

Abstract
Few therapy options exist for patients with advanced papillary and anaplastic thyroid cancer. We and others have previously identified c-Src as a key mediator of thyroid cancer pro-tumorigenic processes and a promising therapeutic target for thyroid cancer. To increase the efficacy of targeting Src in the clinic, we sought to define mechanisms of resistance to the Src inhibitor, dasatinib, to identify key pathways to target in combination. Using a panel of thyroid cancer cell lines expressing clinically relevant mutations in BRAF or RAS, which were previously developed to be resistant to dasatinib, we identified a switch to a more invasive phenotype in the BRAF-mutant cells as a potential therapy escape mechanism. This phenotype switch is driven by FAK kinase activity, and signaling through the p130Cas>c-Jun signaling axis. We have further shown this more invasive phenotype is accompanied by alterations in the secretome through the increased expression of pro-inflammatory cytokines, including IL-1β, and the pro-invasive metalloprotease, MMP-9. Furthermore, IL-1β signals via a feedforward autocrine loop to promote invasion through a FAK>p130Cas>c-Jun>MMP-9 signaling axis. We further demonstrate that upfront combined inhibition of FAK and Src synergistically inhibits growth and invasion, and induces apoptosis in a panel of BRAF- and RAS-mutant thyroid cancer cell lines. Together our data demonstrate that acquired resistance to single-agent Src inhibition promotes a more invasive phenotype through an IL-1β>FAK>p130Cas>c-Jun>MMP signaling axis, and that combined inhibition of FAK and Src has the potential to block this inhibitor-induced phenotype switch.
Keywords: Src; FAK; resistance; invasion; migration; phenotype switch; proliferation; thyroid cancer; dasatinib; PF-562,271; BRAF; RAS
INTRODUCTION
Significant morbidity and mortality remains a problem for advanced thyroid cancer patients with distant metastatic disease, and anaplastic thyroid cancer (ATC), which remains one of the most lethal human cancers (1,2). There has been much interest in targeting the MAP kinase pathway in thyroid cancer due to a high prevalence of BRAF and RAS mutations (3,4), and while BRAF-directed therapies are promising (5–8), there is a need for unique therapeutic approaches that will be effective in both BRAF wild-type and mutant cancers (2,3).
We and others have identified Src as a novel therapeutic target in thyroid cancer due to its role in growth, invasion, and metastasis (9–14). Src regulates pro-tumorigenic functions through multiple downstream pathways, of which Focal Adhesion Kinase (FAK) is a critical cellular substrate (15). FAK regulates growth, survival, migration, and invasion through its dual functions as a kinase and scaffolding protein. FAK autophosphorylation leads to the recruitment of Src, which then phosphorylates additional residues on FAK, mediating the full catalytic activity of FAK, and the phosphorylation of binding sites for downstream effector pathways, including p130Cas and Grb2 (16,17). Activation of the FAK-Src complex then signals to downstream effectors, including c-Jun N-terminal kinase (JNK) and MAPK/ERK, and the transcriptional regulation of pro-invasive genes, including matrix metalloproteinases (MMPs) (18).
Like many single-agent targeted therapies, Src inhibitors have had limited efficacy in the clinic likely due to underlying resistance mechanisms (19,20). Treatment failures generally result from mutations in the kinase that block drug binding and/or the activation of bypass signaling pathways. A change in cellular phenotype is an emerging mechanism of resistance that allows cells to survive and invade in response to therapy (21). Of interest, several mechanisms for phenotype switching in response to therapy are being identified, including activation of the FAK signaling pathway (22–28). In addition, a therapy-induced secretome, consisting of pro-inflammatory cytokines, can stimulate a more invasive phenotype through the regulation of FAK, MAPK, nuclear factor–ҡB pathways, and MMPs (22,29–32).
To more effectively target Src, we previously generated a panel of BRAF- and RAS-mutant thyroid cancer cell lines with acquired resistance to the Src inhibitor, dasatinib (33). In the present study, we identified a Src inhibitor-induced invasive phenotype that is specific to BRAF-mutant dasatinib resistant thyroid cancer cells, and is reliant on a FAK>p130Cas>c-Jun signaling axis. Combined FAK and Src inhibition attenuates the invasive phenotype, and shRNA knockdown of p130Cas or c-Jun phenocopies these results. Chronic Src inhibition alters the tumor cell secretome, resulting in the FAK- and Src-dependent upregulation of IL-1β and MMPs. Blocking IL-1β or MMPs inhibits invasion, phenocopying combined FAK and Src inhibition. Finally, upfront combined inhibition of FAK and Src in a panel of BRAF- and RAS-mutant thyroid cancer cell lines results in the synergistic inhibition of growth, increased cell death, and enhanced inhibition of invasion. Together, these results demonstrate that Src inhibition results in an increased reliance on an IL-1β>FAK>p130Cas>c-Jun>MMP signaling axis, which can be targeted upfront to increase the efficacy of Src targeted therapies.
RESULTS:
Chronic Src inhibition alters cellular morphology and promotes migration and invasion in BRAF-mutant thyroid cancer cells
To enhance the efficacy of Src-directed therapies, we previously established a novel model of acquired resistance to the Src inhibitor, dasatinib, using 2 BRAF-mutant (BCPAP; SW1736) and 2 RAS-mutant (Cal62, C643) thyroid cancer cell lines and a standard dose escalation approach until all cell lines were able to grow in 2 μM dasatinib (33). We previously showed that resistance to Src inhibition in the RAS-mutant dasatinib resistant (DasRes) cell lines (Cal62, C643) is primarily due to genetic mutation of c-Src itself, while no genetic changes were identified in the BRAF-mutant DasRes cells by genome-wide RNA-sequencing (33). We therefore focused on mechanisms of transcriptional reprogramming in the BRAF-mutant cells. In addition, we further observed that the BRAF-mutant cell lines acquired a more fibroblastic, spindle-like phenotype compared to their control, DMSO-treated counterparts, while the RAS-mutant Cal62 and C643 DasRes cells did not (Fig. 1A). We confirmed these observations by analyzing the aspect ratio of these cells (34), which showed that both of the BRAF-mutant DasRes cell lines (BCPAP and SW1736) exhibited an increased aspect ratio or more elongated morphology (Fig. 1A; p≤0.0001), while no changes in the aspect ratio were observed in the RAS-mutant (Cal62, C643) cell lines (Fig. 1A; p=n.s.). Accordingly, the BRAF-mutant DasRes cells are significantly more migratory (1.2–2.0-fold increase; Fig. 1B; BCPAP: p≤0.0001; SW1736: p≤0.001) and invasive (1.5–2.2-fold increase; Fig. 1C; BCPAP: p≤0.05; SW1736: p≤0.001) than their counterpart controls. Increased migration or invasion was not observed in the RAS-mutant cells (not shown). Together, these results demonstrate that the BRAF-mutant DasRes cells have acquired a more motile and invasive phenotype.
Figure 1. Cellular morphology and motility is altered in dasatinib-resistant cells.
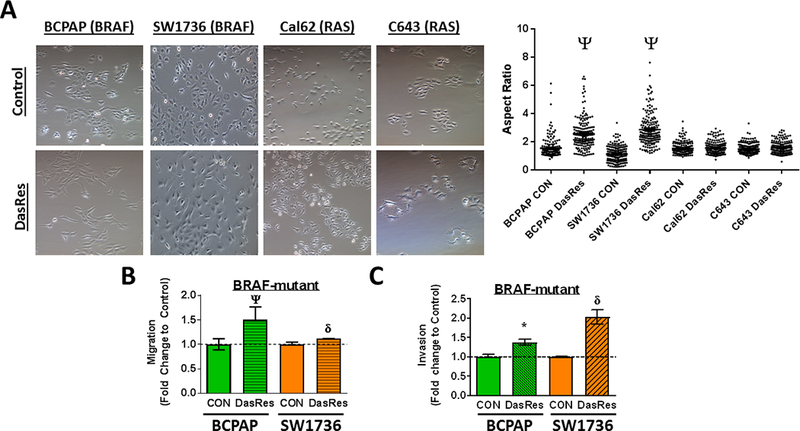
(A) Brightfield images were taken (10x magnification) of Control and DasRes cells (left) and quantification of aspect ratios to measure length versus width was performed using ImageJ. Migration (B) and Invasion (C) of Control and DasRes cells was quantified after 24 hours. DasRes data was normalized to Control set to 1. Data shown are mean ± SEM of 3 independent experiments performed in duplicate. (A-C) All data were analyzed with a paired t-test. DasRes cells were maintained in 2 μM dasatinib. Symbols indicate * p≤0.05, δ p≤0.001, and Ψ p≤0.0001.
Dasatinib-induced phenotype switching is mediated by FAK signaling
We previously identified FAK as a key kinase activated in the DasRes cells (35). Together with the known role of FAK in invasion and migration, we hypothesized that FAK signaling mediates resistance to Src inhibition by promoting a more invasive phenotype. We therefore determined whether inhibition of FAK kinase activity with PF-562,271 alone or in combination with the Src inhibitor, dasatinib, would block invasion of the DasRes cells. Figure 2A shows that FAK inhibition alone does not affect invasion, and as expected, the dasatinib-resistant cells are not inhibited by dasatinib. However, dual inhibition of FAK and Src resulted in a significant decrease in invasion in the BRAF-mutant DasRes cells (BCPAP, SW1736; 2-fold decrease; Fig. 2A; p≤0.01), but not the RAS-mutant DasRes cells (Fig. S1). Inhibition of invasion by combined FAK and Src inhibition was not due to effects on cell growth at this 24-hour time point (not shown).
Figure 2. Combined FAK and Src inhibition blocks invasion in the BRAF-mutant DasRes cells, which correlates with inhibition of FAK, p130Cas, and c-Jun.
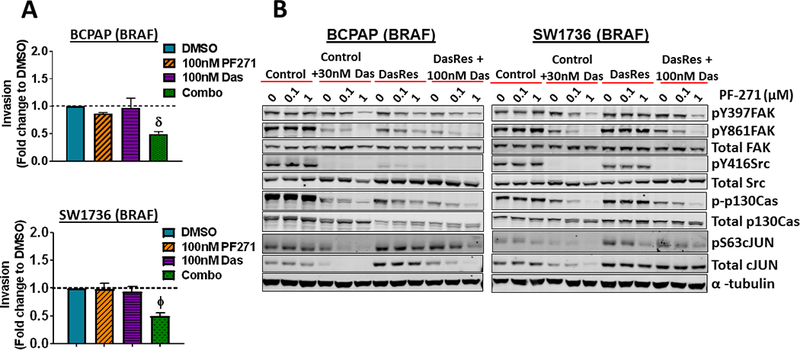
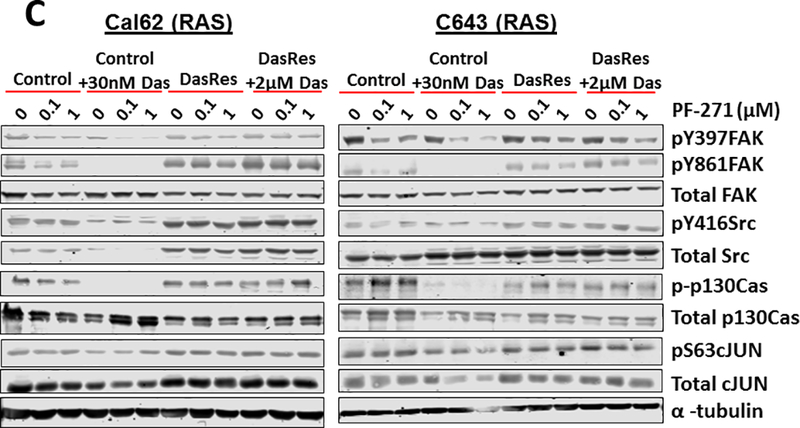
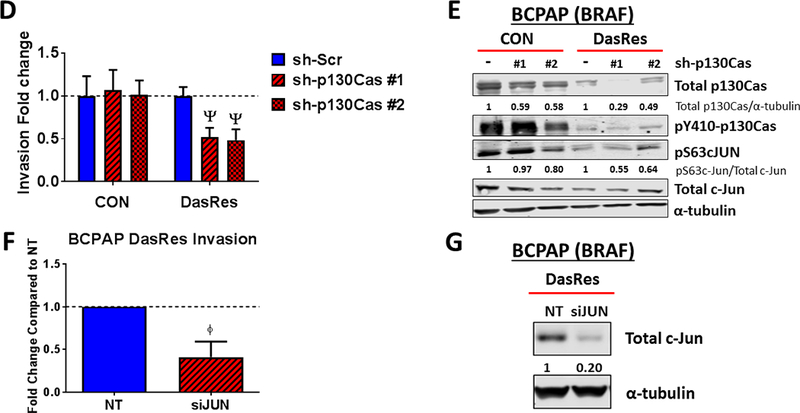
(A) Invasion of DasRes cells after 24 hours in the presence of DMSO, 100 nM PF-562,271, 100 nM dasatinib, or the combination of 100 nM PF-562,271 and 2𝜇M dasatinib was quantified. Results were normalized to DMSO-treated control set to 1. Results shown are mean ± SEM of 3 independent experiments performed in duplicate. Data were analyzed with a paired t-test. (B-C) Western blot analysis was performed on cells treated with the indicated concentrations of PF-562,271 and/or dasatinib for 24 hours. At least 3 independent experiments were performed for each cell line. Representative blots are shown. (D) BCPAP Control and DasRes cells were stably transfected with a scramble (Scr) control shRNA or shRNAs targeting p130Cas (sh-p130Cas #1 and #2). Invasion was performed on Scr and sh-p130Cas-expressing cells and data from each cell line was normalized to Scr expressing cells. Results shown are mean ± SEM of 3 independent experiments performed in duplicate. Data was analyzed by paired t-test. (E) Western blot analysis was performed on 3 independent cell lysates and a representative blot is shown. Quantification was performed on 3 independent experiments and the average is shown. (F) Invasion was performed on BCPAP DasRes cells transfected with nontargeting siRNA (NT) or siRNA targeting c-Jun. Data from each cell line was normalized to Scr expressing cells. Results shown are mean ± SEM of 3 independent experiments performed in duplicate. (G) Western blot analysis was performed on 3 independent cell lysates and a representative blot is shown. Quantification was performed on 3 independent experiments and the average is shown. DasRes cells were maintained in 2 μM dasatinib unless otherwise indicated. Symbols indicate * p≤0.05, Φ p≤0.01, δ p≤0.001, and Ψ p≤0.0001.
We next asked whether FAK kinase activity was elevated in the DasRes cells and found that FAK kinase activity was only significantly increased in the SW1736 DasRes cells, as measured by the autophosphorylation site of FAK, pY397 (1.5-fold, p≤0.05; Fig. 2B; not shown). We therefore evaluated FAK- and Src-dependent signaling responses in the BRAF- and RAS-mutant DasRes cells and compared responses to their counterpart Control cells by Western blot analysis. The BRAF-mutant (Fig 2B; Fig S2A) and RAS-mutant (Fig 2C; Fig S2B) Control cells lines, treated with the FAK inhibitor, PF-562,271, alone (0.1–1 μM) results in a minimal to moderate 15–49% inhibition of FAK kinase activity (pY397FAK), consistent with previous observations (36–39). As expected, inhibition of FAK with PF-562,271 does not significantly affect Src activity, as measured by pY416Src, and the Src-dependent site, pY861FAK (Fig. 2B-2C; Fig. S2). Similarly, inhibition of Src alone does not significantly affect FAK autophosphorylation (pY397FAK), as this is not a Src-dependent site. Notably, combined inhibition of FAK and Src results in enhanced inhibition of pY397FAK and pY861FAK in all of the Control cells (~65–95% inhibition), and ~95% inhibition of pY416Src in the BRAF-mutant cells (Fig. 2B-2C; Fig. S2). Consistent with this, combined inhibition of FAK and Src results in enhanced inhibition of pY397FAK and pY861FAK in the BRAF-mutant DasRes cells (15–65% inhibition PF-562,271 alone vs 60–90% inhibition dasatinib + PF-562,271; Fig. 2B; Fig. S2A). Of note, PF-562,271 inhibited pY861FAK in the BCPAP-DasRes cells, which may reflect a decreased interaction of Src and FAK due to inhibition of pY397FAK. Minimal inhibition of these signaling molecules was observed in the RAS-mutant DasRes cells, likely because these cells acquired the c-Src gatekeeper mutation (Fig 2C; Fig S2B) (33). Accordingly, ectopic expression of the drug-resistant c-Src gatekeeper mutant (c-Src-GK) blocked dasatinib inhibition of the FAK>p130Cas>c-Jun signaling module, indicating c-Src is the key mediator of these responses (Fig. S3). These data further suggest that cells are more dependent on the FAK>p130Cas>c-Jun signaling module in response to short term or long term Src inhibition.
To further understand the signaling mechanisms important for inhibition of invasion, we evaluated the phosphorylation and expression of the molecular scaffold, p130Cas, which can be phosphorylated by FAK or Src, and regulates a variety of signaling molecules, including CRK, and JNK (40). Similar to pFAK and pSrc, baseline levels of pY410-p130Cas did not correlate with resistance to dasatinib (Figure 2B), and combined inhibition of FAK and Src resulted in enhanced inhibition of pY410-p130Cas in the Control and BRAF-DasRes cells, and not the RAS-mutant DasRes cells. We further evaluated the regulation of c-Jun, which is a key downstream component of the FAK>p130Cas pathway. Similarly, baseline c-Jun levels did not correlate with dasatinib resistance, and pS63-c-Jun levels were inhibited in response to combined FAK and Src inhibition, except in the RAS-mutant DasRes cells likely due to the presence of the c-Src gatekeeper mutation (Fig. 2B-2C; Fig. S2). Together, these data indicate that the FAK>p130Cas>c-Jun signaling module represents a shared signaling mechanism in response to early and late Src inhibition.
Given the key role for p130Cas in FAK-mediated invasion (40), and the regulation of p130Cas by FAK and Src observed in our study (Fig. 2B), we tested the role of p130Cas in regulating invasion in the BRAF-mutant DasRes cells. Figure 2D shows an equivalent ~70% knockdown of p130Cas was achieved in the Control and DasRes BCPAP cells. Interestingly, similar to FAK or Src inhibition alone (Fig. 2A), shRNA knockdown of p130Cas does not affect invasion in the Control cells, but results in a ~2-fold decrease in invasion in the DasRes cells, which phenocopies the 2- to 3-fold inhibition of invasion by combined FAK and Src inhibition (Fig. 2A vs. 2D). We further show that phosphorylation of c-Jun is mediated, at least in part, through p130Cas signaling, as knockdown of p130Cas in the BRAF-mutant cells results in decreased pS63 levels (Fig. 2E). Finally, knockdown of c-Jun phenocopies inhibition of p130Cas, resulting in a 2.4-fold inhibition of invasion in the DasRes cells (Fig. 2F-2G). Taken together, our signaling and invasion data indicate that upon resistance to dasatinib, BRAF-mutant thyroid cancer cells adapt through a FAK>p130Cas signaling axis, which regulates c-Jun phosphorylation, resulting in an inhibitor-induced invasive phenotype.
Chronic Src inhibition with dasatinib alters the secretome of BRAF-mutant thyroid cancer cells
We observed enhanced inhibition of c-Jun in response to combined FAK and Src inhibition and knockdown of p130Cas (Fig. 2). Many targets of the c-Jun/AP-1 transcription factor complex encode pro-inflammatory and pro-invasive proteins, which are a part of the cancer secretome (41,42). We therefore performed a transcriptional secretome analysis by extracting a list of known and predicted secreted proteins (Secretome Protein Database; http://spd.cbi.pku.edu.cn/) (43) from our genome-wide RNA-sequencing data (33). Comparing BRAF-mutant DasRes cells to Control cells, 320 secretome genes were upregulated ≥ 2-fold (not shown). Functional annotation clustering indicated the highest enrichment scores were protein kinase activity, transcriptional regulation, and metalloprotease activity (Fig. S4A), suggesting that invasion of the BRAF-mutant DasRes cells may be promoted by altered kinase activity, controlling transcription, and secretion of pro-invasive factors, such as MMPs.
To test whether secreted factors promote invasion, we performed invasion assays using the Control cells in the presence or absence of conditioned media (CM) isolated from Control or DasRes cells. CM from BRAF-mutant Control cells (BCPAP or SW136) had no effect on invasion when compared to a DMSO-treated control media (1% FBS; Fig. 3A; not shown). However, CM isolated from the BRAF-mutant BCPAP DasRes cells significantly increased invasion of Control cells, regardless of mutational status (BRAF: 1.2–3.0-fold increase; Fig. 3A, BCPAP, p≤0.001; RAS: ~2.5-fold increase; Fig. 3B, Cal62, p≤0.001). As expected, CM from RAS-mutant DasRes cells had no effect on invasion (Fig. S5).
Figure 3. Conditioned media from BRAF-mutant dasatinib-resistant cells promotes invasion.

Conditioned media (CM) was collected from BCPAP Control and DasRes cells. (A) Invasion assays of Control cells were performed in the presence of 1% FBS control, BCPAP Control CM, or BCPAP DasRes (DR) CM. Results shown are mean ± SEM of 3 independent experiments performed in duplicate. (B) Invasion of Cal62 (RAS-mutant) Control cells was performed in the presence of BCPAP (BRAF-mutant) Control CM or DasRes (DR) CM. Quantification was performed and data was normalized to CM from Control cells, set to 1. Results shown are mean ± SEM of 3 independent experiments performed in duplicate. (C) ELISAs were performed on conditioned media (CM) from BCPAP Control or DasRes cells. The fold-change of IL-1β in DasRes (DR) CM to Control CM is shown. Results shown are average of 2 replicates ± SD. (D) BCPAP DasRes cells were treated with the indicated inhibitors for 24 h and qRT-PCR of IL-1β was performed. Fold-change of IL1β in the BCPAP DasRes cells compared to the Control cells is shown. Results shown are the mean of 3 experiments ± SD. Data was analyzed by paired t-test. DasRes cells were maintained in 2 μM dasatinib. Symbols indicate * p≤0.05, Φ p≤0.01, δ p≤0.001, and Ψ p≤0.0001.
IL-1β regulates invasion of BRAF-mutant dasatinib-resistant cells through the FAK>p130Cas>c-Jun signaling axis
To identify the secreted factor(s) mediating invasion, we identified 25 secretome genes in common between the BRAF-mutant BCPAP and SW1736 DasRes cells, with interleukin-1 beta (IL-1β) being the top regulated gene (~10-fold upregulation; Fig. S4B). We confirmed IL-1β regulation by ELISA (11.5-fold; p=0.004; Fig. 3C), and qRT-PCR analysis (41.2-fold increase; Fig. 3D). We further showed that combined FAK and Src inhibition results in enhanced inhibition of IL-1β mRNA (2-fold; p=0.03; Fig. 3D). Importantly, IL-1β mRNA or secretion was not increased in the RAS-mutant Cal62 DasRes cells (not shown). We therefore focused on the role of IL-1β, which has been shown to mediate tumorigenesis and resistance to therapy (31,32,44), and asked whether IL-1β blockade would attenuate invasion in the BRAF-mutant DasRes cells. As expected, IL-1β blockade with a neutralizing antibody did not affect invasion of the BCPAP Control cells, as these cells do not have high levels of IL-1β (Fig. 4A). However, invasion was significantly reduced in BCPAP DasRes cells treated with anti-IL-1β (Fig. 4A; ~1.6-fold decrease; p≤0.001). Consistently, anti-IL-1β abrogated invasion promoted by BRAF-mutant DasRes CM (~2.5-fold decrease; p≤0.01; Fig. 4B). These results indicate that chronic treatment with dasatinib induces expression of the pro-inflammatory cytokine IL-1β, contributing to a more invasive phenotype.
Figure 4. IL-1𝛽 regulates invasion through a FAK>p130Cas>c-Jun signaling axis.

(A) Invasion assays of BCPAP Control and DasRes cells were performed in the presence of absence of 0.1μg/mL of IL-1𝛽 neutralizing antibody or an IgG isotype control. Quantification was performed and data from each cell line was normalized to its respective DMSO-treated control. Results shown are mean ± SEM of 3 independent experiments performed in duplicate. (B) Invasion of BCPAP Control cells was performed in the presence of either BCPAP Control conditioned media (CM) or DasRes conditioned media, treated with either DMSO or anti-IL-1𝛽. Quantification was performed and data was normalized to the appropriate conditioned media treated with DMSO. Results shown are mean ± SEM of 3 independent experiments performed in duplicate. (C) Western blot analysis was performed on BCPAP Control and DasRes cells treated with the indicated concentration of anti-IL-1𝛽 or IgG isotype control antibody. Three independent experiments were performed and a representative blot is shown. Quantification was performed and shown as the average of 3 experiments. DasRes cells were maintained in 2 μM dasatinib. δ indicates p≤0.001; Φ indicates p≤0.01.
Previous studies have shown localization of IL-1 receptors to focal adhesions is critical for IL-1 signal transduction (45), and that IL-1β-dependent activation of FAK and Src promotes invasion of breast cancer cells (31). We therefore performed Western blot analysis on BRAF-mutant Control and DasRes cells treated with a neutralizing IL-1β antibody for 48 hours. Figure 4C shows that treatment of the DasRes BCPAP cells with anti-IL-1β inhibits the entire FAK>Src>p130Cas>c-Jun signaling module by 35–69%, whereas only pY397FAK, pY861FAK, pY410p130Cas, and pS63c-Jun are inhibited in the Control cells by 16–32%. Taken together with the regulation of IL-1β expression by FAK and Src (Fig. 3D), these results indicate that IL-1β signals via an autocrine fashion to promote FAK and Src signaling through p130Cas and c-Jun, thus generating a feed-forward loop.
MMP expression and activity are increased in BRAF-mutant dasatinib-resistant cells through FAK/Src activity
Several studies have demonstrated that inhibitor-induced invasion can occur, in part, through regulation of MMPs (22,24). In addition, FAK activity has been shown to regulate MMP expression (31,46–48), and many MMPs are target genes of c-Jun and the AP-1 complex (42,49). Of further interest, functional annotation clustering showed a ~2-fold enrichment score for metalloprotease activity in the BRAF-mutant DasRes cells, with MMP-2 and MMP-9 being upregulated 3- and 7-fold, respectively (Fig. S4). We validated MMP-9 expression in the BCPAP DasRes cells by qRT-PCR, which showed a 27-fold increase compared to Control cells (Fig. 5A; p=0.04). Interestingly, treatment of DasRes cells with either PF-562,271 or dasatinib reduced MMP-9 transcript levels ~4- to 6-fold compared to the DMSO-treated control (p≤0.05), and combined FAK and Src inhibition further inhibited MMP-9 expression (Fig. 5A; ~12-fold; p = 0.01). Consistently, treatment with a selective MMP-2 and MMP-9 inhibitor, SB-3CT, blocked invasion promoted by the DasRes-CM by > 10-fold (Fig. 5B; p≤0.001). A zymography assay showed that both MMP-2 and −9 activity are upregulated in the BCPAP DasRes-CM (2.9- and 2.0-fold increase, respectively), and that SB-3CT treatment reduced MMP-9 activity to levels below that of Control-CM, and minimally affected MMP-2 (Fig. 5C). Together, these results indicate that MMP-9 is the predominant MMP regulating the invasive phenotype in the BRAF-mutant BCPAP DasRes cells.
Figure 5. Expression and activity of MMP-9 are increased in BRAF-mutant dasatinib-resistant cells.
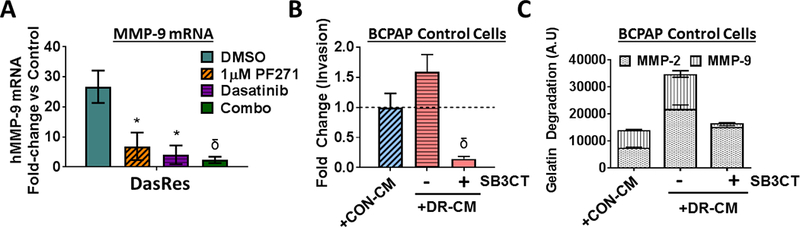
(A) Transcript levels of MMP-9 were evaluated by qRT-PCR. Levels of MMP-9 in the BCPAP DasRes cells compared to the Control cells is shown (~27-fold-increase). DasRes cells were treated with the either DMSO, 1𝜇M PF-562,271, dasatinib (30nM for controls or 2𝜇M for DasRes), or the combination. (B) Invasion of BCPAP Control cells was performed in the presence of Control conditioned media (CM), DasRes CM, or DasRes CM treated with SB-3CT. Quantification was performed and data normalized to Control CM set to 1. Results shown are mean ± SD of 3 independent experiments performed in duplicate. (C) MMP activity was assessed by zymography assay and gelatin-degradation was quantified using ImageJ and averaged over 3 independent experiments. Symbols indicate * p≤0.05; δ p≤0.001.
Combined inhibition of FAK and Src results in synergistic inhibition of growth and enhanced inhibition of invasion.
Our previous studies have demonstrated a key role for Src in regulating thyroid cancer growth (10,13), whereas we have shown FAK expression, but not kinase activity, is important for thyroid cancer adherent growth (36). Based on our data showing combined FAK and Src inhibition results in enhanced inhibition of FAK kinase activity (Fig. 2B-2C), we investigated the ability of combined FAK and Src to block the growth of the Control and DasRes cells. As we have previously reported, inhibition of FAK kinase activity with PF-562,271 has little effect on the growth of the Control cells (36), and as expected, the DasRes cells are resistant to dasatinib (33) (Fig. 6A). However, inhibition of FAK kinase activity with PF-562,271, in the presence of dasatinib resulted in a ~3- to 10-fold enhanced inhibition of growth in the BRAF-mutant DasRes cells (Figs. 6A; 6B). Of note, combined FAK and Src inhibition did not affect the growth of the RAS-mutant DasRes cell lines (Fig. 6A; dashed lines), likely due to the acquisition of the c-Src gatekeeper mutation in the RAS-mutant cells (33). Indeed, expression of the drug-resistant c-Src gatekeeper mutant (c-Src-GK), blocks the growth inhibitory effects of dasatinib in the SW1736 DasRes cells, with an IC50 > 2.5 μM (Fig. 6C). Together, the combined inhibition of FAK kinase activity with PF-562,271 and Src with dasatinib (Fig. 2B), along with the key anti-proliferative rescue mediated by the c-Src gatekeeper mutant (Fig. 6C), provide strong evidence that c-Src and FAK are the primary mediators of cellular growth in response to dasatinib and PF-562,271 treatment. These results further show a key switch in dependency from FAK adaptor/scaffolding function to FAK kinase activity occurs in response to Src inhibition.
Figure 6. Combined FAK and Src inhibition synergistically decreases growth in thyroid cancer cells.
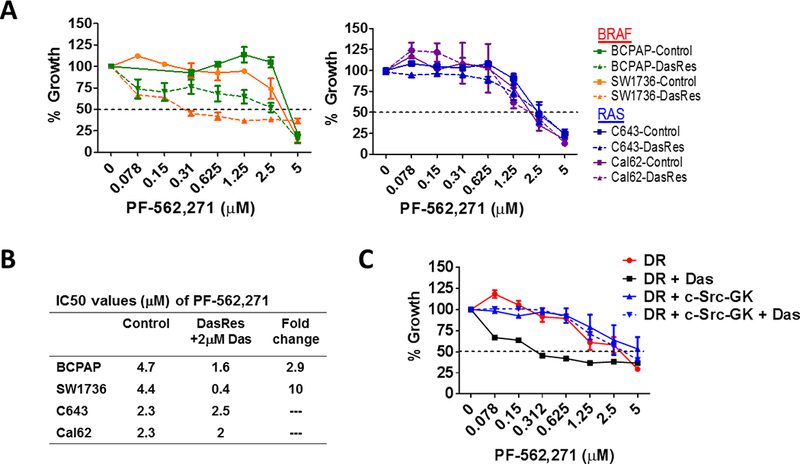
(A) SRB assays were performed to assess cell growth. BRAF-mutant (left) and RAS-mutant (right) Control and DasRes cells were treated with the indicated concentration of PF-562,271. The DasRes cells were maintained in the presence of 2𝜇M dasatinib. Data was normalized to DMSO treated control set to 100%. Results shown are mean ± SEM of 3 independent experiments performed in triplicate. (B) IC50 values of PF-562,271 were calculated for the Control cells or DasRes cells + Das and listed in the table shown. Fold change in IC50 values of the DasRes cells compared to Control cells is shown. (C) SW1736 DasRes (DR) cells were stably transfected with empty vector or a c-Src gatekeeper expression vector (c-Src-GK) and growth sensitivity to PF-562,271 and dasatinib was assessed using SRB growth assays, as in 6A. Results shown are the mean of 3 experiments performed in triplicate ± SEM.
Finally, upfront combination therapies are of interest to synergistically inhibit growth and block mechanisms of resistance from developing (50). We therefore tested a panel of five parental thyroid cancer cell lines expressing clinically relevant mutations in BRAFV600E (BCPAP, SW1736, 8505C) or RAS (Cal62, C643), in order to model responses in cells expressing key oncogenic mutations in thyroid cancer. Cells were treated with increasing concentrations of dasatinib (0.019–1.25 μM), alone or in combination with two clinically relevant doses of FAK inhibitor (100 nM or 1 μM PF-562,271; Fig. 7A; not shown). As expected, treatment with PF-562,271 minimally affects cells growth (average IC50s > 3.4 μM; not shown). Notably, combined FAK and Src inhibition results in a ~2 to almost 11-fold enhanced inhibition of growth, with inhibition beyond the Bliss Additivity scores, demonstrating synergistic response to combined FAK and Src inhibition (Fig. 7A; p<0.01; not shown). Accordingly, combined inhibition of FAK and Src significantly reduced clonogenic growth and survival compared to DMSO or single agent (Fig. 7B; 64.3% inhibition; p≤0.001for SW1736 and 62% inhibition; p<0.001 in C643) in the BRAF-mutant SW1736 and RAS-mutant C643 cell lines. Additionally, combined FAK and Src inhibition resulted in enhanced apoptosis compared to DMSO or single agent (2.8–6.7 fold increase Cleaved Caspase-3/7; Fig. 7C), which correlated with enhanced cell death compared to DMSO or single agent, as measured by Annexin V and propidium iodide staining (~20% cell death induction for SW1736; p<0.001 and 58% cell death induction for C643; p<0.01; Fig. 7D). Finally, we further evaluated the efficacy of upfront combined inhibition of FAK and Src on invasion of a panel of parental thyroid cancer cell lines. Figure 7E shows that combined FAK and Src inhibition results in enhanced inhibition of invasion in all cell lines tested (p≤ 0.05). Together, these data show that the upfront co-targeting of FAK and Src has the potential to inhibit both growth and invasion, and the potential to prevent a phenotype switch and therapy escape.
Figure 7. Upfront combined inhibition of FAK and Src results in synergistic inhibition of growth, enhanced induction of cell death, and inhibition of invasion.
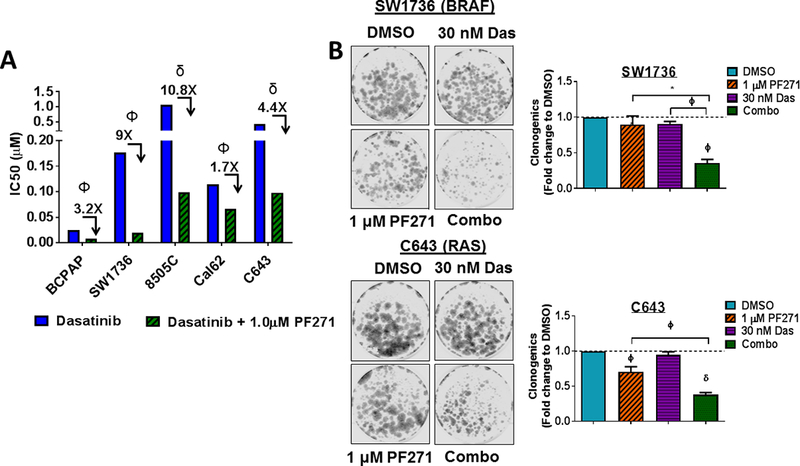
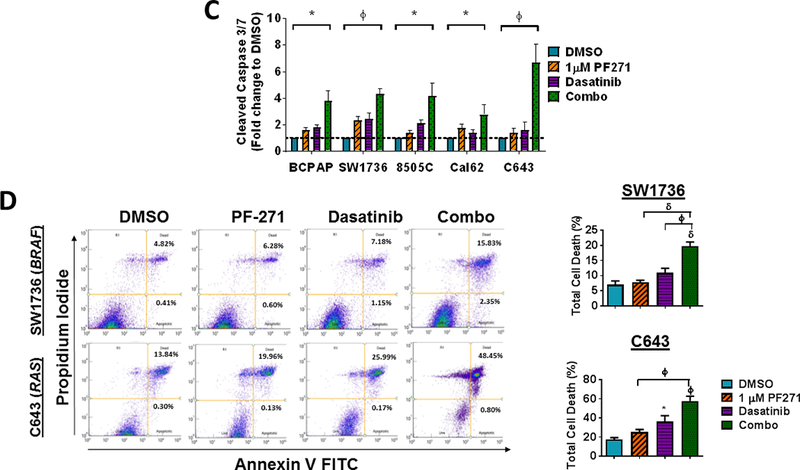
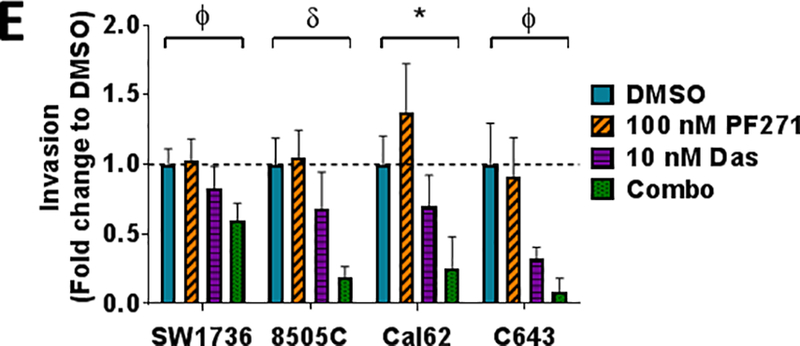
(A) A panel of parental thyroid cancer cell lines were treated with increasing concentrations of dasatinib (0.019–1.25 μM) with or without 1 μM PF-562,271 and analyzed by 3 independent SRB assays performed in triplicate. IC50 values for dasatinib and the combination of dasatinib and 1𝜇M PF-562,271 were calculated. Results shown are mean IC50 values ± SD. Fold changes and p-values were calculated using Students t-test and are noted above the bars. (B) Clonogenic growth was detected by crystal violet staining in the SW1736 or C643 cell lines. 24 h after plating, cell lines were treated with either 30 nM dasatinib, 1 μM PF-562,271, or a combination of dasatinib and 1 𝜇M PF-562,271 for 6 days. Following 6 days of treatment, the cells were released for an additional 7 days. Quantification was performed and data was normalized to DMSO. Results shown are mean ± SD of 3 independent experiments. Statistical analysis was performed using 2-tailed paired student’s t-test. (C) Cleaved caspase 3/7 activity in a panel of thyroid cancer cell lines was measured at 48 hours in the presence of either DMSO, PF-562,271, dasatinib, or the combination, as indicated. BCPAP, SW1736, and Cal62 cells were treated with 30 nM dasatinib. 8505C and C643 cell lines were treated with 50 nM dasatinib. Quantification was performed and data was normalized to DMSO. Results shown are mean ± SD of 2–3 independent experiments performed in duplicate. Statistical analysis was performed using 2-tailed paired student’s t-test. (D) Annexin V FITC propidium iodide assays were performed on SW1736 and C643 cells after 48 hour of treatment with DMSO, 30 nM dasatinib, 1 μM PF-562,271, or a combination of dasatinib and PF-562,271. Representative Annexin V FITC vs propidium iodide plots are shown. Quantification was performed for percent of total cell death as indicated by high annexin V and high propidium iodide levels. Results shown are the mean of 3 experiments ± SD. Statistical analysis was performed using 2-tailed unpaired student’s t-test. (E) Invasion of a panel of thyroid cancer cell lines was performed in the presence of either DMSO, PF-562,271, dasatinib, or the combination, as indicated. Quantification was performed and data was normalized to DMSO. Results shown are mean ± SD of 3 independent experiments performed in duplicate. Statistical analysis was performed using 2-tailed student’s t-test. Symbols indicate * p≤0.05, Φ p≤0.01, and δ p≤0.001.
DISCUSSION:
New therapeutic strategies are needed for patients with advanced thyroid cancer. We and others have demonstrated that Src represents an alternative, clinically relevant pathway in thyroid cancer, and targeting this pathway inhibits thyroid cancer growth, invasion, and metastasis (9,10,13,33). In order to enhance the efficacy of Src-directed therapies, we developed a novel model of acquired resistance to dasatinib to identify targets for upfront combination therapies (33). Herein, we have identified a switch to a more invasive phenotype as a potential therapy escape mechanism (Fig. 1), which is driven by FAK kinase activity, and an increased reliance on the p130Cas>c-Jun signaling axis (Fig. 2). We have further shown that this more invasive phenotype is accompanied by an altered secretome, resulting in the upregulation of IL-1β and MMP-9 (Figs. 3 and 5), and that IL-1β signals via a feed-forward loop (Fig. 4; Fig. 8 model). Finally, upfront combined inhibition of FAK and Src has the potential to block this phenotype switch, and results in the synergistic inhibition of growth, induction of cell death, and invasion (Fig. 7), providing key rationale to pursue this combination therapy in the clinic.
Figure 8. Model depicting how Src inhibition drives increased invasion via FAK>p130Cas>c-Jun.
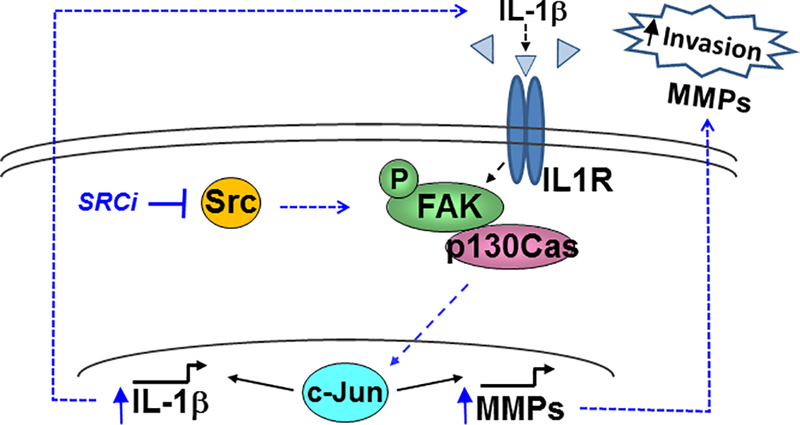
Following Src inhibition (SRCi), c-Jun is activated and IL-1beta is transcriptionally upregulated, along with transcription of MMP-9 and IL-1beta itself, creating a feed-forward loop with chronic SRCi to promote invasion and FAK>p130Cas>c-Jun signaling. This switch is not necessarily mediated by an increase in intrinsic FAK kinase activity, and therefore may be mediated through different protein-protein interactions and/or localization.
Emerging studies in other tumor types are showing therapeutic adaptation can lead to a phenotype switch through an epithelial-to-mesenchymal (EMT) transition, and an increase in tumor invasion and metastasis, which is consistent with our study (22–25,49,51,52). Of particular relevance, the FAK-Src pathway has been shown to mediate EGFR inhibitor resistance and drug-induced EMT in nonsmall cell lung cancer (51), and more recently to BRAF inhibition in colon and melanoma (28,53). Additionally, FAK has been causally associated with dasatinib resistance in lung and colon cancer, and similar to our study, combined FAK and Src inhibition led to synergistic inhibition of pY397FAK and enhanced antitumor activity (54,55). Furthermore, an emerging role for cytokines in promoting cancer cell survival and invasion and therapy resistance is consistent with our studies (22,49,56–60). The role of IL-1β has important clinical implications for thyroid cancer, as macrophages are a major source of IL-1β, and increased tumor-associated macrophages have been associated with a more aggressive and invasive phenotype and decreased patient survival (61,62). Of further interest, only the BRAF-mutant, but not RAS-mutant DasRes cells underwent a phenotype switch in response to Src inhibition. We previously demonstrated that only the RAS-mutant DasRes cells acquired the c-Src gatekeeper as a key mechanism of resistance (33). This differential acquisition of the c-Src gatekeeper may be due to different DNA repair mechanisms in BRAF- vs RAS-mutant cells, and thus render the RAS-mutant cells less dependent on transcriptional reprogramming and therefore less likely to undergo a phenotype switch.
Our studies have also demonstrated the importance of transcriptional reprogramming mechanisms in promoting resistance to targeted therapies. The regulation of c-Jun is of particular interest, as c-Jun levels have been shown to correlate with resistance in other tumor types (49,59,63,64). Here, we show that combined FAK and Src inhibition blocks expression and phosphorylation of c-Jun (Fig. 2B; Fig. S2A), which correlates with enhanced inhibition of IL-1β and MMP-9 expression (Figs. 3 and 5). Thus, upfront combined inhibition of FAK and Src has the potential to block resistance through the inhibition of key transcriptional reprogramming mechanisms.
Finally, we previously reported that the FAK scaffolding/adaptor function, but not FAK kinase activity, is the primary mediator of thyroid cancer pro-tumorigenic processes (36). In contrast, here we demonstrate that FAK kinase activity is critical for growth and invasion in response to Src inhibition. Thus, FAK may function as a molecular rheostat, by switching from its adaptor/scaffolding function(s) to its kinase signaling function, which results in an increased dependency on p130Cas and c-Jun, and together function to promote the growth and survival. This switch is not necessarily mediated by an increase in intrinsic FAK kinase activity (Fig. 2), and therefore may be mediated through different protein-protein interactions and/or localization. In summary, these studies have identified a key role for the IL-1beta>FAK>p130Cas>c-Jun>MMP signaling module in promoting a more invasive phenotype and resistance to therapy, thus informing Src-directed therapies for advanced thyroid cancer and other Src- and FAK-dependent tumors.
MATERIALS AND METHODS:
Reagents
Dasatinib was from LC labs (Woburn, MA), SB-3CT was purchased from MedChem Express (Monmouth Junction, NJ). The human IL-1β neutralizing antibody was from R&D bio-techne. PF-562,271, PF-573,228, and p-aminophenylmurcuric acetate (APMA), for MMP activation, was purchased from Sigma (St. Louis, MO). All drugs were dissolved in dimethyl sulfoxoide (Sigma).
Cell culture
Human thyroid cancer cell lines BCPAP, 8505C, Cal62, SW1736, and C643 cells were grown as previously described (36). Cells resistant to dasatinib (DasRes) were generated as previously described (33). All cell lines were validated using short tandem repeat profiling using the Applied Biosystems Identifiler kit (#4322288) in the Barbara Davis Center BioResources Core Facility, Molecular Biology Unit, at the University of Colorado (10). Cells were tested for Mycoplasma contamination using the Lonza Mycoalert system (Walkersville, MD). Cell lines were passaged no more than 30 times after thawing. Control and DasRes cells were treated with 30 nM, 100 nM, or 2 μM dasatinib unless otherwise indicated.
Generation of stable cell lines and siRNA knockdown
BCPAP DasRes cells were transduced with pBabe-hygro or pBabe-c-Src-Dasatinib-Resistant-T3381 retrovirus (Addgene plasmid 26980) and selected with hygromycin (9). shRNAs targeting p130Cas (Sigma mission TRCN0000115984, TRCN0000115985) or a scrambled control (Sigma mission pLKO.1-puro SHC016) were transduced and selected with puromycin. BCPAP DasRes cells were transfected a gene pool of 5 different siRNAs targeting c-Jun (siJUN) or nontargeting (NT) siRNA (5 nM each) using a final concentration of 0.5% Dharmafect I transfection reagent, according to manufacturer’s protocols Dharmacon (Broomfield, CO).
Cell morphology analysis and aspect ratio
Cell lines (BCPAP, SW1736, Cal62, and C643) were plated in 6-well plates and allowed to adhere for 48 hours. Bright field images were collected at 10X and used to visualize cell shape. For each cell line, 200 cells were quantified by ImageJ (NIH, Bethesda, MD) and the aspect ratio was calculated as a function of length versus width (34).
Generation of conditioned media
Conditioned media was generated as previously described (30). BCPAP (4 × 106), SW1736 (3 × 106), Cal62 (2 × 106), and C643 (3 × 106) Control and DasRes cells were plated in 15-cm dishes. After 24 hours, the media was replaced with media containing 1% FBS. After 72 hours, (~80% cell confluency) the media was collected, centrifuged @ 1000 rpm for 5 minutes, filtered through 0.45 μm pore size, aliquoted, and stored at −80°C.
Invasion and Migration assays
BCPAP (7.5 × 105), SW1736 (1 × 106), Cal62 (1 × 106), and C643 cells (1 × 106) were starved in RPMI with 0.1% FBS and treated with DMSO, dasatinib, PF-562,271, the combination, or the neutralizing antibody anti-IL1β, at the indicated concentrations. For conditioned media experiments, cell media was changed to 1% FBS. After 24 hours, cells were harvested and seeded in the upper chambers of Matrigel-coated transwells (24-well, 8 μM pore size; BD Biosciences, San Jose, CA) in 0.1% FBS RPMI with the appropriate drug/combination. RPMI with 10% FBS and the appropriate drug/combination was added to the lower chamber. For conditioned media experiments, conditioned media with or without drug, was added to either the upper or lower chamber, with 1% FBS with or without drug in the opposite chamber, as indicated. For MMP inhibitor treatments, SB-3CT (6 μM) was added to the upper and lower chamber. Cells were allowed to invade for 24 hours and assessed as previously described (36). Migration assays were performed similarly, except cells were plated in uncoated transwells (24-well, 8.0 μm pore size; Falcon) and allowed to migrate towards 10% FBS.
Western blotting
Western blotting was performed as previously described (36) using the following antibodies: total FAK (BD Bioscience:610087), total p130Cas (BD Bioscience:610271); pY397FAK (Abcam, Cambridge, MA:ab81298); pY416Src (Cell Signaling, Danvers, MA:2101), total Src (Cell Signaling:2109), pY410p130Cas (Cell Signaling:4011), total c-Jun (Cell Signaling:2315), pS63c-Jun (Cell Signaling:2361); pY861FAK (Invitrogen, Carlsbad, CA:44–626G); α-tubulin (CALBIOCHEM, Burlington, MA:CP06). Phosphorylated protein expression was normalized to total protein for quantification using the Odyssey CLx imager (Li-Cor).
Cellular growth assays
Sulforhodamine B (SRB) were performed as previously described (9,35) and treated with the indicated concentrations of dasatinib or PF-562,271. IC50 values were calculated using PRISM software. Synergy was calculated using Bliss independence model (65). For clonogenic growth assays, SW1736 (1 × 103) or C643 cells (1 × 103) were plated in 6-well plates and treated the following day with the indicated concentrations of dasatinib, PF-562,271, or the combination of dasatinib and PF-562,271 and subsequently treated every 3 days for 6 days. On day 6, the cells were washed and released from treatment for an additional 7 days. Cells were fixed with 4% paraformaldehyde, stained with crystal violet, and imaged and analyzed using the Odyssey CLx imager (Li-Cor) (33).
RNA-sequencing and Secretome analysis
For the Secretome analysis, a gene list from the Secreted Protein Database (43) (http://spd.cbi.pku.edu.cn/) was extracted from our previously generated RNA-sequencing data (33) of transcripts with expression levels > 1. Gene changes were determined with a ≥ 2-fold cutoff.
RNA and qRT-PCR
BCPAP (5.0 × 105) and Cal62 (3.0 × 106) cells were treated with the indicated concentrations of dasatinib, PF-562,271, or the combination. Total RNA was isolated after 24 hours using the RNeasy kit (Qiagen, Germantown, MD) and reverse transcribed using the High Capacity c-DNA kit (Applied Biosystems, Foster City, CA). The following primer/probe sequences were used:
Human MMP9 - Forward Primer: 5’-CCCGGACCAAGGATACAGTTT-3’, Reverse Primer: 5’- GAATGATCTAAGCCCAGCGC −3’, TaqMan Probe: 5’- 6FAM-CCTCGTGGCGGCGCATGAG-TAMRA −3’; Human IL1β - Forward Primer: 5’-GGCCCTAAACAGATGAAGTGCT-3’, Reverse Primer: 5’-GTAGCTGGATGCCGCCAT-3’, TaqMan Probe: 5’−6FAM-CCAGGCCCTGGACCTCTGCCCTCT-TAMRA −3’. Quantities of target in test samples were normalized to 18-s rRNA (PE ABI).
Gelatin zymography
MMP activation was profiled by gelatin zymography using equal volumes of conditioned media, mixed 1:4 with 5X non-reducing sample buffer (0.313 M Tris-HCL pH 6.8, 10% SDS, 50% glycerol, 0.05% bromophenol blue), and loaded on 10% gelatin-polyacrylamide gels (ReadyGel®, BioRad, Hercules, CA), and subjected to SDS-polyacrylamide gel electrophoresis. Recombinant MMP-2 and MMP-9 (Biolegend, San Diego, CA) were activated by incubation with 1mM APMA for 15 minutes and 4 hours, respectively, and used as positive controls. SDS was removed from the gels using a 25% Triton X-100 solution and gels were incubated at 37°C for 48 hours in incubation buffer (50 mM Tris-HCl pH7.4, 10 mM CaCl2, 0.02% NaN3). Gels were stained with 0.25% Coomassie Brilliant Blue and slowly destained to observe substrate cleavage. Gels were imaged using an AlphaImager Gel (Protein Simple, San Jose, CA). Densitometric analysis of the bands was performed using ImageJ (NIH, Bethesda, MD).
Apoptosis Assays
For Caspase-3/7 assays, cells were plated 1,000–2,000 cells/well, starved in RPMI with 0.1% FBS, and treated with the indicated concentrations of dasatinib, PF-562,271, or the combination. Apoptosis was measured using the IncuCyte® Caspase-3/7 Green Apoptosis Assay Reagent (Essen BioScience, Ann Arbor, MI) for 48 hours, with 2–4 images per well taken in 4 hour increments, using the IncuCyte® Zoom in the UCCC Protein Production/Mab/Tissue Culture Core. Processing definitions were generated and fold changes in green object counts were normalized to DMSO. For Annexin V apoptosis assays, SW1736 and C643 cells were starved in RPMI with 0.1% FBS and treated with the indicated concentrations of dasatinib, PF-562,271, or the combination for 48 hours. Following 48 hours, cells from supernatant were collected and adherent cells were detached with 3 mM EDTA in PBS. Cells were then stained with Annexin V FITC and propidium iodine according to the eBioscience™ Annexin V Apoptosis Detection Kit FITC. Cells were analyzed using the ZE5™ Cell Analyzer (formerly known as YETI).
Statistical analysis
All the statistical analyses in this study were performed in GraphPad Prism (La Jolla, CA). We analyzed the frequency distribution of the data to test normal distribution, and F-test to test equal variances between groups. Data are presented as mean ± SD (standard deviation) or mean ± SEM (standard error of measure) of at least 2 independent experiments. The difference of mean values between two groups was analyzed by Student’s t two-sided test. A p-value lower than 0.05 was considered as significant. Symbols indicate *p≤0.05, Φp≤0.01, δp≤0.001, Ψp≤0.0001, and n.s=not significant. Sample sizes were based on similar experiments which were reported in the literature and can achieve >80% power for testing medium differences between two groups. Samples were not subject to randomization. The investigators were blinded to images to quantify the aspect ratios, invasion, and migration assays. Investigators were not blinded to group allocation for further experiments. The statistical tests were justified as appropriate for every figure.
Supplementary Material
ACKNOWLEDGMENTS:
We would like to thank Dr. Christopher Korch, UCCC, and Randall Wong at the B. Davis Center BioResources Core Facility for STR profiling of the cell lines. We thank Dr. Rytis Prekeris for assistance with the zymography assays. This work was supported by NIH/NCI grant 1RO1CA164193 (RES), 1R01CA222299 (RES) American Cancer Society RSG-13-060-01-TBE (RES), the Cancer League of Colorado (RES), and the Front Range Cancer Challenge Fellowship (KMM). The UCCC DNA Sequencing, Flow Cytometry, and Protein Production/Mab/Tissue Culture Shared Resources are supported by NCI Cancer Center support grant, P30CA046934.
Financial Support: NIH/NCI grant 1RO1CA164193 (RES), American Cancer Society RSG-13–060-01-TBE (RES), the Cancer League of Colorado (RES), the Front Range Cancer Challenge Fellowship (KMM), and UCCC Support Grant from the NCI (P30CA046934).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
COMPETING INTERESTS: The authors have not competing financial interests to declare.
REFERENCES:
- 1.Sipos JA, Shah MH. Thyroid cancer: emerging role for targeted therapies. Ther Adv Med Oncol. SAGE Publications; 2010;2:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kebebew E, Greenspan FS, Clark OH, Woeber KA, McMillan A. Anaplastic thyroid carcinoma. Treatment outcome and prognostic factors. Cancer 2005;103(7):1330–5 doi 10.1002/cncr.20936.AQ6 [DOI] [PubMed] [Google Scholar]
- 3.Antonelli A, Fallahi P, Ferrari SM, Ruffilli I, Santini F, Minuto M, et al. New targeted therapies for thyroid cancer. Curr Genomics. 2011;12:626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. NIH Public Access; 2012;16:103–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carneiro RM, Carneiro BA, Agulnik M, Kopp PA, Giles FJ. Targeted therapies in advanced differentiated thyroid cancer. Cancer Treat Rev. 2015; [DOI] [PubMed] [Google Scholar]
- 6.Hayes DN, Lucas AS, Tanvetyanon T, Krzyzanowska MK, Chung CH, Murphy BA, et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res. 2012;18:2056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah MH, Wei L, Wirth LJ, Daniels GA, A SJ, Timmers CD. Results of randomized phase II trial of dabrafenib versus dabrafenib plus trametinib BRAF-mutated papillary thryoid carcinoma. J Clin Oncol. 2017;32:6022. [Google Scholar]
- 8.Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600–Mutant Anaplastic Thyroid Cancer. J Clin Oncol. 2017;JCO.2017.73.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan CM, Jing X, Pike L a., Zhou Q, Lim D-JDJ, Sams SB, et al. Targeted inhibition of Src kinase with dasatinib blocks thyroid cancer growth and metastasis. Clin Cancer Res. 2012;18:3580–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schweppe RE, Kerege A a., French JD, Sharma V, Grzywa RL, Haugen BR. Inhibition of Src with AZD0530 reveals the Src-focal adhesion kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancer. J Clin Endocrinol Metab. 2009;94:2199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michailidi C, Giaginis C, Stolakis V, Alexandrou P, Klijanienko J, Delladetsima I, et al. Evaluation of FAK and Src expression in human benign and malignant thyroid lesions. Pathol. Oncol. Res 2010. page 497–507. [DOI] [PubMed] [Google Scholar]
- 12.Owens LV, Xu L, Dent GA, Yang X, Sturge GC, Craven RJ, et al. Focal adhesion kinase as a marker of invasive potential in differentiated human thyroid cancer. Ann Surg Oncol. 1996;3:100–5. [DOI] [PubMed] [Google Scholar]
- 13.Chan D, Tyner JW, Chng WJ, Bi C, Okamoto R, Said J, et al. Effect of dasatinib against thyroid cancer cell lines in vitro and a xenograft model in vivo. Oncol Lett. 2012;3:807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim WG, Guigon CJ, Fozzatti L, Park JW, Lu C, Willingham MC, et al. SKI-606, an Src Inhibitor, Reduces Tumor Growth, Invasion, and Distant Metastasis in a Mouse Model of Thyroid Cancer. Clin Cancer Res. 2012;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolós V, Gasent JM, López-Tarruella S, Grande E, Bolós MV. OncoTargets and Therapy The dual kinase complex FAK-Src as a promising therapeutic target in cancer. [DOI] [PMC free article] [PubMed]
- 16.Almeida EA, Ilić D, Han Q, Hauck CR, Jin F, Kawakatsu H, et al. Matrix survival signaling: from fibronectin via focal adhesion kinase to c-Jun NH(2)-terminal kinase. J Cell Biol. The Rockefeller University Press; 2000;149:741–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ilić D, Almeida EA, Schlaepfer DD, Dazin P, Aizawa S, Damsky CH. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol. The Rockefeller University Press; 1998;143:547–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee BY, Timpson P, Horvath LG, Daly RJ. FAK signaling in human cancer as a target for therapeutics. Pharmacol Ther. 2015;146:132–49. [DOI] [PubMed] [Google Scholar]
- 19.Johnson FM, Bekele BN, Feng L, Wistuba I, Tang XM, Tran HT, et al. Phase II Study of Dasatinib in Patients With Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2010;28:4609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finn RS, Bengala C, Ibrahim N, Roché H, Sparano J, Strauss LC, et al. Dasatinib as a Single Agent in Triple-Negative Breast Cancer: Results of an Open-Label Phase 2 Study. Clin Cancer Res. 2011;17. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Fu L. Mechanisms of acquired resistance to tyrosine kinase inhibitors. Acta Pharm Sin B. 2011;1:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandri S, Faião-Flores F, Tiago M, Pennacchi PC, Massaro RR, Alves-Fernandes DK, et al. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol Res. 2016;111:523–33. [DOI] [PubMed] [Google Scholar]
- 23.Paraiso KHT, Das Thakur M, Fang B, Koomen JM, Fedorenko IV, John JK, et al. Ligand-Independent EPHA2 Signaling Drives the Adoption of a Targeted Therapy-Mediated Metastatic Melanoma Phenotype. Cancer Discov. 2014;5:264–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zubrilov I, Sagi-Assif O, Izraely S, Meshel T, Ben-Menahem S, Ginat R, et al. Vemurafenib resistance selects for highly malignant brain and lung-metastasizing melanoma cells. Cancer Lett. 2015;361:86–96. [DOI] [PubMed] [Google Scholar]
- 25.Chow AK-M, Ng L, Lam CS-C, Wong SK-M, Wan TM-H, Cheng NS-M, et al. The Enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS One. 2013;8:e78675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kemper K, de Goeje PL, Peeper DS, van Amerongen R. Phenotype switching: tumor cell plasticity as a resistance mechanism and target for therapy. Cancer Res. American Association for Cancer Research; 2014;74:5937–41. [DOI] [PubMed] [Google Scholar]
- 27.Taylor KN, Schlaepfer DD. Adaptive Resistance to Chemotherapy, A Multi–FAK-torial Linkage. Mol Cancer Ther. 2018;17:719–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen G, Gao C, Gao X, Zhang DH, Kuan S-F, Burns TF, et al. Wnt/β-Catenin Pathway Activation Mediates Adaptive Resistance to BRAF Inhibition in Colorectal Cancer. Mol Cancer Ther. 2018;17:806–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barderas R, Mendes M, Torres S, Bartolomé RA, López-Lucendo M, Villar-Vázquez R, et al. In-depth characterization of the secretome of colorectal cancer metastatic cells identifies key proteins in cell adhesion, migration, and invasion. Mol Cell Proteomics. American Society for Biochemistry and Molecular Biology; 2013;12:1602–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. Nature Publishing Group; 2015;520:368–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mon NN, Senga T, Ito S. Interleukin-1b activates focal adhesion kinase and Src to induce matrix metalloproteinase-9 production and invasion of MCF-7 breast cancer cells. Oncol Lett. 2017;13:955–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. American Association for the Advancement of Science; 2010;3:cm1. [DOI] [PubMed] [Google Scholar]
- 33.Beadnell TC, Mishall KM, Zhou Q, Riffert SM, Wuensch KE, Kessler BE, et al. The Mitogen Activated Protein Kinase Pathway Facilitates Resistance to the Src Inhibitor, Dasatinib, in Thyroid Cancer. Mol Cancer Ther. 2016;15:1952–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leight JL, Tokuda EY, Jones CE, Lin AJ, Anseth KS. Multifunctional bioscaffolds for 3D culture of melanoma cells reveal increased MMP activity and migration with BRAF kinase inhibition. Proc Natl Acad Sci. 2015;112:5366–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mishall KM, Beadnell TC, Kuenzi BM, Klimczak DM, Superti-Furga G, Rix U, et al. Sustained activation of the AKT/mTOR and MAP kinase pathways mediate resistance to the Src inhibitor, dasatinib, in thyroid cancer. Oncotarget. Impact Journals; 2017;8:103014–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kessler BEBE, Sharma V, Zhou Q, Jing X, Pike LALA, Kerege AAAA, et al. FAK expression, not kinase activity, is a key mediator of thyroid tumorigenesis and protumorigenic processes. Mol Cancer Res. 2016;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen XL, Nam J-O, Jean C, Lawson C, Walsh CT, Goka E, et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell. 2012;22:146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat cell …. 2000;2:249–56. [DOI] [PubMed] [Google Scholar]
- 39.Hauck CR, Hsia DA, Schlaepfer DD. Focal adhesion kinase facilitates platelet-derived growth factor-BB-stimulated ERK2 activation required for chemotaxis migration of vascular smooth muscle cells. J Biol Chem. 2000;275:41092–9. [DOI] [PubMed] [Google Scholar]
- 40.Defilippi P, Di Stefano P, Cabodi S. p130Cas: a versatile scaffold in signaling networks. Trends Cell Biol. 2006;16:257–63. [DOI] [PubMed] [Google Scholar]
- 41.Qiao Y, He H, Jonsson P, Sinha I, Zhao C, Dahlman-Wright K. AP-1 Is a Key Regulator of Proinflammatory Cytokine TNFα-mediated Triple-negative Breast Cancer Progression. J Biol Chem. American Society for Biochemistry and Molecular Biology; 2016;291:5068–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wynn TA, Reddy NM, Zhang W, Reddy SP, Wynn T, Aso Y, et al. Expression profiling of genes regulated by Fra-1/AP-1 transcription factor during bleomycin-induced pulmonary fibrosis. J Exp Med. BioMed Central; 2011;208:1339–50. [Google Scholar]
- 43.Chen Y, Zhang Y, Yin Y, Gao G, Li S, Jiang Y, et al. SPD--a web-based secreted protein database. Nucleic Acids Res. Oxford University Press; 2004;33:D169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, et al. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. Kluwer Academic Publishers-Plenum Publishers; 2006;25:387–408. [DOI] [PubMed] [Google Scholar]
- 45.McCulloch CA, Downey GP, El-Gabalawy H. Signalling platforms that modulate the inflammatory response: new targets for drug development. Nat Rev Drug Discov. 2006;5:864–76. [DOI] [PubMed] [Google Scholar]
- 46.Wu X, Gan B, Yoo Y, Guan J-L. FAK-mediated src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Dev Cell. 2005;9:185–96. [DOI] [PubMed] [Google Scholar]
- 47.Segarra M, Vilardell C, Matsumoto K, Esparza J, Lozano E, Serra-Pages C, et al. Dual function of focal adhesion kinase in regulating integrin-induced MMP-2 and MMP-9 release by human T lymphoid cells. FASEB J. 2005;19:1875–7. [DOI] [PubMed] [Google Scholar]
- 48.Zhang P, Li Y-J, Guo L-Y, Wang G-F, Lu K, Yue E-L. Focal adhesion kinase activation is required for TNF-α-induced production of matrix metalloproteinase-2 and proinflammatory cytokines in cultured human periodontal ligament fibroblasts. Eur J Oral Sci. 2015;123:249–53. [DOI] [PubMed] [Google Scholar]
- 49.Ramsdale R, Jorissen RN, Li FZ, Al-Obaidi S, Ward T, Sheppard KE, et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal. 2015;8:ra82–ra82. [DOI] [PubMed] [Google Scholar]
- 50.Glickman MS, Sawyers CL. Converting Cancer Therapies into Cures: Lessons from Infectious Diseases. Cell. 2012;148:1089–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson C, Nicholes K, Bustos D, Lin E, Song Q, Stephan J-P, et al. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget. 2014;5:7328–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ware KE, Hinz TK, Kleczko E, Singleton KR, Marek LA, Helfrich BA, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis. 2013;2:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, et al. Intravital Imaging Reveals How BRAF Inhibition Generates Drug-Tolerant Microenvironments with High Integrin β1/FAK Signaling. Cancer Cell. 2015;27:574–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu H, Wang L, Gao W, Meng J, Dai B, Wu S, et al. IGFBP2/FAK pathway is causally associated with dasatinib resistance in non-small cell lung cancer cells. Mol Cancer Ther. 2013;12:2864–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Golubovskaya VM, Gross S, Kaur AS, Yang XH, Cance WG. Simultaneous Inhibition of Focal Adhesion Kinase and Src Enhances Detachment and Apoptosis in Colon Cancer Cell Lines Simultaneous Inhibition of Focal Adhesion Kinase and Src Enhances Detachment and Apoptosis in Colon Cancer Cell Lines. Mol cancer Res. 2003;1:755–64. [PubMed] [Google Scholar]
- 56.Stanam A, Gibson-Corley KN, Love-Homan L, Ihejirika N, Simons AL, Stanam A, et al. Interleukin-1 blockade overcomes erlotinib resistance in head and neck squamous cell carcinoma. Oncotarget. Impact Journals; 2016;7:76087–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gelfo V, Teresa Rodia M, Pucci M, Dall’Ora M, Santi S, Solmi R, et al. A module of inflammatory cytokines defines resistance of colorectal cancer to EGFR inhibitors. Oncotarget. 2016;7:72167–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee C-R, Kang J-A, Kim H-E, Choi Y, Yang T, Park S-G. Secretion of IL-1β from imatinib-resistant chronic myeloid leukemia cells contributes to BCR - ABL mutation-independent imatinib resistance. Chang Z, editor. FEBS Lett. 2016;590:358–68. [DOI] [PubMed] [Google Scholar]
- 59.Fallahi-Sichani M, Moerke NJ, Niepel M, Zhang T, Gray NS, Sorger PK, et al. Systematic analysis of BRAF(V600E) melanomas reveals a role for JNK/c-Jun pathway in adaptive resistance to drug-induced apoptosis. Mol Syst Biol. 2015;11:797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, et al. Relief of Feedback Inhibition of HER3 Transcription by RAF and MEK Inhibitors Attenuates Their Antitumor Effects in BRAF-Mutant Thyroid Carcinomas. Cancer Discov. 2013;3:520–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caillou B, Talbot M, Weyemi U, Pioche-Durieu C, Al Ghuzlan A, Bidart JM, et al. Tumor-Associated Macrophages (TAMs) Form an Interconnected Cellular Supportive Network in Anaplastic Thyroid Carcinoma. Agoulnik I, editor. PLoS One. 2011;6:e22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ryder M, Ghossein RA, Ricarte-Filho JCM, Knauf JA, Fagin JA. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr Relat Cancer. 2008;15:1069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Titz B, Lomova A, Le A, Hugo W, Kong X, ten Hoeve J, et al. JUN dependency in distinct early and late BRAF inhibition adaptation states of melanoma. Cell Discov. Nature Publishing Group; 2016;2:16028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fallahi‐Sichani M, Becker V, Izar B, Baker GJ, Lin J, Boswell SA, et al. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de‐differentiated state. Mol Syst Biol. 2017;13:905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao W, Sachsenmeier K, Zhang L, Sult E, Hollingsworth RE, Yang H. A New Bliss Independence Model to Analyze Drug Combination Data. J Biomol Screen. 2014;19:817–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.