

SUMMARY
Hematopoietic stem cell (HSC) quiescence is a tightly regulated process crucial for hematopoietic regeneration, which requires a healthy and supportive microenvironmental niche within the bone marrow (BM). Here we show deletion of Ptpn21, a protein tyrosine phosphatase highly expressed in HSCs, induces stem cell egress from the niche due to impaired retention within the BM. Ptpn21-/- HSCs exhibit enhanced mobility, decreased quiescence, increased apoptosis and defective reconstitution capacity. Ptpn21 deletion also decreased HSC stiffness and increased physical deformability, in part by dephosphorylating Spetin1 (Tyr246), a poorly-described component of the cytoskeleton. Elevated phosphorylation of Spetin1 in Ptpn21-/- cells impaired cytoskeletal remodeling, cortical instability, and decreased cell rigidity. Collectively these findings show that Ptpn21 maintains cellular mechanics, which is correlated with its important functions in HSC niche retention and preservation of hematopoietic regeneration capacity.
Keywords: Biomechanics, Hematopoietic stem cell, Niche, Ptpn21, Protein tyrosine phosphatase
Graphical Abstract
eTOC Blurb
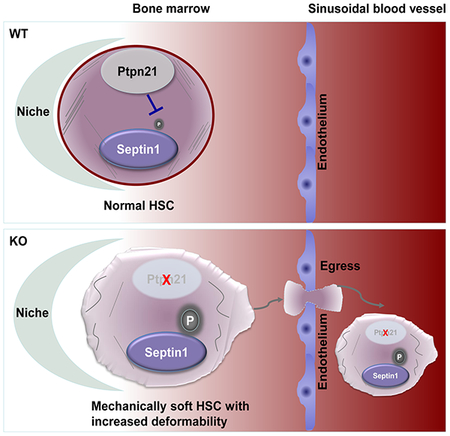
Ptpn21, a protein tyrosine phosphatase highly expressed in HSCs, plays an important role in maintaining cellular mechanics and HSC retention in the niche. Knock-out of Ptpn21 results in decreased cellular stiffness, enhanced deformability/motility, stem/progenitor cell egress, and defective reconstitution capabilities. Mechanistically, Ptpn21 modulates cell mechanics by dephosphorylating Septin1 (Tyr246).
INTRODUCTION
In adult mammals, a majority of hematopoietic stem cells (HSCs) are in a quiescent/dormant state (Cheng et al., 2000; Cheshier et al., 1999). Only a small portion of HSCs get activated, entering the cell cycle to either self-renew or produce progeny (i.e., differentiation) during steady-state hematopoiesis (Wright et al., 2001). Balanced quiescence and activation in this cell reservoir is crucial for maintaining hematopoietic regeneration and long-term hematopoiesis (Nakamura-Ishizu et al., 2014; Orford and Scadden, 2008; Pietras et al., 2011). Loss of stem cell quiescence/dormancy leads to aberrant activation and increased apoptosis, which in the long run can cause stem cell exhaustion and defects in repopulation capabilities. It is believed that HSC quiescence is achieved in part by the localization and retention of HSCs in the specialized healthy and supportive bone marrow (BM) microenvironment (also known as the niche) (Calvi and Link, 2015; Crane et al., 2017; Mendelson and Frenette, 2014; Scadden, 2014). Indeed, homing/engraftment and quiescence of HSCs are critically regulated by their adhesion to their microenvironment (Mendelson and Frenette, 2014; Potocnik et al., 2000). Studies in the last decade have demonstrated cytokine/chemokine signaling, transcriptional, genetic, epigenetic, and metabolic regulation of HSC quiescence. However, our understanding of the mechanisms regulating HSC maintenance and function remains incomplete.
Emerging evidence has linked cell intrinsic mechanics to functional behaviors (Fletcher and Mullins, 2010). The biophysical trait of a single cell is inextricably linked to the cytoskeleton, the interconnected network of filamentous polymers and regulatory proteins. It has become increasingly evident that intrinsic and extrinsic mechanical properties, which describe the resistance to deformation (elasticity) or flow (viscosity) in response to an applied force, regulate cellular behaviors, such as cell morphology, adhesion, migration, and trafficking. Studies of mesenchymal stem cells, embryonic stem cells, and HSCs cultured on matrices of different elasticity have suggested that differentiation of these stem cells is mechanosensitive (Chowdhury et al., 2010; Engler et al., 2006; Gonzalez-Cruz et al., 2012; Holst et al., 2010; McBeath et al., 2004). The effect of cell intrinsic mechanical properties on the function of stem cells, especially HSCs, is not well understood. Recent studies have demonstrated that cell contractile forces, polarized motility, and nuclear deformability are associated with self-renewal and differentiation of HSCs (Shin et al., 2014; Shin et al., 2013). However, the direct relationship between cell intrinsic mechanics and HSC niche retention and mobility in the in vivo setting remains unclear.
Ptpn21, a widely expressed protein tyrosine phosphatase (Moller et al., 1994), is poorly studied. This phosphatase contains an N-terminal sequence homologous to cytoskeletal-associated proteins, including a four-point-one/ezrin/radixin/moesin (FERM) domain, which is a modular structure that mediates interactions with the plasma membrane. Indeed, it has been shown that Ptpn21 is localized along actin filaments and that its FERM domain is required for this association (Carlucci et al., 2008). The catalytic domain of Ptpn21 is positioned at the end of the C terminus, and Ptpn21 catalytic activity is required for actin filament stability (Carlucci et al., 2008). Consistent with its important role in stabilizing actin filaments, Ptpn21 is involved in the regulation of cytoskeleton-associated cellular processes, including cell adhesion and motility (Carlucci et al., 2008). Importantly, missense mutations and frameshift truncating mutations in Ptpn21 have been identified in chronic lymphocytic leukemia (IntOGen - mutational cancer drivers database) and colon cancer (Giannakis et al., 2014; Korff et al., 2008; Seshagiri et al., 2012), respectively. However, the mechanisms by which Ptpn21 regulates these physiological and pathophysiological processes are poorly understood. Our recent gene expression analyses for protein tyrosine phosphatases show that Ptpn21 is highly expressed in HSCs as opposed to CD45+ leukocytes. To further determine the role of Ptpn21 in hematopoietic cell development, we generated Ptpn21 knock-out mice. With this mouse model, we have identified an important role of Ptpn21 in cell mechanics and HSC niche retention.
RESULTS
Knock-out of Ptpn21 Results in HSC Defects and Impaired Hematopoiesis
Our recent gene expression analyses showed that Ptpn21 was highly expressed in HSCs and early progenitors. Levels of Ptpn21 in HSCs (Lin−Sca-1+c-Kit+CD150+CD48−) were >7-fold higher than those in whole BM cells (Figure 1A). To determine the role of Ptpn21 in hematopoietic cell development, we generated Ptpn21 knock-out mice through gene targeting (Figure S1A and S1B). PCR analyses of genomic DNA confirmed that the targeted Ptpn21 DNA fragment was deleted in homozygous knock-out (Ptpn21−/−) blood cells (Figure S1C). Quantitative reverse transcription PCR (qRT-PCR) assays showed that Ptpn21 mRNA was undetectable in Ptpn21−/− early progenitor LSK (Lin−Sca-1+c-Kit+) cells (enriched for HSCs) (Figure S1D). Ptpn21 knock-out mice did not display overt abnormalities during 12 months of follow-up. However, total numbers of BM cells and lineage cells (Mac-1+Gr-1+ myeloid, B220+ B, and CD3+ T cells) in Ptpn21−/− mice were reduced as compared to wild-type (WT) (Ptpn21+/+) control mice (Figure S2A and S2B). Interestingly, Ptpn21−/− mice showed increased white blood cell (WBC) counts in the peripheral blood (PB) (Figure S2C). The absolute number of myeloid (but not B or T lymphoid) cells in the PB increased in these knock-out mice (Figure S2D).
Figure 1. Knock-out of Ptpn21 Results in HSC Defects and Impaired Hematopoiesis.

(A) Lin+, Lin−, LK, and LSK cells and HSCs were sorted from the BM harvested from healthy C57BL/6 mice (n = 5). Total RNA was extracted from these purified cells and whole BM (WBM) cells. Ptpn21 mRNA levels were determined by qRT-PCR. (B and C) BM cells freshly harvested from 6–8 week old Ptpn21−/− and Ptpn21+/+ mice were assayed by multiparameter FACS analyses to determine the numbers of LK cells, LSK cells, and HSCs (n = 6 mice per genotype) (B), and CLPs, CMPs, GMPs, and MEPs (n = 7 mice per genotype) (C). (D) BM cells harvested from Ptpn21−/− and Ptpn21+/+ mice (n = 5 per genotype) were assessed by CFU assays. Hematopoietic colonies (CFU-GEMM, CFUGM, and BFU-E) were enumerated. (E and F) BM cells harvested from Ptpn21−/− and Ptpn21+/+ mice (n = 6 per genotype) were assayed by FACS for the cell cycle status and apoptosis in HSCs. Percentages of HSCs in G0, G1, and S/G2/M phases (E) and apoptotic cells (Annexin V+) in HSCs (F) were quantified. (G) Six to eight-week-old Ptpn21−/− and Ptpn21+/+ mice (n = 10 per genotype) were administrated 5-Fluorouracil (5-FU) [250 mg/kg body weight, intraperitoneal (i.p.)]. White blood cell (WBC) counts in the PB were monitored. (H) Ptpn21−/− (n = 10) and Ptpn21+/+ (n = 11) mice were administrated two doses of 5-FU (150 mg/kg body weight, i.p.) at one week interval. Animal survival rates were determined. (I) BM cells (test cells) harvested from Ptpn21−/− or Ptpn21+/+ mice (CD45.2+) were mixed with BoyJ (CD45.1+) BM cells (1:1 ratio) and transplanted into lethally irradiated BoyJ recipients (n = 10 per genotype). Test cell reconstitution in the whole cell population of the PB at the indicated time points was determined by FACS analyses. (J) Test cell reconstitution in the whole cell population and in each lineage in the BM was determined 4 months after primary transplantation (n= 8 mice per genotype). (K) BM cells harvested from recipients 4 months after primary transplantation were transplanted into lethally irradiated secondary BoyJ recipient mice (n = 9 per genotype). Test cell reconstitution in the PB of secondary recipients was determined at the indicated time points as above. (L) Test cell contributions to the BM HSCs were determined 4 months after primary and secondary transplantation by multiparameter FACS analyses (n = 9 mice per genotype). See also Figures S1, S2, and S3.
To assess the effects of Ptpn21 deficiency on HSCs, we quantified phenotypic stem and progenitor cell populations in Ptpn21 knock-out mice. Both percentages (Figure S2E) and absolute numbers (Figure 1B) of HSCs and LSK cells in the BM significantly decreased in Ptpn21−/− mice as compared to Ptpn21+/+ mice. Frequencies (Figure S2F) and absolute numbers (Figure 1C) of common myeloid progenitors (CMPs, Lin−c-Kit+Sca-1−CD16/32medCD34+), common lymphoid progenitors (CLPs, Linc-Kit lowSca-1lowCD127+), and more committed myeloid progenitors, including granulocyte-monocyte progenitors (GMPs, Lin−c-Kit+Sca-1−CD16/32high CD34+) and megakaryocyte-erythroid progenitors (MEPs, Lin−c-Kit+Sca-1−CD16/32med/lowCD34−) also decreased in Ptpn21−/− mice. Consistent with these phenotypic data, functional colony-forming unit (CFU) assays verified that CFU-granulocyte, erythrocyte, macrophage, and megakaryocyte (CFU-GEMM), CFU-granulocyte and macrophage (CFU-GM), and burst forming unit-erythroid (BFU-E) decreased in Ptpn21−/− BM (Figure 1D). Cell cycle analyses revealed that percentages of quiescent cells (G0 phase) in HSC and LSK cell populations were decreased whereas percentages of HSCs and LSK cells in the G1 or S/G2/M phases were increased in Ptpn21−/− mice (Figure 1E and Figure S2G). Moreover, increased apoptosis was detected in Ptpn21−/− HSCs (Figure 1F), LSK, LK, and Lin−, but not Lin+ cells (Figure S2H). These data suggest that Ptpn21 is important for maintenance of HSCs and early progenitors. Loss of Ptpn21 decreased quiescence and increased apoptosis in stem/early progenitor cells.
Ptpn21-deleted HSCs Are Impaired in Hematopoietic Reconstitution
To determine the impact of Ptpn21 deficiency on the function of HSCs, we treated Ptpn21−/− and WT mice with 5-fluorouracil (5-FU), a cell-cycle-dependent myelotoxic agent that kills proliferating cells including cycling hematopoietic stem/progenitor cells. Following a single dose of 5-FU, WBC counts dropped quickly in both Ptpn21−/− and Ptpn21+/+ mice. The hematopoietic recovery, which depends on replenishment/regeneration from surviving long-term HSCs, was essentially blocked in Ptpn21−/− mice. Compared to WT mice in which WBC counts recovered to the normal level two weeks after 5-FU treatment, WBC counts in Ptpn21−/− mice continued to decrease and remained very low (Figure 1G). Consistent with this data, 100% of Ptpn21−/− mice challenged with two lower doses of 5-FU over one week died of hematopoietic failure while ~80% of WT mice survived (Figure 1H).
To further assess Ptpn21−/− HSC function, we first determined their homing ability, but found no difference between Ptpn21−/− and WT cells (Figure S3A). We then performed competitive repopulation assays. BM cells (test cells) isolated from Ptpn21−/-− or Ptpn21+/+ mice (CD45.2+) were mixed with BM cells (competitor cells) isolated from normal BoyJ mice (CD45.1+) at the ratio of 1:1 and transplanted into lethally irradiated BoyJ mice. Four months after transplantation, BM cells harvested from these mice were transplanted into secondary recipients. In this competitive setting, hematopoietic reconstitution from Ptpn21-deleted donor stem cells was compromised in primary (Figure 1I and 1J) and secondary transplants (Figure 1K). Contributions of Ptpn21 knock-out stem cells to the whole hematopoietic cell population and each lineage in primary and secondary transplants were markedly decreased (Figure 1I–1K, S3B, and S3C). Moreover, in both primary and secondary transplants, self-renewed Ptpn21−/− HSCs were also decreased (Figure 1L). Furthermore, we performed competitive repopulation assays with purified HSCs. Equal numbers of HSCs sorted from Ptpn21−/− or Ptpn21+/+ mice were mixed with the same amount of competitor BoyJ BM cells and transplanted into lethally irradiated BoyJ recipients. Ptpn21 knock-out donor HSCs indeed showed greatly decreased repopulation capabilities compared to Ptpn21+/+ counterparts (Figure S3D–S3F). HSCs derived from Ptpn21−/− donor stem cells in the transplants significantly decreased compared to those derived from Ptpn21+/+ donor stem cells (Figure S3G), verifying that deletion of Ptpn21 impaired HSC maintenance and function.
Loss of Ptpn21 Causes HSC Mobilization and Egress
WBC counts in the PB increased while numbers of stem cells and early progenitors in the BM decreased in Ptpn21 knock-out mice; these seemingly contradictory observations led us to hypothesize that extramedullary hematopoiesis and stem/early progenitor cell egress might occur in these knock-outs. To test this hypothesis, we examined LSK cells in the PB of Ptpn21−/− and Ptpn21+/+ mice. Compared to WT mice, Ptpn21−/− mice exhibited increased frequency and absolute number of these early progenitors in the PB (Figure 2A). Consistent with these findings, hematopoietic progenitors detected by CFU-assays were doubled in Ptpn21−/− PB (Figure 2B). LSK cells and HSCs in the spleen also significantly increased in Ptpn21−/− mice although no splenomegaly was observed (Figure 2C). The significant increase in the numbers of LSK cells in the PB and HSCs in the spleen in Ptpn21−/− mice indicated that stem/early progenitor cells lacking Ptpn21 had enhanced mobility. To further address this possibility, we administered Ptpn21−/− and Ptpn21+/+ mice with granulocyte-colony stimulating factor (G-CSF), a clinically used agent for stem cell mobilization. Compared to control mice, G-CSF-induced mobilization of LSK cells and HSCs to the PB and spleen in Ptpn21−/− mice was substantially increased (Figure 2D and 2E). Consistent with these results, hematopoietic progenitors in the PB detected by CFU assays were also increased (Figure 2F). Moreover, WBC counts increased in knock-out mice in response to G-CSF administration (Figure 2G). The increased sensitivity of Ptpn21−/− stem and progenitor cells to GCSF-induced mobilization indicated that Ptpn21 might be required for maintenance of stem cells in the BM niches.
Figure 2. Loss of Ptpn21 Enhances Mobilization of Stem and Early Progenitor Cells.

(A and B) PB cells freshly collected from 6–8 week-old Ptpn21−/− and Ptpn21+/+ mice were assayed by FACS to determine the frequency and number of LSK cells (n = 9 mice per genotype) (A), and subjected to CFU assays. Hematopoietic colonies were enumerated (n = 5 mice per genotype) (B). (C) Splenocytes isolated from 6–8 week-old Ptpn21−/− and Ptpn21+/+ mice were assayed by FACS to determine the absolute numbers of LSK cells (n = 9 mice per genotype) and HSCs (n = 5 mice per genotype). (D-G) Six to eight-week-old Ptpn21−/− and Ptpn21+/+ mice were administered by intraperitoneal injection with G-CSF (50 μg/day/kg body weight) or PBS for 5 consecutive days. PB was collected and assayed by multiparameter FACS analyses to determine the frequency and absolute number of LSK cells (n = 5 mice per genotype) (D). Mice were sacrificed 2 hours after the last dose of G-CSF. The frequency and absolute number of HSCs in the spleen were determined by multiparameter FACS analysis (n = 5 mice per genotype) (E). PB cells were collected and subjected to CFU assays. Hematopoietic colonies were enumerated (n = 5 mice per genotype) (F). WBC counts in the PB were monitored 0, 2, and 4 days after G-CSF administration (n = 6 mice per genotype) (G).
In situ imaging of HSCs in the BM illustrated that the distance of HSCs from CD31+CD144+ endothelial cells, which comprise endothelial HSC niches (Kiel et al., 2005), was significantly increased (Figure 3A). The percentages of HSCs close to Nestin+ mesenchymal stem/progenitor cells (MSPCs) (Figure 3B), which constitute unique sinusoidal vascular and arteriolar stem cell niches (Kunisaki et al., 2013; Mendez-Ferrer et al., 2010), and CD41+ megakaryocytes, which also serve as niches for a subset of HSCs (Bruns et al., 2014; Zhao et al., 2014) (Figure 3C), were decreased by >half in Ptpn21−/− mice. These imaging data together with the above functional data suggest that deletion of Ptpn21 dislocates HSCs in the BM, leading to their egress into the PB.
Figure 3. Deletion of Ptpn21 Results in HSC Dislocation and Stem/Progenitor Cell Egress in Ptpn21 Global Knock-out Mice through a Cell Intrinsic Mechanism.

Bone sections (one section per femur or tibia) prepared from 6–8 week-old Ptpn21−/− and Ptpn21+/+ mice were immunostained with the indicated antibodies. (A) The distance of HSCs (Lin–CD48–CD41–CD150+) from the closest CD31+CD144+ endothelial cells was determined (n = 5 mice per genotype). (B) The spatial relationship between HSCs (Lin−CD48−CD41−CD150+) and Nestin+ MSPCs was examined. HSCs within <8 μm of MSPCs were considered close to these cells (n = 4 mice per genotype). (C) The spatial relationship between HSCs (Lin−CD41−CD150+) and megakaryocytes (CD41+) was examined. HSCs within <8 μm of megakaryocytes were considered close to megakaryocytes (n = 4 mice per genotype). Representative images are shown. (D-I) BM cells collected from 3-month-old Ptpn21−/− mice (CD45.2+) were transplanted into lethally irradiated WT BoyJ mice (CD45.1+). In addition, BM cells collected from BoyJ mice were transplanted into 3-month-old Ptpn21−/− or Ptpn21+/+ mice (n = 5–7 per group). At 20 weeks following transplant, PB, spleen, and BM cells collected from the recipients were examined by CFU assays for hematopoietic progenitors (D) and FACS analyses for LSK cells and HSCs (E-H). In addition, the cycling status of HSCs in the BM were determined by FACS analyses (I).
Ptpn21 was globally deleted from the knock-out mice; to further determine whether the impaired niche occupancy and egress phenotypes of HSCs in Ptpn21 knock-out mice were caused by a cell-intrinsic effect or a defect in the BM microenvironment, we performed reciprocal transplantation assays. At 20 weeks following transplantation, significantly increased hematopoietic progenitors and LSK cells were detected in the PB and spleen whereas HSCs and LSK cells decreased in the BM of the WT recipients transplanted with Ptpn21−/− cells (Figure 3D–3H), recapitulating the phenotypes of Ptpn21 global knock-out mice. Moreover, Ptpn21 knock-out HSCs in WT recipients also showed enhanced cell cycling (Figure 3I). In contrast, no significant defects in WT donor stem/early progenitor cells in Ptpn21-/- recipient mice were observed (Figure 3D–3H). These results suggest that stem/early progenitor cell egress in Ptpn21 global knock-out mice is caused by a cell-intrinsic mechanism.
Deletion of Ptpn21 Decreases Stiffness, Leading to Increased Deformability in Stem/Early Progenitor Cells
Adhesion of HSCs to microenvironmental cells is known to be important for the retention of HSCs within BM niches (Mendelson and Frenette, 2014; Potocnik et al., 2000). We therefore examined adhesion activities of Ptpn21−/− and WT LSK cells. Knock-out cells showed reduced adhesion to fibronectin-coated slides (Figure 4A), although cell adhesion molecules α4 integrin (CD49d), β1 integrin (CD29), CD44, P-Selectin ligand-1 (Selplg-1), and E-selectin ligand-1 (Esl-1) were comparably expressed in the knock-out and WT cells (Figure S4A). Intriguingly, despite reduced adhesion and unchanged cell size (Figure S4B), knock-out LSK cells migrated through micropores more efficiently than WT cells in chemotaxis transwell assays with 5 μm pores (Figure 4B). When challenged with 3 μm pores (which are considerably smaller than the smallest dimension of hematopoietic cells), Ptpn21 knock-out cells performed much better in migrating through pores than did control cells with or without the chemokine stromal cell-derived factor 1 (SDF-1) (a chemoattractant) (Figure 4B). These results suggest that Ptpn21-deficient cells are more deformable and elastic compared to control cells.
Figure 4. Ptpn21 Deficiency Increases Deformability in Stem and Early Progenitor Cells.

(A and B) LSK cells were sorted from Ptpn21−/− and Ptpn21+/+ mice (n = 4 per genotype). Cell adhesion to fibronectin (Fn) and transmigration were determined by adhesion assays (A) and transwell migration assays (5 μm and 3 μm pores) in the absence or presence of SDF-1 (100 ng/mL) (B). (C) Diagram of the 3D HSC motility assay system. (D) Two layers of collagen hydrogels were prepared. Lower gels contained SDF-1 (100 ng/mL). Upper gels were either soft (0.5 mg/mL collagen), intermediate (1.0 mg/mL collagen), or stiff (5.0 mg/mL collagen). HSCs sorted from Ptpn21−/− and Ptpn21+/+ mice (n = 3 per genotype) were stained with calcein (2 μM) for 30 min, seeded on the top of upper gels, and incubated in PBS in a 37 °C 5% CO2 incubator. Five hours later, 3D stack images of HSCs migrating into the collagen gels were captured using a confocal microscopy. See also Figure S4.
Previous studies have shown that mechanical properties of extracellular matrix can significantly affect stem cell motility (Holst et al., 2010; Shin et al., 2014). To better examine Ptpn21-/- HSCs, we devised a 3 dimensional (3D) HSC motility assay system with two layers of collagen hydrogels (bottom gels contained SDF-1), and determined HSC deformability and motility in upper collagen hydrogels of varying elasticity (the stiffness of the collagen hydrogels increased with the collagen concentrations while the pore size decreased) (Figure 4C). Sorted HSCs were seeded to the top of the upper gels and migration of HSCs was real time tracked. 3D stack images illustrated that more Ptpn21−/− HSCs than Ptpn21+/+ HSCs migrated into the upper gels regardless of gel stiffness or pore sizes (Figure 4D). This was especially the case when stiff gels were used (WT HSCs barely migrated). These results suggest that Ptpn21−/− stem cells have increased deformability/motility independent of matrix substrate mechanical properties.
Cytoskeletal remodeling, particularly changes in actin, is an important process regulating cell adhesion and migration (Parsons et al., 2010). β-actin was constant in Ptpn21 knock-out HSCs (data not shown). We therefore examined filamentous actin (F-actin). Confocal microscopic examination and flow cytometry analyses revealed significantly reduced cortical polymerized actin in Ptpn21-deficient HSCs (Figure 5A and 5B). Differences in F-actin cytoskeletal reorganization might translate into local effects on cell mechanics (Oakes et al., 2009; Raab et al., 2012). We therefore measured stiffness of HSCs, LSK and Lin− cells with an Atomic Force Microscopy (AFM), and found that Ptpn21−/− cells were significantly softer than WT counterparts (Figure 5C). A consequence of reduced cytoskeletal rearrangements that lead to a decrease in cell stiffness can be enhanced cell deformability (Fletcher and Mullins, 2010). To further examine the relative deformability of Ptpn21 knock-out cells, we utilized a microfluidic mode (Figure 5D), an established bioengineering instrument with multiple microchannels [5.9 ± 0.8 μm, which are smaller than the average HSC and lymphocyte diameters (Fay et al., 2016; Rosenbluth et al., 2008)]. Given that the physical deformability of normal human CD34+CD38− cells has been shown to depend on myosin-II and to decrease in differentiation (Shin et al., 2014), we assessed the deformability of normal mouse HSCs, LSK and Lin+ cells. Despite a smaller volume (Figure 5E), stem and early progenitor cells traversed the microfluidic channels much more slowly compared to Lin+ cells, suggesting that stem cells are less deformable (Figure 5F). AFM measurements confirmed that HSCs, LSK cells, and Lin− cells were stiffer than Lin+ cells (Figure 5G). Although no significant difference in cell rigidity between quiescent (in the G0 phase) and activated HSCs (in the G1/S/G2/M phases) was detected (Figure 5H), cell tension quickly decreased upon differentiation in culture (Figure 5I), consistent with the data obtained from human early progenitor cells (Shin et al., 2014). Compared to WT HSCs, Ptpn21-/-HSCs traversed the microfluidic channels remarkably faster and a much lower proportion of Ptpn21−/− cells were stuck/obstructed within the device (>10 sec transit time) (Figure 5J and 5K), validating the important role of Ptpn21 in cell mechanics and motility.
Figure 5. Loss of Ptpn21 Decreases Cell Stiffness and Increases Mobility in HSCs.

(A) HSCs sorted from Ptpn21−/− and Ptpn21+/+ mice (n = 4 per genotype) were serum starved for 4 hours followed by stimulation with SDF-1 (100 ng/mL). The cells were then subjected to fluorescence staining for F-actin followed by counterstaining with DAPI. Confocal microscopy images shown are representative of >100 cells examined for each genotype. (B) BM cells freshly harvested from Ptpn21−/− and Ptpn21+/+ mice (n = 4 per genotype) were immunostained with antibodies recognizing HSC markers. Cells were then fixed, permeabilized, and fluorescence stained for F-actin. Mean fluorescence intensity (MFI) in the gated HSC population was quantified by FACS. (C) Lin− cells, LSK cells, and HSCs isolated from Ptpn21−/− and Ptpn21+/+ mice (n = 3–7 per genotype; 28 cells examined per mouse sample) were measured for stiffness by Atomic-force microscopy (AFM). (D) A microfluidic device composed of small microchannels used to measure relative cell deformability. (E) Relative cell sizes of purified HSCs, CD3+, B220+, and Mac1+Gr-1+ cells were determined by FACS based on forward scatter (FSC-A). (F and G) HSCs, LSK, Lin−, CD3+, B220+, and Mac1+Gr-1+ cells isolated from healthy C57BL/6 mice (n = 4 mice; 26 cells examined per mouse sample) were assessed by microfluidic assays. Transit time through the device was documented (F). Mechanical stiffness of these cells was measured by AFM (G). (H) HSCs in the G0 (quiescent) and G1/S/G2/M (activated) phases were sorted separately and measured for cell stiffness (n = 4 mice; 29 cells examined per mouse sample). (I) HSCs sorted from healthy C57BL/6 mice were cultured in 50 ng/mL SCF, 50 ng/mL Flt3L, 20 ng/mL IL-3, and 20 ng/mL IL-6 containing medium for 12 hours. Cells were then collected and measured for cell stiffness (n = 3 mice; 40 cells examined per mouse sample). (J and K) HSCs isolated from Ptpn21−/− and Ptpn21+/+ mice (n = 4 per genotype) were assayed by the microfluidic system. Cells in transit were recorded using a bright-field microscopy and transit times were manually calculated (I), and average transit times are determined (J). See also Figure S5.
The Role of Ptpn21 in Maintaining Cell Mechanics Depends on the FERM Domain and the Catalytic Activity
Ptpn21 is a protein tyrosine phosphatase and contains the FERM domain (Carlucci et al., 2008; Moller et al., 1994), which has been shown to mediate interactions of FERM-containing proteins with the actin cytoskeleton and their localization to the plasma membrane. We determined the structural basis of Ptpn21 function in maintaining cell mechanics by testing rescue effects of the catalytically-inactive mutant (Ptpn21C1110S) and truncated Ptpn21 lacking the FERM domain (Ptpn21Δ1–307) in Ptpn21 null cells. These mutants along with WT Ptpn21 cDNA were transduced into Ptpn21-/- LSK cells through lentiviral-mediated gene transfer (Figure 6A and 6B). Transwell (3 μm pores) migration, adhesion, and microfluidics assays demonstrated that WT Ptpn21 restored Ptpn21−/− cell functions (Figure 6C–6E), confirming the cell autonomous effect of Ptpn21 deficiency on cellular mechanics. In contrast, neither Ptpn21C1110S nor Ptpn21Δ1–307 mutants showed any rescue effects, suggesting that both catalytic activity and localization to the actin cytoskeleton are required for Ptpn21 to maintain cell mechanical properties.
Figure 6. The FERM Domain and the Catalytic Activity Are Required for Ptpn21 to Maintain Cellular Mechanics.

(A) Schematic representation of WT, catalytically inactive (Ptpn21C1110S), and FERM domain-truncated (Ptpn21Δ1–307) cDNAs. (B-E) LSK cells isolated from Ptpn21+/+ and Ptpn21−/− mice (n = 4 per genotype) were infected with the indicated recombinant lentiviruses. Transduction efficiencies of WT and Ptpn21−/− LSK cells infected with Ptpn21-expressing lentiviruses were assessed 48 hours later by FACS analyses of GFP, which was independently expressed by the vector backbone. Expression levels of Ptpn21 in transduced Ptpn21−/− Lin− cells were examined by immunoblotting (B). Lentiviruses-infected Ptpn21+/+ and Ptpn21−/− LSK cells were assessed by transwell (3 μm pores) migration (C), adhesion (D), and microfluidic (E) assays.
We next sought to determine the mechanisms by which Ptpn21 deficiency compromises cellular mechanics. Previous studies have demonstrated that Ptpn21 is a positive regulator of c-Src signaling (Cardone et al., 2004; Carlucci et al., 2010). We therefore examined c-Src activity in Ptpn21 knock-out Lin− cells. Phosphorylation of Src (Tyr416), which is associated with c-Src activation, was marginally decreased in Ptpn21-deficient cells relative to that in WT cells (Figure S5A). Cytokine-induced activation of Stat5 and Erk was also slightly decreased in the knock-out cells (Figure S5A), consistent with previous findings that Src kinase can activate Stat5 and Erk pathways through non-canonical mechanisms. In addition, we examined activities of the Rho family small GTPases, which play an essential role in cytoskeletal reorganization and cell motility (Gu et al., 2003; Nayak et al., 2013; Yang et al., 2007), with primary embryonic fibroblasts (in order to obtain sufficient cell lysates for the assays) [Ptpn21-/- fibroblasts and hematopoietic cells displayed similar biomechanical defects (data not shown)]. Activities of RhoA or Rac1 were not significantly altered in Ptpn21-/- cells (Figure S5B). Thus, slight changes in cell signaling activities did not likely account for the mechanical defects of Ptpn21 knock-out cells.
Ptpn21 Functions in Cellular Mechanics by Dephosphorylation of Septin1 (Tyr246)
To further define the molecular mechanism by which Ptpn21 modulates cellular mechanics, we aimed to identify the downstream substrate/effector that mediates Ptpn21 function. We performed phosphoproteomics analysis with Lin− cells isolated from Ptpn21−/− and Ptpn21+/+ mice. One of the peptides with >6-fold (Log2) increased tyrosine-phosphorylation (pY) levels from Ptpn21 knock-out cells was Septin1 (Tyr246 as the phosphorylation site) (Table S1) (ProteomeXchange Consortium; Accession number: PXD011381), a GTP-binding protein that can polymerize into hetero-oligomeric protein complexes that form filaments and higher-order structures (Mostowy and Cossart, 2012) [The other positive hit (lamina-associated polypeptide 2) associated with the cytoskeleton was eliminated from our study because we failed to verify that its tyrosine phosphorylation levels were increased in Ptpn21 knock-out cells (data not shown)]. Septin1 caught our attention also because Septins have been newly recognized as important components of the cytoskeleton, and they play a critical role in actin filament organization, regulating the mechanical properties of the cell (Gilden et al., 2012; Mavrakis et al., 2014; Mostowy and Cossart, 2012; Tooley et al., 2009). Indeed, increased tyrosine phosphorylation of Septin1 in Ptpn21−/− BM and Lin− cells was verified by immunoprecipitation/immunoblotting analyses (Figure 7A). Immunostaining examination illustrated that WT HSCs displayed O-shaped staining of Septin1 and that it partially co-localized with F-actin. In contrast, Septin1 was disorganized in Ptpn21−/− HSCs, and its co-localization with F-actin was barely detectable (Figure 7B).
Figure 7. Ptpn21 Maintains Cell Mechanics by Dephosphorylation of Septin1 (Tyr246).

(A) BM cells and Lin− cells freshly isolated from Ptpn21−/− and Ptpn21+/+ mice were lysed and examined by immunoprecipitation followed by immunoblotting analyses. Representative results from at least 3 independent experiments are shown. (B) HSCs isolated from Ptpn21+/+ and Ptpn21−/− mice (n = 4 per genotype) were processed for immunofluorescence staining for Septin1 and F-actin. Representative confocal images and quantification of the percentages of cells with normal F-actin structure and F-actin/Septin1 co-localization (90–120 cells examined per mouse sample) are shown. (C-G) Lin− cells isolated from Ptpn21+/+ and Ptpn21−/− mice (n = 3–4 per genotype) were infected with lentiviruses expressing WT Septin1 or Septin1Y246F. Forty-eight hours later, infected cells were assessed for transduction efficiencies by FACS analyses of GFP and for expression levels of Spetin1 by immunoblotting (C). Lentiviruses-infected Lin− cells were also assessed by transwell (3 μm pores) migration assays in the presence of SDF-1 (D), microfluidic assays (E), AFM measurements of cell stiffness (F), and immunofluorescence staining for Septin1 and F-actin. Representative confocal images and quantification of the percentages of cells with normal F-actin structure and F-actin/Septin1 co-localization (90–120 cells examined per mouse sample) are shown (G). See also Figures S6 and S7.
To further determine whether increased phosphorylation of Septin1 (Tyr246) was responsible for the perturbation of the actin cytoskeleton and membrane cortical instability in Ptpn21 knock-out cells, we generated the Septin1Y246F mutant, and overexpressed Septin1Y246F and WT Septin1 in Ptpn21−/−Lin− cells (Figure 7C). Overexpression of WT Septin1 failed to rescue the defective biomechanics-associated phenotypes. However, interference of endogenous WT Septin1 function by overexpression of Septin1Y246F effectively restored the mechanical properties of Ptpn21 knock-out cells in transwell (3 μm pores) migration and microfluidic assays (Figure 7D and 7E). Mechanical stiffness of Ptpn21−/− cells was also largely restored by overexpression of Septin1Y246F (Figure 7F). Moreover, overexpression of Septin1Y246F but not WT Septin1 in Ptpn21-deficient cells rescued cortical stabilization of polymerized actin (F-actin) and co-localization of Septin1 and F-actin (Figure 7G). The robust rescue effects of the dominant negative mutant Septin1Y246F in Ptpn21−/− cells suggest that the decreased cell stiffness and enhanced deformability caused by Ptpn21 deficiency were mainly attributable to increased phosphorylation of Septin1 (TyrY246).
To determine whether the mechanical alterations induced by Ptpn21 deficiency was responsible for the defects in HSC functions in Ptpn21 knock-out mice, we performed in vivo rescue experiments with the dominant negative Septin1Y246F mutant. Ptpn21−/− Lin− cells were transduced with Septin1Y246F, WT Septin1, or vector through lentiviral-mediated gene transduction (Figure S6A). These cells were transplanted into lethally irradiated congenic mice. Indeed, 16 weeks following transplant, overexpression of Septin1Y246F, but not WT Septin1, effectively rescued Ptpn21−/− stem/early progenitor cell function and corrected their egress in recipient mice (Figure S6B–6D). Moreover, the numbers of LSK cells and HSCs in the BM recovered and the cell cycling status of BM HSCs was largely restored (Figure S6E and S6F). These in vivo rescue data suggest that the hematopoietic defects and stem cell egress caused by Ptpn21 deficiency were attributed to increased phosphorylation of Septin1Y246. Given that the cellular biomechanics were restored by Septin1Y246F (Figure 7D–7G), these functional rescue data also indicate that the impaired HSC niche retention and stem cell egress in Ptpn21 knock-out mice might be caused by defective cell mechanics. Additionally, we treated normal mice with Blebbistatin, a reversible inhibitor of non-muscle myosin II, which decreases hematopoietic cell rigidity due to inhibition of actomyosin contractility (Shin et al., 2014). Blebbistatin administration resulted in stem/early progenitor cell egress in the treated animals in a dose-dependent manner. The numbers of LSK cells in the PB (Figure S7A), and LSK cells and HSCs in the spleen (Figure S7B) were markedly increased while LSK cells and HSCs in the BM decreased in the Blebbistatin-treated mice (Figure S7C). Similar to HSCs in Ptpn21 knock-out mice, HSCs in Blebbistatin-treated mice also showed decreased quiescence and enhanced cycling (Figure S7D).
DISCUSSION
In this study, we report an important role of protein tyrosine phosphatase Ptpn21 in biomechanics and HSC retention in the BM niche. Deletion of Ptpn21, which is highly expressed in HSCs, resulted in defective hematopoiesis due to impaired retention of HSCs within niches. Ptpn21 deleted stem cells showed decreased quiescence, increased apoptosis, and defective reconstitution capabilities. Loss of Ptpn21 produced only marginal impact on cell survival/proliferation signaling, and the Rho family small GTPases were not affected, which does not explain the HSC defects in Ptpn21 knockout mice. Interestingly, Ptpn21 deficient HSCs displayed decreased stiffness and increased physical deformability/mobility due to hyper phosphorylation of the cytoskeleton-associated Septin1 (Tyr246), a downstream target of Ptpn21 identified in this study. The cell mechanical changes might be linked to the stem cell functional defects because overexpression of the Septin1Y246F mutant lacking the Ptpn21 target site, which restored cell mechanical properties, also corrected functional defects in Ptpn21 knock-out HSCs. This notion is also supported by the fact that administration of Blebbestatin, which decreases cell tension, resulted in stem/progenitor cell egress/mobilization in mice, although the stem cell egress phenotype being a consequence of a possible systemic effect of this drug cannot be fully excluded.
Cell mechanics might be important for maintaining HSC attachment to the physical microenvironment (niche), which is critical for preserving HSC quiescence/dormancy and hematopoietic regeneration. A compromise in actin remodeling causing cell cortical cytoskeleton instability, as seen in Ptpn21-deleted HSCs, can affect the cytoskeleton-associated local adhesion, reducing stem cell attachment to the niche. However, impaired cytoskeletal organization may not affect stem cell niche localization merely by reducing cell local adhesion. Our present study with the Ptpn21 knock-out model suggests that reduced intracellular tension (possibly below a certain threshold) might also mechanically cause HSCs to lose their ability to locate and take up residence within their normal microenvironments, resulting in loss of quiescence and defective hematopoiesis. In addition, mechanically softer cells conceivably possess increased physical deformability and trafficability. Indeed, Ptpn21 knock-out HSCs showed increased micropore migration from marrow through the endothelial barrier and into sinusoidal vessels, leading to spontaneous stem cell egress into the circulation. Nevertheless, given that technologies to directly test the role of cell mechanics in the HSC-niche interaction are currently unavailable, the possibility that the cell mechanical changes and the functional defects in Ptpn21 knock-out HSCs might be correlative cannot be completely excluded. Further studies on multiple pathways that similarly affect HSC’s properties and function are needed to unequivocally demonstrate the role of cell mechanics in HSC niche retention and quiescence.
Another interesting finding in this report is that normal HSCs are stiff and less compliant/deformable than lineage cells. Stem cells are the most primitive precursor cells within the hematopoietic system. These immature cells were much more rigid as compared to lineage cells, and their physical deformability was lower, indicating that cytoskeleton rearranges as stem cells differentiate toward specific lineages. Differentiated cells and mature cells are more flexible. This might be because these cells migrate more frequently, and the need to traffic is intricately connected to whole cell stiffness. Indeed, it has been known for a long time that as granulocytes differentiate they become soft to better traffic from marrow through the endothelial barrier and into the circulation (Lichtman, 1970). Ptpn21 appears to play an important role in maintaining mechanical stiffness of hematopoietic cells in all stages. However, the impact of Ptpn21 deletion on cell functions are more severe in stem and early progenitor cells than mature cells, highlighting the significance of differential cellular mechanics for distinct cell populations in the hematopoietic system. Nevertheless, detailed mechanisms underlying the different mechanical properties in different hematopoietic cell populations remain to be further determined.
Last, this study reveals a molecular mechanism by which Septin1 regulates cytoskeletal organization and cellular mechanics. Septins, highly conserved GTP-binding proteins, are newly recognized as important components of the cytoskeleton (Mostowy and Cossart, 2012). These proteins frequently co-localize with actin stress fibers, and bind to plasma membrane. Septin1 is mainly expressed in hematopoietic cells and cells of the central nervous system (Mostowy and Cossart, 2012). Abnormal expression of Septin1 is associated with diseases including leukemia, lymphoma, and Alzheimer’s disease (Mostowy and Cossart, 2012). Some Septin isoforms have been shown to be phosphorylated at serine residues in several organisms and phosphorylation of Septins can affect cell morphology and functions (DeMay et al., 2009; Hernandez-Rodriguez and Momany, 2012; Sinha et al., 2007). Our study in this report has identified that Septin1 can be phosphorylated at a tyrosine residue, specifically Tyr246, in mammalian cells. Balanced phosphorylation and desphosphorylation of Septin1 at this site appear to be essential for maintaining cortical cytoskeleton integrity/stability and cell tension. Elevated phosphorylation levels of Septin1 (Tyr246), as seen in Ptpn21 knock-out stem cells, lead to impaired actin polymerization at the membrane cortex and decreased cell stiffness. Further studies, however, are required to address how increased phosphorylation of Tyr246 affects Septin1 distribution and function in cytoskeletal network remodeling and cell mechanics.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Cheng-Kui Qu (cheng-kui.qu@emory.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse studies
Ptpn21−/− mice were generated by a conventional gene-targeting strategy, in which exons 3–10 were replaced with a neomycin (neo) resistance cassette, flanked by two loxP sites. The targeting construct was electroporated into mouse embryonic stem cells with the C57BL/6 genetic background and clones that underwent homologous recombination were identified using Southern blotting with 5’ probes and 3’ probes following BamH1 or Sac1 digestion of genomic DNA. Heterozygous mice (Ptpn21+/-) were generated following standard procedures after in vivo Cre-mediated excision of the loxP-neo-loxP cassette (Bouabe and Okkenhaug, 2013). Mice were kept under specific pathogen-free conditions in Emory University Division of Animal Resources. They were separated by sex and housed with a maximum of 5 mice per cage prior to the experiments. Mice of the same age (6–8 week-old), sex, and genotype were randomly grouped for subsequent experiments (investigators were not blinded). No gender differences in gene deletion-associated phenotypes were observed in Ptpn21−/− mice. All animal procedures complied with the NIH Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
METHOD DETAILS
RNA Isolation and quantitative reverse transcription PCR (qRT-PCR) analysis
Total cellular RNA was isolated with a Quick-RNA Miniprep Kit. The RNA was then reverse transcribed using the SuperScriptIII First-Strand Synthesis System for RT-PCR. The reverse transcription products were subjected to real-time PCR with Power SYBR Green PCR Master Mix. All primers used were listed in KEY RESOURCES TABLE. All samples prepared from individual animals were run in triplicates. The mean value of the triplicates from each mouse was used for final statistical analyses. Actb was used as an internal normalization control.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE-Mac-1 | eBioscience | Cat# 12-0112-83 RRID: AB_2734870 |
| PE-Gr-1 | eBioscience | Cat# 12-5931-83 RRID: AB_466046 |
| eFIuor 450-Gr-1 | eBioscience | Cat# 48-5931-82 RRID: AB_1548788 |
| PE-B220 | eBioscience | Cat# 12-0452-83 RRID: AB_465672 |
| APC-B220 | eBioscience | Cat# 17-0452-82 RRID: AB_469395 |
| PE-CD3e | BD Biosciences | Cat# 553064 RRID: AB_394597 |
| APC-eFluor 780-CD3e | eBioscience | Cat# 47-0031-82 RRID: AB_11149861 |
| PE-CD45.1 | eBioscience | Cat# 12-0453-82 RRID: AB_465675 |
| FITC-CD45.2 | BD Biosciences | Cat# 553772 RRID: AB_395041 |
| Biotin-Lineage Panel antibody | BioLegend | Cat# 133307 RRID: AB_11124348 |
| PE-Cy7-Sca-1 | BD Biosciences | Cat# 558162, RRID: AB_647253 |
| APC-Fluor 780-c-Kit | eBioscience | Cat# 47-1171-82 RRID: AB_1272177 |
| PE-Cy5-CD150 | BioLegend | Cat# 115911 RRID: AB_493599 |
| PE-CD150 | eBioscience | Cat# 12-1502-80 RRID: AB_1548767 |
| Alexa Fluor 647-CD150 | BioLegend | Cat# 115918 RRID: AB_2239178 |
| FITC-CD48 | eBioscience | Cat# 11-0481-82 RRID: AB_465077 |
| PE-CD48 | BioLegend | Cat# 103405 RRID: AB_313020 |
| Alexa Fluor 647-CD34 | BD Biosciences | Cat# 560230 RRID: AB_1645200 |
| FITC-CD16/32 | BD Biosciences | Cat# 553144 RRID: AB_394659 |
| PE-Cy5-CD127 | eBioscience | Cat# 15-1271-82 RRID: AB_468793 |
| Alexa Fluor 647-CD31 | BioLegend | Cat# 102516 RRID: AB_2161029 |
| APC-CD144 | BioLegend | Cat# 138011, RRID: AB_10679039 |
| PE-CD49d | BD Biosciences | Cat# 557420 RRID: AB_396693 |
| PE-CD29 | eBioscience | Cat# 12-0291-81 RRID: AB_657733 |
| APC-CD44 | BioLegend | Cat# 103012 RRID: AB_312963 |
| FITC-Annexin V | BioLegend | Cat# 640945 RRID: AB_2629519 |
| Alexa Fluor 488-Ki-67 | BioLegend | Cat# 151204 RRID: AB_2566800 |
| Biotin-Ki-67 | eBioscience | Cat# 13-5698-82 RRID: AB_2572794 |
| Mouse anti-Nestin | Millipore | Cat# MAB353 RRID: AB_94911 |
| Rabbit anti-PTPN21 | Abcam | Cat# Ab133812 RRID: N/A |
| Mouse anti-Septin1 | Santa Cruz | Cat# sc-398586 RRID: N/A |
| Mouse anti-phosphotyrosine | Millipore | Cat# 05-321 RRID: AB_309678 |
| Rabbit anti-phospho-Src (Tyr416) | Cell Signaling | Cat# 2101 RRID: AB_331697 |
| Mouse anti-c-Src | Santa Cruz | Cat# sc-8056 RRID: AB_627306 |
| Mouse anti-phospho-STAT5A/STAT5B (Tyr694/Tyr699) | Millipore | Cat# 05-495 RRID: AB_309762 |
| Rabbit anti-STAT5 | Santa Cruz | Cat# sc-835 RRID: AB_632446 |
| Mouse anti-phospho-ERK | Santa Cruz | Cat# sc-7383 RRID: AB_627545 |
| Mouse anti-ERK1/2 | Santa Cruz | Cat# sc-514302 RRID: AB_2571739 |
| Alexa Fluor 488- phalloidin antibody | Thermo Fisher | Cat# A12379 RRID: AB_2315147 |
| Rhodamine- phalloidin antibody | Thermo Fisher | Cat# R415 RRID: AB_2572408 |
| Mouse anti-β-actin | Santa Cruz | Cat# sc-47778 RRID: AB_626632 |
| Bacterial and Virus Strains | ||
| pReciever-Lv215 | Genecopoeia | N/A |
| pReciever-Lv215-Ptpn21 | Generated in this study | N/A |
| pReciever-Lv215- Ptpn21Δ1-307 | Generated in this study | N/A |
| pReciever-Lv215- Ptpn21C1110S | Generated in this study | N/A |
| pJG-IRES-GFP | Emory Integrated Genomics Core |
N/A |
| pJG-IRES-GFP-Septin1 | Generated in this study | N/A |
| pJG-IRES-GFP-Septin1Y246F | Generated in this study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| IMDM culture medium | Thermo Fisher | Cat# 12440053 |
| StemSpan Serum-Free Expansion Medium (SFEM) | STEMCELL Technologies |
Cat # 09600 |
| Fetal bovine serum | Fisher Scientific | Cat# SH3008803IH |
| Recombinant Murine SCF | Pepro Technology | Cat# 250-03 |
| Recombinant Murine Flt3-Ligand | Pepro Technology | Cat# 250-31L |
| Recombinant Murine TPO | Pepro Technology | Cat# 315-14 |
| Recombinant Murine IL-3 | Pepro Technology | Cat# 213-13 |
| Recombinant Murine IL-6 | Pepro Technology | Cat# 216-16 |
| Recombinant Murine SDF-1α (CXCL12) | Pepro Technology | Cat# 250-20A |
| 5-Fluorouracil (5-FU) | Sigma | Cat# F6627 |
| G-CSF(Filgrastim) | Kirin-Amgen Inc. | N/A |
| Power SYBR Green PCR Master Mix | Thermo Fisher | Cat# 4367659 |
| Polybrene (Hexadimethrine bromide) | Sigma | Cat# 107689-10G |
| Target Retrieval Solution | Agilent | Cat# S169984-2 |
| Critical Commercial Assays | ||
| Quick-RNA Miniprep Kit | Zymo | Cat# R1050 |
| SuperScript First-Strand Synthesis System for RT-PCR | Invitrogen | Cat# 11904018 |
| BD Cytofix/Cytoperm™ Fixation/Permeabilization Solution Kit |
BD Biosciences | Cat# BDB554714 |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | Cat# 130-090-858 |
| Lenti-Pac™ HIV Expression Packaging Kit | Genecopoeia | Cat# LT001 |
| Experimental Models: Cell Lines | ||
| HEK293T | Cheng-Kui Qu Lab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ptpn21−/− mice | Generated in this study | N/A |
| Oligonucleotides used for qRT-PCR | ||
| Forward primer for mouse Ptpn21 cDNA: 5’-TCTTTGGAGACTCGGTTGCT -3’ | Synthesized in this study | N/A |
| Reverse primer for mouse Ptpn21 cDNA: 5’-CAGTGGCATCTTCTCCCTTC -3’ | Synthesized in this study | N/A |
| Forward primer for mouse Selplg-1 cDNA: 5’-GAAAGGGCTGATTGTGACCCC -3’ | Synthesized in this study | N/A |
| Reverse primer for mouse Selplg-1 cDNA: 5’-AGTAGTTCCGCACTGGGTACA -3’ | Synthesized in this study | N/A |
| Forward primer for mouse Esl-1 cDNA: 5’-CAAGATGACGGCCATCA -3’ | Synthesized in this study | N/A |
| Reverse primer for mouse Esl-1 cDNA: 5’-TTCCCAAGACGAATGCTGC -3’ | Synthesized in this study | N/A |
| Forward primer for mouse β-actin cDNA: 5’-AGAGGGAAATCGTGCGTGAC -3’ | Synthesized in this study | N/A |
| Reverse primer for mouse β-actin cDNA: 5’-CAATAGTGATGACCTGGCCGT -3’ | Synthesized in this study | N/A |
| Software and Algorithms | ||
| Flowjo v10.1 | https://www.flowjo.com/ | N/A |
| ImageJ v1.51 s | https://imagej.nih.gov/ij/ | N/A |
| Prism version 6 | Prism Graphpad | N/A |
5-Fluorouracil (5-FU) treatment
5-FU was administered intraperitoneally to Ptpn21−/− and Ptpn21+/+ mice (250 mg/kg body weight). Periphery blood (PB) of individual mice was separately collected into heprin-coated capillary tubes to measure white blood cell (WBC) counts 0, 4, 8, 12, and 16 days after 5-FU administration. For the survival assay, 5-FU was administered to mice intraperitoneally at a dose of 150 mg/kg body weight, once a week for 2 weeks, and the survival of the mice was monitored.
Colony-forming unit assay
Freshly harvested BM cells (2×104 cells/mL) from each mouse were plated in triplicates in 0.9% methylcellulose IMDM medium containing 15% fetal bovine serum, glutamine (10−4 M), β-mercaptoethanol (3.3×10−5 M), SCF (50 ng/mL), IL-3 (20 ng/mL), IL-6 (20 ng/mL), and erythropoietin (EPO, 3 Units/mL). After 10 days of incubation at 37 °C in 5% CO2, hematopoietic colonies (CFU-GEMM, CFU-GM, and BFU-E) were counted under an inverted microscope. The mean value of the triplicates from each mouse was used for final statistical analyses.
Flow cytometry analysis and cell sorting
The pool size, cell cycle status, and apoptosis of HSCs and LSK cells were analyzed by multiparameter fluorescent activated cell sorting (FACS) analyses, as previously described (Xu et al., 2011; Yu et al., 2013). For HSC staining, BM cells were harvested, washed, and incubated for 30 min at 4°C in phosphate buffered saline (PBS) wi th 2% fetal bovine serum (FBS) containing the following antibodies: biotin-lineage panel antibody (RRID: AB_11124348), Sca-1-PE-Cy7 (RRID: AB_647253), c-Kit-APC-Fluor 780 (RRID: AB_1272177), CD48-FITC (RRID: AB_465077), and CD150- Alexa Fluor 647 (RRID: AB_2239178). After 30 minutes at 4°C, cells were further stained with streptavidin-PE. HSCs were defined as Lin−Sca-1+c-Kit+CD150+CD48−. For hematopoietic progenitor staining, antibodies used were biotin-lineage panel antibody (RRID: AB_11124348), Sca-1-PE-Cy7 (RRID: AB_647253), c-Kit-APC-Fluor 780 (RRID: AB_1272177), together with CD34-Alexa Fluor 647 (RRID: AB_1645200) and CD16/32-FITC (RRID: AB_394659), and CD127-PE-Cy5 (RRID: AB_468793), followed by streptavidin-PE. Common myeloid progenitors (CMPs) were defined as Lin−c-Kit+Sca-1−CD16/32medCD34+, common lymphoid progenitors (CLPs) as Lin−c-Kit lowSca-1lowCD127+, granulocyte-monocyte progenitors (GMPs) as Lin−c-Kit+Sca-1−CD16/32high CD34+, and megakaryocyte-erythroid progenitors (MEPs) as Lin−c-Kit+Sca-1−CD16/32med/lowCD34−.
For cell cycle analyses, BM cells freshly harvested from femurs and tibias were stained with antibodies labeled with various fluorochromes, fixed and permeabilized using a Cytofix/Cytoperm kit, stained with Ki-67-Alexa Fluor 488 antibody (RRID: AB_2566800), and further incubated with Hoechest 33342 (20 μg/mL). For apoptosis analyses, fresh BM cells were stained for HSCs, and then incubated with Annexin V-FITC (RRID: AB_2629519) (2.5 μg/mL) and 7-amino-actinomycin D (5 μg/mL). Data were collected on BD LSR II Flow Cytometer and analyzed with FlowJo. For cell sorting, BM cells were first lineage-depleted using a lineage depletion kit. Cells were then stained with fluorochrome-labeled antibodies. Sorting of specific cell populations was conducted using BD FACSAria. Cell gating strategies used in FACS analyses and cell sorting are shown in Figure S2I–2K.
In vivo homing assay
The short-term in vivo homing assay was performed as described before (Foudi et al., 2006; Hoggatt et al., 2009). In brief, BM cells (2×107) freshly isolated from Ptpn21−/− and Ptpn21+/+ mice (CD45.2+) were transplanted into lethally-irradiated BoyJ mice (CD45.1+). The numbers of CD45.2+ LSK and CD45.2+ LK cells injected per mouse (N) were determined by FACS. Sixteen hours later, the total numbers of BM cells (B) recovered from the recipients were determined. BM cells were examined for the frequencies (A) of homed donor LSK and LK cells (CD45.2+) by FACS. The percentage of homing = (AxB)/N (%).
Competitive repopulation assay
BM cells (2×106) freshly harvested from Ptpn21−/− or Ptpn21+/+ mice (CD45.2+) were mixed with BM cells (2×106) isolated from BoyJ (CD45.1+) mice and transplanted into lethally irradiated (1,100 rad) BoyJ mice through tail vein injection. In addition, purified HSCs (LSKCD150+CD48−) (200) sorted from Ptpn21−/− or Ptpn21+/+ mice (CD45.2+) were mixed with BM cells (4×105) isolated from BoyJ mice and transplanted into lethally irradiated BoyJ mice as above. Test donor cell reconstitution (CD45.2+) in the whole cell population was determined 1, 2, 3, and 4 months after primary transplantation by FACS analyses of PB and BM cells as we previously described (Xu et al., 2011; Yu et al., 2013). BM cells harvested from recipients 4 months after transplantation were transplanted (1×106 cells/mouse) into lethally irradiated secondary BoyJ recipients. Test cell reconstitution in PB and BM cells was determined by FACS 1, 2, 3, and 4 months after secondary transplantation as above. Long-term reconstitution of BM LSK cells and HSCs in primary and secondary recipients was also assessed as above.
G-CSF mobilization assay
Ptpn21−/− and Ptpn21+/+ mice were injected intraperitoneally with G-CSF (50 -g/kg body weight/day) or PBS for 5 consecutive days. WBC counts were determined 0, 2, 4 days after GCSF or PBS administration. One hundred microliters of PB were subjected to CFU assays after the last injection of G-CSF or PBS. In addition, mice were euthanized 2 hours after the last injection. PB and spleens were collected and analyzed for LSK cells and HSCs by FACS analyses.
Cell adhesion assay
Briefly, non-tissue culture plates were coated with fibronectin (20 μg/mL) or BSA at 4°C overnight. The plates were subsequently blocked with 2% BSA at room temperature for 30 min. A total of 1×105 sorted Ptpn21−/− or Ptpn21+/+ LSK cells in IMDM media containing 10% FBS were allowed to adhere to the coated plates in a humidified 5% CO2 incubator at 37°C for 1 hour. The adherent cells were collected by rinsing the plates with PBS and then counted.
Transmigration assay
Transmigration assays were carried out with 3-μm-pore and 5-μm-pore transwells. Briefly, LSK cells sorted from Ptpn21−/− and Ptpn21+/+ individual mice were incubated in IMDM media containing 20% FBS, 50 ng/mL SCF, 50 ng/mL Flt3L, 20 ng/mL IL-3, and 20 ng/mL IL-6 at 37°C in a humidified 5% CO2 incubator for 18 hours. LSK cells were then washed with IMDM media twice, resuspended in serum-free IMDM media at 1×105/mL. One hundred microliters of cell suspension was loaded to the upper chamber, and 600 μL of IMDM media with or without stromal-derived factor (SDF-1, 100 ng/mL) was added to the lower chamber of the transwell plates. Cells were allowed to migrate across the membrane at 37°C in 5 % CO2, and cells that migrated into the lower chambers were collected and counted 4 hours later.
In vitro 3D HSC motility assay
Two layers of collagen hydrogels were prepared for this assay. To generate a SDF-1 gradient in the hydrogels, the base layer of collagen hydrogel containing SDF-1 was first made by pipetting 50 μL neutralized 1 mg/mL collagen solution containing 100 ng/mL SDF-1 into a 96-well plate and incubating at 37 °C for 20 min. The top layer of collagen hydrogel was then made by pipetting 50 μL neutralized collagen solution (0.5, 1, or 5 mg/ml) onto the base layer and incubating at 37 °C for another 20 min (the elasticity and pore sizes of the upper gels decreased as the concentrations of collagen increased). HSCs sorted from Ptpn21−/− and Ptpn21+/+ mice were stained with 2 μM calcein for 30 min. The cells (2000) suspended in 100 μL PBS were then seeded on the top of collagen hydrogels and incubated in a 37 °C 5% CO2 incubator. Five hours later, 3D stack images of HSCs migrating into the collagen gels were captured using a confocal microscopy (Zeiss LSM 700).
Immunofluorescence staining
HSCs, LSK and Lin− cells sorted from Ptpn21−/− and Ptpn21+/+ mice were fixed in 1% paraformaldehyde at room temperature for 15 min. Cells were then permeabilized with 0.2% Triton X-100 at room temperature for 30 min. To visualize F-actin, cells were stained for 30 min with Alexa Fluor 488 phalloidin. For Septin1 immunostaining, cells were stained with Septin1 primary antibody followed by Alexa Fluor 568-conjugated anti-mouse secondary antibody. Cells were mounted on glass slides with Fluro-gel mounting media and covered with glass coverslips. Images were taken with a confocal microscope using a 63×1.4NA oil objective. For immunofluorescence staining of BM sections, paraffin sections of mouse femurs were deparaffinized and rehydrated following standard protocols. Heat-induced epitope retrieval was applied using Target Retrieval Solution (citrate buffer, pH 6.0) at 98°C for 25 min. Sections were blocked/p ermeabilized with Dulbecco PBS containing 0.2% Triton X-100 and 10% normal goat serum, and then stained overnight at 4°C with primary antibodies. After washing, secondary staining was performed at room temperature for 1 hour (antibodies are listed in KEY RESOURCES TABLE). DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) was used as a nuclear stain. Cell images were captured on a confocal microscope using Confocal Software v2.61 (Leica).
Small GTPase assays
RhoA/Rac1 GTPase activation was determined by the GST-Rhotekin-RBD (for RhoA) or GSTPAK-CRIB (for Rac1) pull-down assays. Cells were lysed on ice for 5 min in RIPA buffer (50 mM Tris-HCl pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 10 μg/mL leupeptin, 10 μg/mL aprotinin, and 1 mM PMSF). Supernatants were collected quickly after centrifugation. An equal volume of whole cell lysates (500 μg) were incubated with 60 -g of GSTRhotekin-RBD or GST-PKA-CRIB glutathione-agarose beads at 4 °C for 3 hours. Proteins bound to glutathione-agarose beads were then resolved by SDS-PAGE and immunoblotted with anti-RhoA or anti-Rac1 antibodies. Total cellular RhoA and Rac1 levels in the whole cell lysates were also determined by immunoblotting.
Atomic force microscopy (AFM) measurement of cellular stiffness
The AFM used in this study was MFP-3D (Asylum Research, Santa Barbara, CA) with a combined Nikon Ti inverted optical microscope (Nikon, Melville, NY), which was used to optically align the probe to the cells. AFM calibration and methodology to measure single cell stiffness were performed as previously described (Fay et al., 2016). Briefly, freshly isolated cells were attached to 0.01% poly-L-lysine-treated glass AFM fluorodishes. To improve cell stability during AFM measurement, a monolayer of poly-L-lysine (MW 300 kDa) was applied to gently attach cells to the glass substrate. Prior to cell measurements, the cantilever was calibrated on the glass bottom of fluorodishes using the thermal vibration method with the resultant thermal spectrum fitted with Lorentzian function to determine the spring constant. Beaded silicon nitride cantilevers were used to indent the center of the cells. Sufficient force was applied to achieve at least 5 μm deformation, which was close to the microfluidic compression. The force versus indentation curves from each measurement were analyzed using a Hertzian contact model to obtain the Young’s modulus of each cell.
Microfluidic assay
A microfluidic device was used to determine cell deformability (Fay et al., 2016; Rosenbluth et al., 2008). For the microfluidic device fabrication, a soft lithography mold was created using SU-8 (Microchem) following the manufacturer’s protocol. Polydixmethylsilane (PDMS; Dow Corning) was poured over the molds and allowed to cure at 60 °C overnight. The PDMS was then bonded to coverslips using plasma cleaner (Harrick Plasma). Devices were then blocked with 10% BSA in PBS for a minimum of 1 hour to prevent non-specific binding. For the microfluidic experiments, freshly isolated cells were diluted to 1×106 cells/mL in PBS and then loaded into the inlet of the microfluidic device. Using a syringe pump (Harvard Apparatus), cells were drawn through the microfluidic device consisting of capillaries (5.9 ± 0.8 μm wide) at 0.5 μL/min. Cells in transit were recorded using a bright field microscopy (Nikon TE2000-U) and a 20 × 0.45A air objective. Transit time was calculated manually.
Lentiviral-mediated gene transduction
To make lentiviral vectors expressing Ptpn21C1110S, Ptpn21Δ1–307, or wild-type (WT) Ptpn21, Ptpn21 mutant cDNAs were generated based on WT cDNA sequence (NM_001146199.1) and subcloned into a lentiviral expression vector pReciever-Lv215 (with the eGFP reporter). Similarly, to make lentiviral vectors expressing Septin1Y246F or WT Septin1, Septin1Y246F mutant cDNA was generated based on WT cDNA sequence (NM_017461.2) and subcloned into a lentiviral vector with the eGFP reporter (pJG-IRES-GFP). Correct cloning of the constructs was confirmed by DNA sequencing. Viral particles were produced in HEK293T cells using the Lenti-Pac™ Lentiviral Packaging System. For lentiviral-mediated cDNA transduction, freshly purified LSK or Lin− cells were resuspended in 2×105/mL of StemSpan serum-free expansion medium, supplemented with 50 ng/mL SCF, 50 ng/mL Flt3L, 20 ng/mL IL-3, 20 ng/mL IL-6, and 100 ng/mL TPO. Lentiviral particles were added to cell suspension with polybrene (8 μg/mL) and spinoculated at 700 g at 22°C for 90 min. Cells were collected for subsequent assays 48 hours after transduction (transduction efficiency was approximately 80% for all groups). In addition, Ptpn21-/- Lin− cells transduced with lentiviral vectors expressing Septin1Y246F or WT Septin1 were transplanted into lethally-irradiated BoyJ mice (2×105 cells/mouse) as described above.
Phosphoproteomics analyses
BM Lin− cells isolated from mice were processed and analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) on an Orbitrap Fusion mass spectrometer (ThermoFisher Scientific, San Jose, CA) at the Emory Integrated Proteomics Core. Briefly, cell pellets were lysed and digested with HALT protease in the presence of a phosphatase inhibitor cocktail (Pierce). The resulting peptide mixtures were separated on a self-packed C18 (1.9 μm) fused silica column (25 cm × 75 μM internal diameter; New Objective, Woburn, MA) by a Dionex Ultimate 3000 RSLCNano and monitored on a fusion mass spectrometer. Elution was performed over a 120 min gradient at a rate of 300 nl/min. The mass spectrometer cycle was programmed to collect at the top speed for 3 sec cycles. The MS scans (400–1500 m/z range, 400,000 AGC, 50 ms maximum ion time) were collected at a resolution of 120,000 at m/z 200 in profile mode and the HCD MS/MS spectra were detected in the ion trap. Dynamic exclusion was set to exclude previous sequenced precursor ions for 20 sec within a 10 ppm window. Precursor ions with +1, and +8 or higher charge states were excluded from sequencing. All raw data files were processed using Proteome Discoverer 2.0 against mouse Uniprot database. Searching parameters included fully tryptic restriction and a parent ion mass tolerance (± 20 ppm). Methionine oxidation (+15.99492 Da), asparagine and glutamine deamidation (+0.98402 Da) and protein N-terminal acetylation (+42.03670) were variable modifications (up to 3 allowed per peptide); cysteine was assigned a fixed carbamidomethyl modification (+57.021465 Da). Percolator was used to filter the peptide spectrum matches to a false discovery rate of 1%.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were conducted with GraphPad Prism (version 6). Statistical significance of the differences between two groups was determined using unpaired Student’s t test (two-sided), unless otherwise specified in figure legends. Normal distribution of samples was assumed on the basis of published studies with analyses similar to ours. Comparison of overall survival rates was performed using the Mantel_Cox log-rank test. In all studies, data are shown as mean ± SD, and the exact numbers of biological replicates or animals per group (denoted by “n”) for each dataset are provided in figure legends. Sample sizes were chosen on the basis of published work in which similar phenotypical characterization were reported. No exclusion of any data or subjects were applied. Statistically significant differences are indicated on figures. *p < 0.05; **p < 0.01; ***p < 0.001. N.S., not significant.
DATA AND SOFTWARE AVAILABILITY
The phosphoproteomics data of the study have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) with the accession number PXD011381.
Supplementary Material
Related to Figure 7. Phosphoproteomics data from Ptpn21 knockout and wild-type control Lin− cells (see a separate Excel file).
Highlights.
Normal HSCs are stiffer and less deformable than mature blood cells.
Deletion of Ptpn21 results in HSC egress due to impaired retention in the niche.
Ptpn21 loss decreases cell stiffness and increases mobility/deformability in HSCs.
Ptpn21 modulates cellular mechanics by dephosphorylation of Septin1 (Tyr246).
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health grants HL130995 and DK092722 (to C.K.Q.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Bouabe H, and Okkenhaug K (2013). Gene targeting in mice: a review. Methods Mol Biol 1064, 315–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, et al. (2014). Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nature medicine 20, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi LM, and Link DC (2015). The hematopoietic stem cell niche in homeostasis and disease. Blood 126, 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone L, Carlucci A, Affaitati A, Livigni A, DeCristofaro T, Garbi C, Varrone S, Ullrich A, Gottesman ME, Avvedimento EV, et al. (2004). Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novel positive regulator of src signaling. Mol Cell Biol 24, 4613–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlucci A, Gedressi C, Lignitto L, Nezi L, Villa-Moruzzi E, Avvedimento EV, Gottesman M, Garbi C, and Feliciello A (2008). Protein-tyrosine phosphatase PTPD1 regulates focal adhesion kinase autophosphorylation and cell migration. J Biol Chem 283, 10919–10929. [DOI] [PubMed] [Google Scholar]
- Carlucci A, Porpora M, Garbi C, Galgani M, Santoriello M, Mascolo M, di Lorenzo D, Altieri V, Quarto M, Terracciano L, et al. (2010). PTPD1 supports receptor stability and mitogenic signaling in bladder cancer cells. J Biol Chem 285, 39260–39270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, and Scadden DT (2000). Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287, 1804–1808. [DOI] [PubMed] [Google Scholar]
- Cheshier SH, Morrison SJ, Liao X, and Weissman IL (1999). In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci U S A 96, 3120–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury F, Na S, Li D, Poh YC, Tanaka TS, Wang F, and Wang N (2010). Material properties of the cell dictate stress-induced spreading and differentiation in embryonic stem cells. Nat Mater 9, 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane GM, Jeffery E, and Morrison SJ (2017). Adult haematopoietic stem cell niches. Nat Rev Immunol 17, 573–590. [DOI] [PubMed] [Google Scholar]
- DeMay BS, Meseroll RA, Occhipinti P, and Gladfelter AS (2009). Regulation of distinct septin rings in a single cell by Elm1p and Gin4p kinases. Mol Biol Cell 20, 2311–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, and Discher DE (2006). Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689. [DOI] [PubMed] [Google Scholar]
- Fay ME, Myers DR, Kumar A, Turbyfield CT, Byler R, Crawford K, Mannino RG, Laohapant A, Tyburski EA, Sakurai Y, et al. (2016). Cellular softening mediates leukocyte demargination and trafficking, thereby increasing clinical blood counts. Proc Natl Acad Sci U S A 113, 1987–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher DA, and Mullins RD (2010). Cell mechanics and the cytoskeleton. Nature 463, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foudi A, Jarrier P, Zhang Y, Wittner M, Geay JF, Lecluse Y, Nagasawa T, Vainchenker W, and Louache F (2006). Reduced retention of radioprotective hematopoietic cells within the bone marrow microenvironment in CXCR4-/- chimeric mice. Blood 107, 2243–2251. [DOI] [PubMed] [Google Scholar]
- Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, Saksena G, Lawrence MS, Qian ZR, Nishihara R, et al. (2014). RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet 46, 1264–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilden JK, Peck S, Chen YC, and Krummel MF (2012). The septin cytoskeleton facilitates membrane retraction during motility and blebbing. J Cell Biol 196, 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Cruz RD, Fonseca VC, and Darling EM (2012). Cellular mechanical properties reflect the differentiation potential of adipose-derived mesenchymal stem cells. Proc Natl Acad Sci U S A 109, E1523–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Filippi MD, Cancelas JA, Siefring JE, Williams EP, Jasti AC, Harris CE, Lee AW, Prabhakar R, Atkinson SJ, et al. (2003). Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science 302, 445–449. [DOI] [PubMed] [Google Scholar]
- Hernandez-Rodriguez Y, and Momany M (2012). Posttranslational modifications and assembly of septin heteropolymers and higher-order structures. Curr Opin Microbiol 15, 660–668. [DOI] [PubMed] [Google Scholar]
- Hoggatt J, Singh P, Sampath J, and Pelus LM (2009). Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood 113, 5444–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst J, Watson S, Lord MS, Eamegdool SS, Bax DV, Nivison-Smith LB, Kondyurin A, Ma L, Oberhauser AF, Weiss AS, et al. (2010). Substrate elasticity provides mechanical signals for the expansion of hemopoietic stem and progenitor cells. Nat Biotechnol 28, 1123–1128. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, and Morrison SJ (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. [DOI] [PubMed] [Google Scholar]
- Korff S, Woerner SM, Yuan YP, Bork P, von Knebel Doeberitz M, and Gebert J (2008). Frameshift mutations in coding repeats of protein tyrosine phosphatase genes in colorectal tumors with microsatellite instability. BMC Cancer 8, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. (2013). Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman MA (1970). Cellular deformability during maturation of the myeloblast. Possible role in marrow egress. N Engl J Med 283, 943–948. [DOI] [PubMed] [Google Scholar]
- Mavrakis M, Azou-Gros Y, Tsai FC, Alvarado J, Bertin A, Iv F, Kress A, Brasselet S, Koenderink GH, and Lecuit T (2014). Septins promote F-actin ring formation by crosslinking actin filaments into curved bundles. Nat Cell Biol 16, 322–334. [DOI] [PubMed] [Google Scholar]
- McBeath R, Pirone DM, Nelson CM, Bhadriraju K, and Chen CS (2004). Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell 6, 483–495. [DOI] [PubMed] [Google Scholar]
- Mendelson A, and Frenette PS (2014). Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nature medicine 20, 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, and Frenette PS (2010). Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller NP, Moller KB, Lammers R, Kharitonenkov A, Sures I, and Ullrich A (1994). Src kinase associates with a member of a distinct subfamily of protein-tyrosine phosphatases containing an ezrin-like domain. Proc Natl Acad Sci U S A 91, 7477–7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostowy S, and Cossart P (2012). Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol 13, 183–194. [DOI] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Takizawa H, and Suda T (2014). The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development 141, 4656–4666. [DOI] [PubMed] [Google Scholar]
- Nayak RC, Chang KH, Vaitinadin NS, and Cancelas JA (2013). Rho GTPases control specific cytoskeleton-dependent functions of hematopoietic stem cells. Immunol Rev 256, 255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes PW, Patel DC, Morin NA, Zitterbart DP, Fabry B, Reichner JS, and Tang JX (2009). Neutrophil morphology and migration are affected by substrate elasticity. Blood 114, 1387–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orford KW, and Scadden DT (2008). Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet 9, 115–128. [DOI] [PubMed] [Google Scholar]
- Parsons JT, Horwitz AR, and Schwartz MA (2010). Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Warr MR, and Passegue E (2011). Cell cycle regulation in hematopoietic stem cells. J Cell Biol 195, 709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potocnik AJ, Brakebusch C, and Fassler R (2000). Fetal and adult hematopoietic stem cells require beta1 integrin function for colonizing fetal liver, spleen, and bone marrow. Immunity 12, 653–663. [DOI] [PubMed] [Google Scholar]
- Raab M, Swift J, Dingal PC, Shah P, Shin JW, and Discher DE (2012). Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J Cell Biol 199, 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth MJ, Lam WA, and Fletcher DA (2008). Analyzing cell mechanics in hematologic diseases with microfluidic biophysical flow cytometry. Lab Chip 8, 1062–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden DT (2014). Nice neighborhood: emerging concepts of the stem cell niche. Cell 157, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, et al. (2012). Recurrent R-spondin fusions in colon cancer. Nature 488, 660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JW, Buxboim A, Spinler KR, Swift J, Christian DA, Hunter CA, Leon C, Gachet C, Dingal PC, Ivanovska IL, et al. (2014). Contractile forces sustain and polarize hematopoiesis from stem and progenitor cells. Cell Stem Cell 14, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JW, Spinler KR, Swift J, Chasis JA, Mohandas N, and Discher DE (2013). Lamins regulate cell trafficking and lineage maturation of adult human hematopoietic cells. Proc Natl Acad Sci U S A 110, 18892–18897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha I, Wang YM, Philp R, Li CR, Yap WH, and Wang Y (2007). Cyclin-dependent kinases control septin phosphorylation in Candida albicans hyphal development. Dev Cell 13, 421–432. [DOI] [PubMed] [Google Scholar]
- Tooley AJ, Gilden J, Jacobelli J, Beemiller P, Trimble WS, Kinoshita M, and Krummel MF (2009). Amoeboid T lymphocytes require the septin cytoskeleton for cortical integrity and persistent motility. Nat Cell Biol 11, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DE, Wagers AJ, Gulati AP, Johnson FL, and Weissman IL (2001). Physiological migration of hematopoietic stem and progenitor cells. Science 294, 1933–1936. [DOI] [PubMed] [Google Scholar]
- Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL, and Qu CK (2011). Nonlineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med 208, 1977–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang L, Geiger H, Cancelas JA, Mo J, and Zheng Y (2007). Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci U S A 104, 5091–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WM, Liu X, Shen J, Jovanovic O, Pohl EE, Gerson SL, Finkel T, Broxmeyer HE, and Qu CK (2013). Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 12, 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, Ahamed J, and Li L (2014). Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nature medicine 20, 1321–1326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Figure 7. Phosphoproteomics data from Ptpn21 knockout and wild-type control Lin− cells (see a separate Excel file).
Data Availability Statement
The phosphoproteomics data of the study have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) with the accession number PXD011381.