

Abstract
Human cytochrome P450 3A4 (CYP3A4) is an important drug metabolizing enzyme involved in a number of drug-drug and food-drug interactions. As such, much effort has been devoted into investigating its mechanism of interaction with ligands. CYP3A4 has one of the highest levels of substrate promiscuity for an enzyme, and can even bind multiple ligands simultaneously. The location and orientation of these ligands depend on the chemical structure and stoichiometry, and are generally poorly understood. In the case of the steroid testosterone, up to three copies of the molecule can associate with the enzyme at once, likely two in the active site and one at a postulated allosteric site. Recently, we demonstrated that steroid bioconjugation at the allosteric site results in an increase in activity of CYP3A4 toward testosterone and 7-benzyloxy-4-trifluoromethylcoumarin oxidation. Here, using the established bioconjugation methodology, we show how steroid bioconjugation at the allosteric site affects the heme spin state, the binding affinity (KS) of CYP3A4 for testosterone, as well as the enzyme coupling efficiency.
Keywords: Cytochrome P450 3A4, Allostery, Progesterone, Testosterone, Coupling, Heme spin-state
1. Introduction
Human cytochrome P450 3A4 (CYP3A4) is responsible for the metabolism of over 50% of existing drugs [1]. It belongs to a large and ubiquitous family of enzymes that is also involved in the biosynthesis of many diverse molecules (e.g. vitamins, steroids, secondary metabolites, bile acids) [1]. The clinical importance of CYP3A4 in drug metabolism and its implication in drug-drug interactions [2,3] are major incentives to fully elucidate the ligand-binding mechanism of this enzyme.
Owing to its large and flexible active site [4–14], CYP3A4 can oxidize substrates varying greatly in size (e.g. ranging from xylene at 106 Da, to cyclosporine at 1202 Da) [15,16], and can even bind multiple ligands simultaneously. Global analysis of multiple kinetic parameters suggests that this monomeric enzyme can bind up to three molecules of testosterone (TST), progesterone, α-naphthoflavone or carbamazepine [17–19]. The latter ability could explain the deviations from simple enzyme kinetics observed for some CYP3A4 reactions [5,20–23].
The presence of an allosteric binding site in CYP3A4 has also been suggested [5,20,24–32]. Noteworthy is a crystal structure of CYP3A4 with a co-crystallized steroid molecule, progesterone, at the periphery of the enzyme, suggesting that this region could be an allosteric pocket (Fig. 1) [5,25]. The relevance of this X-ray structure is debated because the pocket is at the interface of an unnatural dimer, yet the same area of the protein has been identified by mutagenesis [26–28] and by FRET studies as a potential allosteric site [29]. Although ligand interactions with CYP3A4 remain challenging to study, several ligand binding mechanisms have been proposed. In the case of the steroid TST, it is thought that the first binding event (higher affinity) occurs at the allosteric site, while the two subsequent binding events take place in the active site [17]. The first TST binding event induces some high-spin shift in the heme iron, which may promote the transfer of electrons from the redox partner to CYP3A4 without product formation [17,19]. The subsequent active site binding event results in product formation, whereas association of the third TST molecule improves the enzyme coupling efficiency (ratio of oxidized substrate to the reducing equivalents consumed) [17–19]. A similar mechanism has been proposed for other substrates as well [18,19]. Whether the apparent allosteric behavior of CYP3A4 is a true property of the enzyme or an artifact of a complicated substrate binding mechanism remains debated [33].
Fig. 1.
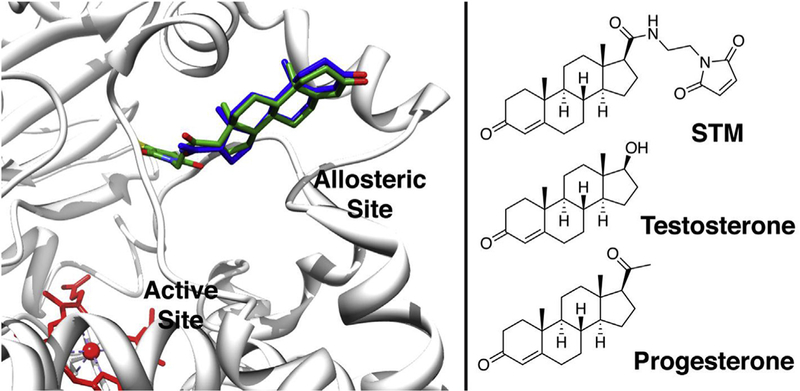
A modified X-ray crystal structure image showing steroid-maleimide (STM) conjugated to C215 of a CYP3A4 mutant. STM (in green) is superimposed on the existing, co-crystallized progesterone (in blue). STM and C215 were built into the allosteric site of CYP3A4 using the USCF Chimera package [35], PDB structure 1W0F [5] and the Persistence of Vision ™ Raytracer. Chemical structures of the steroids and STM are shown for comparison. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
We have recently reported the use of steroid bioconjugation to CYP3A4 as a complementary tool to study the postulated allosteric site and explore its effect on enzyme kinetics [34]. Via bioconjugation of a steroid-maleimide derivative (STM, Fig. 1; previously named PgM) selectively at the allosteric site, we have reported a positive effect on the kinetic activity of CYP3A4 towards two substrates, 7-benzyloxy-4-trifluoromethylcoumarin (BFC) and TST. Here, we build on these results and explore the significance of steroid bioconjugation at the putative allosteric site on substrate affinity and coupling efficiency.
2. Materials and methods
2.1. General information
All reagents were purchased from Sigma-Aldrich Canada Ltd. (Oakville, ON, Canada), Alfa Aesar (Ward Hill, MA, USA), Chem-Impex (Wood Dale, IL, USA), Agilent Technologies Canada Inc. (Mississauga, ON, Canada) or BioRad (Mississauga, ON, Canada). Reagents were used without further purification. CYP3A4 and CPR plasmids were generously provided by Dr. J. R. Halpert (University of Connecticut) and Dr. C. B. Kasper (University of Wisconsin), respectively.
2.2. Expression and purification of wild type and the C215 mutant of CYP3A4
Wild type CYP3A4 and the C215 mutant were expressed and purified as previously described, with minor modifications [34,36]. Wild type refers to the N-terminus-truncated Δ2–12 CYP3A4, which was used as a template for preparation of the C215 mutant containing the following mutations: C58T/C64A/C98S/F215C/C239S/C468G. For protein expression, the cells were harvested at 5000 × g instead of 4000 × g. For lysis, 1.3 μg/mL aprotinin and 1.25% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) were supplemented instead of 1 μg/mL aprotinin and 1% CHAPS, while concentrations of other protease inhibitors were kept the same as published. The lysed sample was split into 3 beakers and sonication was performed at 80% cycle duty. Both the Ni-NTA agarose column and Macro-Prep High S Support column were run at a slower flow rate (0.6 mL/min). The protein concentration was calculated from the reduced CO spectrum only, using a molar extinction coefficient of 91 mM−1 cm−1 between 450 and 490 nm [37].
2.3. Expression and purification of cytochrome P450 reductase (CPR)
CPR was expressed and purified as previously described with minor changes [38]. For CPR expression, the starting culture was generated by inoculating 100 mL of lysogeny broth (LB) with 8 colonies. The overnight culture was then used to inoculate the bulk culture (12 mL in 1 L) for 10 L total volume (10 flasks × 1 L). Following expression, the cells were harvested by centrifugation at 3000 × g and resuspended in 10 mL of 0.1 M potassium phosphate buffer (PBS, pH 7.4) before storage at −80 °C. For protein purification, the original protocol was shortened by condensing the two lysis steps into one step, and the two sonication steps into one step. In this case, the PBS buffer was removed by centrifugation at 3000 × g and the pellet was resuspended in 40 mL TSE buffer (75 mM tris buffer, pH 8, with 250 mM sucrose and 250 mM EDTA), after which lysozyme (1.2 mg in 20 mL TSE buffer) was added and the suspension was incubated for 20 min at 4 °C. The lysate was centrifuged at 3000 × g for 30 min and 4 °C, and the pellet was resuspended in 60 mL lysis buffer (50 mM tris buffer, pH 8, 0.5 mM EDTA, 10 μg/mL aprotinin, 1 mM PMSF). Sonication was performed in 7 cycles at 60% cycle duty, power 8 for 30 s and the homogenate was centrifuged at 73 000 × g for 45 min at 4 °C. The supernatant was supplemented with 1 mM PMSF and purified first by a 2′,5′ADP Sepharose 4B column (GE Healthcare Life Sciences) then a DEAE Sepharose Fast Flow column (GE Healthcare Life Science). The concentration of holo-CPR was determined by measuring its flavin content using an extinction coefficient of 21.2 mM−1 cm−1 at 455 nm, and the activity was measured by following the reduction of cytochrome c [38].
2.4. STM bioconjugation
Mutant C215 was covalently labeled with steroid-maleimide (STM) following our published protocol, except for the removal of excess STM and reducing agents, which was achieved using centrifugal filters instead of desalting columns [34]. The mutant (1 μM, 0.7 nmol) was incubated with TCEP (45 μM, 31.5 nmol, from a 10 mM stock in 0.1 M potassium phosphate buffer, pH 7.4) at room temperature for 20 min on an orbital shaker (50 RPM). The reaction was initiated with the addition of STM (100 μM, 70 nmol from a 20 mM stock solution in anhydrous DMSO) and allowed to proceed at 4 °C, for 2 h with orbital shaking (50 RPM). The reaction was quenched with DTT (2 mM, 1.4 μmol from a 100 mM stock in 0.1 M potassium phosphate buffer, pH 7.4) for 10 min at 4 °C. The excess DTT, TCEP and STM were removed by buffer exchange at 4 °C using Amicon Ultra-4 Centrifugal Filters (50 K cutoff, 3 × 5000 RPM, 30 min, Milllipore). Control reactions (to generate the unlabeled enzyme) were treated in the same manner but with the addition of pure DMSO instead of STM. No remaining STM or DTT-quenched STM was observed by HPLC-MS after the buffer exchange (see Figs. 2 and 3).
Fig. 2. Absorbance spectra of the ligand-free unlabeled, STM-labeled, and PGS-treated F215C mutant of CYP3A4.
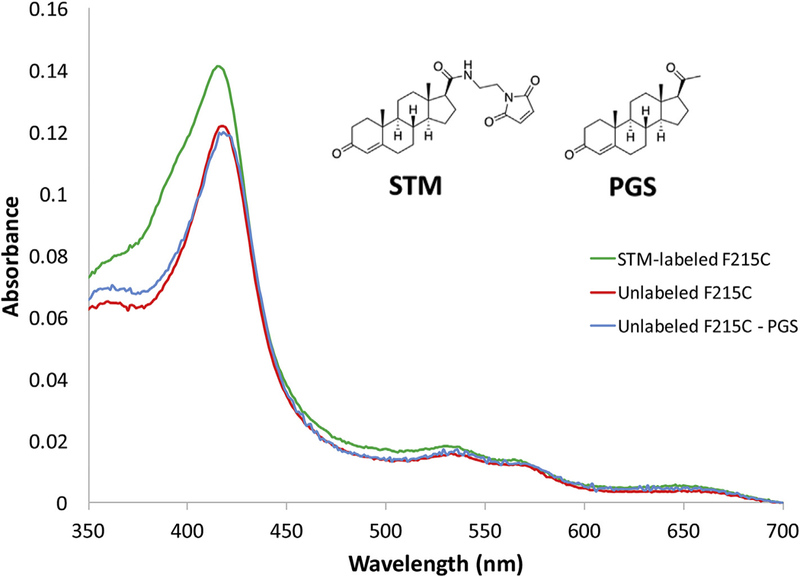
To demonstrate that unconjugated STM was likely removed during the washing step and is not responsible for the high-spin shift (390 nm peak), a mock-bioconjugation control experiment was performed by treating the F215C mutant with non-modified progesterone (PGS, a good mimic of STM) under similar conditions. As evident from the absorbance spectra, the PGS-treated CYP3A4 spectrally resembles the untreated P450. Thus, the high-spin shift in STM-labeled CYP3A4 is induced by the attached steroid rather than the trace amounts of non-conjugated STM that might be present in the sample.
Fig. 3. HPLC-UV chromatograms of the testosterone (TST) oxidation reaction by STM-labeled or unlabeled mutant F215C.
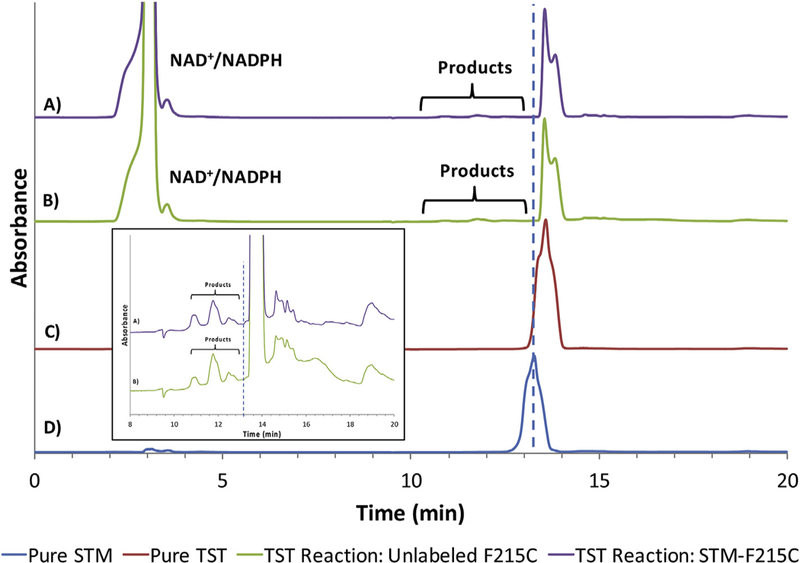
A) Oxidation of TST by STM-labeled mutant F215C consisting of P450, CPR, SOD, excess TST and excess NADPH. The location of the 4 products is shown by the bracket. B) Oxidation of TST by unlabeled mutant F215C consisting of P450, CPR, SOD, excess TST and excess NADPH. The location of the products is shown by the bracket. C) Pure TST with a retention time of 13.4 min. D) Pure STM with a retention time of 13.1 min. The dashed blue line is drawn to demonstrate the expected location of STM in chromatograms A and B. The inset is zoomed in to show the product peaks in samples A and B. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Bioconjugation yields were calculated using an LC system coupled to a MS-QToF. Following bioconjugation, the sample was washed with Milli-Q water (3 × 0.5 mL) using a Spin-X UF concentrator (30 K MWCO, Corning) for 15 min at 15000 × g. The full-length protein was analyzed by LC-MS using a Dionex Ultimate 3000 HPLC system coupled to a Bruker Maxis Impact QToF MS in positive ESI mode. Samples were run through a Poros R2/10 column from Applied Biosystems (10 μm, 2000 Å, 2.1 × 100 mm) using a gradient of 90% mobile phase A (0.1% formic acid in Milli-Q water) and 10% mobile phase B (0.1% formic acid in acetonitrile) to 0% mobile phase A and 100% mobile phase B in 15 min. The data was processed and deconvoluted using the Bruker Data Analysis software version 4.1.
The high spin content in STM-bioconjugated CYP3A4 was estimated based on the absorbance spectra of the ligand-free (100% low-spin) and bromoergocryptine-bound protein (100% high-spin conversion) samples.
2.5. Equilibrium binding studies
All UV–Vis spectra of the enzyme-ligand complexes were obtained on a Cary 300 UV–Vis spectrophotometer (Santa Clara, CA, USA). CYP3A4 (1 μM, 600 pmol) was dissolved in filtered PBS (593 μL, 0.6 M pH 7.5, 1 mM DTT, 20% glycerol) and added to the sample cuvette while only buffer was added to the reference cuvette. An absorbance measurement (the blank) was recorded from 350 to 700 nm before TST was titrated from one of two stocks (5 mM and 10 mM in DMSO) such that the total DMSO concentration never exceeded 7% (v/v), and absorbance was monitored as the TST concentration was increased. The DMSO concentrations reached had no detectable effect on the enzyme absorption spectrum. If precipitation was observed, the samples were spun down (14 000 × g) for 5 min at room temperature before acquiring the spectra. Difference spectra were plotted by subtracting the initial spectrum without TST from the consecutive spectra with TST. The difference in the absorbance between 405 nm and 418 nm (trough) was plotted against substrate concentration to obtain a binding curve. Spectral dissociation constants (KS) were derived from the binding curves using the GraphPad software. The data were fit to Equation (1), where ΔA is the difference in absorbance between the peak and trough, ΔAmax is the maximum reachable value for ΔA at saturating substrate concentration, [S] is the substrate concentration, KS is the substrate concentration at half saturation and n is the Hill coefficient, which is indicative of the cooperativity of a system.
| (1) |
2.6. Coupling of TST oxidation to NADPH consumption
CYP3A4 (5 μM, 1 nmol), CPR (5 μM, 1 nmol) and superoxide dismutase (2.5 μM, 0.5 nmol) were combined in potassium phosphate buffer (0.1 M, pH 7.4), with or without TST (100 μM, 20 nmol, 10 mM stock in DMSO) and incubated at room temperature for 30 min. The reaction was initiated with NADPH (1 mM, 200 nmol, stock in Milli-Q water) and NADPH consumption was monitored by UV–Vis at room temperature until completion. The reaction was diluted in acetonitrile (100 μL), vortexed for 1 min and centrifuged at 21 000 × g for 5 min. The supernatant was diluted in buffer (100 μL) and directly analyzed by HPLC-UV-MS. MS detection was used to identify the products and UV detection was used to quantify each product based on a calibration curve. The separation and quantification of the products was achieved using an Eclipse XDB-C18 (5 μm, 4.6 × 150 mm) column in line with a UV detector (set to monitor at 244 nm) and mobile phases A (Milli-Q water) and B (acetonitrile) at a flow rate of 0.5 mL/min. The elution entailed an isocratic step at 100% phase A for 3 min, followed by an increase to 95% phase B over 12 min before an isocratic step at 95% phase B over 5 min. The retention times were: 13.9 min for the major product 6β-hydroxy-TST, 13.5 min, 14.4 min and 15.0 min for the minor hydroxylated TST products (likely 1β-hydroxy-TST, 2β-hydroxy-TST, 15β-hydroxy-TST as suggested by Krauser [39]), and 16.5 min for TST. The product yield was calculated by converting the sum of the areas under the UV peaks of all products into a concentration (μM) using a calibration curve and TST as the standard. The coupling efficiency was determined using Equation (2).
| (2) |
2.7. Coupling of BFC oxidation to NADPH consumption
To determine the initial rate of NADPH consumption, CYP3A4 (1 μM, 0.2 nmol), CPR (1 μM, 0.2 nmol) and superoxide dismutase (2.5 μM, 0.5 nmol) were combined in PBS (0.1 M, pH 7.4), with or without BFC (100 μM, 20 nmol, 10 mM stock in DMSO) and incubated at room temperature for 30 min. The reaction was initiated with the addition of NADPH (200 μM, 40 nmol, stock in Milli-Q water) and NADPH consumption was monitored at 340 nm and room temperature for 8 min, with the data remaining linear throughout the measurements. The initial rate of NADPH consumption was determined from the change in absorbance over 3 min and converted to concentration (μM) using an extinction coefficient of 6220 M−1cm−1. The concentration of BFC used was selected to be saturating based on the reported KM, which ranges from 26 to 48 μM [34,40,41].
Formation of the fluorescent product, 7-hydroxy-4-trifluoromethyl coumarin (HFC) was assayed under the same experimental conditions using a microtiter plate fluorimeter (SpectraMax i3, Molecular Device). The initial rate of HFC production was determined from the change in fluorescence over 15 min, with the data remaining linear throughout the measurements, and converted to concentration (μM) using a calibration curve. The coupling efficiency was calculated using Equation (3).
| (3) |
3. Results and discussion
To further study the effect of steroids on the ligand binding and functional activity of CYP3A4, a steroid analogue (STM) containing a maleimide functional group was regioselectively bioconjugated to a CYP3A4 mutant, as previously described [34]. Based on structural similarity between STM and the steroids progesterone and TST (Fig. 1), we reason that it could be a good mimic of both molecules. A cysteine depleted mutant (C58T/C64A/C98S/C239S/C468G) with a newly introduced cysteine (F215C) was used to achieve site-selective bioconjugation in high yield (> 84% as estimated by HPLC-MS-QToF). The STM-labeled and unlabeled preparations of CYP3A4 used in this study were treated under similar bioconjugation conditions, except for the lack of STM in the unlabeled sample.
3.1. Spectral differences between the unlabeled and STM-labeled CYP3A4
In the resting state, cytochrome P450 enzymes have a Soret absorption peak at 416–418 nm, corresponding to the hexa-coordinated heme iron (low-spin state) with water as the 6th ligand. Upon substrate binding, the water molecule uncoordinates from the iron, resulting in a penta-coordinated heme iron (high-spin state) which absorbs light at 380–390 nm. Absorbance spectra of both STM-labeled and unlabeled CYP3A4 have their Soret peak at 418 nm (Fig. 4). However, a unique feature of STM-labeled enzyme is a pronounced shoulder at 386 nm due to a larger content of the high-spin state heme (~20%). A low-to-high spin shift usually takes place upon substrate (type I ligand) binding, with the high-spin content depending on the nature of the ligand. To ensure that the high-spin shift is not caused by the residual amounts of non-conjugated STM, a mock-bioconjugation control experiment was performed, where the F215C mutant was treated with PGS under the same conditions. As shown in Fig. 2, there were no visible spectral changes in the PGS-treated CYP3A4.
Fig. 4. Absorbance spectra of the purified STM-labeled and unlabeled CYP3A4 C215 mutant.
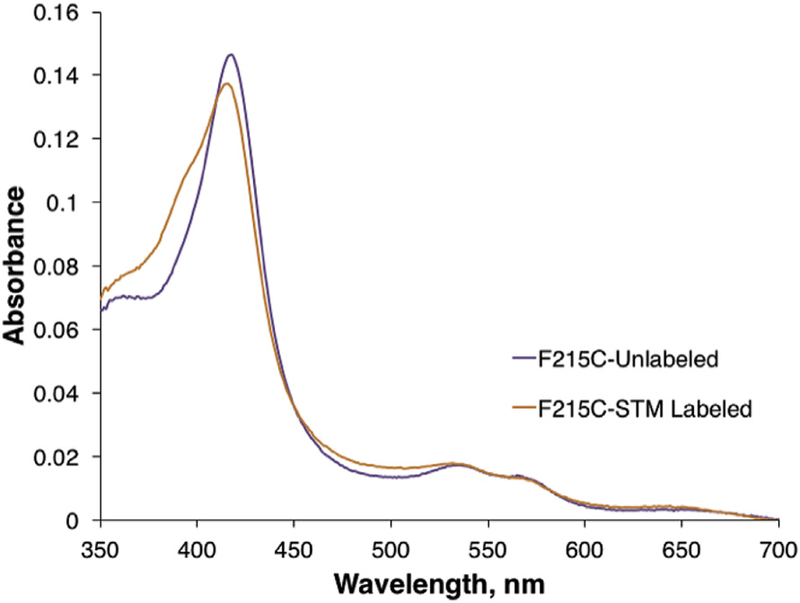
Both enzymes are predominantly in the low-spin state (418 nm peak), but the STM-labeled mutant has the larger high-spin content (a pronounced shoulder at 386 nm). The high spin content in STM-bioconjugated CYP3A4 (~20%) was estimated based on the absorbance spectra of the ligand-free (100% low-spin) and bromoergocryptine-bound protein (100% high-spin conversion) samples.
Without crystallographic evidence, it is difficult to rationalize the structural effect of the covalently-linked progesterone molecule on the spin state of CYP3A4. The high-spin shift may be due to partial reorientation of the linked steroid from the allosteric site into the active site and displacement of the distal water ligand, or due to an allosteric effect on the enzyme structure. Previous reports were not able to detect changes in the spin state for the first progesterone binding event [18,19,42]. However, studies on the CYP3A4-TST interaction have suggested that even the first binding event notably increases the amount of high-spin species (to 25%) [17,19], consistent with what is observed here. Association of the first TST molecule at the allosteric site and the resulting high-spin shift may explain why there is no product formation for the one-bound species despite the increased NADPH oxidation rate [17,19].
3.2. Equilibrium binding titrations
To investigate whether STM bioconjugation at the allosteric site affects ligand binding, the affinity of both the STM-labeled and unlabeled CYP3A4 for TST was determined via equilibrium binding titrations. Spectral changes were monitored in the 405–420 nm region, regularly used for studying substrate binding to CYPs and for determination of the spectral binding constant (KS), a measure of the ligand binding affinity [13,43–48]. A clear difference was observed in the binding affinity of TST for the labeled and unlabeled CYP3A4 (Fig. 5). The KS for the STM-labeled enzyme was ~1.4 × higher than that for the unlabeled species (158 vs 110 μM, respectively), suggesting that attachment of a steroid at the allosteric site reduces the affinity of the enzyme for TST. This is in line with the hypothesis that the peripheral area is a high affinity site that is occupied first, followed by association of the subsequent TST molecules to the lower affinity sites located within the active site pocket [17,19,28]. Since in the labeled CYP3A4 the high affinity site is already occupied, the measured KS value reflects the binding event to the lower affinity sites. In other words, the STM attachment to the allosteric site simplifies the system by decreasing the number of binding events from three to two.
Fig. 5. Titrations of STM-labeled and unlabeled mutant CYP3A4 with TST.
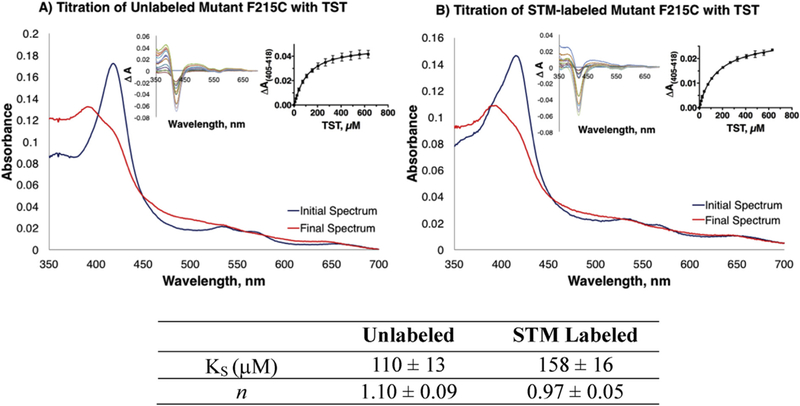
Only the initial and final absorbance spectra recorded during equilibrium titrations of the unlabeled (A) and STM-labeled enzyme (B) with TST are displayed. Insets show the difference spectra for the entire titration (left panels) and the derived titration plots with fitting curves (right panels). The spectral binding constants (KS) and Hill coefficient (n) are listed in the table as an average of three replicates with the standard error.
While the Hill coefficient (n) calculated for the unlabeled enzyme (1.10 ± 0.09, Fig. 5) suggests minimal positive cooperativity for TST binding to CYP3A4, no cooperativity is observed for the STM-labeled species (0.97 ± 0.05). Both results, however, are within error and their differences could also be considered negligible. Due to poor solubility and the negligible spectral changes induced by BFC, its KS for the enzymes could not be determined.
3.3. NADPH consumption
We next looked at the effect of steroid bioconjugation on the rate of NADPH consumption. Table 1 shows the rates of NADPH consumption for both STM-labeled and unlabeled CYP3A4 in the presence and absence of substrate. In the absence of substrate, the STM-labeled CYP3A4 consumed 1.4 × more NADPH (4.5 ± 0.2 μmol/min/μmol) than the unlabeled enzyme (3.2 ± 0.5 μmol/min/μmol). This may be the result of the larger high-spin content in STM-labeled CYP3A4 (Fig. 4), known to promote the inter-protein electron transfer. At saturating TST concentrations, when all binding sites are occupied and the heme iron is almost fully in the high-spin state (Fig. 5), the NADPH oxidation rates for the unlabeled and labeled CYP3A4 were similar: 5.0 and 4.4 μmol/min/μmol, respectively. This is consistent with both enzyme species being saturated with ligand. In the presence of a saturating concentration of BFC, the NADPH consumption rates were also similar (2.0 and 2.3 μmol/min/μmol for the labeled and unlabeled enzyme, respectively) and 2-fold lower than the respective rates measured in the absence of substrate (Table 1). It is unclear at the moment how BFC exerts this negative effect. In any case, the rate of NADPH oxidation by the unlabeled mutant was comparable to that for the wild type enzyme (3.5, 2.0 and 3.6 μmol/min/μmol for no substrate, BFC and TST, respectively) and, therefore, the CYP3A4 mutant could serve as a model for the wild type enzyme.
Table 1.
Coupling parameters for BFC and TST for unlabeled and STM-labeled mutant CYP3A4.a
| CYP3A4 Variant | Substrate | NADPH consumptionb | HFC producedb | TST-OH producedc | Coupling %d |
|---|---|---|---|---|---|
| Unlabeled CYP3A4 | none | 3.2 ± 0.5 | n.a.e | n.a. | n.a. |
| + BFC | 2.0 ± 0.7 | 0.0044 ± 0.0008 | n.a. | 0.2 ± 0.1 | |
| + TST | 5.0 ± 0.1 | n.a. | 8.6 ± 0.7 | 0.9 ± 0.1 | |
| STM-labeled CYP3A4 | none | 4.5 ± 0.2 | n.a. | n.a. | n.a. |
| + BFC | 2.3 ± 0.6 | 0.012 ± 0.002 | n.a. | 0.5 ± 0.1 | |
| + TST | 4.4 ± 0.1 | n.a. | 9.0 ± 1.9 | 0.9 ± 0.2 |
Each result is an average of two experiments performed in duplicates, with the standard deviation shown.
Reported in μmol/min/μmol enzyme.
The amount of all hydroxytestosterone products (TST-OH; in μM) formed upon consumption of 1000 μM NADPH.
With the substrate BFC, the coupling efficiency is determined by comparing the rate of HFC production to that of NADPH consumption. With the substrate TST, the coupling efficiency is determined by comparing the total amount of TST-OH produced (in μM) to the total amount of NADPH consumed (1000 μM).
n.a.: data point is not applicable because it is not related to the substrate of interest.
3.4. Coupling efficiency
CYP3A4 is well known for being a poorly coupled enzyme, meaning that a large portion of the reducing equivalents from NADPH are used to generate by-products (hydrogen peroxide, superoxide and/or water) rather than the oxidized substrate [17–20,42,49,50]. Thus, the effectiveness of CYP3A4 catalysis can be quantified in terms of coupling efficiency, which relates the amount of oxidized product formed to the amount of NADPH consumed. The effect of STM bioconjugation at the allosteric site on the coupling efficiency of CYP3A4 was next investigated for both BFC and TST (Table 1).
BFC is oxidized by CYP3A4 to produce the fluorescent product HFC. The production of HFC was monitored to determine the initial rate of BFC oxidation, which was compared to the initial rate of NADPH consumption to determine the coupling efficiency. It was observed that the STM-labeled mutant metabolizes BFC 2.7 × faster than the unlabeled control, yet the rate of NADPH consumption was similar for both (Table 1). This suggests that the STM-labeled enzyme is better coupled than the unlabeled control (0.5% vs 0.2%, respectively), and that the enhanced coupling is due to a more effective BFC transformation rather than changes in the NADPH consumption rate.
The coupling efficiency with the substrate TST was determined by an end-point assay which compares the total amount of hydroxytestosterone produced (TST-OH, total of all regioisomers produced) and the amount of NADPH consumed (1000 μM). HPLC-UV chromatograms for TST oxidation by the STM-labeled and unlabeled F215C mutant are shown in Fig. 3. It should be noted that neither the mutagenesis, nor the bioconjugation were found to modify the product profile of this enzymatic reaction. The results show similar coupling efficiency for both the STM-labeled and unlabeled species (~0.9%). Moreover, the coupling efficiency of the unlabeled enzyme was comparable to that of the wild type enzyme (0.3% and 1% for BFC and TST, respectively), once again suggesting that the behavior of the mutant may be representative of that of the wild type enzyme.
In summary, we have demonstrated how bioconjugation can be used to simplify studies of enzymes capable of binding multiple ligands simultaneously. By conjugating a steroid to a postulated allosteric site in CYP3A4, we were able to determine its effect on the ligand binding affinity and enzyme coupling efficiency. Our results are consistent with the hypothesis that an allosteric pocket is the higher affinity binding site of TST, and reveal an increased coupling efficiency of the STM-labeled CYP3A4 (at least for BFC) due to an increased rate of substrate oxidation. Moreover, for the first time, we provide direct evidence for the formation of high-spin species upon bioconjugation at the allosteric site, which has been anticipated but only indirectly modelled before [18,19,42].
Acknowledgements
We would like to thank Dr. J. R. Halpert for providing the CYP3A4 plasmid, Dr. C. B. Kasper for the CPR plasmid, Dr. A. S. Wahba for his work on protein LC-MS-QToF. Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Funding sources
This research was funded by the National Science and Engineering Research Council of Canada (NSERC) RGPIN-2017–04107, the Center in Green Chemistry and Catalysis, and the National Institutes of Health Grant ES025767 (I.F.S). V.P. was supported by scholarships from the Dr. Richard H. Tomlinson Foundation, Walter C. Sumner Foundation, the Centre in Green Chemistry and Catalysis, and NSERC.
Abbreviations:
- BFC
7-benzyloxy-4-trifluoromethylcoumarin
- CPR
cytochrome P450 reductase
- CYP3A4
human cytochrome P450 3A4
- DTT
1,4-dithiothreitol
- HFC
7-hydroxy-4-trifluoromethylcoumarin
- NADPH
nicotinamide adenine dinucleotide phosphate reduced
- PGS
progesterone
- STM
a steroid maleimide derivative
- TCEP
tris(2-carboxyethyl)phosphine
- TST
testosterone
- TST-OH
hydroxytestosterone
Footnotes
Disclosure statement
The authors declare no competing financial interest.
References
- [1].Guengerich FP, Human cytochrome P450 enzymes, in: Ortiz de Montellano PR (Ed.), Cytochrome P450, third ed, Springer US, Boston, MA, 2015, pp. 377–530, , 10.1007/0-387-27447-2_10. [DOI] [Google Scholar]
- [2].Guengerich FP, Cytochrome P-450 3A4: regulation and role in drug metabolism, Annu. Rev. Pharmacol. Toxicol 39 (1999) 1–17, 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- [3].Denisov IG, Frank DJ, Sligar SG, Cooperative properties of cytochromes P450, Pharmacol. Ther 124 (2009) 151–167, 10.1016/j.pharmthera.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ekroos M, Sjögren T, Structural basis for ligand promiscuity in cytochrome P450 3A4, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 13682–13687, 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Williams PA, Cosme J, Matak Vinković D, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H, Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone, Science (80-. ) 305 (2004) 683–686, 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- [6].Sevrioukova IF, High-level production and properties of the cysteine-depleted cytochrome P450 3A4, Biochemistry 56 (2017) 3058–3067, 10.1021/acs.biochem.7b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF, The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution, J. Biol. Chem 279 (2004) 38091–38094, 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- [8].Brändén G, Sjögren T, Schnecke V, Xue Y, Structure-based ligand design to overcome CYP inhibition in drug discovery projects, Drug Discov. Today 19 (2014) 905–911, 10.1016/j.drudis.2014.03.012. [DOI] [PubMed] [Google Scholar]
- [9].Sevrioukova IF, Poulos TL, Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4, Proc. Natl. Acad. Sci. Unit. States Am 114 (2017) 486–491, 10.1073/pnas.1616198114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sevrioukova IF, Poulos TL, Structural and mechanistic insights into the interaction of cytochrome P4503A4 with bromoergocryptine, a type I ligand, J. Biol. Chem 287 (2012) 3510–3517, 10.1074/jbc.M111.317081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sevrioukova IF, Poulos TL, Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 18422–18427, 10.1073/pnas.1010693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kaur P, Chamberlin AR, Poulos TL, Sevrioukova IF, Structure-based inhibitor design for evaluation of a CYP3A4 pharmacophore model, J. Med. Chem 59 (2016) 4210–4220, 10.1021/acs.jmedchem.5b01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Poulos TL, Sevrioukova IF, Current Approaches for Investigating and Predicting Cytochrome P450 3A4-ligand Interactions, Springer International Publishing, Cham, 2015, 10.1007/978-3-319-16009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Guo Z, Sevrioukova IF, Denisov IG, Zhang X, Chiu TL, Thomas DG, Hanse EA, Cuellar RAD, Grinkova YV, Langenfeld VW, Swedien DS, Stamschror JD, Alvarez J, Luna F, Galván A, Bae YK, Wulfkuhle JD, Gallagher RI, Petricoin EF, Norris B, Flory CM, Schumacher RJ, O’Sullivan MG, Cao Q, Chu H, Lipscomb JD, Atkins WM, Gupta K, Kelekar A, Blair IA, Capdevila JH, Falck JR, Sligar SG, Poulos TL, Georg GI, Ambrose E, Potter DA, Heme binding biguanides target cytochrome P450-Dependent cancer cell mitochondria, Cell Chem. Biol 24 (2017) 1259–1275, 10.1016/j.chembiol.2017.08.009e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Childers WK, Harrelson JP, Allosteric modulation of substrate motion in cytochrome P450 3A4-mediated xylene oxidation, Biochemistry 53 (2014) 1018–1028, 10.1021/bi401472p. [DOI] [PubMed] [Google Scholar]
- [16].Kelly PA, Wang H, Napoli KL, Kahan BD, Strobel HW, Metabolism of cyclosporine by cytochromes P450 3A9 and 3A4, Eur. J. Drug Metab. Pharmacokinet 24 (1999) 321–328, 10.1007/BF03190040. [DOI] [PubMed] [Google Scholar]
- [17].Denisov IG, Baas BJ, Grinkova YV, Sligar SG, Cooperativity in cytochrome P450 3A4: linkages in substrate binding, spin state, uncoupling, and product formation, J. Biol. Chem 282 (2007) 7066–7076, 10.1074/jbc.M609589200. [DOI] [PubMed] [Google Scholar]
- [18].Denisov IG, Grinkova YV, Baylon JL, Tajkhorshid E, Sligar SG, Mechanism of drug–drug interactions mediated by human cytochrome P450 CYP3A4 monomer, Biochemistry 54 (2015) 2227–2239, 10.1021/acs.biochem.5b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Frank DJ, Denisov IG, Sligar SG, Analysis of heterotropic cooperativity in cytochrome P450 3A4 using alpha-naphthoflavone and testosterone, J. Biol. Chem 286 (2011) 5540–5545, 10.1074/jbc.M110.182055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ueng Y-F, Kuwabara T, Chun Y-J, Guengerich FP, Cooperativity in oxidations catalyzed by cytochrome P450 3A4, Biochemistry 36 (1997) 370–381, 10.1021/bi962359z. [DOI] [PubMed] [Google Scholar]
- [21].Shou M, Mei Q, Ettore MW Jr., Dai R, Baillie TA, Rushmore TH, Sigmoidal kinetic model for two co-operative substrate-binding sites in a cytochrome P450 3A4 active site: an example of the metabolism of diazepam and its derivatives, Biochem. J 340 (1999) 845–853, 10.1042/0264-6021:3400845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hutzler JM, Tracy TS, Atypical kinetic profiles in drug metabolism reactions, Drug Metab. Dispos 30 (2002) 355–362, 10.1124/dmd.30.4.355. [DOI] [PubMed] [Google Scholar]
- [23].Davydov DR, Yang Z, Davydova N, Halpert JR, Hubbell WL, Conformational mobility in cytochrome P450 3A4 explored by pressure-perturbation EPR spectroscopy, Biophys. J 110 (2016) 1485–1498, 10.1016/j.bpj.2016.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Khan KK, He YQ, Domanski TL, Halpert JR, Midazolam oxidation by cytochrome P450 3A4 and active-site mutants: an evaluation of multiple binding sites and of the metabolic pathway that leads to enzyme inactivation, Mol. Pharmacol 61 (2002) 495–506. [DOI] [PubMed] [Google Scholar]
- [25].Sevrioukova IF, Poulos TL, Anion-dependent stimulation of CYP3A4 monooxygenase, Biochemistry 54 (2015) 4083–4096, 10.1021/acs.biochem.5b00510. [DOI] [PubMed] [Google Scholar]
- [26].Domanski TL, He Y-A, Khan KK, Roussel F, Wang Q, Halpert JR, Phenylalanine and tryptophan scanning mutagenesis of CYP3A4 substrate recognition site residues and effect on substrate oxidation and cooperativity, Biochemistry 40 (2001) 10150–10160, 10.1021/bi010758a. [DOI] [PubMed] [Google Scholar]
- [27].Harlow GR, Halpert JR, Alanine-scanning mutagenesis of a putative substrate recognition site in human cytochrome P450 3A4, J. Biol. Chem 272 (1997) 5396–5402, 10.1074/jbc.272.9.5396. [DOI] [PubMed] [Google Scholar]
- [28].Frank DJ, Denisov IG, Sligar SG, Mixing apples and oranges: analysis of heterotropic cooperativity in cytochrome P450 3A4, Arch. Biochem. Biophys 488 (2009) 146–152, 10.1016/j.abb.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Davydov DR, Rumfeldt JAO, Sineva EV, Fernando H, Davydova NY, Halpert JR, Peripheral ligand-binding site in cytochrome P450 3A4 located with fluorescence resonance energy transfer (FRET), J. Biol. Chem 287 (2012) 6797–6809, 10.1074/jbc.M111.325654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Harlow GR, Halpert JR, Analysis of human cytochrome P450 3A4 cooperativity: construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics, Proc. Natl. Acad. Sci. U. S. A 95 (1998) 6636–6641 http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=22578&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Marsch GA, Carlson BT, Guengerich FP, 7,8-benzoflavone binding to human cytochrome P450 3A4 reveals complex fluorescence quenching, suggesting binding at multiple protein sites, J. Biomol. Struct. Dyn (2017) 1–20, 10.1080/07391102.2017.1301270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Woods CM, Fernandez C, Kunze KL, Atkins WM, Allosteric activation of cytochrome P450 3A4 by α-naphthoflavone: branch point regulation revealed by isotope dilution analysis, Biochemistry 50 (2011) 10041–10051, 10.1021/bi2013454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Roberts AG, Campbell AP, Atkins WM, The thermodynamic landscape of testosterone binding to cytochrome P450 3A4: ligand binding and spin state equilibria, Biochemistry 44 (2005) 1353–1366, 10.1021/bi0481390. [DOI] [PubMed] [Google Scholar]
- [34].Polic V, Auclair K, Allosteric activation of cytochrome P450 3A4 via progesterone bioconjugation, Bioconjugate Chem 28 (2017) 885–889, 10.1021/acs.bioconjchem.6b00604. [DOI] [PubMed] [Google Scholar]
- [35].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera - a visualization system for exploratory research and analysis, J. Comput. Chem 25 (2004) 1605–1612, 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- [36].Menard A, Huang Y, Karam P, Cosa G, Auclair K, Site-specific fluorescent labeling and oriented immobilization of a triple mutant of CYP3A4 via C64, Bioconjugate Chem 23 (2012) 826–836, 10.1021/bc200672s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Omura T, Sato R, The carbon monoxide-binding pigment of liver microsomes: 2. Solubilization, purification and properties, J. Biol. Chem 239 (1964) 2379–2385 http://www.jbc.org/content/239/7/2379.short. [PubMed] [Google Scholar]
- [38].Chefson A, Zhao J, Auclair K, Replacement of natural cofactors by selected hydrogen peroxide donors or organic peroxides results in improved activity for CYP3A4 and CYP2D6, Chembiochem 7 (2006) 916–919, 10.1002/cbic.200600006. [DOI] [PubMed] [Google Scholar]
- [39].Krauser JA, Voehler M, Tseng L-H, Schefer AB, Godejohann M, Guengerich FP, Testosterone 1 beta-hydroxylation by human cytochrome P450 3A4, Eur. J. Biochem 271 (2004) 3962–3969, 10.1111/j.1432-1033.2004.04339.x. [DOI] [PubMed] [Google Scholar]
- [40].Donato MT, Jimenez N, Castell JV, Gomez-Lechon MJ, Fluorescence-based assays for screening nine cytochrome P450 (P450) activities in intact cells expressing individual human P450 enzymes, Drug Metab. Dispos 32 (2004) 699–706. [DOI] [PubMed] [Google Scholar]
- [41].Lu P, Lin YUH, Rodrigues AD, Rushmore TH, Baillie TA, Shou M, Testosterone, 7-Benzyloxyquinoline, and 7-benzyloxy-4-trifluoromethyl-coumarin bind to different domains within the active site of cytochrome P450 3A4, Drug Metab. Dispos 29 (2001) 1473–1479. [PubMed] [Google Scholar]
- [42].Denisov IG, Baylon JL, Grinkova YV, Tajkhorshid E, Sligar SG, Drug–drug interactions between atorvastatin and dronedarone mediated by monomeric CYP3A4, Biochemistry 57 (2018) 805–816, 10.1021/acs.biochem.7b01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Davydov DR, Davydova NY, Tsalkova TN, Halpert JR, Effect of glutathione on homo- and heterotropic cooperativity in cytochrome P450 3A4, Arch. Biochem. Biophys 471 (2008) 134–145, 10.1016/j.abb.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ichikawa T, Tsujino H, Miki T, Kobayashi M, Matsubara C, Miyata S, Yamashita T, Takeshita K, Yonezawa Y, Uno T, Allosteric activation of cytochrome P450 3A4 by efavirenz facilitates midazolam binding, Xenobiotica (2017) 1–10, 10.1080/00498254.2017.1412540. [DOI] [PubMed] [Google Scholar]
- [45].Anderson KW, Mast N, Hudgens JW, Lin JB, Turko IV, Pikuleva IA, Mapping of the allosteric site in cholesterol hydroxylase CYP46A1 for efavirenz, a drug that stimulates enzyme activity, J. Biol. Chem 291 (2016) 11876–11886, 10.1074/jbc.M116.723577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sligar SG, Coupling of spin, substrate, and redox equilibriums in cytochrome P450, Biochemistry 15 (1976) 5399–5406, 10.1021/bi00669a029. [DOI] [PubMed] [Google Scholar]
- [47].Isin EM, Guengerich FP, Kinetics and thermodynamics of ligand binding by cytochrome P450 3A4, J. Biol. Chem 281 (2006) 9127–9136, 10.1074/jbc.M511375200. [DOI] [PubMed] [Google Scholar]
- [48].Denisov IG, Mak PJ, Grinkova YV, Bastien D, Bérubé G, Sligar SG, Kincaid JR, The use of isomeric testosterone dimers to explore allosteric effects in substrate binding to cytochrome P450 CYP3A4, J. Inorg. Biochem 158 (2016) 77–85, 10.1016/j.jinorgbio.2015.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Grinkova YV, Denisov IG, McLean MA, Sligar SG, Oxidase uncoupling in heme monooxygenases: human cytochrome P450 CYP3A4 in Nanodiscs, Biochem. Biophys. Res. Commun 430 (2013) 1223–1227, 10.1016/j.bbrc.2012.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Perret A, Pompon D, Electron shuttle between membrane-bound cytochrome P450 3A4 and b5 rules uncoupling mechanisms, Biochemistry 37 (1998) 11412–11424, 10.1021/bi980908q. [DOI] [PubMed] [Google Scholar]