

Abstract
More reliable and cheaper sequencing technologies have revealed the vast mutational landscapes characteristic of many phenotypes. The analysis of such genetic variants has led to successful identification of altered proteins underlying many Mendelian disorders. Nevertheless the simple one‐variant one‐phenotype model valid for many monogenic diseases does not capture the complexity of polygenic traits and disorders. Although experimental and computational approaches have improved detection of functionally deleterious variants and important interactions between gene products, the development of comprehensive models relating genotype and phenotypes remains a challenge in the field of genomic medicine. In this context, a new view of the pathologic state as significant perturbation of the network of interactions between biomolecules is crucial for the identification of biochemical pathways associated with complex phenotypes. Seminal studies in systems biology combined the analysis of genetic variation with protein–protein interaction networks to demonstrate that even as biological systems evolve to be robust to genetic variation, their topologies create disease vulnerabilities. More recent analyses model the impact of genetic variants as changes to the “wiring” of the interactome to better capture heterogeneity in genotype–phenotype relationships. These studies lay the foundation for using networks to predict variant effects at scale using machine‐learning or algorithmic approaches. A wealth of databases and resources for the annotation of genotype–phenotype relationships have been developed to support developments in this area. This overview describes how study of the molecular interactome has generated insights linking the organization of biological systems to disease mechanism, and how this information can enable precision medicine.
This article is categorized under:
Translational, Genomic, and Systems Medicine > Translational Medicine
Biological Mechanisms > Cell Signaling
Models of Systems Properties and Processes > Mechanistic Models
Analytical and Computational Methods > Computational Methods
Keywords: disease mechanism, genetic disease, network analysis, variant interpretation
1. INTRODUCTION
Recent advances in sequencing technologies have significantly reduced the costs of genome sequencing and genetic testing, allowing the detection of genetic variants at scale. In particular for humans, previous studies have aimed to identify genetic variants common to different populations (Abecasis et al., 2010) and nucleotide changes associated with phenotypes (Bush, Oetjens, & Crawford, 2016). Variant data collected in population studies have been used to describe the evolutionary history of humans (Schraiber & Akey, 2015) while, in medical settings, research has aimed to detect disease causing variants (Ku, Naidoo, & Pawitan, 2011) and/or variants that increase susceptibility (MacArthur et al., 2017).
The analysis of genomic data and its relation to phenotypes is a fundamental step for enabling precision medicine (Fernald, Capriotti, Daneshjou, Karczewski, & Altman, 2011). Although in the last decades many studies have uncovered genetic variants associated to diseases (Amberger, Bocchini, Schiettecatte, Scott, & Hamosh, 2015), these discoveries only partially explain the biological complexity of most human diseases (Manolio et al., 2009). This observation is more evident in the case of polygenic disorders where the associated genetic variants are carried by only a fraction of the patients (Bomba, Walter, & Soranzo, 2017). Indeed many common and individually weak alleles have been detected for schizophrenia, bipolar disorder (Purcell et al., 2009) and rheumatoid arthritis (Stahl et al., 2012). The presence in the general population of large numbers of rare variants under strong selection suggests the hypothesis that these variants may contribute to a variety of diseases, potentially affecting many genes and pathways (Eichler et al., 2010). Furthermore, it is well established that genes do not cause disease in isolation but rather encode elements that form a dynamic molecular network in which perturbations may result in different phenotypes (Sahni et al., 2013; Vidal, Cusick, & Barabasi, 2011). As many disease mutations affect protein function or expression, this overview focuses on networks of proteins and their interactions. Indeed, knowledge of the protein–protein interaction network has proven relevant for understanding the mechanisms of many human disorders, including ataxia (Kahle et al., 2011), autism (Sakai et al., 2011), Huntington's disease (Kalathur, Pedro Pinto, Sahoo, Chaurasia, & Futschik, 2017) and breast cancer (Taylor et al., 2009). In addition, analysis of the interactome is important for the identification of cross‐phenotype genetic associations (Hackinger & Zeggini, 2017; Zhong et al., 2009). This phenomenon, referred to as pleiotropy, was introduced more than 100 years ago by Ludwig Plate to describe cases where a mutation affecting the same gene results in clinically distinguishable phenotypes (Stearns, 2010).
These observations sustain the need for more accurate tools for genome interpretation that consider the constellation of variants carried by an individual as possible perturbations of the underlying molecular interaction networks. In this review we summarize available resources, and describe how analysis of the interactome has led to an understanding of how the organization of biological systems leads to disease vulnerabilities. We discuss emerging strategies for predicting the impact of genetic variation using interactome networks and highlight future opportunities for network analysis of variants.
2. KEY CONCEPTS
In principle, genetic variants in the protein coding region of the genome can have a broad array of effects on protein activity, ranging from no consequence to severe alteration to the function and/or structure of a protein. The effect of a variant at the single protein level may not reflect the severity of the associated phenotype. Some loss of function mutations are well tolerated. Instead the severity of perturbation to the complex network of molecular interactions in the cell may more closely capture potential to generate a phenotype. Missense mutations that change only a single amino acid in the protein can significantly affect protein–protein, protein–DNA and enzyme–substrate interactions (Sahni et al., 2015). Studying variants in the context of protein–protein interaction (PPI) networks and biochemical pathways can improve our understanding of the mechanisms underlying genotype–phenotype relationships.
2.1. Graphical modeling for computing on networks
Graphical models provide a mathematical framework for studying the architecture of biological systems. Biological systems can be represented as networks, wherein nodes usually represent biomolecules, and edges represent interactions among them. Graphical modeling then allows quantitative measures to be derived from the network topology for analyzing different aspects of biological systems. The network structure itself can be analyzed, or biological measurement data can be mapped onto network nodes and edges to facilitate integration or interpretation of those measurements in the context of the organization of the underlying system. In the context of the relating genotype to phenotype, genetic alterations are mapped onto their respective proteins to identify the PPIs and biochemical pathways that are potentially affected.
2.2. Network analysis measures
Various network measures have been developed to describe the characteristics of nodes within networks (Newman, 2010). In Figure 1 we summarize a few important network measures used to describe nodes, using the PPI network of the NTRK2 activation pathway as an example. Node degree describes the number of interaction partners, and can be used to designate proteins as hubs or peripheral nodes (Figure 1a). The clustering coefficient of a node describes how connected the immediate network neighborhood of a node is (Figure 1b). Various measures of centrality have been developed to capture the importance of a node to information flow in a network. For example, degree centrality captures how connected a node is to the rest of the network (Figure 1c), and betweenness centrality describes the number of shortest paths that traverse a node (Figure 1d). Nodes can also be assigned to modules within the network using community detection algorithms (Leung, Hui, Lio, & Crowcroft, 2009; Newman, 2006). An example is represented in Figure 1 Panel D where NTRK2, BDNF, NTF4 constitute a module obtained using the Girvan–Newman algorithm (Girvan & Newman, 2002) and NTRK2 is a bottleneck connecting two communities. The network measures described above can be used to identify nodes with key “roles” within the network topology (Guimera & Amaral, 2005).
Figure 1.
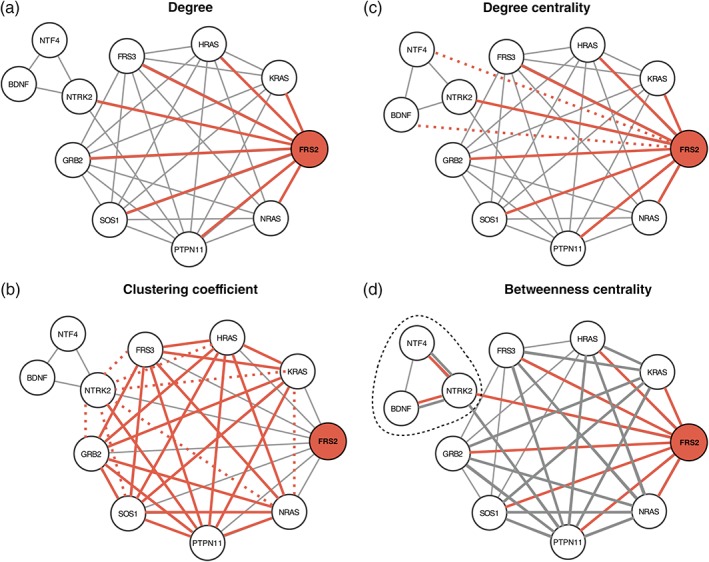
Network analysis measures. PPI network of the NTRK2 activation pathway through FRS2/FRS3. (a) The degree (k) is the number of edges of a node. The degree of FRS2 is 8. The edges are highlighted in red. (b) The clustering coefficient C of a node is calculated as the ratio between the connected triangles (delimited by the solid red and gray lines) and the total number of possible triangles k × (k − 1). The dashed lines represent the unbound triangles. For FRS2, C is 0.75 (21/28). (c) The degree centrality (C D) of a node is the number of edges divided by the total number of possible edges. The C D of FRS2 is 0.8 (8/10). Red dotted lines represent the missing edges. (d) The betweenness centrality (B) is the sum over all the possible pairs of the fraction of shortest path passing through a node (red) divided by the total number of shortest paths. B of FRS2 is 9.167 (18 × 0.5 + 0.167). Gray edges are part of the shortest paths not passing through FRS2. In this example, edge length is determined by the layout algorithm and does not have a quantitative interpretation
3. DATABASES AND RESOURCES
The development of new methods for analyzing and predicting the impact of genetic variants in the context of the PPI network requires the collection of high‐quality data from biological experiments and clinical reports. The primary sources of data needed for such analyses can be divided into three main categories: genetic variants, biological networks and disease association databases.
3.1. Genetic variant databases
There are a growing number of databases of genetic variants available on the internet. The most comprehensive database of small variants is dbSNP (Sherry et al., 2001) which, in the current version (Build 151), includes more than 113 million validated genetic variants. In spite of the name, dbSNP does not contain only single nucleotide polymorphisms (SNPs) but also rare and somatic variants. Considering these different types of genetic variation, single nucleotide variants (SNVs) account for ~90% of small variants. A significant amount of these data are the result of the 1000 Genomes project (Abecasis et al., 2010). The 1000 Genomes Consortium sequenced the whole genomes of more than 2,500 individuals from different populations allowing a more accurate characterization of the landscape of genetic variants in humans and better estimates of the average load of variants per individual (Abecasis et al., 2012).
Several data sources are more focused on collecting information about the phenotypic effects of genetic variants. For example, Clinvar hosts curated information about the health consequences of genetic variants (Landrum et al., 2016). Clinvar includes ~440,000 variants with some supporting evidence of a relationship to human phenotypes (Clinvar release July 16, 2018). Focusing on the variants with clinical significance, Clinvar contains ~81,000 genetic variants classified either as “Pathogenic” or “Benign.” The Pathogenic subset consists of ~52,000 variants from ~3,500 genes which are associated with more than 4,400 Mendelian disorders. Clinvar also includes a small set of disease‐associated variants in intronic and noncoding regions (~4,900), and pathogenic synonymous SNVs (~200). Another important database that contains information about the impact single amino acid variants (SAVs) and their relationship to human phenotype is SwissVar (Mottaz, David, Veuthey, & Yip, 2010). SwissVar curators classify the impact of SAVs either as “Pathogenic” or “Polymorphism,” extracting relevant information from the literature. The current release of SwissVar database (release 18, July 2018) contains ~70,000 SAVs, 42% of which (~29,000) are “Pathogenic” in ~2,750 genes. These pathogenic SAVs are associated with more than 3,450 Mendelian diseases.
Since 2008, the published results of genome‐wide association studies (GWAS) have been systematically reviewed to extract significant association between common variants (SNPs) and complex disorders. This data is hosted by the GWAS Catalog (MacArthur et al., 2017) that contains significant SNP‐Trait associations (p‐value <9e−6) for 50,900 unique genomic locations. About 63% of these loci are mapped to ~11,400 genes while the remaining variants are located in intergenic regions. Among complex diseases, cancer has been the focus of many sequencing studies (International Cancer Genome Consortium, 2010). The analysis of genomic data from cancer patients resulted in the detection of a large number of somatic variants, found by comparing the genetic variants in tumor cells with those in the normal cells from the same individual. The somatic variants detected with this approach are collected in the COSMIC database (Forbes et al., 2017). Version 85 of the COSMIC database (May 2018) contains ~4.4 million variants from ~253,000 tumors samples across 45 primary sites. A small number of the variants reported in COSMIC (Kamburov et al., 2015) are additionally described as causing clinical resistance to pharmaceutical therapies. These 141 mutations affected 21 different genes, but were most prevalent in ABL1, EGFR, and KIT.
3.2. Resources for biological network analysis
Biological networks can be built directly from expert knowledge, or in an unbiased fashion from large experimental screens (Rual et al., 2005) (see Box 1). Perhaps the most intuitive biological network model is a PPI network, where nodes represent proteins and edges indicate physical interactions between proteins. Other common networks model intracellular signaling, mRNA coexpression, gene regulation or metabolic flux. A broad selection of pathways and networks are hosted via online databases such as KEGG (Kanehisa, Furumichi, Tanabe, Sato, & Morishima, 2017), IntAct (Orchard et al., 2014), iRefIndex (Razick, Magklaras, & Donaldson, 2008), Reactome (Fabregat et al., 2018), Pathway Commons (Cerami et al., 2011), BioPlex (Huttlin et al., 2017), DIP (Salwinski et al., 2004) and STRING (Szklarczyk et al., 2017). These databases can be classified according to the type of information collected. Although KEGG (Kyoto Encyclopedia of Genes and Genomes) collects many types of information, it serves as a reference database for biological pathways. The KEGG PATHWAY resource consists of graphical representations of cellular processes, such as metabolism, membrane transport, signal transduction and cell cycle. Recently, the MINT (Licata et al., 2012) and IntAct (Kerrien et al., 2012) databases merged their efforts to provide a curated repository of experimentally determined interactions. The current version of IntAct (August 2018) contains ~546,000 unique PPIs, 35% of which occur between ~23,500 human proteins. IntAct also contains a small set of proteins unlikely to be engaged in an interaction. This negative interaction set represents less than 0.2% of the total number of IntAct interactions. Other resources such as iRefIndex (Razick et al., 2008), Reactome (Fabregat et al., 2018), Pathway Commons (Cerami et al., 2011) integrate pathways and/or PPI data from several primary sources.
BOX 1. INTERACTOME CONSTRUCTION.
PPIs can be detected in a variety of ways. The two most common technologies for high‐throughput PPI screening are yeast two‐hybrid (Y2H) and mass spectrometry (MS). These technologies have advantages and limitations that must be considered when analyzing the resulting interactomes. Y2H can effectively detect binary interactions, but cannot detect multiprotein complexes. MS can characterize the elements of multiprotein complexes, however it does not provide enough information to determine which proteins in the complex are in direct physical contact. Sometimes such complexes are depicted as “cliques” in networks, such that all participating proteins in the complex are linked together by edges. Both technologies have associated false positive (finding an interaction when none exists) and false negative (failing to find an existing interaction) detection rates. Interactions can be also be obtained from low throughput experiments, for example co‐crystal structures obtained via X‐ray crystallography, cryo‐electron microscopy or negative stain electron microscopy. Because many studies that probe protein interactions are not performed as high‐throughput screens, the associated interactions are generally mined from the biomedical literature. Networks generated from literature mining are more susceptible to study bias than networks derived from high‐throughput screens, however networks constructed from the literature tend to contain more interactions and those interactions may be less prone to random error (Chen, Aronow, & Jegga, 2009).
Another widely used resource for PPIs is the STRING (Szklarczyk et al., 2017) database which integrates known and predicted interaction data across multiple organisms. Apart from experimentally verified PPIs, STRING collects data derived from different sources including gene co‐expression analyses, automated text‐mining and computational inference based on gene orthology. For human alone, the latest release of STRING (version 10.5, May 2017) contains more than 691,000 unique associations between ~18,700 genes.
New resources for studying biological networks, such as the Network Data Exchange (NDEx) (Pratt et al., 2015) and the Cell Collective (Helikar et al., 2012), are emerging to provide collaborative platforms for data sharing, analysis and model simulation. In particular NDEx implements a RESTful API which can be programmatically accessed by any application. In the most recent version of NDEx, the curators improved the quality and abundance of biological networks relevant to the cancer research community. Similarly, the Cell Collective platform enables users to build and analyze network models, and use them to run simulations via a web interface. This application can be used to simulate loss/gain of function and test possible scenarios in real time.
High‐throughput experiments for detecting molecular interactions are important for mapping the interactome and are also useful for assessing the quality of available databases, validating the performance of methods that predicting PPIs and selecting sub‐networks obtained from the same experimental technologies. A recent study based on affinity purification mass spectrometry detected more than 56,000 interactions among ~11,000 human proteins (Huttlin et al., 2017). The results of this experiment are accessible through the BioPlex website (http://bioplex.hms.harvard.edu/). Crosslink mass spectrometry is an alternative technique to profile PPIs (Kaake et al., 2014; Zybailov, Glazko, Jaiswal, & Raney, 2013). A popular high‐throughput approach for detecting interactions is by yeast two‐hybrid (Y2H) experiments that detect binary PPIs (Cafarelli et al., 2017; Rolland et al., 2014; Rual et al., 2005). Many such interactions are available through the Human Reference Protein Interaction Mapping Project (HuRI; http://interactome.baderlab.org/).
The large number of available resources for studying biological networks poses a question about the implications of selecting a network for a particular study, including the reliability of particular networks for specific applications (see Box 2). Focusing on the ability to recover disease gene sets, a recent study evaluated 21 human genome‐wide interaction networks (Huang et al., 2018). This analysis showed that performance increased with network size. The STRING database had the best overall performance; however after correcting for size, the smaller network from the Database of Interacting Proteins (DIP) (Salwinski et al., 2004) had the highest per edge performance.
BOX 2. SELECTING THE RIGHT INTERACTOME.
The availability of numerous biological networks constructed from different experimental techniques and literature mining poses a difficult question about the accuracy and reliability of networks for disease studies. Generally speaking the evaluation of the quality of the interaction networks is problem dependent. Focusing on the task of recovering disease‐gene associations based on colocation and connectivity in the interactome, larger networks tend to achieve better overall performance (higher sensitivity) than smaller networks. Contrarily, if the analysis aims to minimize the number of false positive genes recovered, a network with a smaller and well‐curated set of interactions on average scores with higher precision than larger networks (Peng, Wang, Peng, Wu, & Pan, 2017). Interactions that have been detected by multiple technologies are often considered more reliable, thus some studies include interactions with multiple evidences (Chou & Cai, 2006; Sharan et al., 2005; Trivodaliev, Bogojeska, & Kocarev, 2014). However this can throw away real interactions that could be informative for a particular study. As a rule of thumb, PPI data from X‐ray crystallography are more reliable than other types of data. An important limitation for evaluating the quality of PPI networks is the low number of known of noninteracting proteins (negative set). A fair assessment of the quality of a PPI network should include an analysis of the performance on a negative set.
3.3. Disease/phenotype annotation and classification
Pivotal resources for studying the impact of genetic variants in the context biological networks are databases for the annotation and classification of diseases and phenotypes. A systematic classification of diseases and their genetic causes was carried out by McKusick, who developed the primary comprehensive curated repository for genotype/phenotype relationships. Available online since 1987, the Online Mendelian Inheritance in Man (OMIM) database (Amberger et al., 2015) synthesizes and summarizes information extracted from the biomedical literature by careful curation. The OMIM database is freely available upon request for the academic community, and it its current version (August 2018) contains ~5,300 phenotypes with known molecular basis.
Cataloging and description of distinct phenotypes is a limiting step for their analysis and comparison. To overcome this limitation, different standardized vocabularies have been developed to ensure consistent, reusable and sustainable descriptions of human diseases. Initially the U.S. National Library of Medicine (NLM) developed the Unified Medical Language System (UMLS) (Bodenreider, 2004) which includes names, concepts and relationships from different biomedical vocabularies. Similarly, Medical Subject Headings (MeSH) (Rogers, 1963) defines a hierarchically‐organized terminology for indexing and cataloging biomedical information, and the Systematized Nomenclature of Medicine (SNOMED) provides a systematic, computer‐processable collection of medical terms (Ruch, Gobeill, Lovis, & Geissbuhler, 2008).
The medical terms developed by NLM curators are part of specific ontologies for the classification of disease and phenotype such as the Disease Ontology (DO) (Kibbe et al., 2015) and the Human Phenotype Ontology (HPO) (Köhler et al., 2017). DO is a hierarchical disease‐centric ontology collecting additional facts about disease. In the latest version, DO curators expanded the utilities for examination and comparison of genetic variants, phenotypes, proteins, drugs and epitopes. In contrast to DO, the HPO focuses on the analysis of phenotypic abnormalities. The HPO project is divided into three components: the phenotype vocabulary, disease‐phenotype annotations and algorithms that operate on these. With respect to DO, HPO implements a better nomenclature for the description of rare diseases.
There are a number of additional resources for disease‐gene associations such as DisGeNET (Piñero et al., 2017), dSysMap (Mosca et al., 2015) and the Comparative Toxicogenomics Database (CTD) (Davis et al., 2017). In particular, DisGeNET is one of the largest collections of genes and variants associated with human diseases, and integrates data from expert curated repositories, GWAS catalogues, animal models and the scientific literature. The current version of DisGeNET (v5.0, Integrative Biomedical Informatics Group, Barcelona) contains more than 560,000 gene‐disease associations, between ~17,000 genes and more than 20,000 diseases and traits. In terms of variants, it contains more than 135,000 variant‐disease associations, between ~83,000 variants and ~9,200 diseases and phenotypes. Focusing more on protein structure, dSysMap maps human disease‐related mutations onto the structural interactome. In its latest version (April 2018), dSysMap contains ~29,000 mutations in ~2,700 proteins associated to ~3,600 phenotypes. Finally, the CTD is a database that aims to advance the understanding of the effect of environmental exposures on human health. The database is divided in six categories with 11 relationships among them. Apart from gene–disease associations and gene–gene interaction data, CTD collects associations between chemicals and diseases. The current version of the database (June 2018) is composed of ~37,500 curated gene–disease and ~211,500 curated chemical–disease associations. A summary of the resources described above is provided in Table 1.
Table 1.
Selected databases and resources for variant interpretation in the context of biological interactions
| Database | Data | Web address |
|---|---|---|
| Variant databases | ||
| 1000 Genomes | Whole genome and variants of >2,500 individuals | http://www.internationalgenome.org |
| ClinVar | Human variants with clinical significance | https://www.ncbi.nlm.nih.gov/clinvar |
| COSMIC | Catalog of somatic mutations in cancer | https://cancer.sanger.ac.uk/cosmic |
| dbSNP | Small variants from several organisms | https://www.ncbi.nlm.nih.gov/snp |
| GWAS catalog | Disease‐associated variants from published GWAS | https://www.ebi.ac.uk/gwas |
| SwissVar | Annotated single amino acid variants | https://swissvar.expasy.org |
| Network resources | ||
| BioPlex | Human PPIs from AP‐MS | http://bioplex.hms.harvard.edu |
| HuRI | Human PPIs from Y2H | http://interactome.baderlab.org/ |
| IntAct | Manually curated PPIs from literature | https://www.ebi.ac.uk/intact |
| iRefIndex | Integration of PPIs from many databases | http://irefindex.org |
| KEGG | Reference database for biochemical pathways | https://www.genome.jp/kegg |
| NDEx | Platform for sharing and analyzing biological networks | http://www.ndexbio.org |
| Pathway Commons | Human PPIs and pathways from different sources | http://www.pathwaycommons.org |
| Reactome | Integration of PPIs and pathways from many databases | https://reactome.org |
| STRING | Experimental and predicted PPIs | https://string‐db.org |
| Disease/phenotype association and classification | ||
| CTD | Curated gene and chemical‐phenotype associations | http://ctdbase.org |
| Disease ontology | Hierarchical ontology for description of diseases. | http://disease‐ontology.org |
| DisGeNet | Resource of variant and gene association to disease | http://www.disgenet.org |
| dSysMap | Maps of disease mutations on the structural interactome | https://dsysmap.irbbarcelona.org |
| HPO | Ontology for the description of phenotypic abnormalities | https://hpo.jax.org |
| OMIM | Database of genes implicated in Mendelian disorders | https://omim.org |
Note. AP‐MS: affinity purification‐mass spectroscopy; PPI: protein–protein interaction; Y2H: yeast two‐hybrid.
3.4. Computational methods for variant, network and disease annotation
Although not directly relevant to the current review, it is worth noting that a variety of computational tools have been developed to prioritize, annotate and extend the three categories of information required for network analysis of variants. Over the last decade many algorithms have been developed to predict the impact of SNVs (Capriotti, Nehrt, Kann, & Bromberg, 2012; Compiani & Capriotti, 2013), PPIs (Esmaielbeiki, Krawczyk, Knapp, Nebel, & Deane, 2016; Gonzalez & Kann, 2012), and disease‐gene associations (Bromberg, 2013; Tranchevent et al., 2011). In particular many machine learning methods are available to predict deleterious SNVs (Capriotti et al., 2012) and the effect of single amino acid substitutions on protein stability (Compiani & Capriotti, 2013). The prediction of new PPIs can be performed using sequence and/or structure information (Gonzalez & Kann, 2012). Some methods have also been trained to predict the interface residues that mediate the interactions between proteins (Esmaielbeiki et al., 2016). New associations between genes and diseases are frequent in the literature. Thus, methods for mining the literature to recover new disease‐gene associations are essential (Gonzalez, Tahsin, Goodale, Greene, & Greene, 2016) The majority of such tools use algorithms comparing regular text, specific ontologies and biological networks (Bromberg, 2013; Tranchevent et al., 2011). The computational methods described in the above‐cited reviews represent important early attempts to bridge the gap between the vast numbers of catalogued genetic variants and their association with human phenotypes, and are frequently applied to inform mechanistic studies.
4. NETWORK TOPOLOGY AND DISEASE
Networks provide a versatile framework for modeling the architecture of biological systems. Biological network architectures arise through evolution which should select for characteristics that confer a fitness advantage to an organism. For example, protein interaction networks have evolved to be robust to random genetic variation (Albert, Jeong, & Barabasi, 2000; Félix & Barkoulas, 2015; Kaneko, 2012; Payne & Wagner, 2015). As a result, studying mutation rates together with location in PPI networks can provide information about evolutionary constraints on particular proteins. Proteins under stronger evolutionary constraint should be less tolerant to error, and mutations in those proteins should more likely be associated with extreme phenotypes. Thus topology should also be helpful for bridging the gap between genotype and phenotype. Indeed networks that recapitulate the organization of biological systems can be used to study the relevance of the constituent molecules and molecular interactions to fitness or disease (Barabási & Oltvai, 2004; Ideker & Krogan, 2012; Vidal et al., 2011).
4.1. Properties of biological interactome networks and how they relate to phenotype
Studies of PPI network topology have generated multiple insights linking protein location and connectivity within the network to particular phenotypes. The characteristics of PPI network topology that enable function and robustness of biological systems also create certain kinds of vulnerability. PPI networks tend to have a scale‐free topology, such that the number of edges with degree k scales as a power‐law distribution (p(k) ~ k −γ) (Barabasi & Albert, 1999) where the exponent (γ) typically ranges between 2 and 3. As a result, a minority of nodes are hubs with a very large number of interaction partners while most nodes participate in very few interactions. This is thought to render the system robust to random error, since genetic variants at random are more likely to affect a protein with few interactors, and thus cause only a minor perturbation the overall topology of the network. However, this leads to vulnerabilities, as mutations affecting a highly connected hub are likely to have a significant impact on the system (Albert et al., 2000).
In PPIs networks, on average, the shortest path length of edges separating a pair of randomly selected nodes grows proportionally to the logarithm of the number of nodes in the network (small‐world network) (Albert, 2005). The shortest path length is thought to be important for efficient transfer of information and rapid response to perturbation. Redundant paths between nodes may confer robustness to genetic variation (Albert, 2005; Papin & Palsson, 2004). Although small‐world properties would not be expected to generate a modular network topology per se (Gallos, Makse, & Sigman, 2012), biological networks tend to be modular, with densely connected subnetworks that are linked into the global network architecture by a small number of connections (Han et al., 2004). Indeed, there is selection against the formation of new interactions between nodes that are already highly connected. Instead, links between highly connected nodes and nodes with few interactions are favored (Maslov & Sneppen, 2002). The observed balance between modularity and small‐worldness in PPI networks may provide the optimal architecture for information flow (Gallos et al., 2012; Jarman, Steur, Trengove, Tyukin, & van Leeuwen, 2017; Yachie et al., 2016). However this architecture also creates bottlenecks, nodes that bridge more clustered regions of the network, and as such may create additional vulnerabilities (Yu, Kim, Sprecher, Trifonov, & Gerstein, 2007).
Integrating protein topology with other data layers, such as gene expression or protein structure can reveal more detail about how the elements of the network function together. Hubs can be further divided into party and date hubs, dependent on whether interaction partners are all co‐expressed with the hub protein, or co‐expressed at different times or locations (Han et al., 2004). Incorporating protein structure into analysis of topology, Kim, Lu, Xia, and Gerstein, (2006) observed that hubs can be grouped according to whether interactors used different interfaces, allowing multiple simultaneous interactions, or used only a single interface, in which case interactions would be mutually exclusive. Interestingly, multi‐interface hubs corresponded to party hubs, whereas hubs that interacted with multiple partners via a single interface were more likely to be date hubs. The differences in co‐expression and interface usage by date and party hubs suggest that different evolutionary constraints may act on these subsystems. Indeed, in studies of the Saccharomyces cerevisiae PPI, date hubs were found to evolve more rapidly than party hubs, and their removal had a more extreme effect on the average path length of the network (Bertin et al., 2007; Han et al., 2004; Kim et al., 2006). Similarly, multi‐interface hubs were reported to evolve more slowly than single interface hubs (Biswas, Acharya, Podder, & Ghosh, 2018). Bottlenecks are also reportedly significantly less coexpressed with other network nodes, suggesting they may play a more dynamic role in biological systems (Yu et al., 2007).
4.2. Network topology determines the potential of genes to drive phenotypes
Given the clear evidence that nodes with distinct characteristics in the network support different aspects of the function of biological systems and are under different evolutionary constraints, it makes sense to evaluate the implications for fitness‐related phenotypes. Many studies have taken advantage of network measures to examine different classes of gene. Essential genes encode proteins that are required for organismal survival, such that loss of the gene is lethal. Essential genes are reported to have higher degree (Jeong, Mason, Barabási, & Oltvai, 2001), higher clustering coefficients (Feldman, Rzhetsky, & Vitkup, 2008; Said, Begley, Oppenheim, Lauffenburger, & Samson, 2004) and higher betweenness centrality in the PPI network (Yu et al., 2007). Grouping high degree nodes according to their status as party and date hubs further revealed that party hubs are more often essential than date hubs (Han et al., 2004). Cancer genes were also found to be enriched at hubs by several studies (Garcia‐Alonso et al., 2014; Goh et al., 2007; Jonsson & Bates, 2006; Sun & Zhao, 2010). In contrast, Mendelian disease genes were found to be less central in the network than essential and cancer genes (Garcia‐Alonso et al., 2014; Goh et al., 2007), particularly when essential Mendelian genes are excluded from the analysis (Goh et al., 2007). Interestingly, disease genes associated with dominant disorders had higher degree in the network than genes associated with recessive disorders (Feldman et al., 2008). In contrast, gene deletion at the periphery of the network was less frequently associated with an essential or disease phenotypes (Garcia‐Alonso et al., 2014; Khurana, Fu, Chen, & Gerstein, 2013). Figure 2 shows an example of analyzing network feature distributions for different classes of gene.
Figure 2.
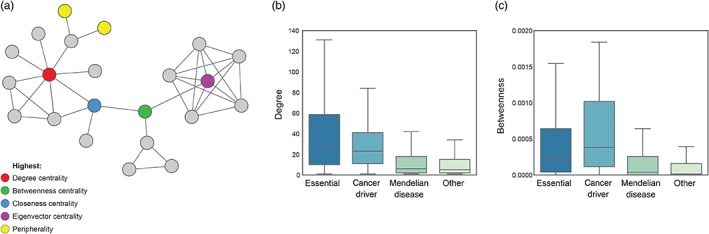
Exploring network topology as a determinant of gene–phenotype relationships. Topological location within the network has implications for biological function. (a) Nodes can be described with respect to particular characteristics in the network, including high degree hubs (red), nodes at the periphery (yellow) and nodes with the highest centrality according to four popular measures of centrality. We calculated network measures including (b) degree and (c) betweenness centrality for four groups of genes: 1,371 essential genes (Hart et al., 2015), 125 cancer genes (Vogelstein et al., 2013), 2,921 Mendelian disease genes (Stenson et al., 2017), and 7,099 other genes based on the latest release of STRING (Szklarczyk et al., 2017) to illustrate the types of observation that have been revealed by systematic studies of genes with respect to location in the interactome
The relationship between network location, gene essentiality and disease raises the possibility of inferring the importance of a gene using network measures. For example, Xu and Li (2006) used a k‐nearest neighbors approach to implicate genes with similar network characteristics as likely Mendelian disease genes. Such approaches have also been generalized to predicting drug targets and toxicities (Kotlyar, Fortney, & Jurisica, 2012; Sun, Zhu, Zheng, & Xu, 2015). Kotlyar et al. (2012) found that the centrality of genes regulated by a drug target was correlated with the toxicity of the drug.
The modular organization of the PPI network has been useful for implicating disease genes. According to the disease module hypothesis, genes related to a particular disease or symptom are likely to reside in the same region of the interactome (Bauer‐Mehren et al., 2011; Goh et al., 2007; Menche et al., 2015). A variety of community detection algorithms are available to identify tightly clustered groups of nodes that are more likely to be functionally related. Approaches have included random walk‐based algorithms (Rosvall & Bergstrom, 2008) and nonnegative matrix factorization (Zhang, Wang, & Zhang, 2007). Other algorithms use modularity to implicate disease genes for various classes of genetic disease. For example, the HetRank method uses networks to rank candidate genes for monogenic diseases exhibiting locus heterogeneity (Dand et al., 2015). In the setting of complex multigenic risk for disorders such as obesity, heart disease or diabetes, disease modularity has been used to uncover shared biological mechanisms underlying diseases by mapping distant risk variants, such as are identified by genome wide association studies, to genes that are close together in the network. Under the assumption that risk genes for the same disorder are more likely to be functionally related, Taşan et al. (2015) used a network of functionally associated genes to prioritize genes in disease‐associated genomic regions.
Efforts to catalog mutations in thousands of tumor genomes have uncovered substantial genetic heterogeneity in cancer as well; despite their phenotypic similarities (cancer cells display certain hallmark behaviors; Hanahan & Weinberg, 2011), individual tumors rarely share the same mutations (Tian, Basu, & Capriotti, 2015). Although there is very little overlap in the genes that are mutated between tumors, the genes affected by causal driver mutations tend to converge on a limited set of pathways (Ali & Sjöblom, 2009; Garraway & Lander, 2013; Vogelstein et al., 2013). Since genes within a pathway also tend to cluster in biological networks, mutations can be mapped onto a network in order to identify sub‐networks of genes that are enriched for alterations (Leiserson et al., 2015; Vandin, Upfal, & Raphael, 2011; Zhang, Zhang, Wang, & Zhang, 2013) or assess tumor similarity using the set of pathways or network regions mutated in common (Hofree, Shen, Carter, Gross, & Ideker, 2013; Zhong, Yang, Zhao, Shyr, & Li, 2015). A more in depth discussion of network analysis for tumor genomes is provided by Ozturk, Dow, Carlin, Bejar, and Carter (2018).
A phenotype of great medical interest is synthetic lethality. In the setting of synthetic lethality, mutations impairing one gene render loss of function at another gene lethal to the cell (Shen & Ideker, 2018). The most extensive studies of synthetic lethality have been performed by knocking out all genes individually and in pairwise combination is S. cerevisiae (Costanzo et al., 2010). Synthetic lethality occurs when cells are robust to knock out of each gene independently but sensitive to the loss of both. This raises the possibility of designing therapies that exploit preexisting mutations in cells to selectively eliminate diseased cells, a strategy that has been successfully used to combat cancer (Bryant et al., 2005; Farmer et al., 2005; Lord, Tutt, & Ashworth, 2015). Analysis using the interactome network topology revealed that synthetic lethal genes pairs were frequently clustered and coded for functionally related proteins that shared interaction partners, implicating protein interactions as a major source of synthetic lethality (Talavera, Robertson, & Lovell, 2013). More recently, CRISPR‐Cas9 was used to analyze the consequences of pairwise gene knockout in mammalian cells. Synthetic lethal pairs overlapped across three cell lines, but also showed significant differences, suggesting that lethality may vary considerably across cell types and conditions (Shen et al., 2017; Shen & Ideker, 2018).
5. NETWORK‐INFORMED VARIANT INTERPRETATION
From the observed relationship between network topology and essential or disease genes, it follows that the potential of genetic variants to cause a phenotype is determined by the location of the altered protein within the network (Carter, Hofree, & Ideker, 2013; Feldman et al., 2008; Vidal et al., 2011). Analysis of loss of function variants with and without pathogenic consequences confirms that interactome topology is a determinant of genotype to phenotype relationships. Garcia‐Alonso et al. reported that loss of function variants in healthy individuals were more frequently observed in genes located near the periphery of the interactome. In contrast, loss of function variants with pathological phenotypes was more central (Garcia‐Alonso et al., 2014). Piñero, Berenstein, Gonzalez‐Perez, Chernomoretz, and Furlong (2016) found that network centrality was inversely correlated with tolerance to mutation and that this could be observed at different scales, global and local, within the interactome. Of note, the association between network centrality and tolerance to loss of function variants was found to hold for PPI and regulatory networks, but not metabolic networks (Khurana et al., 2013). These studies suggest that the topological location of a variant within the network may be helpful in determining its pathogenicity.
5.1. Modeling variants as network perturbations
Most genetic variants are not loss of function events, but rather result in more subtle changes to protein sequences, and mutations within the same protein can have very different effects on its function (Taipale, 2018). It has been shown experimentally that most nonsynonymous Mendelian disease mutations generate stable proteins, supporting that mutation effects on specific protein activities rather than absence of protein drives disease phenotypes (Sahni et al., 2015). Mapping variants to nodes in the network cannot capture such subtle differences in variant effect, however the integration of information about protein structure and functional sites with protein interactions has made it possible to better discriminate variants in some cases by mapping them to network edges. Das et al. (2014), Sahni et al. (2013) and Zhong et al. (2009) introduced the concept of “edgetics” to describe the potential of mutations to perturb distinct interactions in which a protein participates. Under this model, variants mapping to the core have the potential to eliminate all interactions by destabilizing the protein, while variants mapping to interaction interfaces have the potential to perturb specific subsets of interaction (Figure 3).
Figure 3.
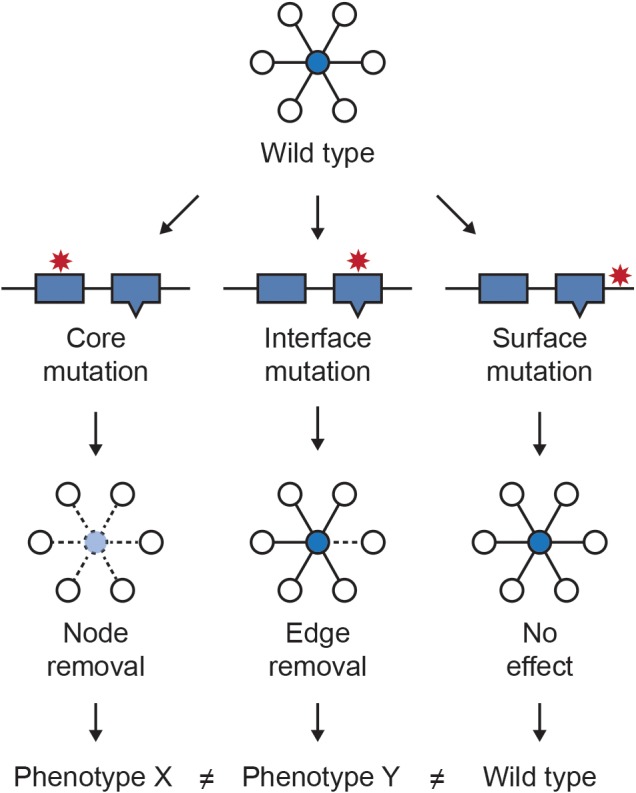
Conceptual framework of edgetics. Location of variants within a protein has implications for their phenotypic consequences. Variants that map to the core of the protein are more likely to destabilize it, resulting in a loss of all interactions in which the protein participates. In contrast, mutations at protein interaction interfaces are more likely to perturb specific interactions. Variants mapping to the protein surfaces outside of binding interfaces are less likely to create a phenotype than core or interface variants
Studies of edgetic effects require information about the three‐dimensional structure of protein complexes, so that amino acid residues can be labeled according to their location in the protein core, on the surface or at an interface between interacting proteins (Figure 4). The framework of edgetics thus allows variants to be studied not only in the context of their location in the network, but also according to their direct impact on network topology. Structurally resolved interactome networks, which integrate information about the domains or amino acid residues that physically interact, are increasingly available to explore the mechanisms by which mutations cause disease at scale (Betts et al., 2015; Das et al., 2014; Meyer, Das, Wang, & Yu, 2013; Mosca, Céol, & Aloy, 2013; Vázquez, Valencia, & Pons, 2015). Multiple studies using structurally resolved networks revealed a statistical excess of known disease mutations at protein interaction interfaces (David, Razali, Wass, & Sternberg, 2012; Guo et al., 2013; Wang et al., 2012), with in‐frame disease mutations enriched at interfaces relative to truncating mutations (Wang et al., 2012), confirming the utility of such networks for systematically investigating disease mechanism.
Figure 4.
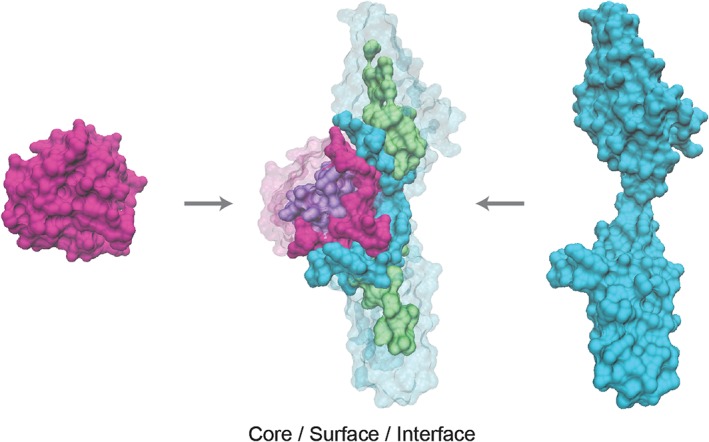
Mapping amino acid position to potential to interfere with protein interactions. Protein structures of FGF2 and FGFR1 are shown on the left and right respectively, and as a complex in the center (protein data Bank structure ID: 1CVS). In the complex, residues are colored according to location in the protein core (purple and green), at the interface (pink and blue) or at the surface outside of the interface (transparent pink and blue) on the two proteins respectively
Early edgetic analyses focused on Mendelian mutations and revealed several key advantages to the edgetic model. Zhong et al. (2009) proposed that edgetics had the potential to describe aspects of genetic disease that could not be captured by topological location of a protein alone (Figure 5). These aspects include: (a) allelic heterogeneity (or pleiotropy), in which a single gene is associated with multiple phenotypes, (b) locus heterogeneity, where a single disorder is caused by a mutation in one of several genes, (c) variable penetrance, wherein not all individuals with a variant have a disease, and (d) variable expressivity, wherein individuals with a disease are not affected equally. Indeed, classic examples of pleiotropy and locus heterogeneity could be explained by edgetics. Mutations in the WASP protein associated with Wiskott–Aldrich syndrome versus X‐linked Neutropenia were found to map to distinct interfaces, while mutations associated with hemolytic uremic syndrome in the C3 and CFH proteins were found to map to reciprocal interfaces on the two proteins (Wang et al., 2012). Guo et al. (2013) further investigated the relationship between edgetic effects and the inheritance mode of diseases caused by particular mutations, finding that recessive disease mutations affecting reciprocal interfaces were more likely to cause the same phenotype than similar mutations associated with dominant effects. They described cases where dominant truncating mutations removed some interfaces while preserving others, such as was found for TRIM27 mutations in ovarian cancer, supporting that truncating variants may also frequently have edgetic effects (Guo et al., 2013).
Figure 5.
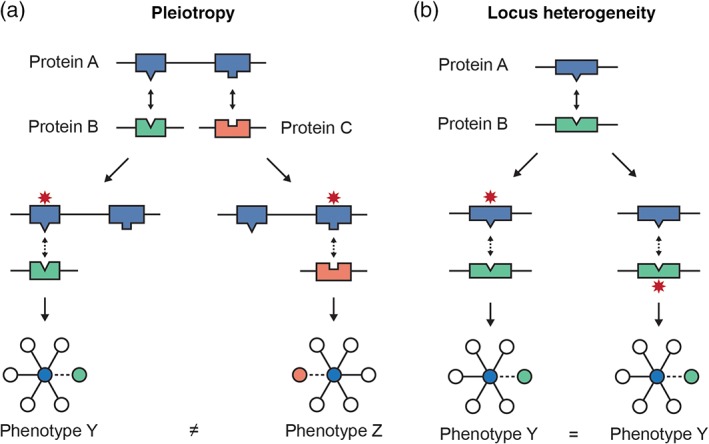
Modeling the edgetic effects of genetic variants supports exploration of disease mechanisms. (a) Pleiotropy can result when different variants in the same gene affect different interactions in which a protein participates. (b) Variants at reciprocal interfaces of interacting proteins can contribute to locus heterogeneity
Edgetics have also provided mechanistic insights for complex diseases including autism and cancer. For example, a study of 1,733 de novo missense mutations from autism spectrum disorder exomes demonstrated a significant enrichment of missense mutations affecting protein interactions in probands relative to unaffected siblings (Chen et al., 2018). Experimental assessment using the CloneSeq pipeline to test the effect of mutations on binary protein interactions (Wei et al., 2014) found that 74 of 361 (20%) of tested interactions were altered in probands versus only 21 of 208 (10%) in unaffected siblings. Several studies have demonstrated that somatic mutations found in tumor genomes are overrepresented at protein interaction interfaces (Engin, Kreisberg, & Carter, 2016; Kamburov et al., 2015; Porta‐Pardo, Garcia‐Alonso, Hrabe, Dopazo, & Godzik, 2015; Raimondi et al., 2016), suggesting that perturbations of protein interactions frequently contribute to tumor development. The Cancer Cell Map Initiative is now systematically experimentally mapping the impact of driver mutations on the interactome in cancer to further reveal the links between network rewiring and tumorigenesis (Krogan, Lippman, Agard, Ashworth, & Ideker, 2015).
5.2. Genetic variants perturbing network topology
While most edgetic variants would be expected to perturb existing interactions, it is also possible for variants to generate new interactions. For examples, the R273H amino acid substitution in TP53 was found to create a binding site for NRD1, and this novel interaction was found to promote cellular invasion in the setting of cancer (Coffill et al., 2012; Sahni et al., 2013). Such novel interactions may be difficult to predict, and it remains unclear how frequently amino acid substitutions generate new interfaces. The simplest way for novel interactions to emerge is likely through modified specificity at existing binding sites, and such events may be most prevalent in proteins families with high functional similarity. For example, many mutations in cancer are thought to alter the specificity of kinases for their substrates (Creixell et al., 2015; Reimand, Wagih, & Bader, 2013). Frequent cancer mutations were reported at acetylation and ubiquitination sites as well (Narayan, Bader, & Reimand, 2016). Experimental evidence also suggested that many disease mutations affect the motif specificity of transcription factors, with many mutations reducing the specificity of DNA binding sites, thereby allowing more promiscuous binding (Sahni et al., 2015).
Nonsynonymous variants and small insertions and deletions have been the focus of most edgetic studies, however there are other types of alteration that have the potential to “rewire” the interactome. Alternative splicing generates different proteins from the same gene, and dependent on which exons are included, these protein isoforms can include different binding interfaces. Yang et al. (2016) isoforms used experimental approaches to map the interactome of 1,423 protein, and subsequently Ghadie, Lambourne, Vidal, and Xia (2017)) used in silico analysis of interface containing domain usage by various splice isoforms to construct an isoform‐specific interactome. Both studies found that patterns of protein interaction with different isoforms were associated with divergent disease phenotypes, creating additional opportunity for pleiotropy (Ghadie et al., 2017; Yang et al., 2016). In cancer, aberrant splicing was enriched at domain families that mediate protein interactions which also frequently harbored other types of mutation (Climente‐González, Porta‐Pardo, Godzik, & Eyras, 2017).
Not all genes create equal opportunity for “rewiring” biological networks. In cancer, mutations often affect genes capable of causing large changes in the network. For example, cancer mutations frequently affect genes involved in chromatin remodeling, resulting in widespread changes to gene expression (Garraway & Lander, 2013). Fusion proteins are another common event in tumor genomes that can result in network rewiring. A recent pan‐cancer analysis found that fusions often involved genes with high degree in the network and that did not interact prior to the fusion event (Latysheva et al., 2016). Some recurrent cancer fusions involve genes that regulate the activity of multiple targets such as transcription factors, (e.g., RUNX1, ERG) or kinases (e.g., ABL1, NTRK1). Mutations with large impacts on network architecture are likely to be uncommon in inherited disease, as their consequences may be too severe to support early development in multicellular organisms. This hypothesis is consistent with reports that Mendelian disease genes are less central in the PPI network and participate in fewer protein interactions than cancer genes (Garcia‐Alonso et al., 2014).
5.3. Studying variant accumulation at network edges instead of nodes
Studying proteins in the context of their location in the network focuses analysis on node‐level properties. In contrast, in the framework of edgetics, variants are viewed based on their ability to perturb network edges. From this perspective it becomes possible to analyze the accumulation of variation affecting network edges rather than network nodes in order to better understand disease mechanism. Many cases of mutations affecting interacting genes and leading to similar phenotypes such as Branchiootic syndrome, Charcot–Marie–Tooth disease and Polycystic kidney disease have been reported (Oti, Snel, Huynen, & Brunner, 2006). Several groups have developed scoring strategies to prioritize network edges that are enriched for disease mutation. This requires first mapping variants to interface residues or interacting domains of proteins. Some methods evaluate the ratio of observed to expected variants in interface regions controlling or the size of the region relative to the size of the protein (Porta‐Pardo et al., 2015) and or the size and the amino acid composition (Raimondi et al., 2016). An alternative approach uses the nonsynonymous to synonymous (dN/dS) ratio at interfaces, a signature of selective pressure that has been used to identify cancer genes (Greenman et al., 2007), to evaluate whether interfaces are unexpectedly biased toward functional variants (Engin et al., 2016). Mechismo evaluates the consequences of amino acid substitutions at interfaces in terms of their impact on the pair potentials relative to what is expected (Betts et al., 2015), enabling an assessment of the likely effect of the variant on the interface. Other analyses used methods such as FoldX (Schymkowitz et al., 2005) to estimate the impact of the amino acid substitution on the stability of the protein complex.
5.4. Using networks to predict the phenotypic consequences of variants
Since networks influence the potential of variants to have a phenotypic effect, and variants result in phenotypic effects by perturbing biological networks in different ways, network measures should be informative for computational tasks relating to variant prioritization and interpretation. Some methods have begun to incorporate network information into classification tasks. Khurana et al. (2013) combined network and evolutionary properties to build a classifier capable of predicting gene tolerance to loss of function mutations, enabling automated prioritization of loss of function events according to their potential to have phenotypic consequences. Location of a variant at a protein interface was found to be one of the most informative features for discriminating driver missense mutations from neutral passenger mutations in analysis of tumor genomes (Tokheim & Karchin, 2018). Gao et al. (2018) used features derived from gene regulatory networks to predict the functional consequences of noncoding variants under the assumption that the causal effects of gene‐expression altering variants must be transmitted through this network. There may be various ways to derive features from interactome networks for the purpose of predicting variant effects. Yu et al. (2016) derived “ontotypes,” a signature of nodes reached by a gene in an S. cerevisiae‐specific hierarchical interactome network, to predict the impact of specific gene knockouts on colony growth.
Several approaches have used the structure of the network more directly in predicting variant effects. To capture the potential for variants in the same gene to have different effects on biological processes, Engin, Hofree, and Carter (2015) mapped cancer mutations affecting different interfaces on the same protein onto different perturbed network architectures. Network propagation was applied to identify downstream subnetworks specifically affected by each mutation (Figure 6). These subnetworks could then be analyzed for functional enrichment to suggest mechanisms by which mutations perturbing distinct activities of a protein could result in different phenotypes. In a different study, Poole, Leinonen, Shmulevich, Knijnenburg, and Bernard (2017) evaluated the statistical association of clusters of somatic mutations within selected proteins to pathway level changes in gene expression in tumors, raising the possibility of aggregating mutations with common edgetic effects across samples and simultaneously analyzing them using networks.
Figure 6.
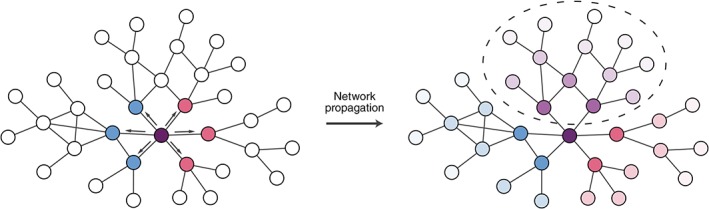
Propagating variant effects on networks. Variants can be used as signal sources for network propagation in order to identify network neighborhoods affected by variants. Edgetic effects can be used to constrain network propagation according to the effects of variants on specific protein interactions. On the left side of this schematic, two variants to the purple node affect interactions with different subsets of partners (indicated by blue and pink nodes respectively). Network propagation can be used to implicate network regions likely to be affected by each variant, and these can be contrasted to identify regions perturbed by both variants that could explain shared phenotypes (right network, circled purple shaded nodes), or regions affected specifically by each variant (right network, blue and pink shaded regions) which could help explain pleiotropic effects
Recently, Ma et al. (2018) developed DeepCell wherein the hierarchical interactome was used to constrain the architecture of a deep convolutional neural network that was trained to predict growth effects from genotype in S. cerevisiae. DeepCell was found to effectively simulate growth phenotypes observed in the laboratory directly from genotype. Furthermore, the weights learned by the neural network could be used to develop testable hypotheses about the mechanisms by which variants generated growth phenotypes, with some variants displaying Boolean‐type effects on specific subsystems. Thus DeepCell mines the multiscale, hierarchical organization of the interactome to extract novel information linking genotype to phenotype.
In summary, multiple strategies have been developed to annotate variants according their consequences for biological networks to support their interpretation in the context of various phenotypes. These works lay the foundation for the next generation of automated variant interpretation tools that will integrate information about the architecture of the biological system and the potential of variants to perturb it. Strategies for quantifying variant effects on the network, or using network information to predict variant activity have thus far focused largely on single variants, however we note that multiple variants can simultaneously be mapped to network effects. This raises the possibility of using networks to predict the joint effects of combinations of variants, such as occur in complex polygenic diseases.
6. CHALLENGES AND FUTURE DIRECTIONS
While early works integrating networks with variant information to understand the mechanisms driving genetic disease show great promise, many challenges remain to be addressed. At times, analyses of network topologies have generated contradictory findings (Coulomb, Bauer, Bernard, & Marsolier‐Kergoat, 2005; Gandhi et al., 2006; Ivanic, Yu, Wallqvist, & Reifman, 2009; Jordan, Wolf, & Koonin, 2003; Yu et al., 2008), suggesting that careful consideration must be given to the limitations of the available data and the implications of choices made in constructing and analyzing network models when drawing biological conclusions.
Data availability and quality is an important consideration for network analyses. PPI networks remain incomplete and may contain many false positive connections. In addition, networks assembled from various published experiments may exhibit literature bias; proteins associated with certain phenotypes may be more studied and as a result, may appear more connected in the network, giving the illusion that higher degree nodes are associated with phenotypes of interest (Chen et al., 2009; Feldman et al., 2008; Xu & Li, 2006). Choice of network may thus influence the biological conclusions drawn from network analysis. Indeed, Huang et al. (2018) showed that network performance at recovering known disease genes varied according to the disease, and no single network performed best for all diseases. To partially address this issue, several methods for selecting high‐quality PPI datasets and score the reliability of an interaction have been developed (Peng et al., 2017).
Models frequently focus on specific aspects of biology while ignoring others. For example, many molecular interactome networks ignore regulatory interactions by which distant proteins in the network can influence each other, and often do not account for posttranslational modifications. Hybrid networks that incorporate both physical and regulatory interactions can be constructed (Rioualen et al., 2017) but statistical analysis may be complicated by the inclusion of different types of relationship. More sophisticated applications of natural language processing may support direct inference of more complex network models directly from the literature (Gyori et al., 2017). Many of the approaches discussed in this overview treat proteins as static network nodes and do not attempt to incorporate protein levels. Integrating interactome networks with more quantitative modeling techniques that rely on differential equations (Schnoerr, Sanguinetti, & Grima, 2017) or agent based models (Abar, Theodoropoulos, Lemarinier, & O'Hare, 2017) could present a pathway for including quantitative and dynamic information about protein levels, however these methods are frequently computationally intensive and may not be practical for large networks.
Structure is not available for the majority of human proteins, limiting the investigation of variants affecting protein interactions. The extent to which networks themselves are complete also remains poorly understood. Many conclusions have been drawn based on the architecture of the human interactome, however some estimates suggest that at most 20% of interactions have been experimentally measured (Menche et al., 2015). In addition, gene expression patterns differ widely across cell types, suggesting that for accurate inference, network architectures need to be cell‐type specific. Indeed, disease network modules tend to include genes that are coexpressed in specific tissues (Kitsak et al., 2016), and several groups have now constructed tissue‐specific networks to study disease variation (Greene et al., 2015; Pierson et al., 2015). Many of the analyses described here have yet to be revisited in a tissue‐ or cell‐type specific setting. Finally, network representations are usually static, whereas the biological networks that they represent are dynamic and conditional. Protein interactions often require particular localization or posttranslational modification. Novel technologies such as APEX, a proximity labeling technique recently developed to enable spatially resolved analysis of protein interaction networks (Lobingier et al., 2017), may provide a solution to further resolve cell‐type specific interactions and subcellular location thereof. Distinguishing between constitutive and transient interactions, and cell‐state specific interactions may be important for further understanding the potential of variants to generate relevant phenotypes (Perkins, Diboun, Dessailly, Lees, & Orengo, 2010). Thus, the Stable Isotope Labeling by Amino acids in Cell culture (SILAC) method, a mass spectrometry technique able to detect differences in protein abundance among samples using nonradioactive isotopic labeling, has been used to measure stability of PPI (Budayeva & Cristea, 2014).
New technologies are emerging that can accelerate the pace of interaction profiling and that will create more complete networks and new opportunities for analysis. Next generation sequencing‐based interaction screening technologies such as BFG‐Y2H (Yachie et al., 2016) and CrY2H‐seq (Trigg et al., 2017) allow higher throughput screening for binary interaction. Combining such technologies with techniques like deep mutational scanning (Fowler & Fields, 2014) could allow more systematic profiling of the edgetic effects of variants. Technologies such as Perturb‐seq also allow high‐throughput profiling of the effects of variants on single cell transcriptional profiles, creating opportunities to quantify the broader effects of variants at the resolution of specific cell types (Dixit et al., 2016). Interactome networks have the potential to play a key role in the interpretation of such assays.
7. CONCLUSION
A rapidly growing body of research has demonstrated the utility of network analysis for understanding disease mechanism. Many early studies relied on interactome networks derived from model organisms, such as S. cerevisiae, but the less complete human interactome has been instrumental for studying the relationships between genetic variation, genes and disease. Many insights have been gained from the study of genes with clear phenotypes, including essential genes, Mendelian disease genes and cancer genes. Interactome networks have also been successfully used to identify drug targets and study mechanisms underlying toxicities. However, a significant proportion of human genetic disease remains poorly understood, particularly for complex multigenic disorders.
As new high‐throughput experimental assays emerge, databases of genetic variation and network models will become increasingly available and more complete. Large consortia are generating invaluable multilayer datasets that can create new opportunities for integrated analysis for variant interpretation. For example, the eGTEx project will add epigenetic measurements to complement the library of tissue‐specific expression data in GTEx (GTEx Consortium, 2013), enabling integration of epigenetic factors and tissue specific gene regulation into models for interpreting disease variants (eGTEx Project, 2017; Li et al., 2017). These new methodologies and datasets create opportunities for the development of computational models that support network‐informed inference to predict the phenotypic consequences of genetic variation and reveal the mechanisms by which variants contribute to complex human diseases.
Recent technological advances are enabling the development of emerging research fields such as the Molecular Pathological Epidemiology (MPE) incorporating interpersonal heterogeneity of a disease process into epidemiology (Hamada, Keum, Nishihara, & Ogino, 2017). In this framework the integration of data capturing the complex combination of genetic heterogeneity (endogenous factors) and the environment (exogenous factors) is providing novel insights underlying etiologic mechanisms of different cancer types (Ogino, Nowak, Hamada, Milner Jr, & Nishihara, 2018) and define new therapeutic strategies (Ogino et al., 2018).
Although these early efforts show great promise, new technological and algorithmic advances will be required to realize the full potential of networks to inform variant interpretation and precision medicine.
CONFLICT OF INTEREST
The authors have declared no conflicts of interest for this article.
RELATED WIREs ARTICLES
Computational approaches for the identification of cancer genes and pathways
Integration of proteomics into systems biology of cancer
Protein–protein interaction networks and subnetworks in the biology of disease
ACKNOWLEDGMENTS
This work was supported by NIH grants DP5‐OD017937 and a CIFAR fellowship to H.C. EC is supported by an FFABR grant from the Ministry of Education, Universities and Research (MIUR). EC acknowledges the CINECA award under the ISCRA initiative, for the availability of high performance computing resources and support. HC and EC acknowledge the support from the University of Bologna through the funding of the UNIBO‐UCSD International Cooperation Project.
Capriotti E, Ozturk K, Carter H. Integrating molecular networks with genetic variant interpretation for precision medicine. WIREs Syst Biol Med. 2019;11:e1443. 10.1002/wsbm.1443
Contributor Information
Emidio Capriotti, Email: emidio.capriotti@unibo.it.
Hannah Carter, Email: hkcarter@ucsd.edu.
References
FURTHER READING
- Bartocci, E. , & Lió, P. (2016). Computational modeling, formal analysis, and tools for systems biology. PLoS Computational Biology, 12(1), e1004591 10.1371/journal.pcbi.1004591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, D. Y. , Kim, Y. A. , & Przytycka, T. M. (2012). Chapter 5: Network biology approach to complex diseases. PLoS Computational Biology, 8(12), e1002820 10.1371/journal.pcbi.1002820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller, D. , & Friedman, N. (2009). Probabilistic graphical models: Principles and techniques. Cambridge, MA: MIT Press. [Google Scholar]
- Mezlini, A. M. , & Goldenberg, A. (2017). Incorporating networks in a probabilistic graphical model to find drivers for complex human diseases. PLoS Computational Biology, 13(10), e1005580 10.1371/journal.pcbi.1005580 [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES
- Abar, S. , Theodoropoulos, G. K. , Lemarinier, P. , & O'Hare, G. M. P. (2017). Agent based modelling and simulation tools: A review of the state‐of‐art software. Computer Science Review, 24, 13–33. [Google Scholar]
- Abecasis, G. R. , Altshuler, D. , Auton, A. , Brooks, L. D. , Durbin, R. M. , Gibbs, R. A. , … Consortium, G. P. (2010). A map of human genome variation from population‐scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecasis, G. R. , Auton, A. , Brooks, L. D. , DePristo, M. A. , Durbin, R. M. , Handsaker, R. E. , … Consortium, G. P. (2012). An integrated map of genetic variation from 1,092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert, R. (2005). Scale‐free networks in cell biology. Journal of Cell Science, 118, 4947–4957. [DOI] [PubMed] [Google Scholar]
- Albert, R. , Jeong, H. , & Barabasi, A. L. (2000). Error and attack tolerance of complex networks. Nature, 406, 378–382. [DOI] [PubMed] [Google Scholar]
- Ali, M. A. , & Sjöblom, T. (2009). Molecular pathways in tumor progression: From discovery to functional understanding. Molecular BioSystems, 5, 902–908. [DOI] [PubMed] [Google Scholar]
- Amberger, J. S. , Bocchini, C. A. , Schiettecatte, F. , Scott, A. F. , & Hamosh, A. (2015). OMIM.org: Online Mendelian inheritance in man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Research, 43, D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi, A. L. , & Albert, R. (1999). Emergence of scaling in random networks. Science, 286, 509–512. [DOI] [PubMed] [Google Scholar]
- Barabási, A. L. , & Oltvai, Z. N. (2004). Network biology: Understanding the cell's functional organization. Nature Reviews. Genetics, 5, 101–113. [DOI] [PubMed] [Google Scholar]
- Bauer‐Mehren, A. , Bundschus, M. , Rautschka, M. , Mayer, M. A. , Sanz, F. , & Furlong, L. I. (2011). Gene‐disease network analysis reveals functional modules in mendelian, complex and environmental diseases. PLoS One, 6, e20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin, N. , Simonis, N. , Dupuy, D. , Cusick, M. E. , Han, J. D. , Fraser, H. B. , … Vidal, M. (2007). Confirmation of organized modularity in the yeast interactome. PLoS Biology, 5, e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts, M. J. , Lu, Q. , Jiang, Y. , Drusko, A. , Wichmann, O. , Utz, M. , … Russell, R. B. (2015). Mechismo: Predicting the mechanistic impact of mutations and modifications on molecular interactions. Nucleic Acids Research, 43, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas, K. , Acharya, D. , Podder, S. , & Ghosh, T. C. (2018). Evolutionary rate heterogeneity between multi‐ and single‐interface hubs across human housekeeping and tissue‐specific protein interaction network: Insights from proteins' and its partners' properties. Genomics, 110, 283–290. [DOI] [PubMed] [Google Scholar]
- Bodenreider, O. (2004). The unified medical language system (UMLS): Integrating biomedical terminology. Nucleic Acids Research, 32, D267–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomba, L. , Walter, K. , & Soranzo, N. (2017). The impact of rare and low‐frequency genetic variants in common disease. Genome Biology, 18, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg, Y. (2013). Chapter 15: Disease gene prioritization. PLoS Computational Biology, 9, e1002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, H. E. , Schultz, N. , Thomas, H. D. , Parker, K. M. , Flower, D. , Lopez, E. , … Helleday, T. (2005). Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature, 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Budayeva, H. G. , & Cristea, I. M. (2014). A mass spectrometry view of stable and transient protein interactions. Advances in Experimental Medicine and Biology, 806, 263–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush, W. S. , Oetjens, M. T. , & Crawford, D. C. (2016). Unravelling the human genome‐phenome relationship using phenome‐wide association studies. Nature Reviews. Genetics, 17, 129–145. [DOI] [PubMed] [Google Scholar]
- Cafarelli, T. M. , Desbuleux, A. , Wang, Y. , Choi, S. G. , De Ridder, D. , & Vidal, M. (2017). Mapping, modeling, and characterization of protein–protein interactions on a proteomic scale. Current Opinion in Structural Biology, 44, 201–210. [DOI] [PubMed] [Google Scholar]
- Capriotti, E. , Nehrt, N. L. , Kann, M. G. , & Bromberg, Y. (2012). Bioinformatics for personal genome interpretation. Briefings in Bioinformatics, 13, 495–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, H. , Hofree, M. , & Ideker, T. (2013). Genotype to phenotype via network analysis. Current Opinion in Genetics & Development, 23, 611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami, E. G. , Gross, B. E. , Demir, E. , Rodchenkov, I. , Babur, O. , Anwar, N. , … Sander, C. (2011). Pathway Commons, a web resource for biological pathway data. Nucleic Acids Research, 39, D685–D690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Aronow, B. J. , & Jegga, A. G. (2009). Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinformatics, 10, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Fragoza, R. , Klei, L. , Liu, Y. , Wang, J. , Roeder, K. , … Yu, H. (2018). An interactome perturbation framework prioritizes damaging missense mutations for developmental disorders. Nature Genetics, 50, 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, K. C. , & Cai, Y. D. (2006). Predicting protein–protein interactions from sequences in a hybridization space. Journal of Proteome Research, 5, 316–322. [DOI] [PubMed] [Google Scholar]
- Climente‐González, H. , Porta‐Pardo, E. , Godzik, A. , & Eyras, E. (2017). The functional impact of alternative splicing in Cancer. Cell Reports, 20, 2215–2226. [DOI] [PubMed] [Google Scholar]
- Coffill, C. R. , Muller, P. A. , Oh, H. K. , Neo, S. P. , Hogue, K. A. , Cheok, C. F. , … Gunaratne, J. (2012). Mutant p53 interactome identifies nardilysin as a p53R273H‐specific binding partner that promotes invasion. EMBO Reports, 13, 638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compiani, M. , & Capriotti, E. (2013). Computational and theoretical methods for protein folding. Biochemistry, 52, 8601–8624. [DOI] [PubMed] [Google Scholar]
- Costanzo, M. , Baryshnikova, A. , Bellay, J. , Kim, Y. , Spear, E. D. , Sevier, C. S. , … Prinz, J. (2010). The genetic landscape of a cell. Science, 327, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulomb, S. , Bauer, M. , Bernard, D. , & Marsolier‐Kergoat, M. C. (2005). Gene essentiality and the topology of protein interaction networks. Proceedings of the Biological Sciences, 272, 1721–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creixell, P. , Schoof, E. M. , Simpson, C. D. , Longden, J. , Miller, C. J. , Lou, H. J. , … Linding, R. (2015). Kinome‐wide decoding of network‐attacking mutations rewiring cancer signaling. Cell, 163, 202–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dand, N. , Schulz, R. , Weale, M. E. , Southgate, L. , Oakey, R. J. , Simpson, M. A. , & Schlitt, T. (2015). Network‐informed gene ranking tackles genetic heterogeneity in exome‐sequencing studies of monogenic disease. Human Mutation, 36, 1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, J. , Fragoza, R. , Lee, H. R. , Cordero, N. A. , Guo, Y. , Meyer, M. J. , … Yu, H. (2014). Exploring mechanisms of human disease through structurally resolved protein interactome networks. Molecular BioSystems, 10, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, A. , Razali, R. , Wass, M. N. , & Sternberg, M. J. (2012). Protein–protein interaction sites are hot spots for disease‐associated nonsynonymous SNPs. Human Mutation, 33, 359–363. [DOI] [PubMed] [Google Scholar]
- Davis, A. P. , Grondin, C. J. , Johnson, R. J. , Sciaky, D. , King, B. L. , McMorran, R. , … Mattingly, C. J. (2017). The comparative Toxicogenomics database: Update 2017. Nucleic Acids Research, 45, D972–D978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit, A. , Parnas, O. , Li, B. , Chen, J. , Fulco, C. P. , Jerby‐Arnon, L. , … Adamson, B. (2016). Perturb‐Seq: Dissecting molecular circuits with scalable single‐cell RNA profiling of pooled genetic screens. Cell, 167, 1853–1866.e1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- eGTEx Project . (2017). Enhancing GTEx by bridging the gaps between genotype, gene expression, and disease. Nature Genetics, 49, 1664–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler, E. E. , Flint, J. , Gibson, G. , Kong, A. , Leal, S. M. , Moore, J. H. , & Nadeau, J. H. (2010). Missing heritability and strategies for finding the underlying causes of complex disease. Nature Reviews. Genetics, 11, 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin, H. B. , Hofree, M. , & Carter, H. (2015). Identifying mutation specific cancer pathways using a structurally resolved protein interaction network. Pacific Symposium on Biocomputing, 84–95. [PMC free article] [PubMed] [Google Scholar]
- Engin, H. B. , Kreisberg, J. F. , & Carter, H. (2016). Structure‐based analysis reveals cancer missense mutations target protein interaction interfaces. PLoS One, 11, e0152929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmaielbeiki, R. , Krawczyk, K. , Knapp, B. , Nebel, J. C. , & Deane, C. M. (2016). Progress and challenges in predicting protein interfaces. Briefings in Bioinformatics, 17, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat, A. , Jupe, S. , Matthews, L. , Sidiropoulos, K. , Gillespie, M. , Garapati, P. , … D'Eustachio, P. (2018). The Reactome pathway knowledgebase. Nucleic Acids Research, 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, H. , McCabe, N. , Lord, C. J. , Tutt, A. N. , Johnson, D. A. , Richardson, T. B. , … Martin, N. M. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Feldman, I. , Rzhetsky, A. , & Vitkup, D. (2008). Network properties of genes harboring inherited disease mutations. Proceedings of the National Academy of Sciences of the United States of America, 105, 4323–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félix, M. A. , & Barkoulas, M. (2015). Pervasive robustness in biological systems. Nature Reviews. Genetics, 16, 483–496. [DOI] [PubMed] [Google Scholar]
- Fernald, G. H. , Capriotti, E. , Daneshjou, R. , Karczewski, K. J. , & Altman, R. B. (2011). Bioinformatics challenges for personalized medicine. Bioinformatics, 27, 1741–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes, S. A. , Beare, D. , Boutselakis, H. , Bamford, S. , Bindal, N. , Tate, J. , … Campbell, P. J. (2017). COSMIC: Somatic cancer genetics at high‐resolution. Nucleic Acids Research, 45, D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler, D. M. , & Fields, S. (2014). Deep mutational scanning: A new style of protein science. Nature Methods, 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallos, L. K. , Makse, H. A. , & Sigman, M. (2012). A small world of weak ties provides optimal global integration of self‐similar modules in functional brain networks. Proceedings of the National Academy of Sciences of the United States of America, 109, 2825–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi, T. K. , Zhong, J. , Mathivanan, S. , Karthick, L. , Chandrika, K. N. , Mohan, S. S. , … Mishra, G. (2006). Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nature Genetics, 38, 285–293. [DOI] [PubMed] [Google Scholar]
- Gao, L. , Uzun, Y. , Gao, P. , He, B. , Ma, X. , Wang, J. , … Tan, K. (2018). Identifying noncoding risk variants using disease‐relevant gene regulatory networks. Nature Communications, 9, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Alonso, L. , Jiménez‐Almazán, J. , Carbonell‐Caballero, J. , Vela‐Boza, A. , Santoyo‐López, J. , Antiñolo, G. , & Dopazo, J. (2014). The role of the interactome in the maintenance of deleterious variability in human populations. Molecular Systems Biology, 10, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway, L. A. , & Lander, E. S. (2013). Lessons from the cancer genome. Cell, 153, 17–37. [DOI] [PubMed] [Google Scholar]
- Ghadie, M. A. , Lambourne, L. , Vidal, M. , & Xia, Y. (2017). Domain‐based prediction of the human isoform interactome provides insights into the functional impact of alternative splicing. PLoS Computational Biology, 13, e1005717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvan, M. , & Newman, M. E. (2002). Community structure in social and biological networks. Proceedings of the National Academy of Sciences of the United States of America, 99, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh, K. I. , Cusick, M. E. , Valle, D. , Childs, B. , Vidal, M. , & Barabási, A. L. (2007). The human disease network. Proceedings of the National Academy of Sciences of the United States of America, 104, 8685–8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, G. H. , Tahsin, T. , Goodale, B. C. , Greene, A. C. , & Greene, C. S. (2016). Recent advances and emerging applications in text and data Mining for Biomedical Discovery. Briefings in Bioinformatics, 17, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, M. W. , & Kann, M. G. (2012). Chapter 4: Protein interactions and disease. PLoS Computational Biology, 8, e1002819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene, C. S. , Krishnan, A. , Wong, A. K. , Ricciotti, E. , Zelaya, R. A. , Himmelstein, D. S. , … Troyanskaya, O. G. (2015). Understanding multicellular function and disease with human tissue‐specific networks. Nature Genetics, 47, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman, C. , Stephens, P. , Smith, R. , Dalgliesh, G. L. , Hunter, C. , Bignell, G. , … Stratton, M. R. (2007). Patterns of somatic mutation in human cancer genomes. Nature, 446, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium . (2013). The genotype‐tissue expression (GTEx) project. Nature Genetics, 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimera, R. , & Amaral, L. A. (2005). Cartography of complex networks: Modules and universal roles. Journal of Statistical Mechanics, 2005.2, P02001‐1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y. , Wei, X. , Das, J. , Grimson, A. , Lipkin, S. M. , Clark, A. G. , & Yu, H. (2013). Dissecting disease inheritance modes in a three‐dimensional protein network challenges the "guilt‐by‐association" principle. American Journal of Human Genetics, 93, 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori, B. M. , Bachman, J. A. , Subramanian, K. , Muhlich, J. L. , Galescu, L. , & Sorger, P. K. (2017). From word models to executable models of signaling networks using automated assembly. Molecular Systems Biology, 13, 954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackinger, S. , & Zeggini, E. (2017). Statistical methods to detect pleiotropy in human complex traits. Open Biology, 7, 170125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada, T. , Keum, N. , Nishihara, R. , & Ogino, S. (2017). Molecular pathological epidemiology: New developing frontiers of big data science to study etiologies and pathogenesis. Journal of Gastroenterology, 52, 265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J. D. , Bertin, N. , Hao, T. , Goldberg, D. S. , Berriz, G. F. , Zhang, L. V. , … Vidal, M. (2004). Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature, 430, 88–93. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hart, T. , Chandrashekhar, M. , Aregger, M. , Steinhart, Z. , Brown, K. R. , MacLeod, G. , … Moffat, J. (2015). High‐resolution CRISPR screens reveal fitness genes and genotype‐specific Cancer liabilities. Cell, 163, 1515–1526. [DOI] [PubMed] [Google Scholar]
- Helikar, T. , Kowal, B. , McClenathan, S. , Bruckner, M. , Rowley, T. , Madrahimov, A. , … Rogers, J. A. (2012). The cell collective: Toward an open and collaborative approach to systems biology. BMC Systems Biology, 6, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofree, M. , Shen, J. P. , Carter, H. , Gross, A. , & Ideker, T. (2013). Network‐based stratification of tumor mutations. Nature Methods, 10, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. K. , Carlin, D. E. , Yu, M. K. , Zhang, W. , Kreisberg, J. F. , Tamayo, P. , & Ideker, T. (2018). Systematic evaluation of molecular networks for discovery of disease genes. Cell Systems, 6, 484–495 e485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin, E. L. , Bruckner, R. J. , Paulo, J. A. , Cannon, J. R. , Ting, L. , Baltier, K. , … Harper, J. W. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature, 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker, T. , & Krogan, N. J. (2012). Differential network biology. Molecular Systems Biology, 8, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Cancer Genome Consortium . (2010). International network of cancer genome projects. Nature, 464, 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanic, J. , Yu, X. , Wallqvist, A. , & Reifman, J. (2009). Influence of protein abundance on high‐throughput protein–protein interaction detection. PLoS One, 4, e5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarman, N. , Steur, E. , Trengove, C. , Tyukin, I. Y. , & van Leeuwen, C. (2017). Self‐organisation of small‐world networks by adaptive rewiring in response to graph diffusion. Scientific Reports, 7, 13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, H. , Mason, S. P. , Barabási, A. L. , & Oltvai, Z. N. (2001). Lethality and centrality in protein networks. Nature, 411, 41–42. [DOI] [PubMed] [Google Scholar]
- Jonsson, P. F. , & Bates, P. A. (2006). Global topological features of cancer proteins in the human interactome. Bioinformatics, 22, 2291–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, I. K. , Wolf, Y. I. , & Koonin, E. V. (2003). No simple dependence between protein evolution rate and the number of protein–protein interactions: Only the most prolific interactors tend to evolve slowly. BMC Evolutionary Biology, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaake, R. M. , Wang, X. , Burke, A. , Yu, C. , Kandur, W. , Yang, Y. , … Huang, L. (2014). A new in vivo cross‐linking mass spectrometry platform to define protein–protein interactions in living cells. Molecular & Cellular Proteomics, 13, 3533–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle, J. J. , Gulbahce, N. , Shaw, C. A. , Lim, J. , Hill, D. E. , Barabasi, A. L. , & Zoghbi, H. Y. (2011). Comparison of an expanded ataxia interactome with patient medical records reveals a relationship between macular degeneration and ataxia. Human Molecular Genetics, 20, 510–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalathur, R. K. R. , Pedro Pinto, J. , Sahoo, B. , Chaurasia, G. , & Futschik, M. E. (2017). HDNetDB: A molecular interaction database for network‐oriented investigations into Huntington's disease. Scientific Reports, 7, 5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov, A. , Lawrence, M. S. , Polak, P. , Leshchiner, I. , Lage, K. , Golub, T. R. , … Getz, G. (2015). Comprehensive assessment of cancer missense mutation clustering in protein structures. Proceedings of the National Academy of Sciences of the United States of America, 112, E5486–E5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. , Furumichi, M. , Tanabe, M. , Sato, Y. , & Morishima, K. (2017). KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research, 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko, K. (2012). Phenotypic plasticity and robustness: Evolutionary stability theory, gene expression dynamics model, and laboratory experiments. Advances in Experimental Medicine and Biology, 751, 249–278. [DOI] [PubMed] [Google Scholar]
- Kerrien, S. , Aranda, B. , Breuza, L. , Bridge, A. , Broackes‐Carter, F. , Chen, C. , … Hermjakob, H. (2012). The IntAct molecular interaction database in 2012. Nucleic Acids Research, 40, D841–D846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana, E. , Fu, Y. , Chen, J. , & Gerstein, M. (2013). Interpretation of genomic variants using a unified biological network approach. PLoS Computational Biology, 9, e1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbe, W. A. , Arze, C. , Felix, V. , Mitraka, E. , Bolton, E. , Fu, G. , … Parkinson, H. (2015). Disease Ontology 2015 Update: An expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Research, 43, D1071–D1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, P. M. , Lu, L. J. , Xia, Y. , & Gerstein, M. B. (2006). Relating three‐dimensional structures to protein networks provides evolutionary insights. Science, 314, 1938–1941. [DOI] [PubMed] [Google Scholar]
- Kitsak, M. , Sharma, A. , Menche, J. , Guney, E. , Ghiassian, S. D. , Loscalzo, J. , & Barabási, A. L. (2016). Tissue specificity of human disease module. Scientific Reports, 6, 35241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler, S. , Vasilevsky, N. A. , Engelstad, M. , Foster, E. , McMurry, J. , Aymé, S. , … Robinson, P. N. (2017). The Human Phenotype Ontology in 2017. Nucleic Acids Research, 45, D865–D876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlyar, M. , Fortney, K. , & Jurisica, I. (2012). Network‐based characterization of drug‐regulated genes, drug targets, and toxicity. Methods, 57, 499–507. [DOI] [PubMed] [Google Scholar]
- Krogan, N. J. , Lippman, S. , Agard, D. A. , Ashworth, A. , & Ideker, T. (2015). The cancer cell map initiative: Defining the hallmark networks of cancer. Molecular Cell, 58, 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku, C. S. , Naidoo, N. , & Pawitan, Y. (2011). Revisiting Mendelian disorders through exome sequencing. Human Genetics, 129, 351–370. [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , Brown, G. , Chao, C. , Chitipiralla, S. , … Maglott, D. R. (2016). ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Research, 44, D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latysheva, N. S. , Oates, M. E. , Maddox, L. , Flock, T. , Gough, J. , Buljan, M. , … Babu, M. M. (2016). Molecular principles of gene fusion mediated rewiring of protein interaction networks in Cancer. Molecular Cell, 63, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson, M. D. , Vandin, F. , Wu, H. T. , Dobson, J. R. , Eldridge, J. V. , Thomas, J. L. , … Lawrence, M. S. (2015). Pan‐cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nature Genetics, 47, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, I. X. , Hui, P. , Lio, P. , & Crowcroft, J. (2009). Towards real‐time community detection in large networks. Physical Review. E, Statistical, Nonlinear, and Soft Matter Physics, 79, 066107. [DOI] [PubMed] [Google Scholar]
- Li, X. , Kim, Y. , Tsang, E. K. , Davis, J. R. , Damani, F. N. , Chiang, C. , … Montgomery, S. B. (2017). The impact of rare variation on gene expression across tissues. Nature, 550, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata, L. , Briganti, L. , Peluso, D. , Perfetto, L. , Iannuccelli, M. , Galeota, E. , … Cesareni, G. (2012). MINT, the molecular interaction database: 2012 update. Nucleic Acids Research, 40, D857–D861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobingier, B. T. , Huttenhain, R. , Eichel, K. , Miller, K. B. , Ting, A. Y. , von Zastrow, M. , & Krogan, N. J. (2017). An approach to spatiotemporally resolve protein interaction networks in living cells. Cell, 169, 350–360 e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord, C. J. , Tutt, A. N. , & Ashworth, A. (2015). Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annual Review of Medicine, 66, 455–470. [DOI] [PubMed] [Google Scholar]
- Ma, J. , Yu, M. K. , Fong, S. , Ono, K. , Sage, E. , Demchak, B. , … Ideker, T. (2018). Using deep learning to model the hierarchical structure and function of a cell. Nature Methods, 15, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur, J. , Bowler, E. , Cerezo, M. , Gil, L. , Hall, P. , Hastings, E. , … Parkinson, H. (2017). The new NHGRI‐EBI catalog of published genome‐wide association studies (GWAS catalog). Nucleic Acids Research, 45, D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio, T. A. , Collins, F. S. , Cox, N. J. , Goldstein, D. B. , Hindorff, L. A. , Hunter, D. J. , … Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature, 461, 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov, S. , & Sneppen, K. (2002). Specificity and stability in topology of protein networks. Science, 296, 910–913. [DOI] [PubMed] [Google Scholar]
- Menche, J. , Sharma, A. , Kitsak, M. , Ghiassian, S. D. , Vidal, M. , Loscalzo, J. , & Barabási, A. L. (2015). Disease networks. Uncovering disease‐disease relationships through the incomplete interactome. Science, 347, 1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, M. J. , Das, J. , Wang, X. , & Yu, H. (2013). INstruct: A database of high‐quality 3D structurally resolved protein interactome networks. Bioinformatics, 29, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca, R. , Céol, A. , & Aloy, P. (2013). Interactome3D: Adding structural details to protein networks. Nature Methods, 10, 47–53. [DOI] [PubMed] [Google Scholar]
- Mosca, R. , Tenorio‐Laranga, J. , Olivella, R. , Alcalde, V. , Ceol, A. , Soler‐Lopez, M. , & Aloy, P. (2015). dSysMap: Exploring the edgetic role of disease mutations. Nature Methods, 12, 167–168. [DOI] [PubMed] [Google Scholar]
- Mottaz, A. , David, F. P. , Veuthey, A. L. , & Yip, Y. L. (2010). Easy retrieval of single amino‐acid polymorphisms and phenotype information using SwissVar. Bioinformatics, 26, 851–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan, S. , Bader, G. D. , & Reimand, J. (2016). Frequent mutations in acetylation and ubiquitination sites suggest novel driver mechanisms of cancer. Genome Medicine, 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, M. E. (2006). Modularity and community structure in networks. Proceedings of the National Academy of Sciences of the United States of America, 103, 8577–8582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, M. E. J. (2010). Networks: An introduction. Oxford, England: Oxford University Press. [Google Scholar]
- Ogino, S. , Nowak, J. A. , Hamada, T. , Milner, D. A., Jr. , & Nishihara, R. (2018). Insights into pathogenic interactions among environment, host, and tumor at the crossroads of molecular pathology and epidemiology. Annual Review of Pathology, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino, S. , Nowak, J. A. , Hamada, T. , Phipps, A. I. , Peters, U. , Milner, D. A., Jr. , … Song, M. (2018). Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut, 67, 1168–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard, S. , Ammari, M. , Aranda, B. , Breuza, L. , Briganti, L. , Broackes‐Carter, F. , … Duesbury, M. (2014). The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Research, 42, D358–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oti, M. , Snel, B. , Huynen, M. A. , & Brunner, H. G. (2006). Predicting disease genes using protein–protein interactions. Journal of Medical Genetics, 43, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozturk, K. , Dow, M. , Carlin, D. E. , Bejar, R. , & Carter, H. (2018). The emerging potential for network analysis to inform precision Cancer medicine. Journal of Molecular Biology, 430, 2875–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papin, J. A. , & Palsson, B. O. (2004). Topological analysis of mass‐balanced signaling networks: A framework to obtain network properties including crosstalk. Journal of Theoretical Biology, 227, 283–297. [DOI] [PubMed] [Google Scholar]
- Payne, J. L. , & Wagner, A. (2015). Mechanisms of mutational robustness in transcriptional regulation. Frontiers in Genetics, 6, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, X. , Wang, J. , Peng, W. , Wu, F. X. , & Pan, Y. (2017). Protein–protein interactions: Detection, reliability assessment and applications. Briefings in Bioinformatics, 18, 798–819. [DOI] [PubMed] [Google Scholar]
- Perkins, J. R. , Diboun, I. , Dessailly, B. H. , Lees, J. G. , & Orengo, C. (2010). Transient protein–protein interactions: Structural, functional, and network properties. Structure, 18, 1233–1243. [DOI] [PubMed] [Google Scholar]
- Pierson, E. , Koller, D. , Battle, A. , Mostafavi, S. , Ardlie, K. G. , Getz, G. , … Dermitzakis, E. T. (2015). Sharing and specificity of co‐expression networks across 35 human tissues. PLoS Computational Biology, 11, e1004220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñero, J. , Berenstein, A. , Gonzalez‐Perez, A. , Chernomoretz, A. , & Furlong, L. I. (2016). Uncovering disease mechanisms through network biology in the era of next generation sequencing. Scientific Reports, 6, 24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñero, J. , Bravo, À. , Queralt‐Rosinach, N. , Gutiérrez‐Sacristán, A. , Deu‐Pons, J. , Centeno, E. , … Furlong, L. I. (2017). DisGeNET: A comprehensive platform integrating information on human disease‐associated genes and variants. Nucleic Acids Research, 45, D833–D839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole, W. , Leinonen, K. , Shmulevich, I. , Knijnenburg, T. A. , & Bernard, B. (2017). Multiscale mutation clustering algorithm identifies pan‐cancer mutational clusters associated with pathway‐level changes in gene expression. PLoS Computational Biology, 13, e1005347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta‐Pardo, E. , Garcia‐Alonso, L. , Hrabe, T. , Dopazo, J. , & Godzik, A. (2015). A Pan‐Cancer catalogue of Cancer driver protein interaction interfaces. PLoS Computational Biology, 11, e1004518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt, D. , Chen, J. , Welker, D. , Rivas, R. , Pillich, R. , Rynkov, V. , … Ideker, T. (2015). NDEx, the Network Data Exchange. Cell Systems, 1, 302–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell, S. M. , Wray, N. R. , Stone, J. L. , Visscher, P. M. , O'Donovan, M. C. , Sullivan, P. F. , … Consortium, I. S. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature, 460, 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi, F. , Singh, G. , Betts, M. J. , Apic, G. , Vukotic, R. , Andreone, P. , … Russell, R. B. (2016). Insights into cancer severity from biomolecular interaction mechanisms. Scientific Reports, 6, 34490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razick, S. , Magklaras, G. , & Donaldson, I. M. (2008). iRefIndex: A consolidated protein interaction database with provenance. BMC Bioinformatics, 9, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand, J. , Wagih, O. , & Bader, G. D. (2013). The mutational landscape of phosphorylation signaling in cancer. Scientific Reports, 3, 2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioualen, C. , Da Costa, Q. , Chetrit, B. , Charafe‐Jauffret, E. , Ginestier, C. , & Bidaut, G. (2017). HTS‐net: An integrated regulome‐interactome approach for establishing network regulation models in high‐throughput screenings. PLoS One, 12, e0185400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, F. B. (1963). Medical subject headings. Bulletin of the Medical Library Association, 51, 114–116. [PMC free article] [PubMed] [Google Scholar]
- Rolland, T. , Taşan, M. , Charloteaux, B. , Pevzner, S. J. , Zhong, Q. , Sahni, N. , … Vidal, M. (2014). A proteome‐scale map of the human interactome network. Cell, 159, 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosvall, M. , & Bergstrom, C. T. (2008). Maps of random walks on complex networks reveal community structure. Proceedings of the National Academy of Sciences of the United States of America, 105, 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual, J. F. , Venkatesan, K. , Hao, T. , Hirozane‐Kishikawa, T. , Dricot, A. , Li, N. , … Vidal, M. (2005). Towards a proteome‐scale map of the human protein–protein interaction network. Nature, 437, 1173–1178. [DOI] [PubMed] [Google Scholar]
- Ruch, P. , Gobeill, J. , Lovis, C. , & Geissbuhler, A. (2008). Automatic medical encoding with SNOMED categories. BMC Medical Informatics and Decision Making, 8(Suppl 1), S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni, N. , Yi, S. , Taipale, M. , Fuxman Bass, J. I. , Coulombe‐Huntington, J. , Yang, F. , … Vidal, M. (2015). Widespread macromolecular interaction perturbations in human genetic disorders. Cell, 161, 647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni, N. , Yi, S. , Zhong, Q. , Jailkhani, N. , Charloteaux, B. , Cusick, M. E. , & Vidal, M. (2013). Edgotype: A fundamental link between genotype and phenotype. Current Opinion in Genetics & Development, 23, 649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said, M. R. , Begley, T. J. , Oppenheim, A. V. , Lauffenburger, D. A. , & Samson, L. D. (2004). Global network analysis of phenotypic effects: Protein networks and toxicity modulation in Saccharomyces cerevisiae . Proceedings of the National Academy of Sciences of the United States of America, 101, 18006–18011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, Y. , Shaw, C. A. , Dawson, B. C. , Dugas, D. V. , Al‐Mohtaseb, Z. , Hill, D. E. , & Zoghbi, H. Y. (2011). Protein interactome reveals converging molecular pathways among autism disorders. Science Translational Medicine, 3, 86ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salwinski, L. , Miller, C. S. , Smith, A. J. , Pettit, F. K. , Bowie, J. U. , & Eisenberg, D. (2004). The Database of Interacting Proteins: 2004 update. Nucleic Acids Research, 32, D449–D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoerr, D. , Sanguinetti, G. , & Grima, R. (2017). Approximation and inference methods for stochastic biochemical kinetics—A tutorial review. Journal of Physics A: Mathematical and Theoretical, 50, 093001. [Google Scholar]
- Schraiber, J. G. , & Akey, J. M. (2015). Methods and models for unravelling human evolutionary history. Nature Reviews. Genetics, 16, 727–740. [DOI] [PubMed] [Google Scholar]
- Schymkowitz, J. , Borg, J. , Stricher, F. , Nys, R. , Rousseau, F. , & Serrano, L. (2005). The FoldX web server: An online force field. Nucleic Acids Research, 33, W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan, R. , Suthram, S. , Kelley, R. M. , Kuhn, T. , McCuine, S. , Uetz, P. , … Ideker, T. (2005). Conserved patterns of protein interaction in multiple species. Proceedings of the National Academy of Sciences of the United States of America, 102, 1974–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. P. , & Ideker, T. (2018). Synthetic lethal networks for precision oncology: Promises and pitfalls. Journal of Molecular Biology, 430, 2900–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. P. , Zhao, D. , Sasik, R. , Luebeck, J. , Birmingham, A. , Bojorquez‐Gomez, A. , … Mali, P. (2017). Combinatorial CRISPR‐Cas9 screens for de novo mapping of genetic interactions. Nature Methods, 14, 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry, S. T. , Ward, M. H. , Kholodov, M. , Baker, J. , Phan, L. , Smigielski, E. M. , & Sirotkin, K. (2001). dbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl, E. A. , Wegmann, D. , Trynka, G. , Gutierrez‐Achury, J. , Do, R. , Voight, B. F. , … Plenge, R. M. (2012). Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nature Genetics, 44, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns, F. W. (2010). One hundred years of pleiotropy: A retrospective. Genetics, 186, 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson, P. D. , Mort, M. , Ball, E. V. , Evans, K. , Hayden, M. , Heywood, S. , … Cooper, D. N. (2017). The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Human Genetics, 136, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, J. , & Zhao, Z. (2010). A comparative study of cancer proteins in the human protein–protein interaction network. BMC Genomics, 11(Suppl 3), S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, J. , Zhu, K. , Zheng, W. , & Xu, H. (2015). A comparative study of disease genes and drug targets in the human protein interactome. BMC Bioinformatics, 16(Suppl 5), S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk, D. , Morris, J. H. , Cook, H. , Kuhn, M. , Wyder, S. , Simonovic, M. , … von Mering, C. (2017). The STRING database in 2017: Quality‐controlled protein–protein association networks, made broadly accessible. Nucleic Acids Research, 45, D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale, M. (2018). Disruption of protein function by pathogenic mutations: Common and uncommon mechanisms. Biochemistry and Cell Biology, 1–12. [DOI] [PubMed] [Google Scholar]
- Talavera, D. , Robertson, D. L. , & Lovell, S. C. (2013). The role of protein interactions in mediating essentiality and synthetic lethality. PLoS One, 8, e62866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taşan, M. , Musso, G. , Hao, T. , Vidal, M. , MacRae, C. A. , & Roth, F. P. (2015). Selecting causal genes from genome‐wide association studies via functionally coherent subnetworks. Nature Methods, 12, 154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, I. W. , Linding, R. , Warde‐Farley, D. , Liu, Y. , Pesquita, C. , Faria, D. , … Wrana, J. L. (2009). Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nature Biotechnology, 27, 199–204. [DOI] [PubMed] [Google Scholar]
- Tian, R. , Basu, M. K. , & Capriotti, E. (2015). Computational methods and resources for the interpretation of genomic variants in cancer. BMC Genomics, 16(Suppl 8), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokheim, C. , & Karchin, R. (2018). Enhanced context reveals the scope of somatic missense mutations driving human cancers. bioRxiv, 313296 10.1101/313296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranchevent, L. C. , Capdevila, F. B. , Nitsch, D. , De Moor, B. , De Causmaecker, P. , & Moreau, Y. (2011). A guide to web tools to prioritize candidate genes. Briefings in Bioinformatics, 12, 22–32. [DOI] [PubMed] [Google Scholar]
- Trigg, S. A. , Garza, R. M. , MacWilliams, A. , Nery, J. R. , Bartlett, A. , Castanon, R. , … Ecker, J. R. (2017). CrY2H‐seq: A massively multiplexed assay for deep‐coverage interactome mapping. Nature Methods, 14, 819–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivodaliev, K. , Bogojeska, A. , & Kocarev, L. (2014). Exploring function prediction in protein interaction networks via clustering methods. PLoS One, 9, e99755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandin, F. , Upfal, E. , & Raphael, B. J. (2011). Algorithms for detecting significantly mutated pathways in cancer. Journal of Computational Biology, 18, 507–522. [DOI] [PubMed] [Google Scholar]
- Vázquez, M. , Valencia, A. , & Pons, T. (2015). Structure‐PPi: A module for the annotation of cancer‐related single‐nucleotide variants at protein–protein interfaces. Bioinformatics, 31, 2397–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal, M. , Cusick, M. E. , & Barabasi, A. L. (2011). Interactome networks and human disease. Cell, 144, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein, B. , Papadopoulos, N. , Velculescu, V. E. , Zhou, S. , Diaz, L. A. , & Kinzler, K. W. (2013). Cancer genome landscapes. Science, 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Wei, X. , Thijssen, B. , Das, J. , Lipkin, S. M. , & Yu, H. (2012). Three‐dimensional reconstruction of protein networks provides insight into human genetic disease. Nature Biotechnology, 30, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, X. , Das, J. , Fragoza, R. , Liang, J. , Bastos de Oliveira, F. M. , Lee, H. R. , … Lipkin, S. M. (2014). A massively parallel pipeline to clone DNA variants and examine molecular phenotypes of human disease mutations. PLoS Genetics, 10, e1004819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , & Li, Y. (2006). Discovering disease‐genes by topological features in human protein–protein interaction network. Bioinformatics, 22, 2800–2805. [DOI] [PubMed] [Google Scholar]
- Yachie, N. , Petsalaki, E. , Mellor, J. C. , Weile, J. , Jacob, Y. , Verby, M. , … Roth, F. P. (2016). Pooled‐matrix protein interaction screens using barcode fusion genetics. Molecular Systems Biology, 12, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Coulombe‐Huntington, J. , Kang, S. , Sheynkman, G. M. , Hao, T. , Richardson, A. , … Vidal, M. (2016). Widespread expansion of protein interaction capabilities by alternative splicing. Cell, 164, 805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. , Braun, P. , Yildirim, M. A. , Lemmens, I. , Venkatesan, K. , Sahalie, J. , … Vidal, M. (2008). High‐quality binary protein interaction map of the yeast interactome network. Science, 322, 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. , Kim, P. M. , Sprecher, E. , Trifonov, V. , & Gerstein, M. (2007). The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Computational Biology, 3, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, M. K. , Kramer, M. , Dutkowski, J. , Srivas, R. , Licon, K. , Kreisberg, J. , … Ideker, T. (2016). Translation of genotype to phenotype by a hierarchy of cell subsystems. Cell Systems, 2, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Zhang, S. , Wang, Y. , & Zhang, X. S. (2013). Identification of mutated core cancer modules by integrating somatic mutation, copy number variation, and gene expression data. BMC Systems Biology, 7(Suppl 2), S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Wang, R. S. , & Zhang, X. S. (2007). Uncovering fuzzy community structure in complex networks. Physical Review. E, Statistical, Nonlinear, and Soft Matter Physics, 76, 046103. [DOI] [PubMed] [Google Scholar]
- Zhong, Q. , Simonis, N. , Li, Q. R. , Charloteaux, B. , Heuze, F. , Klitgord, N. , … Swearingen, V. (2009). Edgetic perturbation models of human inherited disorders. Molecular Systems Biology, 5, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, X. , Yang, H. , Zhao, S. , Shyr, Y. , & Li, B. (2015). Network‐based stratification analysis of 13 major cancer types using mutations in panels of cancer genes. BMC Genomics, 16(Suppl 7), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybailov, B. L. , Glazko, G. V. , Jaiswal, M. , & Raney, K. D. (2013). Large scale chemical cross‐linking mass spectrometry perspectives. Journal of Proteomics and Bioinformatics, 6, 001. [DOI] [PMC free article] [PubMed] [Google Scholar]