellent regio- and diastereo-selectivity. Some base-sensitive carbon stereogenic centers were tested without racemization, which highlighted the broader reaction scope with mild reaction conditions. Six ligands-based natural products were synthesized successfully in gram-scale, which illustrated the strong potential of this method in complex molecule synthesis (Scheme 9).
Scheme 9.

Ni-catalyzed cyclization and Negishi cross-coupling [22].
More recently, Zhang and co-workers further extended this terpyridine nickel complex strategy into the synthesis of fluoro- containing compounds [14]. The nickel catalyst was found to selectively react with the C-Br bond in gem-difluoropropargyl bromides. With the incorporation of unactivated alkylzinc reagents, the desired difluoro compounds were prepared in good to excellent yields with high efficiency. Similar to all other tpy-Ni promoted transformations, a radical process was proposed, which was supported by radical trapping experiments. More mechanistic studies suggested a Ni(I/III) catalytic cycle instead of a Ni(0/II) as stated by the authors (Scheme 10).
Scheme 10.

Synthesis of fluoro-containing compounds [14].
Last year, Nevado and co-workers reported the Ni-catalyzed, three-component, β,β-disubstituted vinyl sulfone formation though sulfonyl radical under mild condition [15]. In these catalytic systems, terpyridine was found to be crucial when compared with other nitrogen-containing ligands. This reaction showed high regioselectivity, giving trisubstituted vinyl sulfones as the major product (Scheme 11).
Scheme 11.

Ni-catalyzed intermolecular carbosulfonylation of alkynes [15].
3.3. Tpy-copper catalysis
Similar to nickel, copper could also initiate single electron processes in the catalytic cycle with the existence of multiple inter-convertible oxidation states (CuI, CuII and CuIII). Thus, the catalytic cycle could be enhanced with stabilized ligands, such as terpyridine. Unlike the nickel cation, copper readily reacts with a terminal alkyne to form the copper-acetylide intermediate. Thus, it is plausible to extend metal terpyridine complex reactivity into a potential new group of substrates. In 2015, Liang and co-workers reported the Cu-catalyzed trifluoromethylcyanation of a terminal alkyne using Togni’s reagents as the CF3 radical source [64]. As stated by the authors, trapping vinyl copper intermediate with TMSCN gave the β-trifluoromethylated acrylonitriles in one pot with good yields. Alternatively, reacting the intermediate with TMSN3 resulted in formation of trifluoromethyl-substituted 2H- azirines (Scheme 12). Terpyridine was found as a crucial ligand for these transformations, giving the best results comparing with all other tested nitrogen-based ligands. Notably, reaction with TMSN3 did not give the azidotrifluoromethylation products as expected. The interesting 2H-azirines were obtained instead even without tpy ligand. The yield was further improved with the addition of NaOAc and using higher temperature. A series of CF3-containing pyridines and pyrazines were successfully synthesized from the 2H-azirines with good yields.
Scheme 12.

Copper-catalyzed difunctionalization of alkyne [64].
Besides terminal alkyne, isocyanides (another sp carbon nucleophile) can also be activated with the same strategy. The Liu group recently reported the tpy-copper catalyzed CF3 radical addition to isocyanide with the presence of TMSCN, giving CF3 and CN substi-tuted imine in modest to good yields (Scheme 13) [62]. The product could be reduced using borane to produce the highly valued trifluoropropan-1,2-diamines. Control experiments with TEMPO and EPR confirmed the formation of copper radical intermediate, as expected.
Scheme 13.

Cu-catalyzed cyanotrifluoromethylation of isocyanides [62].
Very recently, Lalic et al. achieved C(sp)-C(sp3) bond coupling with photo-initiation [63]. As shown in Scheme 14, terpyridine plays a crucial role, giving good yield of the coupling product while other tested alternative ligands gave only poor overall performance. Various substrates were suitable for this transformation, including primary, secondary, tertiary iodides and alkyl, aryl and heteroaryl alkyne. Authors believed that besides the stability effect, terpyridine successfully prevented substrate polymerization, leading to the observed good yields and diverse reaction scope. Again, TEMPO control experiment confirmed the radical process in this reaction.
Scheme 14.

Photoinduced Cu-catalyzed C-C coupling [63].
3.4. Tpy-ruthenium catalysis
Metal hydride is one very important functional group in organometallic chemistry and catalysis. Depending on metal electronegativity, metal hydride can react as proton, hydrogen radical or hydride. With the interesting electronic nature, terpyridine ligand could certainly assist catalytic process involving M-H. Recently, the Beller group reported tpy-Ru catalyzed direct nitro group reduction to form amines [71]. As shown in Scheme 15, [tpy-RuH2], formed from isopropanol hydride transfer, could effectively reduce an aryl nitro group to the amine in good to excellent yields. This transformation was highly practical since it did not require any sensitive and expensive phosphine ligands. Good chemoselectivity was also observed with substrates containing other reducible functional groups, such as esters, amides and alkenes.
Scheme 15.

Tpy-Ru catalyzed transfer hydrogenation nitro reduction [71].
Encouraged by this tpy-Ru promoted transfer dehydrogenation, Szymczak and co-workers developed selected enone reduction using modified terpyridine ligands [72]. As shown in Scheme 16A, the hydroxyl modified tpy produces an electron rich ruthenium center under basic conditions. Selective carbonyl reduction (over alkene) was then achieved due to H-bonding directing effect. This substrate-ligand interaction not only gave rise to the observed excellent chemoselectivity, but also greatly enhanced the reaction kinetics by converting the key step into an intramolecular fashion. More recently, the same group further extended the work and developed NHMes modified tpy [70]. With the improved electron density on the ligand, effective alcohol oxidation to carboxylic acid was achieved at even low catalyst loading (0.2 mol%). This new ligand also showed significantly improved stability, which allowed effective catalyst recycling (three times) without significant reduction of catalyst reactivity (Scheme 16B).
Scheme 16.

3.5. Tpy-palladium catalysis
Palladium is one important metal in coupling reactions due to its versatile reactivity. Usually, Pd(ll) adopted four coordination with square planar geometry. This limited the application of tridentate ligands in catalysis; however, recently, Jiang and co-workers reported a very interesting example using tpy-Pd in promoting the oxidation of alkene to 1,2-diacetate (Scheme 17) [65].
Scheme 17.

B2pin2-mediated tpy-Pd catalyzed alkene diacetoxylation [65].
ln this case, Pd-catalyzed acetate addition to alkene gave the Pd intermediate. B2pin2 was used for transmetalation to the Pd intermediate to afford C-Bpin bond, which undergoes oxidative cleavage into the acetate product. O2 was considered as a sole oxidant during the process, giving the desired products with wide functional group tolerability and in moderate to excellent yields. Typically, a challenge in Pd-catalyzed olefin oxidation was the regeneration of Pd(II) from Pd(0) due to palladium black precipitation. ln this transformation, B2pin2 was employed to assist the reformation of Pd(II) under only 1 atm of oxygen. lnterestingly, while terpyridine ligand gave excellent yields, other tested bidentate and tri-dentate ligands gave significantly poorer results.
In 2016, Ritter’s group reported tpy-Pd catalyzed fluorination of arylboronic acid derivatives [66]. Given the importance of fluorine containing compounds in medicinal research, effective fluorination is always welcome for the synthetic community. Fluorination using Pd-catalyzed arylboronic acid derivatives via a cross-coupling reaction generally suffered from slow transmetalation and undesired protodeborylation. ln this approach, fluorination occurred through Pd (II-IV) cycle with selectfluor as both a fluoride source and oxidant. This radical process could avoid the undesired boronic acid homo-coupling pathway under traditional Pd(0/II) catalytic cycle. Meanwhile, tridentate terpyridine is crucial in facilitating intermediate Pd cation stability. With this method, some of the fluorination products were prepared up to 10 g scale under mild conditions (Scheme 18).
Scheme 18.

Pd-catayzed fluorination of arylboronic acid derivatives [SB].
To further improve the reaction efficiency, the same research group later developed a direct C-H fluorination as shown in Scheme 19 [68]. ln this case, a new tpy-Pd-phen palladium catalyst was developed with combination of terpyridine and bidentate phen ligand. With selectfluor as the fluoride source and oxidant, the desired fluorination products were obtained in good yields under very mild conditions.
Scheme 19.

Pd-catalyzed electrophilic aromatic C-H fluorination [68].
According to the authors, using the terpyridine ligand is key to the successful transformation under this mild condition. Unlike other typical Pd-catalyzed coupling mechanisms, the terpyridine ligand prevents the formation of Pd-Ar intermediate. Instead, the Pd(IV)-F intermediate was formed through a single electron transfer (SET) process while treating tpy-Pd(II) with selectfluor (Scheme 20). Interaction between the resulting Pd-F intermediate with arene gave the final aromatic fluorination product. Considering the mild conditions and excellent functional group tolerability, this method provides an effective new protocol for the preparation of aromatic C-F bonds.
Scheme 20.

Proposed tpy-Pd fluorination mechanism. Figure was reproduced from Ref. [68] with permission of the copyright holders.
3.6. Tpy-rhodium catalysis
With the d9 electron configuration, Rh(II) is known to produce di-Rh complexes through the formation of a Rh-Rh bond. For example, complex [Rh(OAc)2]2 is one of the most effective catalyst for diazo activation, opening a new era in carbene chemistry [78].
In 2016, the Wang and Xiao groups reported the preparation of terpyridine coordinated di-rhodium complexes, as shown in Scheme 21 [74,76]. Simply treating primary and secondary alcohols with the catalyst under basic condition gave carboxylic acid (primary alcohol) or ketone (secondary) in gram scale. Furthermore, treating aldehyde with alcohol will yield ester product through direct dehydrogenation. The overall transformation tolerated a wide group of substrates. Preliminary mechanistic study showed that the dimeric Rh(tpy) catalyst was the active catalysts which played an important role to achieve the cross-coupling. Moreover, the catalyst was reusable. Even after 19 reaction cycles, the catalyst could effectively promote the reaction with little decrease of reactivity. These unique features made this tpy-Rh a highly efficient catalytic system, which was operational under green conditions.
Scheme 21.

[Rh(tpy)]2 catalyzed alcohol oxidation and dehydrogenation [74,76].
3.7. Tpy-iron catalysis
With the d6 electron configuration, Fe2+ could form stable octa-hedral complexes by coordinating with up to six coordination ligands. Although it is expected that terpyridine will greatly increase iron cation stability at a low oxidation state, application of this ligand system in iron coordination suffered from a potential problem on how to deal with formation of both mono-tpy (4 or 5 coordination) and bis-tpy (octahedral 6 coordination) complexes. In 2012, the Nakazawa group reported the investigations on hydrosilylation of alkenes using terpyridine coordinated Fe(II) catalysts [24]. Nakazawa and coworkers evaluated various terpyridine derivatives. As shown in Scheme 22, with no substitution on terpyridine 6 and 6’ positions, iron complexes with Cl or Br gave no reaction, suggesting the formation of unreactive octahedral bistpy-Fe complexes. To prevent this problem, ligands with bulky substituted groups at the terpyridine 6,6’ position were prepared and provided positive results. Significantly improved results were obtained with bromide iron salts, giving better yields than chloride salts. Notably, anti-Markovnikov products were obtained exclusively in all cases, which was consistent with previous reports [79].
Scheme 22.

Fe-catalyzed hydrosilylation of alkene [24].
3.8. Tpy-manganese catalysis
Encouraged by the interesting reactivity initiated by terpyridine, other first row base metals have also been tested. Recently, Zhang and coauthors successfully synthesized the terpyridine coordinated Mn complexes [Mn(tpy)(CH2SiMe3)2] [31]. Using these new complexes, successful alkene hydroboration was achieved under mild conditions (25 °C) with excellent yields and high efficiency (1% loading). With aromatic alkene, typical Markov- nikov products were observed with good yields and excellent regioselectivity. For aliphatic alkenes, poor regioselectivity was obtained, slightly favoring anti-Markovnikov products. These results hinted at a radical nature of the overall transformation instead of the typical BH/C=C four-member-ring transition state. Aldehydes and ketones were also tested via this protocol and excellent reactivity was observed. Competition experiments suggested ketone reduction over alkene as expected for typical hydroboration reactions (Scheme 23).
Scheme 23.

Tpy-Mn catalyzed alkene and ketone hydroboration [31].
3.9. Tpy-cobalt catalysis
The Chirik group recently reported their investigations on ligand effects for the Co(I)-catalyzed alkene isomerization- hydroboration [28]. A series of bidentate and tridentate ligands were evaluated toward various substituted alkenes. According to the authors, most of the tested bidentate ligands could not promote this transformation with low conversion and yields (Scheme 24A). Terpyridine-Co(I) complexes successfully catalyzed this transformation for less hindered alkenes with high efficiency (1% loading). Notably, as a radical reaction pathway, hydrogen migration occurred, giving products with Bpin at the terminal carbon. This is consistent with good steric hindrance provided by the terpyridine ligand at the 6 and 6’ position.
Scheme 24.

An air-stable [Co(tpy)(OAc)2] catalyst was later developed by the same group and applied as pre-catalyst in promoting direct aryl C-H borylation as shown in Scheme 24B [27]. The LiOMe additive was crucial for successful transformation by helping the removal of HBPin byproduct, which was likely to cause deactivation of Co catalyst. The desired borylated products were formed in moderate to good yield with good substrate scope.
Very recently, Teo reported the application of Co complexes in promoting Suzuki-Miyaura coupling reaction [26]. Various bidentate and tridentate ligands were tested and modified terpyridine gave the optimal results, which provided another example for the unique reactivity offered by this Pincer NNN ligand. As shown in Scheme 25, various halo-pyridines and analogues were suitable for this transformation. The exact mechanism whether this reaction undergoes Co(0/ll) or Co (I/III) catalytic cycle is currently unclear.
Scheme 25.

Tpy-Co catalyzed Suzuki-Miyaura cross-coupling [26].
ln summary, terpyridine, a Pincer ligand, provides three coordination sites with strong and stable σ-donating and π-recepting ability towards transition metal core. The low-lying LUMO of pyridine rings allows the redox chemistry to occur on ligand instead of metal core. Utilizing this advantage, the tpy ligand can serve as a non-innocent ligand to stabilize low oxidation state of transition metal. For example, tpy-Ru and tpy-Rh demonstrated metal- hydride reactivity. In addition, tpy-Mn, tpy-Fe, and tpy-Co complexes can catalyze the C-C multiple bond hydrofunctionalization and cross-coupling. Furthermore, the tpy-Ni, tpy-Cu, and tpy-Co show the ability to assist a single electron process. The tpy-Ni catalysts have been employed to generate diverse C-C bond, while the tpy-Cu complexes can assist sp carbon activation. Besides the excellent performances that terpyridine has demonstrated in organometallic chemistry, it also arouses great attention in the application in designing and establishing sophisticated supramolecular frameworks through hierarchical self-assembly, which is summarized in the next section.
4. Supramolecular chemistry
In the last decades, appreciation of numerous supramolecular architectures from artistic and mathematical point of view has drawn increasing attentions. However, it remains a challenging task to impose effective control over the self-assembly process to obtain a well-defined giant supermolecule containing multiple species through non-covalent interactions. Combination of the highly directional transition metal with pre-organized ligand can facilitate the precise formation of a desired supramolecular motif under conformational control. Many research groups have successfully discovered novel coordination patterns to synthesis a variety of representative structures, such as Star of David, Knock snowflake, Sierpmski triangle, etc.
Shapes of a supermolecule exhibit two crucial features: 1) to mimic and simplify the biological system. As the biological structure is too sophisticated to be achieved by a linear synthesis, the self-assembly offers a convenient pathway to reach that goal. 2) The artificial shapes of supermolecules allow their applications in chemical sensing, molecular imaging, drug delivery, metal extraction, etc [80]. Based on the “bottom-up” approach, chemists have reported increasing numbers of complicated supramolecular structures to extend the borders of self-assembly area and explore their potential applications.
To set up a vertex angle of supramolecular motif, two major methods are employed. The first approach is to build the vertex with metal core. By taking advantage of the geometry of transition metal complexes, the vertex angle is determined by the binding angle between two empty coordination sites. This approach is straightforward and ubiquitously used, however the resulted supermolecules could often suffer from isomerization. In Scheme 26, as the orientation of Pd(II) complex was not fixed (swaying between 60° and 90°), square and triangle supramolecular structures could be achieved, based on different conditions [81]. It is reported that triangle structures could be obtained under elevated temperature, whereas square structures were more favorable at a higher concentration. To set up a certain angle of the supermolecule component to form a stable and discrete structure, another method using a fixed covalent bond is necessary. This method is utilizing the orientation of an organic framework such as the bond angle between two substituents on an aromatic ring. The more fixed vertex angle of a supramolecular structure, the higher the selectivity of the shape of supermolecule will be. For example, 4,4”-bisterpyridine-carbazole has a firm corner angle due to the rigid aromatic system. Connected by “closed-shell” 180° linker, terpyridne, a pure metallopentacycles could be exclusively achieved (Scheme 27) [82].
Scheme 26.

Equilibrium between square and triangle structure [81].
Scheme 27.

The formation of metallopentacycles [82].
As a tridentate ligand, terpyridine can form linear <tpy-M2+- tpy> complexes, which serve as 180° linker for the supramolecular structure. Comparing a pyridine and bipyridine ligand, the terpyridine ligand contains more coordination sites, which results in more rigid metal complexes. Based on different binding ability of various transition metals, sequential self-assembly of modulated tpy and metal can be realized to construct a range of remarkable supramolecular architectures. Therefore, tpy components of different sizes and shapes can be amalgated into complicated and discrete structures through precisely controlled modular design.
Many excellent terpyridine-based supramolecular related reviews have summarized this area and further offered valuable understanding of the role of terpyridine in construction of complex architectures [83–85]. For example, Newkome provided a comprehensive review of the development of terpyridine-based supramo- lecule very recently [52]. Series of well-established terpyridine modules using <tpy-M-tpy> connectivity and their application in supramolecular construction were clearly described. Many useful strategies to build complicated supramolecular structures were summarized, including triangle-based framework, hexagonal fractal designs and flexible polyterpyridine linkers.
Herein, we sought to demonstrate a summary of the most recent developments in the field of supramolecular chemistry with conformational controlled self-assembly of terpyridine (tpy) with emphasis on the iteration of terpyridine-based supramolecular structure in 2D and 3D perspectives. So far, only triangle and hexagon structures can be iterated to several different generations in various directions on 2D level. Naturally, several interesting fractal supramolecular architectures based on triangle and hexagon shape will be included in this review. Similarly, only hexagon structures have been observed and developed to 3D level. Thus, in the following two sections, several selected systems of 2D and 3D supramolecular shaped iteration will be herein described.
4.1. Iteration on 2D level
4.1.1. Three generations of Sierpinski triangles
As mentioned above, the terpyridine-based supermolecules could be iterated on 2D level. In the last decade, three generations of Sierpinski triangle have been achieved by utilizing the coordination-driven self-assembly strategy. Based on fractal geometry, the first-generation Sierpinski triangle have only one triangle-shaped layer; the second generation is composed of four small triangles; the third generation exhibits more complicated structures containing more small triangles.
After the self-assembly of the predesigned terpyridine-based “V” type ligand and Ru2+ or Fe2+, the first-generation Sierpinski triangle could be obtained (Fig. 1) [86]. In addition, the author found these complexes showed unique photophysical properties and ability to form the nanofibers.
Fig. 1.

The first-generation Sierpiński triangle. Figure was reproduced from Ref. [86] with permission of the copyright holders.
In 2014, the Newkome group reported the self-assembly of the second-generation Sierpinski triangle [87]. Through direct self-assembly of “V” type ligand, “K” type ligand, and Cd2+ in ratio of 1:1:3, the second-generation Sierpinski triangle could be obtained in nearly quantitative yield (Fig. 2)
Fig. 2.
One step synthesis of the second-generation Sierpiński triangle. Figure was reproduced from Ref. [87] with permission of the copyright holders.
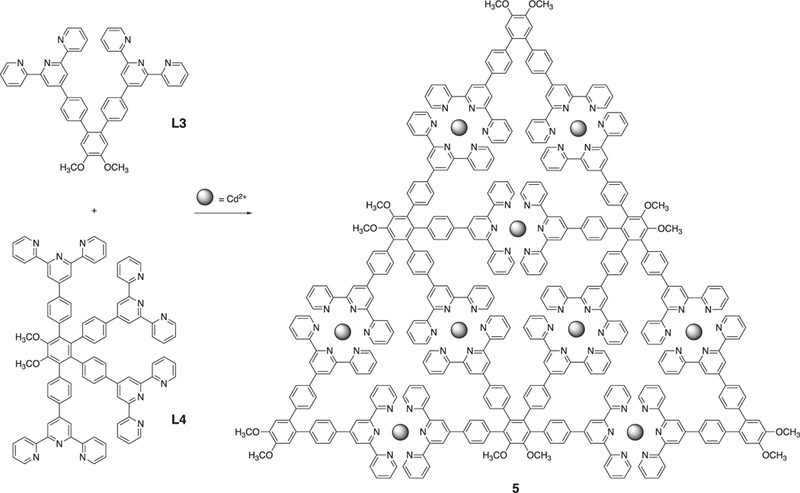
Alternatively, the Li group very recently adopted another method to achieve the second-generation Sierpinski triangle by using modular strategy and step-wise synthesis to avoid the formation of small metallocycles or supramolecular polymer (Fig. 3) [88]. As the <tpy-Ru2+-tpy> complexes are stable towards the palladium-catalyzed coupling reaction and can survive from column chromatography, the author successfully synthesized the key intermediate L6 after seven-step-synthesis. With the L6 ligand in hand, the second-generation Sierpiński triangle was achieved by the self-assembly of ligand L6 and Zn2+ in one pot reaction with nearly quantitative yield.
Fig. 3.
The formation of second-generation modular Sierpiński triangle using modular strategy and step-wise synthesis. Figure was reproduced from Ref. [88] with permission of the copyright holders.
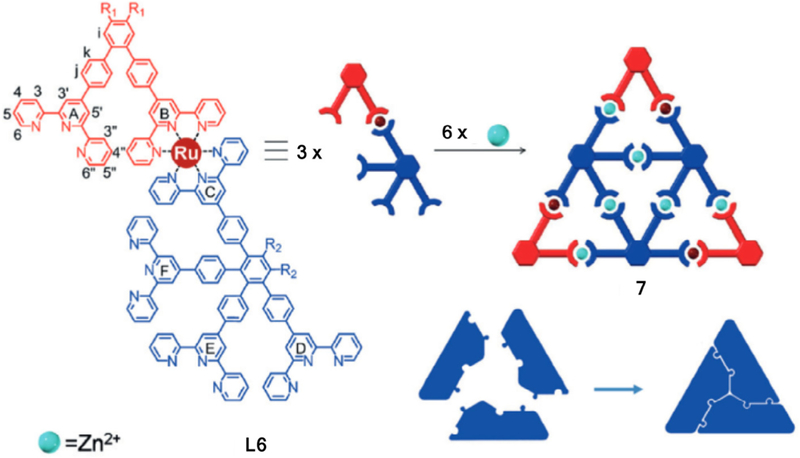
After the successful synthesis of the second generation, the author applied the same strategy towards the self-assembly of the third generation [88]. The author divided the synthesis into two steps as shown in Fig. 4:1) Formation of “A” type Ru complex. The “V” type of vertex corner was self-assembled with “K” type of tetraterpyridine ligand treating with metal Ru(II) to form the building block. 2) Formation of triangle 10. One star-shaped ligand L9 and the three “A” type of complex were mixed together to generate the third-generation triangle 10 in high yield. This is an inspiring example to achieve supramolecular structure though self-assembly of three different ligands.
Fig. 4.

Self-assembly of the third-generation Sierpiński triangle. Figure was reproduced from Ref. [88] with permission of the copyright holders
4.1.2. Four generations of hexagonal ring-in-ring structures
Like the Sierpinski triangle, researchers have developed four generations terpyridine based hexagon supramolecular ring-in- ring structure. The first-generation structure (one layer) is the most common structure. ln 1999, Newkome reported the synthesis of one layer terpyridine based supramolecular hexagon structure (Fig. 5) [89]. Through self-assembly of bisterpyridine ligand and Ru3+ with N-ethylmorpholine as the reductant, the first-generation hexagon supermolecule (G1) was achieved in 40% yield.
Fig. 5.
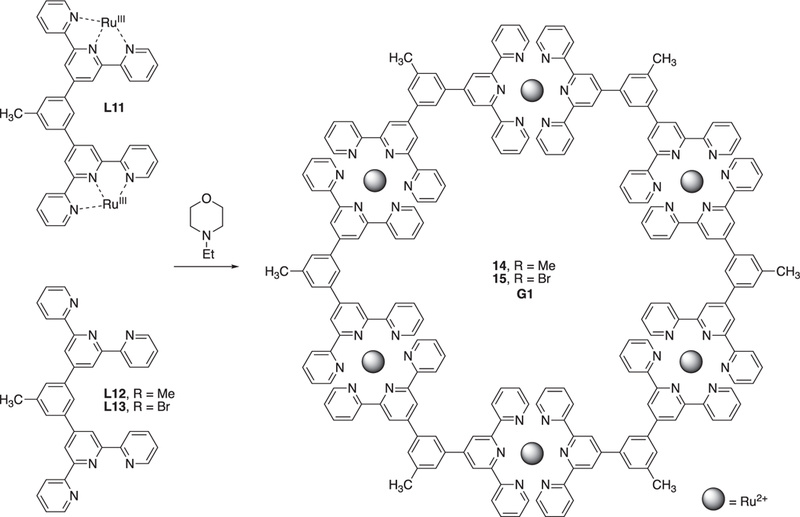
Self-assembly of first-generation hexagon supermolecule. Figure was reproduced from Ref. [89] with permission of the copyright holders.
The second-generation hexagon ring-in-ring structures were developed by Li’s group in 2016 (Fig. 6) [90]. As the self-assembly of one layer structure always resulted in supramolecular mixtures, the authors chose to increase the coordination sites of ligand to precisely control the shape of supermolecule. Self-assembly of various tetratertopic terpyridine ligands and zinc (II) generated a series of second-generation supramolecular hexagonal ring-in-ring structures (G2). Notably, these ligands were synthesized through the condensation of pyridinium salts and primary amines with good yields, providing a facile synthesis for tetratopic terpyridine ligands. Self-assembly studies were carried out by mixing terpyridine ligands and zinc salts in CHCl3/MeOH followed by addition of NH4PF6 to form the precipitate with almost quantitative yield. As long carbon chain improved the complex solubility significantly, the structure was achieved and confirmed by 1H NMR and MALDI-TOF mass spectrometry. These terpyridine ligands of varied carbon length have been shown to self- assemble into organized nanoribbons at the solid/liquid interface, which was further confirmed by STM imaging besides NMR and MS.
Fig. 6.
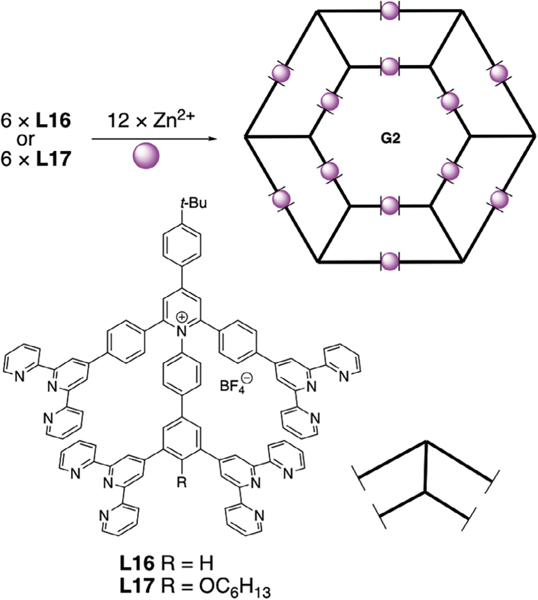
Self-assembly of the second-generation of hexagon ring-in-ring supermolecule. Figure was reproduced from Ref. [90] with permission of the copyright holders.
Encouraged by previous success, the same group later disclosed discrete third- and fourth-generation hexagon ring-in-ring structure through the self-assembly using a series of terpyridine- based monomers, as the building blocks (Fig. 7) [91]. A similar method (condensation between pyrylium salts and primary amines) was adopted for the synthesis of multitopic terpyridine ligands. Employing these ligands plus highly directional and rigid coordination into self-assembling, the third and fourth generation (G3 and G4) of hexagons were achieved. In these self-assembly studies, Cd(II) instead of previously used Zn(II) was chosen to coordinate with terpyridine due to the higher reversibility of Cd(II) complexes to ensure a thermodynamically favorable self-assembly products though a dynamic equilibrium. In addition, they found these hexagons tend to form a tubular-like structures due to the π-π stacking in the solvent mixture, such as DMSO and H2O. These supramolecular structures were confirmed by NMR, ESl-MS and TWlM-MS, etc. Furthermore, these structures exhibited highly antimicrobial activity and negligible toxicity to eukaryotic cells. In contrast, the corresponding ligands failed to show similar properties.
Fig. 7.

Self-assembly of the third and fourth hexagon ring-in-ring supermolecule [89].
4.1.3. Three generations of supramolecular rosettes
Other than ring-in-ring architectures, the hexagon can also be iterated to form three generations rosette-like structures. The first-, second- and third-generation structures have numbers of corresponding layers. Recently, the Li group published three generations of supramolecular rosettes architectures utilizing coordination-driven self-assembly with increasing complexity (Fig. 8) [92]. In this work, by using the self-assembling of the ditopic terpyridine ligand and Cd2+, a mixture of macrocycles was achieved. In contrast, the tetratopic and hexatopic terpyridine ligands exhibited high geometric selectivity, giving only thermodynamic product (double-layer and triple-layer rosettes respectively) under precisely controlled self-assembly process, because of additional coordination sites provided by these ligands. Interestingly, the hexatopic terpyridine ligand could form a very rare triple-layer heptamer structure, owing to the geometry feature of the hexatopic ligand. Furthermore, since tetraphenylethylene (TPE) was introduced into these ligands, based on the aggregation- induced emission (AIE), these complexes showed varied emissive properties. Especially for second-generation structure (G2), it presented pure white light in different solvents.
Fig. 8.

Three generations of rosettes [92].
4.1.4. From hexagon supermolecule to Sierpiński hexagonal gasket
After successful synthesis of the first-generation hexagon supermolecule, Newkome and co-authors reported the formation of Sierpiński hexagon gasket (Fig. 9) [46]. This structure was achieved by self-assembly of six hexagon sub-supermolecule with metal. As the bisterpyridine ligand provided a stable enough vertex angle, the authors used this ligand along with Ru3+ and reductant to generate the key module 28 by step-wise self-assembly. After self-assembling with Fe2+, the self-similar fractal Sierpiński hexagon gasket 29 was constructed with excellent inherent stability.
Fig. 9.

Self-assembly of Sierpinski hexagon gasket. Figure was reproduced from Ref. [46] with permission of the copyright holders.
4.1.5. Five generations of fractal supermolecule
According to the definition, the “fractal structure” is a kind of structure that could not be classified into any basic geometry but shows self-similarity to some extend at different level. Very recently, the Li group reported the self-assembly of five generations of fractal structures, based on the coordination-driven self-assembly of terpyridine ligands (Fig. 10) [94]. By utilizing the stable coordination of <tpy-M-tpy> complexes, modular strategy, step-wise synthesis and high coordination sites density (CODs), all the five generations structures were achieved in high yield with increasing complexity. The first-generation structure was constructed by direct self-assembly of two bisterpyridine ligands with Zn2+, and the fourth generation was obtained though the similar method. While the second, third, and fifth generations were synthesized by step-wise assembly. The key intermediates L31, L32 and L34 were obtained by coordination of Ru2+ and multitopic terpyridine ligand. After self-assembly of corresponding intermediates with Zn2+, the second, third, and fifth generations could be constructed. Geometrically, the first generation is the repeating unit and all the other generations are constitutive of several repeating units. What’s more, all the structures from the first to the fourth generation can be found in the fifth generation as a part of the structure.
Fig. 10.
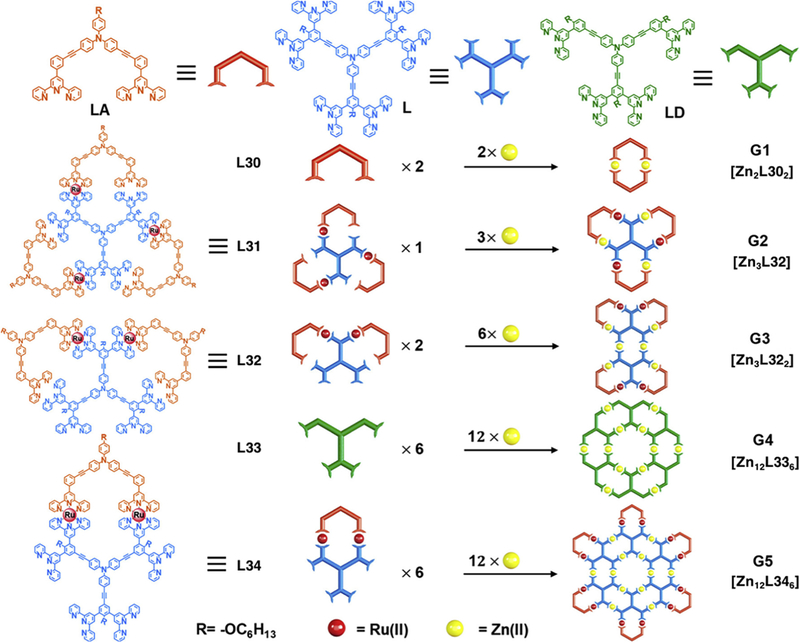
Self-assembly of five generations fractal supermolecule. Figure was reproduced from Ref. [93] with permission of the copyright holders.
4.2. Iteration on 3D level
The iteration of supramolecular shape occurs not only on the 2D level, but also on the 3D level. For example, from the ring-in-ring to sphere-in-sphere, 2D spoked wheel to double- and triple-decker spoked wheel, and 2D spoked wheel to π-π stacking 3D molecular wheel
In 2015, the Li group reported two 2D hexagon ring-in-ring structures (Fig. 11) [94]. In this work, these architectures were achieved by self-assembly of tetratopic terpyridine ligand L35 and 180° diplatinum (II) acceptors L36 or L37. As rigid organic framework and high coordination sites density (CODs) strategy were employed in this system, these architectures were formed in good yield with excellent stability. In the same paper, the authors also developed the 3D sphere-in-sphere supramolecular architecture, by using the Pd(II) square planar geometry. In addition, the authors also proposed a method to calculate the density of coordination sites to evaluate the stability of different supramolecular structures [94]. Furthermore, the 2D structures could self-assemble to the ordered nanostructures and supramolecular metallogels.
Fig. 11.

Self-assembly of 2D ring-in-ring and 3D sphere-in-sphere structures. Figure was reproduced from Ref. [94] with permission of the copyright holders.
The 2D spoked wheel, 3D double- and triple-decker spoked wheel and π-π stacking 3D molecular wheel were successively achieved afterwards. Hexaterpyridine core L9 with its inherent hexagonal binding angles can be used as the star-shape core to coordinate with tritopic terpyridine-type ligands L38 for the construction of supramolecular wheel structures 39. In 2011, New-kome and co-workers reported this novel supramolecular achitecture by one-step self-assembly of the hexaterpyridine core L9, rim L38 and metal Zn(II) or Cd(II) with 94% yield and high selectivity (Fig. 12) [95].
Fig. 12.

Self-assembly of 2D supramolecular spoked wheel. Figure was reproduced from Ref. [95] with permission of the copyright holders.
Recently, Wang and co-workers disclosed a giant 3D supramolecular double- and triple-decker hexagon structure by using covalent link to vertically coordinate two or three layers of plane spoked wheels together based on <tpy-M2+–tpy> connectivity (Fig. 13) [96]. The ligand was synthesized through several Suzuki cross-coupling reaction. After coordinating with Cd2+ under standard condition, supramolecular structure, which contained up to 72+ ions pairs, was achieved in nearly quantitative yield. These supramolecular structures were characterized by NMR, ESI-MS, TWIM-MS, DOSY, TEM and AFM imaging.
Fig. 13.

Self-assembly of triple-decker hexagon supramolecular structure. Figure was reproduced from Ref. [96] with permission of the copyright holders.
Besides the covalent link strategy, another method towards the synthesis of 3D spoked wheel is setting up a π-π stacking core for the supermolecule. In 2014, inspired by the 2D terpyridine type supramolecular “wheel”, the Newkome group reported the self-assembly of the 3D spoked wheels by using 1,3,5-tristerpyridine L46 (“Y” tpye) or 1,2,3-triterpyridine L38 (“W” type) as wheel center to substitute for the hexaterpyridine core L9 (Fig. 14) [45]. When six of L38 ligands were used as rims and two of L46 ligands were employed as the stacking center, 3D supramolecular “bicyclic wheel” 47 was obtained. However, when treated with eight equivalents of L38 ligands and twelve equivalents of Zn2+ in the self-assembly system, a three-dimensional supermolecule 48 was achieved in one step. It is noteworthy that ligand L38 was used for two purposes: six of ligand L38 formed the outside rim and two of ligand L38 staggered at 180° to generate the bisrhomboidal core.
Fig. 14.

Self-assembly of supramolecular 3D wheel. Figure was reproduced from Ref. [45] with permission of the copyright holders.
5. Conclusions and outlook
In summary, 2,2’:6’,2”-terpyridine has unique advantages as a ligand including: 1) three strong nitrogen coordination sites, which provide tight chelation with various metal cations as an NNN Pincer ligand; 2) three electron-deficient pyridine cycles make terpyridine not only a strong σ-donor but also a very good π-receptor; 3) the low LUMO of terpyridine ligand allows it to participate in the redox reaction as a non-innocent ligand. Therefore, terpyridine and its derivatives have been employed into various catalytic reactions. The superior metal-binding ability of terpyridine ligands is also evidenced by its broad application in supramolecular construction. Due to the formation of “closed-shell” <tpy-M2+-tpy> complexes, it has been widely applied as a stable linear linkage in supramolecular chemistry. By combining this linear linker with other rigid organic frameworks, a series of attractive supramolecular architectures have been achieved. However, some challenges, such as unsatisfactory overall synthetic yield and low solubility of tpy ligand, still exist and need to be solved.
Acknowledgments
We are grateful to the NSF (CHE-1665122), NIH (1R01GM120240–01), NSFC (21228204) and the Development Project of the Pharmaceutical Industry of Jilin Province (20150311070YY; 20170307024YY) for financial support.
References
- [1].Hartwig JF, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books 2010 [Google Scholar]
- [2].Liu Z, Sadler PJ, Acc. Chem. Res. 47 (2014) 1174–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Luca OR, Crabtree RH, Chem. Soc. Rev. 42 (2013) 1440–1459. [DOI] [PubMed] [Google Scholar]
- [4].Peris E, Crabtree RH, Coord. Chem. Rev. 248 (2004) 2239–2246. [Google Scholar]
- [5].Vougioukalakis GC, Grubbs RH, Chem. Rev. 110 (2010) 1746–1787. [DOI] [PubMed] [Google Scholar]
- [6].Chi Y, Chou PT, Chem. Soc. Rev. 39 (2010) 638–655. [DOI] [PubMed] [Google Scholar]
- [7].Deiters A, Martin SF, Chem. Rev. 104 (2004) 2199–2238. [DOI] [PubMed] [Google Scholar]
- [8].Fache F, Schulz E, Tommasino ML, Lemaire M, Chem. Rev. 100 (2000) 2159–2231. [DOI] [PubMed] [Google Scholar]
- [9].Chelucci G, Thummel RP, Chem. Rev. 102 (2002) 3129–3170. [DOI] [PubMed] [Google Scholar]
- [10].Winter A, Newkome GR, Schubert US, ChemCatChem 3 (2011) 1384–1406. [Google Scholar]
- [11].Morgan GT, Burstall FH, J. Chem. Soc. (1932) 20–30. [Google Scholar]
- [12].Armspach D, Constable EC, Housecroft CE, Neuburger M, Zehnder M, J. Organomet. Chem. 550 (1998) 193–206. [Google Scholar]
- [13].Constable EC, Chem. Soc. Rev. 36 (2007) 246–253. [DOI] [PubMed] [Google Scholar]
- [14].An L, Xu C, Zhang XG, Nature Comm. 8 (2017). [Google Scholar]
- [15].Garcia-Dominguez A, Mueller S, Nevado C, Angew. Chem. Int. Ed. 56 (2017) 9949–9952. [DOI] [PubMed] [Google Scholar]
- [16].Hie L, Baker EL, Anthony SM, Desrosiers JN, Senanayake C, Garg NK, Angew. Chem. Int. Ed. 55 (2016) 15129–15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huihui KMM, Shrestha R, Weixs DJ, Org. Lett. 19 (2017) 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Joshi-Pangu A, Ganesh M, Biscoe MR, Org. Lett. 13 (2011) 1218–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Moragas T, Cornella J, Martin R, J. Am. Chem. Soc. 136 (2014) 17702–17705. [DOI] [PubMed] [Google Scholar]
- [20].Ni SY, Zhang WZ, Mei HB, Han JL, Pan Y, Org. Lett. 19 (2017) 2536–2539. [DOI] [PubMed] [Google Scholar]
- [21].Prinsell MR, Everson DA, Weix DJ, Chem. Commun. 46 (2010) 5743–5745. [DOI] [PubMed] [Google Scholar]
- [22].Shekhar KC, Basnet P, Thapa S, Shrestha B, Giri R, J. Org. Chem. 83 (2018) 2920–2936. [DOI] [PubMed] [Google Scholar]
- [23].Shrestha R, Weix DJ, Org. Lett. 13 (2011) 2766–2769. [DOI] [PubMed] [Google Scholar]
- [24].Kamata K, Suzuki A, Nakai Y, Nakazawa H, Organometallics 31 (2012) 3825–3828. [Google Scholar]
- [25].Tondreau AM, Atienza CCH, Darmon JM, Milsmann C, Hoyt HM, Weller KJ, Nye SA, Lewis KM, Boyer J, Delis JGP, Lobkovsky E, Chirik PJ, Organometallics 31 (2012) 4886–4893. [Google Scholar]
- [26].Duong HA, Wu WQ, Teo YY, Organometallics 36 (2017) 4363–4366. [Google Scholar]
- [27].Leonard NG, Bezdek MJ, Chirik PJ, Organometallics 36 (2017) 142–150. [Google Scholar]
- [28].Palmer WN, Diao TN, Pappas I, Chirik PJ, ACS Catal. 5 (2015) 622–626. [Google Scholar]
- [29].Wu J, Zeng HS, Cheng J, Zheng SP, Golen JA, Manke DR, Zhang GQ, J. Org. Chem. 83 (2018) 9442–9448. [DOI] [PubMed] [Google Scholar]
- [30].Limburg J, Vrettos JS, Chen HY, de Paula JC, Crabtree RH, Brudvig GW, J. Am. Chem. Soc. 123 (2001) 423–430. [DOI] [PubMed] [Google Scholar]
- [31].Zhang GQ, Zeng HS, Wu J, Yin ZW, Zheng SP, Fettinger JC, Angew. Chem. Int. Ed. 55 (2016) 14369–14372. [DOI] [PubMed] [Google Scholar]
- [32].Kaes C, Katz A, Hosseini MW, Chem. Rev. 100 (2000) 3553–3590. [DOI] [PubMed] [Google Scholar]
- [33].Ludlow JM, Guo Z, Schultz A, Sarkar R, Moorefield CN, Wesdemiotis C, Newkome GR, Eur. J. Inorg. Chem. 2015 (2015) 5662–5668. [Google Scholar]
- [34].Lu W, Mi BX, Chan MCW, Hui Z, Che CM, Zhu NY, Lee ST, J. Am. Chem. Soc. 126 (2004) 4958–4971. [DOI] [PubMed] [Google Scholar]
- [35].Sakamoto R, Katagiri S, Maeda H, Nishihara H, Coord. Chem. Rev. 257 (2013) 1493–1506. [Google Scholar]
- [36].Sauvage JP, Collin JP, Chambron JC, Guillerez S, Coudret C, Balzani V, Barigelletti F, Decola L, Flamigni L, Chem. Rev. 94 (1994) 993–1019. [Google Scholar]
- [37].Andres PR, Schubert US, Adv. Mater. 16 (2004) 1043–1068. [Google Scholar]
- [38].Eryazici I, Moorefield CN, Newkome GR, Chem. Rev. 108 (2008) 1834–1895. [DOI] [PubMed] [Google Scholar]
- [39].Hofmeier H, Schubert US, Chem. Soc. Rev. 33 (2004) 373–399. [DOI] [PubMed] [Google Scholar]
- [40].Schubert US, Eschbaumer C, Angew. Chem. Int. Ed. 41 (2002) 2892–2926. [DOI] [PubMed] [Google Scholar]
- [41].Chakraborty S, Sarkar R, Endres K, Xie TZ, Ghosh M, Moorefield CN, Saunders MJ, Wesdemiotis C, Newkome GR, Eur. J. Org. Chem. (2016) 5091–5095. [Google Scholar]
- [42].Chen MZ, Wang J, Liu D, Jiang ZL, Liu QQ, Wu T, Liu HS, Yu WD, Yan J, Wang PS, J. Am. Chem. Soc. 140 (2018) 2555–2561. [DOI] [PubMed] [Google Scholar]
- [43].Klosterman JK, Veliks J, Frantz DK, Yasui Y, Loepfe M, Zysman-Colman E, Linden A, Siegel JS, Org. Chem. Front. 3 (2016) 661–666. [Google Scholar]
- [44].Li YM, Jiang ZL, Wang M, Yuan J, Liu D, Yang XY, Chen MZ, Yan J, Li XP, Wang PS, J. Am. Chem. Soc. 138 (2016) 10041–10046. [DOI] [PubMed] [Google Scholar]
- [45].Lu XC, Li XP, Wang JL, Moorefield CN, Wesdemiotis C, Newkome GR, Chem. Commun. 48 (2012) 9873–9875. [DOI] [PubMed] [Google Scholar]
- [46].Newkome GR, Wang PS, Moorefield CN, Cho TJ, Mohapatra PP, Li SN, Hwang SH, Lukoyanova O, Echegoyen L, Palagallo JA, Iancu V, Hla SW, Science 312 (2006) 1782–1785. [DOI] [PubMed] [Google Scholar]
- [47].Schultz A, Li XP, Barkakaty B, Moorefield CN, Wesdemiotis C, Newkome GR, J. Am. Chem. Soc. 134 (2012) 7672–7675. [DOI] [PubMed] [Google Scholar]
- [48].Wang C, Hao XQ, Wang M, Guo CL, Xu BQ, Tan EN, Zhang YY, Yu YH, Li ZY, Yang HB, Song MP, Li XP, Chem. Sci. 5 (2014) 1221–1226. [Google Scholar]
- [49].Xie TZ, Guo K, Guo ZH, Gao WY, Wojtas L, Ning GH, Huang MJ, Lu XC, Li JY, Liao SY, Chen YS, Moorefield CN, Saunders MJ, Cheng SZD, Wesdemiotis C, Newkome GR, Angew. Chem. Int. Ed. 54 (2015) 9224–9229. [DOI] [PubMed] [Google Scholar]
- [50].Xie TZ, Guo K, Huang MJ, Lu XC, Liao SY, Sarkar R, Moorefield CN, Cheng SZD, Wesdemiotis C, Newkome GR, Chem. Eur. J. 20 (2014) 11291–11294. [DOI] [PubMed] [Google Scholar]
- [51].Xie TZ, Liao SY, Guo K, Lu XC, Dong XH, Huang MJ, Moorefield CN, Cheng SZD, Liu X, Wesdemiotis C, Newkome GR, J. Am. Chem. Soc. 136 (2014) 8165–8168. [DOI] [PubMed] [Google Scholar]
- [52].Chakraborty S, Newkome GR, Chem. Soc. Rev. 47 (2018) 3991–4016. [DOI] [PubMed] [Google Scholar]
- [53].Schubert US, Schmatloch S, Precup AA, Des. Monomers Polym. 5 (2002) 211–221. [Google Scholar]
- [54].Heller M, Schubert US, Eur. J. Org. Chem. (2003) 947–961. [Google Scholar]
- [55].Constable EC, Lewis J, Liptrot MC, Raithby PR, Inorg. Chim. Acta 178 (1990) 47–54. [Google Scholar]
- [56].Constable EC, Thompson A, J. Chem. Soc. Perkin. Trans. (1992) 3467–3475. [Google Scholar]
- [57].Newkome GR, He EF, J. Mater. Chem. 7 (1997) 1237–1244. [Google Scholar]
- [58].Schubert US, Eschbaumer C, Hien O, Andres PR, Tetrahedron Lett. 42 (2001) 4705–4707. [Google Scholar]
- [59].Constable EC, Ward MD, J. Chem. Soc., Dalton Trans. (1990) 1405–1409. [Google Scholar]
- [60].Fallahpour RA, Synthesis (2003) 155–184. [Google Scholar]
- [61].Fallahpour RA, Neuburger M, Zehnder M, NewJ. Chem. 23 (1999) 53–61. [Google Scholar]
- [62].Chen S, Feng DF, Li DY, Liu PN, Org. Lett. 20 (2018) 5418–5422. [DOI] [PubMed] [Google Scholar]
- [63].Hazra A, Lee MT, Chiu JF, Lalic G, Angew. Chem. Int. Ed. 57 (2018) 5492–5496. [DOI] [PubMed] [Google Scholar]
- [64].He YT, Wang Q, Zhao JH, Liu XY, Xu PF, Liang YM, Chem. Commun. 51 (2015) 13209–13212. [DOI] [PubMed] [Google Scholar]
- [65].Huang JZ, Ouyang L, Li JX, Zheng J, Yan WX, Wu WQ, Jiang HF, Org. Lett. 20 (2018) 5090–5093. [DOI] [PubMed] [Google Scholar]
- [66].Mazzotti AR, Campbell MG, Tang PP, Murphy JM, Ritter T, J. Am. Chem. Soc. 135 (2013) 14012–14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mondal M, Joji J, Choudhury J, Chem. Commun. 53 (2017) 3185–3188. [DOI] [PubMed] [Google Scholar]
- [68].Yamamoto K, Li JK, Garber JAO, Rolfes JD, Boursalian GB, Borghs JC, Genicot C, Jacq J, van Gastel M, Neese F, Ritter T, Nature 554 (2018) 511–514. [DOI] [PubMed] [Google Scholar]
- [69].Aiki S, Taketoshi A, Kuwabara J, Koizumi TA, Kanbara T, J. Organomet. Chem. 696 (2011) 1301–1304. [Google Scholar]
- [70].Dahl EW, Louis-Goff T, Szymczak NK, Chem. Commun. 53 (2017) 2287–2289. [DOI] [PubMed] [Google Scholar]
- [71].Jagadeesh RV, Wienhofer G, Westerhaus FA, Surkus AE, Junge H, Junge K, Beller M, Chem. Eur. J. 17 (2011) 14375–14379. [DOI] [PubMed] [Google Scholar]
- [72].Moore CM, Bark B, Szymczak NK, ACS Catal. 6 (2016) 1981–1990. [Google Scholar]
- [73].Moore CM, Szymczak NK, Chem. Commun. 49 (2013) 400–402. [DOI] [PubMed] [Google Scholar]
- [74].Cheng JJ, Zhu MJ, Wang C, Li JJ, Jiang X, Wei YW, Tang WJ, Xue D, Xiao JL, Chem. Sci. 8 (2017) 6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Li JJ, Liu YX, Tang WJ, Xue D, Li CQ, Xiao JL, Wang C, Chem. Eur. J. 23 (2017) 14445–14449. [DOI] [PubMed] [Google Scholar]
- [76].Wang XW, Wang C, Liu YX, Xiao JL, Green Chem. 18 (2016) 4605–4610. [Google Scholar]
- [77].Kurahashi T, Matsubara S, Acc. Chem. Res. 48 (2015) 1703–1716. [DOI] [PubMed] [Google Scholar]
- [78].Xiao Q, Zhang Y, Wang J, Acc. Chem. Res. 46 (2013) 236–247. [DOI] [PubMed] [Google Scholar]
- [79].Konkol M, Kondracka M, Voth P, Spaniol TP, Okuda J, Organometallics 27 (2008) 3774–3784. [Google Scholar]
- [80].Kolesnichenko IV, Anslyn EV, Chem. Soc. Rev. 46 (2017) 2385–2390. [DOI] [PubMed] [Google Scholar]
- [81].Weilandt T, Troff RW, Saxell H, Rissanen K, Schalley CA, Inorg. Chem. 47 (2008) 7588–7598. [DOI] [PubMed] [Google Scholar]
- [82].Hwang S-H, Wang P, Moorefield CN, Godinez LA, Manriquez J, Bustos E, Newkome GR, Chem. Commun. (2005) 4672–4674. [DOI] [PubMed] [Google Scholar]
- [83].Constable EC, Housecroft CE, Neuburger M, Schonle J, Zampese JA, Dalton Trans. 43 (2014) 7227–7235. [DOI] [PubMed] [Google Scholar]
- [84].Gao Y, Rajwar D, Grimsdale AC, Macromol. Rapid Commun. 35 (2014) 1727–1740. [DOI] [PubMed] [Google Scholar]
- [85].Winter A, Schubert US, Chem. Soc. Rev. 45 (2016) 5311–5357. [DOI] [PubMed] [Google Scholar]
- [86].Hwang S-H, Moorefield CN, Fronczek FR, Lukoyanova O, Echegoyen L, Newkome GR, Chem. Commun. (2005) 713–715. [DOI] [PubMed] [Google Scholar]
- [87].Sarkar R, Guo K, Moorefield CN, Saunders MJ, Wesdemiotis C, R Newkome G, Angew. Chem. lnt. Ed. 53 (2014) 12182–12185. [DOI] [PubMed] [Google Scholar]
- [88].Jiang Z, Li Y, Wang M, Liu D, Yuan J, Chen M, Wang J, Newkome GR, Sun W, Li X, Wang P, Angew. Chem. lnt. Ed. 56 (2017) 11450–11455. [DOI] [PubMed] [Google Scholar]
- [89].Newkome GR, Cho TJ, Moorefield CN, Baker GR, Cush R, Russo PS, Angew. Chem. lnt. Ed. 38 (1999) 3717–3721. [PubMed] [Google Scholar]
- [90].Wang M, Wang K, Wang C, Huang M, Hao X-Q, Shen M-Z, Shi G-Q, Zhang Z, Song B, Cisneros A, Song M-P, Xu B, Li X, J. Am. Chem. Soc. 138 (2016) 9258–9268. [DOI] [PubMed] [Google Scholar]
- [91].Wang H, Qian X, Wang K, Su M, Haoyang W-W, Jiang X, Brzozowski R, Wang M, Gao X, Li Y, Xu B, Eswara P, Hao X-Q, Gong W, Hou J-L, Cai J, Li X, Nature Comm. 9 (2018) 1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yin G-Q, Wang H, Wang X-Q, Song B, Chen L-J, Wang L, Hao X-Q, Yang H-B, Li X, Nature Comm. 9 (2018) 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wang L, Liu R, Gu J, Song B, Wang H, Jiang X, Zhang K, Han X, Hao X-Q, Bai S, Wang M, Li X, Xu B, Li X, J. Am. Chem. Soc. 140 (2018) 14087–14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Sun B, Wang M, Lou Z, Huang M, Xu C, Li X, Chen L-J, Yu Y, Davis GL, Xu B, Yang H-B, Li X, J. Am. Chem. Soc. 137 (2015) 1556–1564. [DOI] [PubMed] [Google Scholar]
- [95].Wang J-L, Li X, Lu X, Hsieh l.F., Cao Y, Moorefield CN, Wesdemiotis C, Cheng SZD, Newkome GR, J. Am. Chem. Soc. 133 (2011) 11450–11453. [DOI] [PubMed] [Google Scholar]
- [96].Liu D, Chen M, Li Y, Shen Y, Huang J, Yang X, Jiang Z, Li X, Newkome GR, Wang P, Angew. Chem. lnt. Ed. 57 (2018) 14116–14120. [DOI] [PMC free article] [PubMed] [Google Scholar]