

Abstract
Multiple studies have been published emphasizing the significant role of nanoparticle (NP) carriers in antigenic peptide-based subunit vaccines for the induction of potent humoral and cellular responses. Various design parameters of nanoparticle subunit vaccines such as linker chemistry, the proximity of antigenic peptide to NPs, and the density of antigenic peptides on the surface of NPs play an important role in antigen presentation to dendritic cells (DCs) and in subsequent induction of CD8+ T cell response. In this current study, we evaluated the role of peptide antigen proximity and density on DC uptake, antigen cross-presentation, in vitro T cell proliferation, and in vivo induction of CD8+ T cells. To evaluate the role of antigen proximity, CSIINFEKL peptides were systematically conjugated to poly(ethylene glycol) (PEG) hydrogels through N-hydroxysuccinimide–PEG–maleimide linkers of varying molecular weights: 2k, 5k, and 10k. We observed that the peptides conjugated to NPs via the 2k and 5k PEG linkers resulted in higher uptake in bone marrow-derived DCs (BMDCs) and increased p-MHC-I formation on the surface of bone marrow-derived DCs (BMDCs) as compared to the 10k PEG linker formulation. However, no significant differences in vitro T cell proliferation and induction of in vivo CD8+ T cells were found among linker lengths. To study the effect of antigen density, CSIINFEKL peptides were conjugated to PEG hydrogels via 5k PEG linkers at various densities. We found that high antigen density NPs presented the highest p-MHC-I on the surface of BMDCs and induced higher proliferation of T cells, whereas NPs with low peptide density resulted in higher DC cell uptake and elevated frequency of IFN-γ producing CD8+ T cells in mice as compared to the medium- and high-density formulations. Altogether, findings for these experiments highlighted the importance of linker length and peptide antigen density on DC cell uptake, antigen presentation, and induction of in vivo CD8+ T cell response.
Introduction
The three key elements in developing an effective vaccine are an antigen to elicit an adaptive immune response, an immunopotentiator to stimulate the innate immune system, and a delivery system to ensure targeting of an antigen and immunopotentiator to antigen presenting cells (APCs). Nanoparticles (NPs) are often used as a delivery system to target APCs. In developing PRINT NP subunit vaccines, antigens and adjuvants are conjugated to the surface of the NPs. At high loadings, this results in a decrease in particles stability. Nanoparticles (NPs) are often modified with poly(ethylene glycol) (PEG) to increase the particle stability. Various molecular weights (MW) of PEG linkers have been studied to understand the effect of PEG chain length and density on PEG surface conformations (brush vs mushroom).1 Although PEG densities higher than those required for forming the mushroom conformation are necessary for increased circulation half-life of NPs, high PEG densities are unfavorable for intracellular delivery of cargos as the PEG shield limits cellular binding and uptake.2−6
Lessons learned from these studies are being applied to understand the role of PEG linker length (or proximity of targeting ligand) for cellular targeting. Improved cellular targeting has been observed for NPs modified with low MW PEG linkers. Stefanick et al. reported enhanced cellular uptake by ∼9-fold and ∼100-fold in breast cancer and multiple myeloma cells, respectively, when cells were treated with liposomes conjugated to homing peptide by shorter ethylene glycol linker (EG12) as compared to a longer linker (EG45).7 Micelles prepared from linear poly(ethylene imine) (l-PEI) grafted to shorter PEG (500 Da) linkers for plasmid DNA delivery showed higher colloidal stability and transfection efficiency in vitro and in vivo as compared to micelles prepared by l-PEI–PEG (2000 Da).8 Since nanovaccines target immune cells including phagocytic dendritic cells (DCs) for antigen presentation and generation of adaptive immunity, we were interested in evaluating how PEG linker length can influence cargo display on nanovectors, as well as cellular uptake, and the downstream effects on the immune response.
The density of antigens on the surface of nanoparticle scaffolds can also affect the quality and magnitude of an immune response.9 For example, virus like particles displaying conjugated antigen at high densities induced stronger IgG responses.10 Chen et al. found that influenza antigen protein conjugated to glutathione transferase S-fusion proteins at a higher density resulted in higher average avidity for M2e-specific mAb and induced poly clonal antibody with enhanced affinity.11 On the contrary, Brewer et al. showed that NP decorated with viral antigens such as Env or HA at lower density elicited higher titers of antigen-specific serum IgG in mice as well as an increased frequency of antigen-specific antibody secreting cells.12 These findings demonstrate that the density of antigen on a NP scaffold is a critical determinant of the humoral immune response.13−15 Compared to monovalent binding, as in the case of single soluble antigens, antigen display on NPs at an optimum density may enhance multivalent binding to B cell receptors, better stimulate the downstream molecular events, and induce stronger immune response.16
On the other hand, how the T cell response is influenced by the antigen density has not been well studied. Priming of T cells requires uptake by antigen presenting cells (APCs) and processing of antigens into peptides that can be presented by major histocompatibility complex (MHC) class I or II as a peptide/MHC complex (pMHC) on the cell surface. Different from whole protein antigens, synthetic antigenic peptides can bind directly to the cell surface MHC class I or II or be internalized and processed to be presented on the cell surface as pMHC. Peptide cancer vaccines target antigen presenting DCs to induce potent antitumor T cell response and have been explored in preclinical and clinical studies. We have shown that loading T cell antigen peptides on NPs can greatly improve antigen presentation and T cell priming and proliferation.17,18 However, very little is known about how peptide antigen proximity and density on a NP surface affect cellular uptake, DC stimulation, and the induction of in vivo T cell response. Toward that end, we systemically explored the effect of antigen proximity by conjugating MHC-I peptide epitope to NP surface via various molecular weights of PEG linkers (PEG2k, PEG5k, PEG10k) and evaluated their effect on cellular uptake, antigen cross-presentation, in vitro T cell proliferation, and in vivo induction of CD8+ T cells. After selecting the optimum linker length, we conjugated MHC-I peptide epitope at varied densities and evaluated its effect on in vitro and in vivo immune responses. Findings in this work highlight the importance of linker length and peptide antigen density on DC uptake, antigen presentation, and induction of in vivo CD8+ T cell response.
Results and Discussion
Conjugation of CSIINFEKL to PEG Hydrogels
We have synthesized acrylate monomer and PEG cross-linker-based hydrogels and incorporated aminoethyl methacrylate to introduce reactive handle on the surface of hydrogel NPs. Hydrogel NPs (80 × 80 × 320 nm3) were fabricated via particle replication in nonwetting template (PRINT) technology.19 PRINT hydrogel particles were modified with CSIINFEKL utilizing a similar conjugation process as described previously, where amine groups on the NP surface were used to conjugate to the cysteine via SCM–PEG–maleimide linkers (2k, 5k, or 10k molecular weights).18 Briefly, particles were modified with equal moles of linkers to ensure similar linker densities and particle surface charge. After modification with the PEG linkers, NPs were incubated overnight with CSIINFEKL resulting in peptide conjugation to the NPs via Michael addition. To control antigen density, the peptide was charged at various amounts, and peptide loading per mg of NP was quantified utilizing a bicinchoninic acid assay (BCA). This was then converted to peptide density (peptides/nm2). CSIINFEKL-modified NPs were further modified with CpG oligodeoxynucleotides (ODN) by incubation with thiol containing CpG ODN to yield the final formulation of NPs with co-conjugated CSIINFEKL and CpG. However, for medium- and high-density peptide formulations, the addition of CpG leads to particle aggregation. To avoid particle aggregation, two sets of particles were fabricated, one set modified with CSIINFEKL, and one set modified with CpG ODN, particles where then mixed prior to in vivo inoculation. For in vitro cellular uptake studies, CSIINFEKL peptide was conjugated to fluorescein-labeled PEG hydrogels. Size, polydispersity index (PDI), and ζ-potential (ZP) of fluorescein-labeled PEG hydrogels were found to be similar to unlabeled (no fluorescein) NPs (data not shown). PEG hydrogels modified with linker only were also prepared to serve as a control group for the in vitro studies. All final NP formulations used in vitro and in vivo studies were evaluated for endotoxin level and confirmed to have endotoxin <0.1 EU/mg NP.
Nanoparticle surface charge can influence cellular uptake and in vivo biodistribution.20−22 Cationic nanoparticles are known to have higher uptake by cells and can facilitate cytosolic delivery of cargo by facilitating endosomal escape.20,23 Therefore, our overall objective during formulation development was to keep the surface charge of NP positive to achieve optimum uptake by DCs and further induction of CD8+ T cell response.
Physical characterizations of nanoparticles used in cell studies are shown in Table 1. Surface charges of NPs modified with PEG2k and PEG5k were 41 and 47 mV, respectively. When PEG10k was conjugated to NPs, surface charge decreased to 32 mV. This could be due to the charge shielding effect of longer chain PEG10k. This decrease in surface charge was also observed when the CSIINFEKL peptide was conjugated to the PEG2k and PEG5k formulations; however, for PEG10k formulations, the change in ZP was minimal. Similar decreases in ZP were observed when NPs were modified with low, medium, and high antigen density peptides, as seen in Table 1.
Table 1. Physical Characterization of NPs for in Vitro Studies.
| formulations | size (nm) | PDI | ZP (mV) | μg SIINFEKL/mg NP | number of antigen/nm2 |
|---|---|---|---|---|---|
| NP–PEG2k | 299 | 0.159 | 41 | NA | NA |
| NP–PEG5k | 273 | 0.170 | 47 | NA | NA |
| NP–PEG10k | 281 | 0.166 | 32 | NA | NA |
| NP–PEG2k–CSIINFEKL | 275 | 0.164 | 36 | 109 | 1.20 |
| NP–PEG5k–CSIINFEKL | 264 | 0.107 | 35 | 109 | 1.20 |
| NP–PEG10k–CSIINFEKL | 288 | 0.156 | 30 | 102 | 1.13 |
| NP–PEG5k–CSIINFEKLlow | 243 | 0.062 | 31 | 33 | 0.36 |
| NP–PEG5k–CSIINFEKLmedium | 257 | 0.139 | 27 | 171 | 1.89 |
| NP–PEG5k–CSIINFEKLhigh | 308 | 0.180 | 23 | 386 | 3.76 |
Physical characterizations of NPs used in vivo studies are shown in Tables 2 and 3. As shown in Table 2, when CpG ODN was conjugated to PRINT hydrogels, due to its negative charge, the NP surface charge became neutral. Size and PDI slightly increased, possibly due to the stearic crowding of biomolecules (CSIINFEKL and CpG ODN) on the surface of PRINT hydrogels.24,25 However, as shown in Table 3, when CpG and CSIINFEKL were conjugated separately for antigen density studies, ZP of NPs remained positively charged.
Table 2. Physical Characterization of NPs for in Vivo Linker Length Study.
| formulations | size (nm) | PDI | ZP (mV) | μg SIINFEKL/mg NP | μg CpG/mg NP | antigen/nm2 |
|---|---|---|---|---|---|---|
| NP–PEG2k–CSIINFEKL–CpG | 416 | 0.361 | 2 | 109 | 27.0 | 1.20 |
| NP–PEG5k–CSIINFEKL–CpG | 257 | 0.138 | 2 | 109 | 27.8 | 1.20 |
| NP–PEG10k–CSIINFEKL–CpG | 354 | 0.311 | 0 | 102 | 27.4 | 1.13 |
Table 3. Physical Characterization of NPs for in Vivo Density Study.
| formulations | size (nm) | PDI | ZP (mV) | μg SIINFEKL/mg NP | μg CpG/mg NP | antigen/nm2 |
|---|---|---|---|---|---|---|
| NP–PEG5k–CpG | 301 | 0.074 | 30 | NA | 25 | NA |
| NP–PEG5k–CSIINFEKLlow | 265 | 0.074 | 32 | 99 | NA | 0.96 |
| NP–PEG5k–CSIINFEKLmedium | 272 | 0.140 | 31 | 230 | NA | 2.24 |
| NP–PEG5k–CSIINFEKLhigh | 301 | 0.140 | 30 | 377 | NA | 3.67 |
Effect of the Linker Length and Peptide Antigen Density on NP Uptake by BMDCs
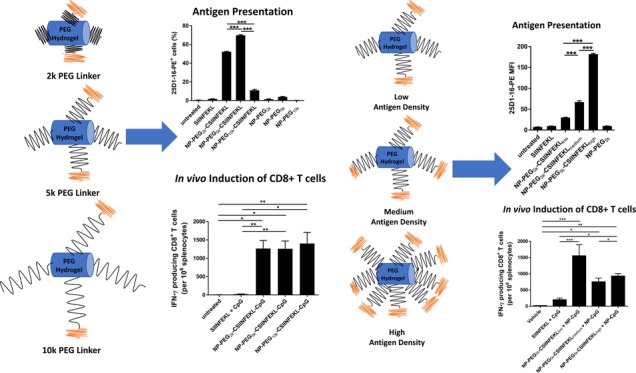
We evaluated the effect of the linker length on cellular uptake of fluorescein-labeled NPs using bone marrow-derived dendritic cells (BMDCs), as DCs are the most efficient antigen presenting cells for T cell priming and DCs are efficient in the uptake of particulate matter through phagocytosis. As vaccines are generally administered based on the dose of antigen, we dosed BMDCs with 5 μg/mL of SIINFEKL, free or associated with NPs (Figure 1A,B). Cells were then washed and examined by flow cytometry for the population that was positive for NP fluorescence. BMDCs dosed with NPs conjugated to CSIINFEKL through 2k PEG and 5k PEG linkers had much higher percentage of uptake (and mean fluorescence intensity (MFI)) as compared to the 10k PEG formulation (Figures 1A and S1A), suggesting that the “stealthiness” generated by 10k PEGs on the particle surface interfered with the interactions between the particles and cells. On the other hand, BMDCs dosed with the low-density CSIINFEKL–NPs at a dose of 5 μg/mL peptide displayed higher NP uptake (and MFI) as compared to high- or medium-density CSIINFEKL NPs (Figures 1B and S1B). Since less NPs were dosed on cells for the higher peptide density NPs at any given antigen dose, the differential cell uptake could be due to the variance in NP mass. Therefore, we also examined the NP uptake with constant NP mass at 50 μg/mL (Figure 1C). Interestingly, an increase in cell uptake was observed with increasing peptide density on NPs (Figure 1C). This increase was even more dramatic when shown as mean fluorescence intensity of cells (Figure S1C).
Figure 1.

Effect of the linker length and peptide antigen density on the uptake of NPs by BMDCs. BMDCs on day 6 were treated with NPs labeled with fluorescein isothiocyanate (FITC) for 4 h, washed and further incubated at 37 °C for 20 h. Cells with NP fluorescence were analyzed by flow cytometry. (A) Linker length comparison at 5 μg/mL peptide dose; (B) antigen density comparison at 5 μg/mL peptide dose; (C) antigen density comparison at 50 μg/mL NP dose. Results are shown as mean ± SEM, n = 3. Data were analyzed by one-way analysis of variance (ANOVA), Tukey’s multiple comparison test, *p < 0.05, **p < 0.01 and ***p < 0.001.
Effect of the Linker Length and Peptide Antigen Density on p-MHC-I Complex Presentation on BMDCs
We hypothesized that antigen proximity would affect the uptake of NPs and thus the antigen presentation by BMDCs. To determine these effects, we investigated the presentation of SIINFEKL–MHC-I complex on BMDCs as a function of antigen proximity. To conduct these studies, two sets of BMDCs (in triplicate) were either untreated or treated with soluble SIINFEKL, NP–PEG2k–CSIINFEKL, NP–PEG5k–CSIINFEKL, or NP–PEG10k–CSIINFEKL for 4 h at 5 μg/mL peptide. After 4 h, BMDCs were washed and treated with citrate buffer (pH 3.0) to remove the surface-bound p-MHC complex. One set of cells was fixed with paraformaldehyde, and another set of cells was further incubated at 37 °C for another 20 h. The purpose of further incubation was to evaluate whether after internalization, if the nanoparticle-bound CSIINFEKL was released and presented on p-MHC-I complex.
Intracellular processing and cross-presentation of exogenous antigen by dendritic cells (DCs) to T cells via MHC-I and MHC-II proteins are relatively well established.26 Particulate antigens are taken up by DCs via receptor-mediated endocytosis or phagocytosis27,28 and are either degraded in endocytic compartments (for class II presentation) or escape the endosome/phagosome and are degraded in the cytosol (for class I presentation).26 The resulting peptide fragments can then be loaded onto MHCs for cell surface presentation as pMHC.26,29 In this study, CSIINFEKL peptide was conjugated to PRINT-based hydrogel via thioether bond, which allows controlled and sustained release of peptides over days under reducing conditions (e.g., cytosol), as previously reported.18 Moreover, enzymes present in various intracellular compartments may also contribute to the cleavage and release of antigenic peptides from PRINT hydrogel nanoparticles.30 Therefore, we think that the antigen bound to PRINT hydrogel via thioether linker is possibly getting released from the NPs in reductive intracellular compartments of DCs. Once CSIINFEKL is released in its native form or bound with thioether linker, it is then cross-presented to T cells via MHC-I pathway.
To quantify antigen cross-presentation, BMDCs were stained with the 25-D1.16 antibody, which recognizes SIINFEKL/H-2Kb on APCs. Signals from the 4 h-treated BMDCs were subtracted from 4 h + 20 h-treated BMDCs, and the data is represented as % of cells positive for p-MHC-I complex or MFI of PE. As shown in the dot plot analysis (Figure 2A), BMDCs treated with linker-modified NPs had no antigen presentation. BMDCs treated with all three formulations, NP–PEG2k–CSIINFEKL, NP–PEG5k–CSIINFEKL, and NP–PEG10k–CSIINFEKL had higher p-MHC-I complex staining intensity as compared to soluble SIINFEKL and untreated cells after 24 h (Figure 2B,C). When BMDCs were treated with NP–PEG10k–CSIINFEKL, staining of p-MHC-I complex was only 12.2% as compared to 52.8 and 72.3% when treated with NP–PEG2k–CSIINFEKL and NP–PEG5k–CSIINFEKL, respectively. CSIINFEKL conjugated to NPs via PEG5k outperformed all other formulations in their antigen presentation efficiency in BMDCs, as shown in Figure 2A,B.
Figure 2.

Effect of the linker length and the antigen density on antigen presentation in BMDCs. Two sets of BMDCs were dosed with different formulations (at a dose of 5 μg/mL peptide, free or associated with NPs) for 4 h at 37 °C, washed and treated with citrate buffer of pH 3.0 to remove surface-bound p-MHC-I complex. One set of BMDCs was further incubated at 37 °C for another 20 h. At the end of the experiment, cells were stained with 25-D1.16 antibody which recognizes SIINFEKL/H-2Kb on APCs and analyzed via flow cytometry. Signals from 4 h-treated BMDCs were subtracted from 4 h + 20 h-treated BMDCs and data are shown as % of cells positive for p-MHC-I complex for (A) linker length and (C) antigen density or shown as MFI of PE for (B) linker length and (D) antigen density. Representative flow cytometry histograms are shown in panel (E). Results are shown as mean ± SEM, n = 3. Data were analyzed by one-way ANOVA, Tukey’s multiple comparison test, **p < 0.01, **p < 0.001 and ****p < 0.0001. Only comparisons among the three NPs groups are shown in (A–D), and all three NP groups are at least *p < 0.05 compared to untreated, SIINFEKL or blank NPs.
To evaluate the multivalence effect (or density of peptide on NP surface), BMDCs were also treated with either NP–PEG5k–CSIINFEKLlow or NP–PEG5k–CSIINFELmedium or NP–PEG5k–CSIINFEKhigh. At same antigen concentrations, we found that BMDCs treated with medium- and high-density CSIINFEKL NPs had higher % of p-MHC-I and MFI as compared to BMDCs dosed with lower antigen density carrying NPs. In other words, BMDCs dosed with the least amount of particles with higher antigen molecules on the surface were able to present the highest amount of p-MHC-I complex as compared to BMDCs treated with the highest amount of particles with low antigen density on their surface. The MFI of BMDCs was found to be doubled when treated with high antigen density NPs as compared to medium-density NPs. This indicates that at a high antigen density, NP–CSIINFEKL presented more p-MHC-I complexes on the surface of BMDC, likely due to stronger multivalent binding and internalization of NP–CSIINFEKL. Future experiments will be designed to gain more insight into processing and cross-presentation of CSIINFEKL-bound PRINT hydrogels in DCs.
Effect of the Linker Length and the Antigen Density on in Vitro Proliferation of OT-I T Cells
In previous studies, we observed that CSIINFEKL conjugated to NPs via 5k PEG linkers had higher cellular uptake as well as higher presentation of p-MHC-I complex via BMDCs. Therefore, we hypothesized that the conjugation of CSIINFEKL to NPs via 5k PEG linker would result in higher activation and proliferation of CD8+ T cells. CD8+ T cells derived from OT-I T cell receptor transgenic mice were used to evaluate SIINFEKL-specific T cell response for NP and soluble vaccine formulations. BMDCs were dosed with soluble SIINFEKL, NP–PEG2k–CSIINFEKL, NP–PEG5k–CSIINFEKL, or NP–PEG10k–CSIINFEKL at either 1 or 10 μg/mL peptide for 4 h at 37 °C. Cells were then washed to remove NPs (soluble SIINFEKL was not removed) and co-cultured with CTV-labeled OT-I T cells for 88 h at 37 °C. At the end of incubation, cells were stained with anti-CD3, CD8, and CD11c antibodies and examined by flow cytometry.
As shown in Figure 3A, soluble SIINFEKL-pulsed BMDCs stimulated OT-I T cell proliferation at 1 μg/mL. BMDCs treated with NP–CSIINFEKL at a peptide concentration of 1 μg/mL resulted in similar T cell growth as compared to BMDCs treated with soluble SIINFEKL (Figure 3B). No significant difference was observed among the three NP formulations evaluated.
Figure 3.

Effect of the linker length and the antigen density on in vitro proliferation of CD8+ OT-I T cells. BMDCs were dosed with various NP formulations at 1 μg/mL concentration of peptide for 4 h at 37 °C. Cells were then washed to remove NPs and co-cultured with CTV-labeled OT-I T cells for 72 h at 37 °C. At the end of the incubation period, cells were stained with anti-CD3, CD8, and CD11c antibodies and examined by flow cytometry. Representative flow cytometry histograms are shown in panel (A). Percentage cell growths of CD8+ OT-I T cells for various linker lengths and antigen density are compared in (B) and (C), respectively. Results are shown as mean ± SEM, n = 3. Data were analyzed by one-way ANOVA, Tukey’s multiple comparison test, *p < 0.05, **p < 0.01, and ***p < 0.001.
To evaluate the multivalency effect of the peptide-decorated NP surface, BMDCs were treated with either NP–PEG5k–CSIINFEKLlow, NP–PEG5k–CSIINFELmedium, or NP–PEG5k–CSIINFEKhigh for 4 h at 37 °C. Consistent with the p-MHC-I presentation results, NPs with the higher antigen density induced higher OT-I T cell proliferation (Figure 3C).
These data sets indicate that differences in antigen proximity observed during cell uptake studies and the antigen presentation assay for 5k PEG linker did not translate into higher proliferation of CD8+ T cells in vitro. However, high antigen density on NPs that displayed higher uptake and p-MHC-I presentation in BMDCs was able to translate into higher CD8+ T cell priming and proliferation.
Effect of the Linker Length and the Antigen Density on Antigen-Specific IFN-γ Producing CD8+ T Cell Response
In an effort to determine if linker length would affect the induction of CD8+ T cell response in mice, the frequency of antigen-specific IFN-γ producing T cells in the spleens of mice was evaluated ex vivo by ELISPOT. To enhance the immunogenicity of the antigenic peptide, CpG was co-conjugated to the NPs with peptides displayed on three linkers to facilitate induction of T cell response (Table 2). As shown in Figure 4A, C57BL6/J mice were vaccinated on day 0 and 13 with either soluble SIINFEKL + soluble CpG, NP–PEG2k–CSIINFEKL–CpG, NP–PEG5k–CSIINFEKL–CpG, or NP–PEG10k–CSIINFEKL–CpG. After 7 days of the boost dose, splenocytes were harvested, and IFN-γ producing T cells were evaluated by ex vivo ELISPOT assay. As shown in Figure 4B, vaccination with soluble peptide and CpG admix induced minimal response similar to untreated mice, even though CpG is a strong immunostimulator. This is consistent with the poor immunogenicity of soluble antigenic peptides reported in the literature that is mostly due to their low stability, tissue retention, and cell uptake,31 which bolsters the need for particulate delivery systems for co-delivering peptide antigens with adjuvants.17 On the other hand, all three nanoparticle formulations induced a high frequency of IFN-γ producing CD8+ T cells (>1000 spots/million splenocytes on average) in mice. However, no significant difference was observed among the three NP formulations with different PEG linker lengths tested in this animal study. The higher particle uptake and p-MHC-I complex presentation of the 5k PEG linker did not translate to any significant improvement in this effector T cell function in vivo.
Figure 4.

Effect of the linker length and the antigen density on IFN-γ producing CD8+ T cells in spleen. (A) Experimental design. On day 0 and 13, mice were immunized with various samples containing the equivalent of 100 μg peptide and/or 20 μg CpG via s.c. injection in the left flank. After 1 week, splenocytes were isolated and restimulated with 5 μg/mL of SIINFEKL for 22 h and analyzed for IFN-γ production by an ELISPOT assay. (B) Effect of the linker length. (C) Effect of antigen density. Results are shown as mean ± SEM, n = 5–6 (except for vehicle control in (B) which is n = 3). Data were analyzed by one-way ANOVA followed by Tukey’s test, *p < 0.05, **p < 0.01, and ***p < 0.001.
As discussed earlier, physicochemical properties of NPs such as size and surface charge affect their uptake by APCs, intracellular trafficking, and in vivo biodistribution.20−22 Generally, NPs with a diameter of <100 nm can traffic directly to lymph nodes by diffusion and convection, whereas nanoparticle with a diameter of ∼200 nm or greater traffic to lymph nodes via cell-mediated transport or through hydrodynamic swelling caused by the injection itself.32,33 We think that the neural surface charge and higher hydrodynamic diameter of these NPs (Table 2) could have resulted in to their variable uptake by APCs and lymphatic trafficking, and thus translated to no significance difference in induction of CD8+ T cells.
We had also observed enhanced particle uptake, MHC-I presentation, and T Cell proliferation with increased peptide density in vitro. Therefore, we evaluated whether changing antigen density on NPs would result in an enhanced induction of CD8+ T cell response in vivo. As shown in Table S1, when medium and high densities of CSIINFEKL were co-conjugated with CpG on NPs, the size of the NPs increased drastically indicating particle instability and aggregation, likely due to the neutralization of the positive particle charge. To eliminate NP size as an influencing factor in the study of antigen density, CSIINFEKL and CpG were conjugated onto separate NPs (through PEG5k only), which were then mixed together for vaccination. As shown in Table 3, these particles were stable and monodisperse. The same dosing schedule in Figure 4A was followed for mice treated with either vehicle, soluble SIINFEKL + CpG, NP–PEG5k–CSIINFELlow + NP–CpG, NP–PEG5k–CSIINFEKmedium + NP–CpG, or NP–PEG5k–CSIINFEKhigh + NP–CpG.
As shown in Figure 4C, all NP constructs induced a high frequency of IFN-γ producing CD8+ T cells in mice as compared to the soluble components. Interestingly, the lowest antigen density resulted in the highest frequency of IFN-γ producing CD8+ T cells, about 1500 T cells per 106 splenocytes compared to 800–1000 for the other two peptide density groups. This is contradictory to the results obtained in the in vitro p-MHC-I presentation study as well as OT-I T cells induction study where NPs with higher antigen density performed significantly better as compared to other densities. However, enhanced particle uptake was observed for low peptide density NPs. In addition, given the same amount of antigen dosed in mice, more particles were administered in the low-density NP sample, which may help overcome the in vivo tissue hindrance and accessibility to immune cells. Therefore, perhaps particle uptake and trafficking play a larger role in predicting in vivo efficacy; however, further investigation needs to be done to evaluate in vivo particle uptake by DCs and trafficking to lymph nodes.
Conclusions
In summary, we investigated the effect of antigen proximity and density on NP uptake and antigen presentation by BMDCs, in vitro T cell proliferation, and induction of IFN-γ producing CD8+ T cells in mice. Although the 5k PEG linker showed higher particle uptake and antigen presentation in BMDCs, in vitro proliferation of OT-I T cells and induction of IFN-γ producing CD8+ T cells were similar to other linkers. NPs with high antigen density resulted in higher antigen presentation and proliferation of OT-I T cells. However, low antigen density PEG hydrogels induced the highest CD8+ T cell response in mice, indicating that more particles with fewer antigens could be better at inducing an immune response in vivo as compared to fewer particles with higher antigen surface density. Taken together, this study sheds light on the importance of MHC-I peptide antigen proximity and density on cellular uptake and antigen presentation by DCs and the induction of a CD8+ T cell response.
Materials and Methods
Materials
Poly(ethylene glycol) diacrylate (Mn 700) (PEG700DA), 2-aminoethyl methacrylate hydrochloride (AEM), diphenyl(2,4,6-trimethylbenzoyl)-phosphine oxide (TPO), thiol-modified CpG 1826 (C6-S–S-C6-tccatgacgttcctgacgtt), DNase, RNase free sterile water were purchased from Sigma-Aldrich. Tetraethylene glycol monoacrylate (HP4A) was synthesized in house. Cysteine-modified OVA257–264 (CSIINFEKL) was purchased from Peptide 2.0. Maleimide PEG succinimidyl N-hydroxysuccinimide (NHS) esters (2k, 5k, and 10k) were purchased from Creative PEG works. PRINT molds (80 × 320 nm2) were obtained from Liquidia Technologies. DNA grade NAP-10 columns were purchased from GE Healthcare. RPMI 1640 medium, penicillin and streptomycin, l-glutamine, fetal bovine serum (FBS) were all from Life Technologies. BCA protein assay kit, Pierce LAL Chromogenic Endotoxin Quantitation Kit, RPMI 1640, fetal bovine serum (FBS), and penicillin–streptomycin (10 000 U/mL) were purchased from Thermo Fisher Scientific. GM-CSF and IL-4 were purchased from R&D Systems. CD8 T cell isolation kit and CD11c DC microbeads mouse were purchased from Miltenyi Biotec. CellTrace Violet Cell Proliferation Kit was obtained from Invitrogen and CD8-PerCP-eFluor710, CD3-FITC, CD11c-PE, CD11c-APC, and 25-D1.16-PE (anti-SIINFEKL/H-2Kb complex) antibodies were purchased from eBioscience. IFN-γ ELISPOT kit was purchased from BD Biosciences.
Methods
PRINT Nanoparticle Fabrication
The PRINT particle fabrication process is described previously.18 Briefly, the preparticle solution was prepared by dissolving 3.5 weight percent (wt %) of the various reactive monomers in isopropyl alcohol. Preparticle solution was composed of 69 wt % HP4A, 10 wt % PEG700DA, 20 wt % AEM, and 1 wt % TPO. Using # 3 mayer rod, a thin film of preparticle solution was drawn on to corona-treated PET using roll-to-roll lab line (Liquidia Technologies) running at 12 feet per minute. The solvent was evaporated by heat guns. Then, 80 × 80 × 320 nm3, the cylinder-shaped mold was laminated to the delivery sheet and passed through nip (80 psi, 12 feet per minute). After delamination, filled molds were cured by passing through UV LED lamp (λmax = 395 nm, 30 psi N2, 12 feet per minute; Phoseon). After cross-linking the hydrogels inside the mold cavities, the filled mold was laminated against PVA harvesting sheet and passed through the heated nip (140 °C, 80 psi, 12 feet per minute). Particles were removed from the mold by splitting the harvesting sheet from the mold. Particles were then harvested by dissolving sacrificial harvesting layer of PVA into water (2 mL of water per 10 feet of harvesting sheet). Particle suspensions were passed through 2 μm filter to remove additional scum layer. To remove excess of PVA, particles were spun down at 14 000 rpm (Eppendorf Thermomixer R) for 25 min and resuspended into sterile water. This purification procedure was repeated 3 times.
Thermogravimetric Analysis (TGA)
Concentrations of particles were determined by thermogravimetric analysis (TGA) using a TA Instrument’s Discovery TGA. Aluminum sample pans were tarred before loading the sample. The stock nanoparticle solutions (20 μL) were loaded on to the pan. Samples suspended in water were heated at 30 °C/min to 130 °C, followed by a 10 min isotherm at 130 °C. All samples were then cooled at 30 °C/min to 30 °C, followed by a 2 min isotherm at 30 °C.
Dynamic Light Scattering (DLS)
Particle size and ζ-potential (ZP) were measured in sterile water by dynamic light scattering (DLS) on a Zetasizer Nano ZS (Malvern Instruments, Ltd.).
Conjugation of Linker to NPs
We utilized amine groups from AEM to conjugate NPs to NHS–PEG–Mal. Equal moles of NHS–PEG2k–Mal (1.56 mg), NHS–PEG5k–Mal (3.89 mg), and NHS–PEG10k–Mal (7.78 mg) were dissolved in dimethylformamide (DMF). The volume of DMF was kept constant at 160 μL regardless of different linker masses used. NPs were modified with NHS–PEG–Mal, as mentioned here in ref (18). Briefly, 1 mg of NPs was continuously agitated for 2 h at 1400 rpm (Eppendorf Thermomixer R) in the presence of linker. The total volume of the reaction was 1 mL. After 2 h, the unconjugated linker was removed from the NPs via 2 centrifugation washes (Eppendorf Centrifuge 5417g) with sterile water at a speed of 13 000g.
Conjugation of CSIINFEKL to Linker-Modified NPs
Conjugation reaction of CSIINFEKL to NPs via NHS–PEG–Mal was performed, as mentioned in ref (18). After modifying NPs with the linker, CSIINFEKL peptide was conjugated by performing the reaction in 1× phosphate-buffered saline (PBS) buffer of pH 7.0. Briefly, an equal amount of peptide (0.200 mg) was charged to the reaction chemistry to conjugate a similar amount of peptide by various linkers. After 2 h, unconjugated peptide was removed from the NPs via 2 centrifugation washes (Eppendorf Centrifuge 5417g) with sterile water at a speed of 13 000g. To conjugate different densities of peptides to NPs, particles were modified by NHS–PEG5k–Mal. Different amounts of CSIINFEKL, 0.050, 0.500, and 1.0 mg, were charged to the conjugation recipe for low-, medium-, and high-density NP, respectively. At the end of the conjugation process, peptide loading was evaluated by performing standard BCA assay (Thermo Fisher Scientific) on particle-conjugated peptides. BCA absorbance reading of linker-modified particles was subtracted from the reading of peptide-conjugated NPs. All formulations were evaluated for endotoxin level using Pierce LAL Chromogenic Endotoxin Quatitation Kit using 1 mg/mL of NP.
Reduction and Purification of CpG
Reduction and purification of CpG were performed, as previously described.17 Briefly, C6 S–S-C6 CpG 1826 was reduced with 100 mM dithiothreitol solution in sodium phosphate buffer (pH 8.0) and purified by gel filtration chromatography using Sephadex NAP-10 column. The concentration of CpG was measured by evaluating absorption at 260 nm by using NanoDrop 2000 Spectrophotometer.
Conjugation of Thiol-CpG 1826 to NPs
After reduction and purification of C6-CpG ODN, conjugation of CpG ODN to NPs via different PEG linkers was done following the literature procedure.17 Briefly, CSIINFEKL-conjugated NPs were incubated with thiolated CpG overnight. NPs were washed 3 times with 10× PBS to remove electrostatically bound CpG. All supernatants were collected. Evaluation of CpG in supernatants was done via UV/vis spectroscopy at 260 nm by using NanoDrop 2000 Spectrophotometer. The amount of conjugated CpG was determined by subtracting the amount of unconjugated CpG in supernatants.
Animals
Female C57BL/6J mice were purchased from Jackson Laboratory and used at age 6–12 weeks. OT-I transgenic mice were purchased from Jackson Laboratory and bred at UNC. All experiments involving mice were carried out in accordance with an animal use protocol approved by the University of North Carolina Animal Care and Use Committee.
Preparation of BMDCs
BMDCs were prepared and cultured as in ref (17). Briefly, bone marrow was collected from mouse femurs and tibias. After lysing erythrocytes by ACK lysis buffer, cells were grown in RPMI 1640 supplemented with 10% FBS, 50 μM 2-mercaptoethanol, 10 ng/mL GM-CSF, and 10 ng/mL of IL-4 (R&D Systems). On day 6, BMDCs were harvested and further purified CD11c+ DCs with Mouse CD11c Microbeads (Miltenyi Biotec) following manufacturing instructions.
Cell Uptake Study
To test cellular uptake of particles by BMDCs, particles with FITC incorporated in the matrix were utilized to track them. Day 6 BMDCs were dosed with samples for 4 h, washed, and further incubated at 37 °C for 20 h. Cells were then examined with Cyan ADP followed by analysis with the Summit software.
OT-I T Cell Proliferation Study
Day 6 BMDCs were dosed with SIINFEKL or NP samples for 4 h, washed, and further incubated at 37 °C for 23 h. OT-I CD8+ T cells were isolated from spleens of OT-I mice using mouse CD8 T cell isolation kit (Miltenyi Biotec) and labeled with CellTrace Violet Cell Proliferation Kit (Invitrogen) for 10 min at 37 °C. Labeled OT-I cells were then added to treated BMDCs and co-incubated for 72 h at 37 °C. After the incubation, cells were stained with CD8-PerCP-eFluor710, CD3-FITC, and CD11c-PE (eBioscience) and examined for T cell proliferation with Cyan ADP followed by analysis with Summit software.
Antigen Presentation Assay in BMDCs
Day 6 BMDCs (3 × 105 cells) were either untreated or treated with various formulations or soluble CSIINFEKL peptide at a dose of 5 μg/mL for 4 h. After 4 h of incubation, cells were either washed with citrate-phosphate buffer (pH 3.0) for 3 min on ice to strip off the MHC-I–peptide complex or NP-peptide/H-2Kb complex from the cell surface. Additionally, cells were reincubated at 37 °C for an additional 20 h postwashes and then stained with CD11c-APC and 25-D1.16-PE (anti-SIINFEKL/H-2Kb complex) antibodies (eBioscience), followed by flow cytometry analysis on Cyan ADP (Dako).
Immunization Study
Immunization studies were performed, as in ref (17). Briefly, all formulations were prepared 24 h before injections, resuspended in isotonic 9.25% sucrose/H2O, and subcutaneously administered in the left flank. SIINFEKL was given at a dose of 100 μg, and CpG was injected at a dose of 20 μg. Prime and boost doses were administered on day 0 and 13, respectively. On day 20, mice were sacrificed, and spleens were harvested for further analysis, as described here previously.18
IFN-γ ELISPOT Assay
The frequency of antigen-specific IFN-γ producing T cells in the spleen was evaluated using IFN-γ ELISPOT kit (BD Biosciences). Immobilon-P hydrophobic PVDF plates (Millipore) were briefly treated with 35% ethanol, washed 2 times with PBS, and coated overnight with anti-mouse IFN-γ antibody at 4 °C. The following day, plates were blocked with 200 μL R-10 medium with for 2 h at room temperature. 100 000 splenocytes in R-10 medium were plated in each well with or without restimulation with 10 μg/mL of SIINFEKL peptide overnight at 37 °C. Spots were then developed following the manufacturer’s instructions. Unstimulated control showed minimum numbers of spot. Data presented herein are obtained using n = 4–6 animals (except for vehicle control in linker length study which was n = 3).
Acknowledgments
The authors thank Dr Ashish Pandya for the synthesis of HP4A and the University of North Carolina Animal Studies Core for their assistance with animal experiments. This work was supported by Liquidia Technologies, the Carolina Center for Cancer Nanotechnology Excellence (U54CA151652), and by the University Cancer Research Fund.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03391.
Physical characterization of particles for linker length study, effect of the linker length and the peptide antigen density on the uptake of NPs by BMDCs (PDF)
Author Present Address
# Department of Biomedical Engineering, University of Delaware, Newark, Delaware 19716, United States (C.H.K.).
The authors declare the following competing financial interest(s): The research reported in this paper received partial financial support from Liquidia Technologies, Inc., which Joseph M. DeSimone co-founded. Currently he has personal financial interests in Liquidia Technologies, Inc.
Supplementary Material
References
- Pozzi D.; Colapicchioni V.; Caracciolo G.; Piovesana S.; Capriotti A. L.; Palchetti S.; De Grossi S.; Riccioli A.; Amenitsch H.; Lagana A. Effect of polyethyleneglycol (PEG) chain length on the bio-nano-interactions between PEGylated lipid nanoparticles and biological fluids: from nanostructure to uptake in cancer cells. Nanoscale 2014, 6, 2782–2792. 10.1039/c3nr05559k. [DOI] [PubMed] [Google Scholar]
- Mosqueira V. C.; Legrand P.; Morgat J. L.; Vert M.; Mysiakine E.; Gref R.; Devissaguet J. P.; Barratt G. Biodistribution of long-circulating PEG-grafted nanocapsules in mice: effects of PEG chain length and density. Pharm. Res. 2001, 18, 1411–1419. 10.1023/A:1012248721523. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Jones S. W.; Parker C. L.; Zamboni W. C.; Bear J. E.; Lai S. K. Evading immune cell uptake and clearance requires PEG grafting at densities substantially exceeding the minimum for brush conformation. Mol. Pharm. 2014, 11, 1250–1258. 10.1021/mp400703d. [DOI] [PubMed] [Google Scholar]
- Fang C.; Shi B.; Pei Y. Y.; Hong M. H.; Wu J.; Chen H. Z. In vivo tumor targeting of tumor necrosis factor-alpha-loaded stealth nanoparticles: effect of MePEG molecular weight and particle size. Eur. J. Pharm. Sci. 2006, 27, 27–36. 10.1016/j.ejps.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Bazile D.; Prud’homme C.; Bassoullet M. T.; Marlard M.; Spenlehauer G.; Veillard M. Stealth Me.PEG-PLA nanoparticles avoid uptake by the mononuclear phagocytes system. J. Pharm. Sci. 1995, 84, 493–498. 10.1002/jps.2600840420. [DOI] [PubMed] [Google Scholar]
- Perry J. L.; Reuter K. G.; Kai M. P.; Herlihy K. P.; Jones S. W.; Luft J. C.; Napier M.; Bear J. E.; DeSimone J. M. PEGylated PRINT nanoparticles: the impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano Lett. 2012, 12, 5304–5310. 10.1021/nl302638g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanick J. F.; Ashley J. D.; Kiziltepe T.; Bilgicer B. A systematic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide-targeted liposomes. ACS Nano 2013, 7, 2935–2947. 10.1021/nn305663e. [DOI] [PubMed] [Google Scholar]
- Williford J. M.; Archang M. M.; Minn I.; Ren Y.; Wo M.; Vandermark J.; Fisher P. B.; Pomper M. G.; Mao H. Q. Critical Length of PEG Grafts on lPEI/DNA Nanoparticles for Efficient in Vivo Delivery. ACS Biomater. Sci. Eng. 2016, 2, 567–578. 10.1021/acsbiomaterials.5b00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer T. J.; Zmolek A. C.; Irvine D. J. Beyond antigens and adjuvants: formulating future vaccines. J. Clin. Invest. 2016, 126, 799–808. 10.1172/JCI81083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegerlehner A.; Storni T.; Lipowsky G.; Schmid M.; Pumpens P.; Bachmann M. F. Regulation of IgG antibody responses by epitope density and CD21-mediated costimulation. Eur. J. Immunol. 2002, 32, 3305–3314. . [DOI] [PubMed] [Google Scholar]
- Liu W.; Chen Y.-H. High epitope density in a single protein molecule significantly enhances antigenicity as well as immunogenicity: a novel strategy for modern vaccine development and a preliminary investigation about B cell discrimination of monomeric proteins. Eur. J. Immunol. 2005, 35, 505–514. 10.1002/eji.200425749. [DOI] [PubMed] [Google Scholar]
- Brewer M. G.; DiPiazza A.; Acklin J.; Feng C.; Sant A. J.; Dewhurst S. Nanoparticles decorated with viral antigens are more immunogenic at low surface density. Vaccine 2017, 35, 774–781. 10.1016/j.vaccine.2016.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltgrad E.; O’Reilly M. K.; Liao L.; Han S.; Paulson J. C.; Finn M. G. On-virus construction of polyvalent glycan ligands for cell-surface receptors. J. Am. Chem. Soc. 2008, 130, 4578–4579. 10.1021/ja077801n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeg J.; Axmann M.; Matic J.; Liapis A.; Depoil D.; Afrose J.; Curado S.; Dustin M. L.; Spatz J. P. T cell activation is determined by the number of presented antigens. Nano Lett. 2013, 13, 5619–5626. 10.1021/nl403266t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson M. C.; Abraham W.; Crespo M. P.; Chen S. H.; Liu H.; Szeto G. L.; Kim M.; Reinherz E. L.; Irvine D. J. Liposomal vaccines incorporating molecular adjuvants and intrastructural T-cell help promote the immunogenicity of HIV membrane-proximal external region peptides. Vaccine 2015, 33, 861–868. 10.1016/j.vaccine.2014.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Sagaseta J.; Malito E.; Rappuoli R.; Bottomley M. J. Self-assembling protein nanoparticles in the design of vaccines. Comput. Struct. Biotechnol. J. 2016, 14, 58–68. 10.1016/j.csbj.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapadia C. H.; Tian S.; Perry J. L.; Luft J. C.; DeSimone J. M. Reduction Sensitive PEG Hydrogels for Codelivery of Antigen and Adjuvant To Induce Potent CTLs. Mol. Pharm. 2016, 13, 3381–3394. 10.1021/acs.molpharmaceut.6b00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapadia C. H.; Tian S.; Perry J. L.; Sailer D.; Christopher Luft J.; DeSimone J. M. Extending antigen release from particulate vaccines results in enhanced antitumor immune response. J. Controlled Release 2018, 269, 393–404. 10.1016/j.jconrel.2017.11.020. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Nunes J. K.; Gratton S. E. A.; Herlihy K. P.; Pohlhaus P. D.; DeSimone J. M. Fabrication of multiphasic and regio-specifically functionalized PRINT particles of controlled size and shape. New J. Phys. 2009, 11, 075018 10.1088/1367-2630/11/7/075018. [DOI] [Google Scholar]
- Fromen C. A.; Rahhal T. B.; Robbins G. R.; Kai M. P.; Shen T. W.; Luft J. C.; DeSimone J. M. Nanoparticle surface charge impacts distribution, uptake and lymph node trafficking by pulmonary antigen-presenting cells. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 677–687. 10.1016/j.nano.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S. A.; Feng S. S. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm. Res. 2013, 30, 2512–2522. 10.1007/s11095-012-0958-3. [DOI] [PubMed] [Google Scholar]
- Foged C.; Brodin B.; Frokjaer S.; Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Yue Z. G.; Wei W.; Lv P. P.; Yue H.; Wang L. Y.; Su Z. G.; Ma G. H. Surface charge affects cellular uptake and intracellular trafficking of chitosan-based nanoparticles. Biomacromolecules 2011, 12, 2440–2446. 10.1021/bm101482r. [DOI] [PubMed] [Google Scholar]
- Shemetov A. A.; Nabiev I.; Sukhanova A. Molecular Interaction of Proteins and Peptides with Nanoparticles. ACS Nano 2012, 6, 4585–4602. 10.1021/nn300415x. [DOI] [PubMed] [Google Scholar]
- Lin A. Y.; Almeida J. P.; Bear A.; Liu N.; Luo L.; Foster A. E.; Drezek R. A. Gold nanoparticle delivery of modified CpG stimulates macrophages and inhibits tumor growth for enhanced immunotherapy. PLoS One 2013, 8, e63550 10.1371/journal.pone.0063550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joffre O. P.; Segura E.; Savina A.; Amigorena S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- Yameen B.; Choi W. I.; Vilos C.; Swami A.; Shi J.; Farokhzad O. C. Insight into nanoparticle cellular uptake and intracellular targeting. J. Controlled Release 2014, 190, 485–499. 10.1016/j.jconrel.2014.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt C. D.; Ma J. K.; Chalouni C.; Ebersold M.; Bou-Reslan H.; Carano R. A. D.; Mellman I.; Delamarre L. Mature dendritic cells use endocytic receptors to capture and present antigens. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 4287–4292. 10.1073/pnas.0910609107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savina A.; Amigorena S. Phagocytosis and antigen presentation in dendritic cells. Immunol. Rev. 2007, 219, 143–156. 10.1111/j.1600-065X.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- Saito G.; Swanson J. A.; Lee K. D. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv. Drug Delivery Rev. 2003, 55, 199–215. 10.1016/S0169-409X(02)00179-5. [DOI] [PubMed] [Google Scholar]
- Skwarczynski M.; Toth I. Peptide-based synthetic vaccines. Chem. Sci. 2016, 7, 842–854. 10.1039/C5SC03892H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine D. J.; Hanson M. C.; Rakhra K.; Tokatlian T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem. Rev. 2015, 115, 11109–11146. 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz M. A.; Hirosue S.; Hubbell J. A. Engineering approaches to immunotherapy. Sci. Transl. Med. 2012, 4, 148rv9 10.1126/scitranslmed.3003763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.