

SUMMARY
In cells, dedicated AAA+ ATPases deposit hexameric, ring-shaped helicases onto DNA to initiate chromosomal replication. To better understand the mechanisms by which helicase loading can occur, we used cryo-EM to determine sub-4 Å-resolution structures of the E. coli DnaB•DnaC helicase•loader complex with nucleotide in pre- and post-DNA engagement states. In the absence of DNA, six DnaC protomers latch onto and crack open a DnaB hexamer using an extended N-terminal domain, stabilizing this conformation through nucleotide-dependent ATPase interactions. Upon binding DNA, DnaC hydrolyzes ATP, allowing DnaB to isomerize into a topologically-closed, pre-translocation state competent to bind primase. Our data show how DnaC opens the DnaB ring and represses the helicase prior to DNA binding, and how DnaC ATPase activity is reciprocally regulated by DnaB and DNA. Comparative analyses reveal how the helicase loading mechanism of DnaC parallels and diverges from homologous AAA+ systems involved in DNA replication and transposition.
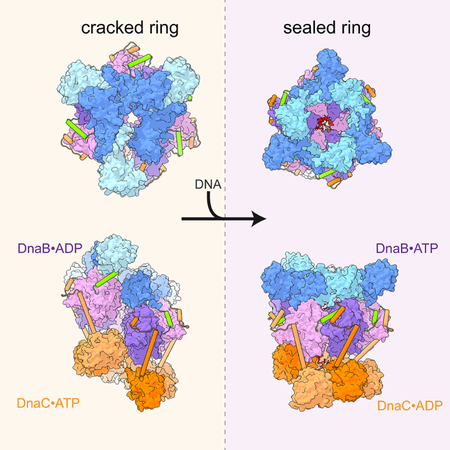
Graphical Abstract
INTRODUCTION
DNA replication is central to the proliferation of all living organisms. In cells, replication is carried out by a large multi-subunit complex termed the replisome, which coordinates disparate molecular activities to increase the fidelity and speed of strand synthesis (MacNeill, 2011; Yao and O’Donnell, 2016). Replisomes do not form spontaneously but are assembled in a tightly regulated manner in accord with cell cycle cues (Katayama et al., 2010; Soultanas, 2012). A key step in replisome formation is the loading of a ring-shaped, hexameric replicative helicase onto origin DNA that promotes the unwinding of parental template DNA strands.
The replicative helicase used by bacteria is a protein known as DnaB (DnaC in Gram-positive organisms) (Arai and Kornberg, 1981; LeBowitz and McMacken, 1986; Wickner and Hurwitz, 1975). To gain access to origin DNA, two copies of DnaB must first be loaded onto a single-stranded DNA (ssDNA) bubble produced during the initiation of replication. Because a DNA bubble is topologically inaccessible to the binding of a closed-ring helicase, many DnaB proteins require the action of a dedicated deposition factor, called DnaC (DnaI in Gram-positives), to catalyze this loading event (Funnell et al., 1987; Kobori and Kornberg, 1982a, 1982b; Soultanas, 2002).
The DnaBC system has long served as a textbook model for understanding how ring-shaped hexameric helicases can be deposited onto DNA to support replisome formation (e.g., see Craig et al., 2014; Nelson and Cox, 2012). However, despite decades of research, many unanswered questions still persist regarding how DnaB loading physically takes place. DnaC is a member of the ATPases Associated with various cellular Activities (AAA+) superfamily, a group of proteins that control diverse cellular processes ranging from DNA replication and repair to gene expression, nucleosome positioning, proteolytic breakdown, ribosome biogenesis, and vesicle trafficking (Erzberger and Berger, 2006; Neuwald et al., 1999). The AAA+ fold of DnaC, which belongs to a specific ‘initiator’ clade of AAA+ enzymes that includes replication initiator proteins (bacterial DnaA and archaeal/eukaryotic Cdc6/Orc1) and the DNA transposition co-factor IstB (Iyer et al., 2004; Koonin, 1992), is augmented by an N-terminal extension that binds to DnaB (Chodavarapu et al., 2016; Ioannou et al., 2006; Loscha et al., 2009; Ludlam et al., 2001; Tsai et al., 2009) (Figure 1A). Although DnaC has been reported to form a cracked-ring homo-oligomer that physically opens DnaB rings to support loading (Arias-Palomo et al., 2013), the physical basis by which helicase opening and DNA engagement occurs is not known. The role of ATP in DnaC function likewise is unresolved; although ATP binding to DnaC allows the loader to self-assemble and block both ATP turnover and DNA unwinding by DnaB (Mott et al., 2008; Wahle et al., 1989a), whether DnaC itself is a functional ATPase (and the purpose of such an activity) has been debated (Davey et al., 2002; Koonin, 1992; Makowska-Grzyska and Kaguni, 2010; Roychowdhury et al., 2009).
Figure 1. Structure of DNA-free E. Coli DnaBC. See also Figure S1 and S2.

(A) Primary structure of E. coli DnaC and DnaB. Color scheme is maintained throughout all figures.
(B) Dilated conformation of DnaB (PDB 2R6A (Bailey et al., 2007)).
(C) Constricted conformation of DnaB (PDB 4NMN (Strycharska et al., 2013)).
(D) Two orthogonal views of E. coli DnaBC. Insets depict the unsharpened EM density to highlight the gap in the structure. In the upper panel, an entry pathway for ssDNA has been highlighted.
(E) View of E. coli DnaBC highlighting domain structure and organization.
(F) Detailed view of the boxed area in (E) showing the interaction network between DnaB and DnaC. Density contoured at a threshold value of 0.05.
Numerous questions reciprocally persist around how DnaB responds to and acts upon ssDNA, DnaC, and ATP during loading. In the context of a DnaB hexamer, the N-terminal domain (NTD) of the helicase forms homodimers that can adopt two markedly different conformations, termed ‘dilated’ and ‘constricted,’ in response to substrate and helicase loader binding (Arias-Palomo et al., 2013; Strycharska et al., 2013) (Figure 1A–C). In the dilated form, two helical hairpins that dimerize the NTDs are offset ~120˚ apart from each other and run tangential to the helicase pore; contacts formed between the globular regions of the NTD complete a circuit that circumscribes a large (>30 Å) triangular aperture that sits atop the RecA domains. In the constricted state, the NTD dimers swivel inward, repositioning the globular regions of three NTDs close to central axis of the hexamer and creating a triskelion-type arrangement with a substantially narrower (15 Å) hole. DnaC stabilizes a constricted form of the NTD collar (Arias-Palomo et al., 2013), but how the loader accomplishes this task is unknown. Although it has been suggested that transitions in the DnaB collar may permit the release of a domain-swapped linker element that is important for topologically sealing the helicase ring around ssDNA (Arias-Palomo et al., 2013; Chodavarapu et al., 2016), this event has not been confirmed experimentally. How DnaB senses ssDNA, and whether this interaction alters either the functional state of the helicase or the mechanism by which it engages DnaC is similarly unknown.
In the current study, we have determined near-atomic resolution structures of the 480 kDa DnaBC complex, together with the ATP-mimetic, ADP•BeF3, in the absence and presence of ssDNA by single-particle cryo-EM. The structures reveal how DnaC latches onto and opens the DnaB ring prior to DNA binding, and demonstrate that the loader maintains this grip following ssDNA engagement. We confirm that DnaC is indeed a functional ATPase; inspection of the EM models explains why ATP turnover by the loader requires the presence of both DnaB and ssDNA, and further reveals that ATP hydrolysis weakens but does not abrogate DnaC’s hold on the helicase. These findings explain how DnaC in turn regulates the activity and conformational status of DnaB, and unexpectedly show that ssDNA remodels DnaB into a structural state proposed to reflect to a translocation intermediate, even though the loader remains bound to the helicase. Collectively, our work provides the first high-resolution view of how replicative helicase loading occurs in bacteria and explains how this mechanism both parallels and diverges from homologous hexameric helicase and DNA polymerase clamp loader systems.
RESULTS
Mechanism of DnaB recognition by DnaC
We first set out to determine the structure of a nucleotide-bound, DNA-free DnaBC complex (or ‘apo’ state) using high-resolution cryo-EM. E. coli DnaB and DnaC were overexpressed independently, purified, and mixed together before purifying over an analytical gel filtration column equilibrated in buffer containing ADP•BeF3 (Methods). Peak fractions were applied to open-hole grids coated with a carbon film (pretreated with poly-lysine to increase orientation variability), vitrified in liquid ethane, and imaged on a Titan Krios electron microscope. Following particle 2D and 3D classification and three-dimensional refinement with a soft-edged mask, the reconstruction converged at an overall resolution of 3.9 Å (Figure 1D–E, Figure S1, Table 1). The quality of the cryo-EM reconstruction readily permitted rebuilding of docked homology models for DnaB and the AAA+ region of DnaC, and additionally allowed for de novo modeling of the amino acid register for the DnaC N-terminus (Figure 1F).
Table 1.
Data collection, reconstruction, and model refinement statistics.
| apo DnaBC | DnaBC-dT36 | |
|---|---|---|
| Data collection and processing | ||
| Microscope | Titan Krios (U. Leeds) | Titan Krios (DLS) |
| Voltage (kV) | 300 | 300 |
| Electron dose (e/A2) | 61 | 58 |
| Dose rate (e/A2/fraction) | 1.53 | 1.45 |
| Detector | K2 Summit | K2 Summit |
| Defocus range (μm) | 1.2–2.7 | 1.2–2.7 |
| Calibrated pixel size (Å) | 1.07 | 1.05 |
| Micrographs | 3161 | 1858 |
| Total extracted particles (no.) | 551,641 | 473,888 |
| Final particle images (no.) | 104,913 | 229,097 |
| Symmetry imposed | C1 | C1 |
| Resolution (per 0.143 FSC) (Å) | 3.9 | 3.4 |
| Applied B-factor (Å2) | −158 | −125 |
| Refinement | ||
| RMS deviations | ||
| Bond lengths (Å) | 0.009 | 0.011 |
| Bond angles (°) | 1.208 | 1.288 |
| Ramachandran (%) | ||
| Outliers | 0.00 | 0.00 |
| Allowed | 12.89 | 9.49 |
| Favored | 87.11 | 90.51 |
| Rotamer outliers (%) | 0.49 | 0.54 |
| MolProbity score | 1.81 | 1.78 |
| Clashscore | 4.03 | 4.72 |
The global shape of the apo DnaBC reconstruction is similar to a low-resolution model (~25 Å) reported previously from negative-stain EM (Arias-Palomo et al., 2013). The higher-resolution maps confirm that the helicase-loader complex is dodecameric, with six subunits each of DnaB and DnaC. The two proteins assemble into a three-tier, right-handed spiral organized around a central channel coincident with the helical axis of the particle (Figure 1D–E). A clear crack is evident along one side of the particle that laterally breaches the interior channel of the complex to solvent. The dimensions and almost linear configuration of the gap, together with the flexibility found in the neighboring regions, indicates that the break in the ring can allow ssDNA passage into the central cavity. The lowest tier of the complex is composed of the AAA+ ATPase domains of DnaC. The middle tier comprises the RecA ATPase folds of six DnaB subunits, while the upper is formed by three pairs of DnaB N-terminal domain (NTD) dimers. As seen in other DnaB structures (Bailey et al., 2007; Bazin et al., 2015; Itsathitphaisarn et al., 2012; Liu et al., 2013; Lo et al., 2009; Strycharska et al., 2013; Wang et al., 2008), the NTD of each subunit is domain-swapped with the RecA region of an adjoining partner protomer through an a-helical connection element (the ‘linker helix’) (Figure 1E). The one exception to this domain-swapped configuration is the uppermost DnaB subunit of the spiral, in which the linker helix is disengaged from its lowermost partner at the point where the helicase ring is cracked.
The DnaC ATPase regions do not contact DnaB directly. Instead, the N-terminus of each loader subunit forms an α-helical extension that reaches up along the exterior side of a single RecA fold of DnaB to engage the helicase like the arm of a grappling hook (Figure 1E–F). At the tip of the DnaC N-terminal arm, the extension breaks into a small helix-loop-helix motif that contacts the DnaB linker helix; this interaction occurs with five of the six DnaC protomers but is absent for the lowermost subunit that resides at the crack in the complex. Significantly, the build for the DnaC NTD comports with prior genetic and biochemical data identifying helicase/loader contact points (Chodavarapu et al., 2016; Ludlam et al., 2001). For example, Arg10 and Trp32 of DnaC were identified as being important for binding to DnaB (Ludlam et al., 2001); these amino acids can be seen to contact Asp187, and Arg278, and Arg296 of the helicase. Similarly, hydrogen-deuterium exchange studies reported that residues 8–11 and 31–44 of DnaC were likely to interact with amino acids 295–304 and 431–435 of DnaB (Chodavarapu et al., 2016), a prediction both confirmed and expanded upon by the structure.
In accord with a prior, low-resolution EM model (Arias-Palomo et al., 2013), the act of DnaC binding shifts the DnaB NTDs into a constricted (cracked-ring) state. However, the higher resolution data obtained here afford new insights into this conformation that were not evident from previous studies. In particular, the N-terminal region of the loader alters how the linker helix of one subunit contacts its neighboring protomer, wedging the interaction apart (Figure S2A). The binding of DnaC NTD also causes the N-terminal domain dimers of DnaB pivot with respect to their partner RecA domains (Figure S2B), which themselves undergo an upward swiveling motion (Movie 1). These transitions indicate that DnaC loosens the grip of the DnaB linker helices, which help to maintain the DnaB ring in a topologically-closed state, and that loader binding also disrupts interactions between DnaB NTD dimers that would otherwise promote ring planarity. This combined action allows the helicase ring to detach at one point, allowing the motor domains of the helicase to adopt an extended-spiral configuration seen in other RecA-superfamily proteins (Story et al., 1992; Wang et al., 2008). Interestingly, this cracked conformation appears to be sustained by interactions between DnaB NTD dimers that are now tripartite (rather than bipartite) in nature, with the globular head of one NTD touching the head and helical hairpin of a second NTD and engaging the N-terminal helical hairpin of a third (chain C with chains B and A, respectively; chain E with chains D and C) (Figure S2C). Moreover, in two instances (chains I and K), the first α-helix of the DnaC NTD also appears to help stabilize the constricted configuration through its interactions with head region of the DnaB NTD (chains B and D) (Figure S2D). Taken together, these set of contacts, in combination with inter-AAA+ domain interactions in DnaC, would appear to preclude movement of the helicase subunits, thereby explaining how the loader maintains DnaB in an inactive state (Wahle et al., 1989a).
DnaB licenses, but does not activate, ATP turnover by DnaC
Both DnaB and DnaC bind ATP (Arai and Kornberg, 1981; Davey et al., 2002; Galletto et al., 2000; Reha-Krantz and Hurwitz, 1978; Wahle et al., 1989a). In the DnaBC assembly, five of the six nucleotide-binding sites of DnaC adopt a ‘canonical’ oligomeric configuration for AAA+ proteins (Erzberger and Berger, 2006; Ogura and Wilkinson, 2001) in which the Walker A and B signature sequence motifs of one subunit juxtapose with a positively-charged amino acid the ‘arginine finger’ from a neighboring protomer (Figure S3A). One DnaC subunit lacks this interaction due to the crack in the complex. Despite the moderate resolution of the reconstruction, inspection of the electron density around the nucleotide-binding sites for DnaC shows that the loader occupies an ATP-like (ADP•BeF3-bound) configuration, with the exception of the terminal-most subunit, which only engages ADP (Figure S3A and S3B). By contrast, the active sites of all six DnaB protomers are seen to coordinate ADP (Figure S3C and S3D). The ability of ADP•BeF3 to support both ATP- and ADP-like configurations has been seen before in structural studies of other ring-ATPases such as the E. coli Rho transcription termination factor and the phage T4 gp44/62 clamp loader (Kelch et al., 2011; Thomsen and Berger, 2009; Thomsen et al., 2016).
Given that an ATP-like state is observed for DnaC when bound to DnaB, we set out to resolve whether the loader is indeed a functional ATPase using a coupled spectrophotometric assay (Methods). We first incubated DnaC with a catalytically-inactive variant of DnaB and either included or omitted ssDNA (M13 plasmid). In agreement with prior work from the O’Donnell group (Davey et al., 2002), we detected a clear ATPase signature from DnaC, and only when both DnaB and ssDNA were present (Figure 2A). Inspection of a catalytically-inactive Walker-A mutant of DnaC showed only background levels of activity, confirming that DnaC is responsible for the observed turnover of nucleotide (Figure 2A). Assessment of initial rates at varying ATP concentrations shows that DnaC follows apparent Michaelis-Menten behavior and works at maximal velocity of ~3.5 molecules of ATP/min per molecule of DnaC (Figure 2B).
Figure 2. DnaC is a functional AAA+ ATPase. See also Figure S3.

(A) DnaC ATPase activity requires both DnaB and ssDNA. Rates for ATP-hydrolysis were determined for WT and K112R Walker-A mutant using a coupled ATP-hydrolysis assay. Data are represented as mean ± SEM.
(B) Kinetics of ATP-hydrolysis by DnaC. A coupled ATP-hydrolysis assay was performed using wild type DnaC to calculate hydrolysis rates at various ATP concentrations. Rates were fitted to the Michaelis-Menten equation. Error bars represent the variation over three independent experiments (mean ± SEM).
(C) The helical pitch and curvature of the DnaC hexamer in the complex with the helicase is more expanded than that observed for the isolated DnaC AAA+ domains (PDB 3ECC (Mott et al., 2008)).
(D)The loop adjacent to the Walker-B motif in E. coli DnaC adopts a different configuration from that of A. aeolicus DnaC.
If DnaC is an active ATPase, and if it forms oligomeric interactions that generate a composite AAA+ active site in the context of DnaB, then why does the helicase by itself fail to trigger nucleotide hydrolysis by the loader? Part of the answer is that the overall pitch and curvature of the hexameric spiral formed by DnaC in the presence of DnaB differs substantially from that seen for the isolated DnaC ATPase domains alone (Figure 2C), which have been imaged as an oligomer with an ideally organized ATPase site (Mott et al., 2008). Although the resolution of electron density maps obtained for the apo DnaBC complex is insufficient to accurately model all side chain rotamers in the catalytic center, this change in tilt between subunits likely leads to a misalignment of catalytic groups. In addition, the EM reconstruction shows that the configuration a loop adjacent to a critical signature sequence motif the Walker-B element, which is known to be required for ATP hydrolysis in AAA+ ATPases (Erzberger and Berger, 2006; Wendler et al., 2012) from that seen in a DnaC oligomer formed by its isolated ATPase domains (Figure 2D) (Mott et al., 2008). This reorganization likely helps maintain the catalytic residues of the loader in a configuration incompatible with ATP hydrolysis.
Mechanism of ssDNA binding by DnaC and its impact on loader structure and function
Examination of the 3D reconstruction of the nucleotide-bound DnaBC complex did not initially explain either why DnaB persists in an ADP state when bound to DnaC alone, or how ssDNA triggers DnaC ATPase activity. We therefore determined the three-dimensional structure of the DnaBC complex bound to both nucleotide and ssDNA. Purified DnaB and DnaC were first mixed together with a dT36 nucleic acid in presence of ADP•BeF3, deposited onto open-hole grids coated with a thin carbon layer (and pre-incubated with poly-lysine), flash-frozen, and examined by cryo-electron microscopy. Data collection, image processing, and model building were then performed as for apo DnaBC. The resultant 3D reconstruction, determined to an average resolution of 3.4 Å, shows clear density for ssDNA bound to both the helicase and loader (Figures 3, S4; Table 1).
Figure 3. Molecular architecture of E. coli DnaBC bound to ssDNA. See also Figure S4.

(A) Top view of the E. coli DnaBC complex bound to dT36. The nucleic acid (red) can be seen in the central pore.
(B) Side view of the DnaBC structure.
(C) Density for ssDNA bound to the central pore of the helicase and loader hexamers. The most frontal DnaB and DnaC monomers have been removed to visualize the nucleid acid. Threshold value of 0.05 used for rendering.
The structure of the complex reveals key insights into how DnaC both binds ssDNA and responds to this substrate in the presence of DnaB. Consistent with the observation that DNA binding promotes nucleotide hydrolysis, the DnaC protomers now occupy an ADP-bound configuration (in which there is also no observable density for the coordinating Mg2+ ion), and the Arginine-finger (Arg220) and Sensor II (Arg237) residues of each subunit has disengaged from the active site of its partner (Figure 4A). Concomitantly, the global conformation of the six loader subunits shifts into a more narrowed, cracked-ring hexamer that encircles the nucleic acid substrate (Figure 4B, Movie 2). The loop connecting each DnaC ATPase domain to its corresponding N-terminal extensions also undergoes a large (>90˚) outward rotation, repacking the base of the connector helix into a concave surface formed by a distinguishing element of the initiator/helicase loader clade of AAA+ proteins (the initiator specific motif, or ‘ISM’) (Figure 4C–D and Movie 3). Five of the six DnaC AAA+ ATPase domains (chains L, G, H, I, and J) can be seen engage ssDNA; the nucleic acid is both kinked and less well-ordered at the junction between DnaC and DnaB, and it is at least partially detached from the uppermost loader subunit in the spiral (chain K). Where DnaC does bind DNA, each protomer engages two nucleotides in a cleft formed between the extended loop of the Walker-B element and the ISM (the latter of which undergoes a disorder-to-order transition from the DNA-free to DNA-bound state). One amino acid in particular Phe146, which is located at the tip of the DnaC ISM acts as a wedge to separate the ssDNA strand into dinucleotide segments (Figure 4E). Another residue, Tyr179, sits at the junction where the Walker-B loop transitions into an α-helix, forming van der Waals contacts with the ribose region of one of the two nucleotides. An adjoining amino acid in the same loop, Ser177, forms a hydrogen bond with the neighboring phosphate (Figure 4E).
Figure 4. Molecular basis for ssDNA recognition by and activation of DnaC.

(A) DNA-bound DnaC associates with ADP. The Arg finger and sensor-II residues are disengaged from the nucleotide. Density contoured at a threshold value of 0.05.
(B) The DnaC ATPase hexamer constricts around DNA.
(C) DnaC NTDs pivot between apo and ssDNA-bound states.
(D) The junction between the DnaC NTD and AAA+ domain is fulcrum that accommodates ATPase movements upon ssDNA binding.
(E) Detailed view of DnaC residues involved in engaging bound ssDNA. Density contoured at a threshold value of 0.05.
(F) ssDNA binding triggers a conformational change in the extended Walker-B loop.
Interestingly, the set of interactions between DnaC and ssDNA revert the conformation of the extended Walker-B loop into a configuration reminiscent to that seen in the oligomer of the isolated DnaC ATPase domain (Figure 4F) (Mott et al., 2008). We therefore examined whether these protein-nucleic acid contacts are important for DnaC activity. Coupled ATPase assays show that Phe146, Ser177, and Tyr179 which are chemically conserved across DnaC homologs are all essential for nucleotide hydrolysis by DnaC (Figure 5A). In addition, the mutants proved partially to severely deficient for binding to a single-stranded, FAM-labeled dT25 oligonucleotide in presence of ATP, as measured using fluorescence anisotropy (Figure 5B). Finally, the ATP-dependent loading of DnaB loading was assessed, using a single-stranded phage M13 substrate and resolving the complex on a large pore Bio-Gel column; analysis of the fraction of DnaB that associates with ssDNA showed that loss of any of the three residues in DnaC leads to a defect in helicase loading (Figures 5C, S5). Collectively, these findings confirm that the interactions seen between ssDNA and DnaC in the structure of the complex are important for helicase loader function.
Figure 5. ssDNA binding by DnaC is essential for ATP hydrolysis and helicase loading. See also Figure S5.

(A) Coupled ATP-hydrolysis assays show that mutations to residues involved in binding ssDNA impair ATPase activity. Data are represented as mean ± SEM.
(B) Fluorescence anisotropy assays show that the mutation of residues seen to interact with ssDNA lead to binding defects. Binding reactions contained 10 nM fluorescein-labeled dT25 oligonucleotide titrated against either WT or specified DnaC mutant protein. Error bars represent the variation over three independent experiments (mean ± SEM).
(C) DnaC-DNA interactions are critical for DnaB loading. Data presented are from three independent experiments (mean ± SEM).
ssDNA binding triggers a substantial conformational rearrangement of DnaB.
The structure of the ssDNA-DnaBC complex shows that the nucleic acid substrate also interacts with the helicase. Clear density is evident for 14 nucleotides in the central channel of DnaB that extend from the DNA bound by DnaC. Only 11 of the observed nucleotides interact directly with the helicase. Binding occurs predominantly through the so-called ‘L2’ loop that emanates from the RecA ATPase fold of DnaB; a small set of additional contacts are also seen with the tip of an α-helix that borders the ‘L1’ loop region. The ssDNA is coiled into a right-handed spiral that closely approximates the geometry of a single DNA strand from an A-form helix, tracking the phosphodiester backbone along six successive subunits the DnaB hexamer (Figure 3C, 6A). The backbone is recognized largely through van der Waals interactions from the L2 loop main-chain, although additional van der Waals contacts also occur with the side chains of Thr358, Arg403, and Glu404. Only three hydrogen-bonding/salt bridge interactions are seen for DnaB and ssDNA, between the non-bridging oxygens of the phosphodiester linkages and Asn386, Arg387, and Arg403.
Figure 6. DNA remodels the molecular organization of DnaB.

(A) Close-up view DnaB residues involved in DNA binding. Density contoured at a threshold value of 0.05. (Inset) View of ssDNA bound to the RecA domains of DnaB.
(B) The DnaB RecA domains adopt a more planar, crack-ring configuration in the presence of nucleic acid.
(C) ADP•BeF3 is associated with the active sites of ssDNA-bound DnaB. Density contoured at a threshold value of 0.05.
(D) A terminal DnaB subunit relocates from one end of the cracked helicase ring to the other during a conformation transition induced by ssDNA binding. The open crack in the DnaB ring in absence of DNA is sealed off by the linker helix following DNA engagement, topologically linking the helicase around DNA. (Inset) Top-down views showing the conformational transition in the DnaB collar and the shift in the position of the crack in the helicase ring following the transition. Asterisks denote the position of the crack in the DnaB ring.
(E) The configuration of the helicase in the ssDNA-bound DnaBC complex is highly reminiscent to that seen for the free helicase bound to nucleic acid and GDP•AlFx (PDB 4ESV (Itsathitphaisarn et al., 2012)).
Notably, DnaB itself undergoes a large structural rearrangement upon binding ssDNA. The ATPase modules tilt downward by ~25˚ as they engage the nucleic acid segment, adopting a less-pronounced spiral conformation than seen in the DNA-free state (Figure 6B). Interestingly, five of the six nucleotide-binding sites for DnaB now contain ADP•BeF3 (the one solvent-exposed catalytic center is empty) (Figure 6C), indicating that nucleotide has been exchanged during domain reorganization. The rotation/compression of the RecA domains is further accompanied by an ~60˚ rotational shift in their associated NTDs to a dilated (but still cracked) configuration (Figure 6D, Movie 4). This change requires that one of the terminal subunits in the split-ring hexamer detach from one partner and re-engage another; although either subunit flanking the crack in the DNA-free state is conceivably free to move, a transition of the uppermost subunit in the DnaB spiral toward the bottommost protomer coincides with the known 5’➔3’ directionality of helicase movement along ssDNA (Movie 5). This subunit movement in turn allows the linker helix of the transiting subunit, the entirety of which is visible in unsharpened maps of the ssDNA-bound complex, to topologically seal the break in the split hexameric ring (Figure 6D). Remarkably, the resultant DnaB conformation that follows DNA binding is a near identical match to that of a putative translocation intermediate determined for G. stearothermophilus DnaB (Itsathitphaisarn et al., 2012) (Figure 6E).
Discussion
Mechanism of DnaB ring opening and closing, and control of DnaC function, during helicase loading
The findings presented here resolve many long-standing questions concerning the mechanism by which bacterial replicative helicases can be loaded onto DNA. In the presence of nucleotide, the N-terminal collar of DnaB accesses two distinct conformational states dilated and constricted which differ in the relative orientation of NTD dimers (Figure 1B–C) (Strycharska et al., 2013). DnaC promotes the formation of a constricted state of DnaB, while at the same time driving the helicase ring into an extended spiral conformation (Arias-Palomo et al., 2013). Inspection of the DNA-free state of the DnaBC complex shows that DnaC locally remodels inter-domain and inter-subunit interactions in DnaB to induce the formation of an open-ring state in which the linker helix of one subunit fully disengages from its partner to provide an entryway for single DNA into the interior of the complex (Figure 1D). The structure also demonstrates that loader subunits promote the formation of a constricted collar configuration by using their extended N-terminal arms to engage DnaB NTD dimers through contact surfaces that are inaccessible when the helicase collar is dilated (Figure S2B). The contacts observed in the DnaBC complex are consistent with prior biochemical and genetic data (Chodavarapu et al., 2016; Ludlam et al., 2001), and together with ATP-dependent interactions formed between neighboring DnaC ATPase domains, work in concert to enforce an ADP-bound, stretched-spiral state upon DnaB. The associations between DnaC ATPase regions and between DnaC and DnaB would appear to restrain helicase subunit movement, accounting for early observations showing that ATP-bound DnaC inhibits DNA unwinding by DnaB (Wahle et al., 1989a).
Although DnaC has long been known to possess many of the hallmarks of an ATPase (Koonin, 1992), there has been some question as to whether DnaC possesses a capacity to hydrolyze nucleotide. Our biochemical studies confirm early observations from the O’Donnell group (Davey et al., 2002), showing that DnaC can turnover ATP, but only when both ssDNA and DnaB are present. The structure of the DnaBC complex bound to ssDNA explains this synergy, showing how the loader switches from an ATP to ADP state following nucleic acid binding. Examination of the DNA-free DnaBC structure reveals that DnaB licenses DnaC ATPase activity by templating oligomeric interactions between loader subunits that are required for turnover, but that the helicase does not in and of itself impose a perfectly positioned set of catalytic contacts needed to drive hydrolysis; instead, DnaC is blocked from attaining its catalytic configuration by a slight misalignment between neighboring AAA+ folds and a remodeling of the Walker-B loop (Figure 2C and 2D), a region important for nucleotide hydrolysis in AAA+ ATPases and P-loop NTPases in general. ssDNA overcomes this inactive conformation at least in part by associating with Ser177 and Tyr179 of DnaC, which form the terminal part of the Walker-B loop (Figure 4E and 4F). DNA binding also changes the conformation of the DnaC ISM, which allows the loader ATPase elements to adopt a new, tighter split-ring conformation that favors inter-subunit interactions necessary to facilitate ATP turnover (Figure 2A and 4B). The mutation of three amino acids in DnaC seen to interact with ssDNA leads to a direct loss of the loader’s DnaB- and ssDNA-dependent ATPase and helicase loading activities (Figure 5), confirming the role of these contacts in supporting DnaC function. One of these residues, Phe146, additionally has been shown previously to be important for cell viability (Mott et al., 2008). Interestingly, the mechanism by which DnaC binds DNA differs substantially from its two closest paralogs, the replication initiator DnaA and the IS21 transposase regulator IstB, as well as from more distantly-related orthologs, such as archaeal/eukaryl Orc/Cdc6 replication initiators (Figure S6). In particular, the ISM of DnaC’s AAA+ domain binds only two nucleotides of ssDNA, compared to the three nucleotides bound by DnaA; both IstB and Orc/Cdc6 proteins bind also DNA duplexes, rather than single-stranded substrates, and DnaC binds DNA using a different face of its ISM than either IstB or ORC•Cdc6. That just one region of the AAA+ fold of a closely-related clade of enzymes can adopt such different modes of substrate engagement reflects the tremendous plasticity of this ATPase superfamily.
The change in the ATPase status of DnaC following DNA binding releases the helicase loader’s restraint on DnaB. The structure of the ssDNA-bound DnaBC complex shows that as the loader ATPase spiral narrows (Figure 4B), the split ring of the helicase also partly compresses (Figure 6B), exchanging ADP for ATP as its six RecA ATPase domains wrap around 12 nucleotides of a helical ssDNA coil (2 nt/subunit). Concomitant with these changes in the ATPase region of DnaB, the associated NTD dimers of the helicase rotate substantially to isomerize into a dilated, cracked-ring state (Figure 6D). This transition necessitates that one DnaB subunit detach from one of the end of the helicase spiral and reengage with a partner protomer at opposite end, a movement that causes the linker helix of the mobile subunit to bridge the gap in ring, topologically coupling DnaB to DNA (Figure 6D). Interestingly, this conformational change mirrors the transition proposed to take place during DNA unwinding by the helicase (O’Donnell and Li, 2018). Moreover, the resultant configuration for DnaB matches that of a proposed translocation state of helicase bound to ssDNA and an ATPase transition state inhibitor, GDP•AlFx (Itsathitphaisarn et al., 2012) (Figure 6E). This congruency indicates either that the loading of DnaB by DnaC results in a translocation-competent form of the helicase, or that the prior ssDNA-bound structure of DnaB corresponds to a post-loading intermediate rather than a true translocating form. Regardless, the new NTD conformation assumed by DnaB is now compatible with binding to the DnaG primase protein (Bailey et al., 2007; Liu et al., 2013), which is required to initiate strand synthesis by replicative polymerases (Frick and Richardson, 2001; Kuchta and Stengel, 2010).
It is unclear at present how DnaC dissociates from DnaB. Although early work initially suggested that ATP is needed for DnaC to form a complex with DnaB (Wahle et al., 1989b), subsequent studies have shown that neither ATP nor ADP are necessary for this interaction to occur (Davey et al., 2002; Galletto et al., 2003; Mott et al., 2008). The cryo-EM reconstructions presented here demonstrate that, with the exception of localized contacts at the tip of the DnaC NTD, a majority of the interactions between the helicase and loader remain essentially unchanged between DNA-free and DNA-bound states. Thus, although an interaction with DnaB is critical for DnaC’s ATPase activity (Davey et al., 2002) (Figure 2A), the molecular interactions with the helicase itself are not affected by the nucleotide binding state of the loader. Once DNA binds, however, DnaC converts to an ADP state, which would be expected to form less stable oligomeric interactions between loader subunits. Consistent with this notion, we find that the two ‘terminal’ ATPase domains which flank the crack in the DnaC hexamer are less well ordered in the ssDNA-bound form of the complex compared to the apo state (Figure S4). In addition, two of the six reconstructions obtained from the 3D classification step performed during image processing show that a fraction of the population of the nucleic acid-bound molecules contain weak density for only one terminal subunit of the loader (Figure S4B). Thus, while the act of hydrolysis releases a set of contacts that help cooperatively anchor DnaC to DnaB, this event is not sufficient in and of itself to promote complete DnaC dissociation. Primer synthesis by DnaG has been reported to trigger DnaC release (Makowska-Grzyska and Kaguni, 2010); how this occurs mechanistically remains to be determined.
Mechanistic insights into ring-loading reactions occurring during replication
All cells use two classes of protein rings that encircle DNA to support replication hexameric helicases and DNA polymerase processivity clamps (Kelch et al., 2012; Pomerantz and O’Donnell, 2007). The encirclement of helicases and clamps around DNA requires the action of dedicated loading factors, which use nucleotide to promote the formation of conformational transitions in client rings that permit their stable loading (Kelch et al., 2012; O’Shea and Berger, 2014). Helicase loaders share a common AAA+ ATPase fold with clamp loaders, but have been classified into a different evolutionary lineage based on the presence of an extra a-helical element (the ISM) in their ATP-binding domain (Erzberger and Berger, 2006; Iyer et al., 2004). Given the shared ancestry of their ATPase elements, the mechanisms used by replicative helicase and processivity clamp loaders are often thought to parallel one another. However, the present work, along with recent data from other groups, shows that there are actually intriguing and important differences both between and within the two types of loading factors.
For the clamp loaders found in cells, these factors work by forming a pentameric array of AAA+ subunits (Figure 7A). The binding of ATP stimulates the adoption of a cracked-ring conformational state that can engage and physically open a client clamp (Kelch et al., 2011). DNA binding, which can occur because the clamp and loader rings are both breached, triggers ATP hydrolysis by the loader, which allows the clamp to close around DNA and the clamp loader to dissociate from the DNA-tethered clamp. A key feature of this cycle is that the ATPase domains of the loader directly engage the clamp, using shape and pitch of the oligomeric AAA+ assembly to template the formation of an open clamp.
Figure 7. Comparison of ring-loading reactions occurring during replication (see text for details). See also Figure S6 and S7.

(A) Diagram of the DNA polymerase clamp loader reaction.
(B) Schematic of the helicase loading reaction catalyzed by DnaC.
(C) Overview of MCM2–7 loading by ORC and Cdc6. Although DNA is encircled by ORC (Li et al., 2018), the complex is not topologically closed and readily dissociates from DNA (Duzdevich et al., 2015); Cdc6 binding to ORC topologically entraps DNA (Bleichert et al., 2018; Yuan et al., 2017). Recent studies have shown that DNA is bent substantially upon both ORC and ORC•Cdc6 binding (Bleichert et al., 2018; Li et al., 2018).
DnaC-family replicative helicase loaders of the type imaged here are also ‘ring-breakers’ (Davey and O’Donnell, 2003). However, unlike clamp loaders, which form a stable pentameric complex on their own, oligomeric interactions between DnaC subunits are templated by their association with a pre-formed DnaB hexamer (Arias-Palomo et al., 2013) (Figure 7B). Here, we show that helicase opening explicitly results from interactions with the N-terminal arm of DnaC, not with its ATPase elements. This finding accords with prior biochemical data showing that the DnaC N-terminus is sufficient by itself to promote helicase loading (Arias-Palomo et al., 2013). The DnaC ATPase domains appear to be used to stabilize the open-ring helicase state, accounting for why DnaB loading appears more efficient in the presence of full-length DnaC than with the N-terminal arm alone (Arias-Palomo et al., 2013). As with the clamp loaders, DNA binding triggers ATP turnover by DnaC, but the helicase loader differs in that it does not dissociate from its client ring, instead remaining stably associated with both DnaB and ssDNA. Remarkably, DnaC maintains this connection with DnaB even though the helicase itself undergoes a substantial conformational reorganization that involves the translocation of one subunit from one end of the cracked ring to the other and the commensurate closure of the DnaB ring (Movie 6).
If the mechanism by which DnaC catalyzes a ring-loading reaction only partly parallels the approach used by clamp loaders, it diverges even more sharply from the other major class of replicative helicase loader with which it shares a closer phylogenetic relationship, namely, archaeal and eukaryotic Orc/Cdc6 proteins. In eukaryotes, ORC serves double duty both as a factor that recognizes replication origins and that loads the replicative MCM2–7 helicase onto these sites. Five of ORC’s six subunits (Orc1-Orc5), along with a sixth partner (Cdc6), contain AAA+ ATPase folds that are necessary for loading to occur (Bowers et al., 2004; Neuwald et al., 1999; Randell et al., 2006; Siddiqui and Stillman, 2007; Speck and Stillman, 2007; Speck et al., 2005). As with DnaC, Orc1–5 and Cdc6 possess the ISM element that distinguishes them phylogenetically from clamp loaders (Erzberger and Berger, 2006; Iyer et al., 2004); however, unlike both DnaC and the clamp loaders, ORC and Cdc6 first bind to and encircle DNA before they engage their client Mcm2–7 ring (Bleichert et al., 2017; Li et al., 2018), thus topologically linking the loader to DNA before its target helicase can arrive (Figure 7C). At first glance, this mechanism seems counterintuitive, as DNA would sterically block the helicase ring from engaging the face of the encircling ORC•Cdc6 complex. MCMs solve this problem by pre-adopting an open-ring state (Costa et al., 2011; Frigola et al., 2017; Lyubimov et al., 2012; Zhai et al., 2017), which can slot over the DNA as the helicase engages ORC•Cdc6. In this regard, ORC•Cdc6 acts more as a ‘ring-sealer’ than as ring-breakers such as DnaC and clamp loaders (Bleichert et al., 2017; Yardimci and Walter, 2014).
It is interesting to note that there are still additional mechanistic differences between helicase and clamp loaders of close phylogeny, as well as parallels between more distantly related systems. For example, the phage T4 clamp loader can stabilize a T4 clamp that is pre-opened (Alley et al., 1999, 2000; Millar et al., 2004; Trakselis et al., 2001), and it can also pre-bind DNA before loading such opened clamps (Trakselis et al., 2003). In this regard, a bacteriophage clamp loader system would seem to act in a manner akin to ORC•Cdc6 during MCM2–7 loading. By contrast, the DnaI family of replicative helicase loaders, which are close evolutionary cousins of DnaC, has been reported to assemble helicase protomers around DNA (Soultanas, 2002; Velten et al., 2003), thereby acting through a distinct type of ‘ring-maker’ mechanism (Davey and O’Donnell, 2003), while the mechanism of helicase deposition by a third class of loader, DciA, is unknown (Brézellec et al., 2016). Although we expect that the molecular approach by which DnaC and DnaI loaders engage their client helicases will be analogous, sequence alignments between the NTDs of these two loader families reveals little similarity (Figure S7). Future studies will be needed to establish the extent to which DnaC and DnaI operate in a similar manner and how the ATPase cycles of disparate helicase- and clamp-loader systems are differentially channeled into supporting diverse reaction mechanisms.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James M. Berger (jmberger@jhmi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For recombinant expression the Escherichia coli strain C41 (Lucigen) was used. Cells were grown at 37°C as described in the STAR Methods Details section.
METHOD DETAILS
Protein expression and purification
E. coli dnaC was cloned as a 6xHis-MBP fusion protein into a pET28b-derived plasmid, while E. coli dnaB was cloned into a different pET28b background without an affinity tag. All constructs were sequence verified. Wild-type proteins were expressed in strain C41 (Lucigen) by induction at mid-log phase (OD600~0.4) with 1 mM IPTG at 37°C for 3h (DnaB) and 2.5h (DnaC). Following induction, cells were harvested by centrifugation, resuspended in lysis buffers supplemented with protease inhibitors (1 mM PMSF, 1 µg/mL Pepstatin A, and 1 µg/mL Leupeptin) (for DnaB 20 mM HEPES-KOH (pH 7.5), 500 mM NaCl, 10% glycerol, 10 mM MgCl2, 0.1 mM ATP, 1 mM β-mercaptoethanol,; for DnaC 50 mM HEPES-KOH (pH 7.5), 1 M KCl, 10% glycerol, 30 mM imidazole, 10% MgCl2, 0.1 mM ATP, 1 mM β-mercaptoethanol) Resuspended cells were frozen for later use, thawed, and lysed by sonication.
Wild-type DnaB was purified by ammonium sulfate precipitation (30% wt/v), followed by resuspension of the pellet in 100 mM NaCl Q Buffer(100 mM NaCl, 20 mM Hepes pH 7.5, 10 % glycerol, 10 mM MgCl 2, 0.01 mM ATP, 1 mM BME, and protease inhibitors) and passage over an HiTrap Q HP anion exchange column (GE Healthcare). Peak fractions were pooled, concentrated in Amicon Ultra Centrifugal Filters 30,000 MWCO (Millipore) and further purified by gel filtration on a HiPrep 16/60 Sephacryl S-300 column (GE Healthcare) in buffer containing 20 mM Tris-HCl (pH 8.5), 800 mM NaCl, 10% glycerol, 5 mM MgCl2, 1 mM β-mercaptoethanol, 0.1 mM ATP, 1 mM PMSF, 1 µg/mL Pepstatin A, and 1 µg/mL Leupeptin. Peak fractions were then concentrated and applied to a second sizing column run in buffer containing 100 mM NaCl rather than 800 mM NaCl.
Purification for WT DnaC was performed using Ni-Sepharose (GE Healthcare) affinity resin, followed by TEV protease incubation (to remove the affinity tag) and a second Ni-Sepharose step. For structural studies, an additional amylose (New England Biolabs) affinity step was added before the TEV cleavage. The flow-through from this step was concentrated in an Amicon Ultra Centrifugal Filters 10,000 MWCO (Milipore) and applied to a HiPrep 16/60 Sephacryl S-200 gel filtration column (GE Healthcare) in buffer containing 50 mM HEPES (pH 7.5), 500 mM KCl, 10% glycerol, 10 mM MgCl2, 0.1 mM ATP, 1 mM β-mercaptoethanol, 1 mM PMSF, 1 µg/mL Pepstatin A, and 1 µg/mL Leupeptin.
To overcome apparent toxicity, DnaB and DnaC mutants were expressed in the periplasm. Both genes were cloned into 6xHis-MBP fusions with an N-terminal periplasmic localization signal and the point mutations were introduced by QuickChange site-directed mutagenesis. All constructs were sequence verified. Mutant proteins were expressed in strain C41 (Lucigen) by induction at mid-log phase (OD600 ~0.4) with 0.5 mM IPTG overnight at 16˚ C. Following induction, cells were harvested by centrifugation, resuspended in lysis buffers similar to wild type proteins. Resuspended cells were flash frozen and stored at −80˚ C for later use. Purification procedures followed those of wild-type proteins, except that the DnaBKAEA double mutant was purified using sequential Ni-Sepharose (GE Healthcare) and amylose (New England Biolabs) affinity resins, followed by TEV protease incubation (to remove the affinity tag) and a second Ni-Sepharose step. All buffers are similar to the wild type DnaB purification.
The purity of all DnaB and DnaC proteins throughout different purification steps was assessed by SDS-PAGE. The concentration of the purified factors was determined using ND-1000 UV-Vis Spectrophotometer to measure absorbance at 280 nm. Following purification, proteins were aliquoted, flash frozen in liquid nitrogen, and stored at –80° C.
DnaC-ssDNA binding assays
DnaC interaction assays with ssDNA were performed as previously described (2008, Mott M., et. al.). 0–50 µM DnaC was first mixed in a high salt buffer (50 mM HEPES (pH 7.5), 500 mM KCl, 10% (v/v) glycerol, 10 mM MgCl2) before being rapidly diluted four-fold into binding buffer containing 50 mM HEPES (pH 7.5), 10% (v/v) glycerol, 5 mM MgCl2, 2 mM ATP, 2 mM DTT, 0.1 mg/ml BSA, and 10 nM fluorescein-labeled dT25 oligonucleotide (IDT). Dilution of the protein from the high salt buffer in the binding reaction brought the final salt concentration to 125 mM KCl. Aliquots of the samples were transferred to prewarmed 384-well, black small volume polystyrene plates (Greiner Bio-One). The plates were incubated for 10 min and then read in a CLARIOstar (BMG Labtech) microplate reader at 37° C. Averaged delta-anisotropy values were plotted against increasing DnaC concentrations. Data were fit to Hill equation for specific binding using Prism. The experimental data shown reflect the average of 3 independent experiments.
Loading assay
An M13 ssDNA-based assay was used to assess the efficiency of DnaB loading (Fang et al., 1999). Purified DnaB was mixed with equimolar DnaC (WT, F146A, S177D or Y179A) to a final volume of 100 `L and a final concentration of 10 `M in presence of 40 mM HEPES (pH 7.5), 10 mM MgCl2, 5 mM DTT, 0.1 mg/ml BSA, 5 mM ATP, and 1 `g M13 DNA. Reactions were incubated for 15 min at 30 °C and then applied to a Bio-Gel A-1.5m (Bio-Rad) packed in a 0.7X15cm Flex-Column (Kimble). 250 `l fractions were collected by hand at 20˚ C, mixed with SDS-PAGE gel loading dye, and resolved on 12% Tris-Glycine SDS-PAGE gels. Gels were silver-stained and the amount of DnaB in each fraction was estimated using ImageJ (Schneider et al., 2012). Helicase loading efficiency was calculated as the ratio of DnaB that elutes with ssDNA compared to total DnaB. The loading efficiency of each DnaC mutant was normalized to WT DnaC. The experimental data shown reflect the average of 3 independent experiments.
ATP-hydrolysis assay
Coupled ATP-hydrolysis assays were performed in a buffer containing 40 mM HEPES (pH7.5), 10 mM MgCl2, 5 mM DTT, and 0.1 mg/ml BSA. A single Walker-A DnaB mutant (K236A) proved to have high residual ATPase activity background (not shown), so a double mutant of was made in conjunction with catalytic glutamate substitution (E262A, referred to as ‘DnaBKAEA’). For assessing DnaC activity, 5 mM of purified protein was mixed with 5.25 mM DnaBKAEA and 2.1 mg M13 ssDNA, 0.5 mM NADH, and 15 mM PEP in presence of a 4% v/v pyruvate kinase/lactate dehydrogenase enzyme mix (Sigma). Reactions were initiated by the addition of ATP to a final concentration range spanning 25 mM 6.4 mM, and the absorbance of the reaction was then monitored at 340 nm in a 96-well, half-area plate (Corning) in a CLARIOstar (BMG Labtech) microplate reader at 37° C. An NADH standard curve was used to convert A340 to NADH concentration, which in turn was used to calculate the rate of NADH loss that linearly correlates to ATP-hydrolysis; initial rates were then calculated and plotted against ATP-concentration. The resultant data were fit to Michaelis-Menten equation and kcat and Km values estimated using Prism. The experimental data shown reflect the average of 3 independent experiments.
Sample preparation for electron microscopy
For cryo-EM studies, a 1.2-fold molar excess of wild-type DnaC was mixed with wild-type DnaB and dialyzed over-night at 4 °C (Slide-A-lyzer MINI, 10K MWCO, Thermo Scientific) against 20 mM HEPES (pH 7.5), 150 mM KCl, 5 mM MgCl2, 1mM DTT, and 500 µM ADP•BeF3. The complex was then further purified over a Superdex 200 5/150 GL analytical gel filtration column (GE Healthcare) equilibrated in dialysis buffer. We initially characterized the DnaB•DnaC complex in the presence of ATP by negative stain EM to confirm that, at low resolution, the configuration of the complex is identical to the one found in the presence of ADP•BeF3 (not shown), but we elected to use the non-hydrolyzable ATP analog for structural studies as a safeguard to increase the stability of the complex and minimize heterogeneity. For the DNA-bound sample, peak fractions were mixed with a 1.5 molar excess of an HPCL-grade dT 36mer (1.5 dT36 1 DnaBC complex). Samples were then diluted into a final buffer concentration of 20 mM HEPES (pH 7.5), 150 mM KCl, 5 mM MgCl2, 1mM DTT, 500 µM ADP•BeF3, and 0.015% NP-40. From this mix, 3 µL (corresponding to 52 nM DnaBC complex concentration) were applied to glow-discharged C-flat grids (Protochips,Inc) coated with a second layer of homemade, thin continuous carbon film. To obtain different particle orientations, the glow-discharged grids were first floated on 40 µl drops of a 0.1% (w/v) aqueous solution of polylysine hydrobromide (Mw 80 kDa; Polysciences, Inc.) for two min, blotted, and briefly rinsed with distilled water prior to sample application. After 1 min of incubation with sample, grids were blotted for 3.5 sec (using a blot force of 5 on an FEI Vitrobot Mark IV) at room temperature and plunged into liquid ethane. Flash-frozen grids were subsequently stored in liquid nitrogen.
Electron microcopy data acquisition
Cryo-EM grids were pre-screened in either a JEOL 1230 and FEI Talos Artica microscope equipped with a TemCam-F416 (TVIPS, Gauting, Germany) or Falcon III (FEI) camera, respectively. High-resolution data were collected on a Titan Krios electron microscope operated at 300 kV, and images were acquired using a Gatan K2 Summit direct electron detector, operated in electron counting mode. For the apo sample, EPU (FEI) was used to record 3161 micrographs at a nominal defocus range of −1.2 to −2.7 μm, with a magnification of 1.07 Å/px at the specimen level, and a total dose of 61 e-/A2 accumulated over 10 seconds and fractionated across 40 frames. For the DnaBC-ssDNA complex, 1858 micrographs were collected at a nominal defocus range of −1.2 to −2.7 μm, with a magnification of 1.047 Å/px at the specimen level, and a total dose of 58 e-/A2 accumulated over 12 seconds and fractionated across 40 frames.
Image processing
For both the apo and ssDNA-bound datasets, frames were aligned using MOTIONCOR2 (Zheng et al., 2017), which also generated drift-corrected summed images with and without electron-dose weighting. Omission of the first frame gave slightly better densities likely due to a reduction of early beam-induced movement. Micrographs were manually inspected to remove pictures that contained crystalline ice or other forms of visible contamination, and CTF parameters were estimated with GCTF (Zhang, 2016) using non dose-weighted images. A subset of the micrographs was picked with GAUTOMATCH, binned by two, and subjected to 2D classification using RELION 2.1 (Kimanius et al., 2016; Scheres, 2012). The resulting 2D averages were then used as templates to pick an entire dataset with GAUTOMATCH to generate initial particle stacks (551,641 apo and 473,888 ssDNA-bound particles). After one round of 2D classification, only averages showing high-resolution features were retained, which resulted in 182,394 and 287,226 particles for the apo and DNA-bound samples respectively. For the apo sample, a 3D reconstruction obtained from negative stain EM (Arias-Palomo et al., 2013) was low-pass filtered to 40 Å resolution and used as an initial model for 3D refinement with RELION2.1. In the case of the DNA-bound complex, a simple three-tier toroid was used as starting model to minimize the possibility of introducing model bias. X and Y shifts from the refinements were used to re-extract unbinned and centered particles, which were then subjected to a round of 3D refinement followed by a 3D classification step. For the apo complex, five of the resultant six models only showed low-resolution features; these particles were excluded for further processing. The remaining 104,913 particles were used as input for a subsequent 3D refinement run, using a soft-edged mask that followed the contour of the particle, and resulted in a map with an estimated resolution of 3.9Å by gold-standard FSC at 0.143 (Rosenthal and Henderson, 2003). In the case of the ssDNA-bound sample, four out of six models showed high-resolution features, while the remaining two lacked one DnaC subunit and were excluded from further refinement. The remaining stack of 229,097 particles was subjected to 3D refinement with a soft-edged mask that resulted in a 3.4 Å reconstruction according to gold-standard FSC. Local resolution and the B-factors used to sharpen the maps were determined automatically using RELION 2.1 (Kimanius et al., 2016; Rosenthal and Henderson, 2003).
Model building and refinement
Homology models for E. coli DnaB and the DnaC AAA+ domain were generated using the Phyre2 server (top ranking models for DnaB and DnaC were PDB IDs 4NMN and 3ECC, respectively) as a starting point for building (Kelley et al., 2015). For DnaC, the higher resolution DNA-bound map was used for constructing an initial model of the loader. The ATPase region was rigid-body fitted into one of the best-defined monomers (chain H) whereas the N-terminal domain was built de novo using COOT (Emsley and Cowtan, 2004). The full-length DnaC structure was subjected to iterative rounds of model building and real-space refinement with COOT and PHENIX using Ramachandran, rotamer, geometry, and secondary structure restraints (Adams et al., 2010; Emsley and Cowtan, 2004). The resulting model was docked into density for the remaining protomers for the DNA-bound and apo EM densities and then subjected to additional rounds of rebuilding and refinement.
For the DnaB RecA domain, the model was built, refined and optimized similarly to the loader. The N-terminal domains, however, proved to be better defined in the apo DnaBC reconstruction due to more extensive interactions between NTD dimers and contacts these regions make with the C-terminal RecA folds. Accordingly, the apo EM map was used to build and refine the first N-terminal dimer of the helicase, which was subsequently copied and docked into density for the remaining regions to generate a complete helicase hexamer. Specific parts of the reconstructions, such as linker elements and portions of the DnaB N-terminal domains, contain some degree of flexibility and thus became less interpretable following the application of a fixed B-factor sharpening value. In these instances, the initial placement of the atomic coordinates was performed into unsharpened maps, where the density allowed for an unambiguous fit.
For ssDNA region bound to DnaB, a crystal structure of the helicase bound to nucleic acid was used as initial model (Itsathitphaisarn et al., 2012). The DNA bases contacting DnaC were modeled de novo. Once full models were generated, they were manually inspected with COOT, and refined with PHENIX with additional base stacking and NCS restraints. Figures were generated using CHIMERA and PYMOL (DeLano, 2002; Pettersen et al., 2004).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data for DNA binding, loading and ATPase assays are mean values of at least three replicates. More details can be found in the STAR Methods Detail section and figure captions.
Supplementary Material
Linear morphing of a DnaB dimer from a closed-ring/constricted state to the open-ring/constricted configuration found in the apo DnaBC complex.
Linear morphing of DnaC ATPase domains from apo to the DNA-bound configuration.
Linear morphing of DnaC N-terminal domain from apo to the DNA-bound configuration.
Linear morphing of DnaB domains within DnaBC from the apo to the nucleic acid-bound state.
Linear morphing of DnaB domains within DnaBC from the apo to the nucleic acid-bound state.
Linear morphing of DnaBC movements accompanying helicase binding, opening, and ssDNA loading.
ACKNOWLEDGEMENTS
The authors are grateful to: the EM platforms from Johns Hopkins University, the University of Virginia, Vanderbilt University, the New York Structural Biology center, the University of Leeds, and Israel Sanchez Fernandez (Columbia University) for sample characterization and data analysis; and the EM facilities of the CIB and CNB (CSIC) for support in grid screening and preparation. This work was supported by the NIGMS (to JMB, R37-071747), and by the Spanish MINECO/AEI, the ERDF the ESF, (to EAP, BFU2017-89143-P and RYC-2015-19059). We acknowledge Diamond for access and support of the Cryo-EM facilities at the UK national electron bio-imaging centre (eBIC, proposal EM19454), funded by the Wellcome Trust, MRC and BBSRC. Access to eBIC was supported by iNEXT, grant number 653706, funded by the Horizon 2020 programme of the European Commission.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTEREST
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the cryo-EM densities and atomic coordinates for the apo and ssDNA-bound structures are: EMD-4537 and EMD-4538, and PDB IDs 6QEL and 6QEM, respectively.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley SC, Shier VK, Abel-Santos E, Sexton DJ, Soumillion P, and Benkovic SJ (1999). Sliding Clamp of the Bacteriophage T4 Polymerase Has Open and Closed Subunit Interfaces in Solution. Biochemistry 38, 7696–7709. [DOI] [PubMed] [Google Scholar]
- Alley SC, Abel-Santos E, and Benkovic SJ (2000). Tracking sliding clamp opening and closing during bacteriophage T4 DNA polymerase holoenzyme assembly. Biochemistry 39, 3076–3090. [DOI] [PubMed] [Google Scholar]
- Arai K, and Kornberg A (1981). Mechanism of dnaB protein action. II. ATP hydrolysis by dnaB protein dependent on single- or double-stranded DNA. J. Biol. Chem 256, 5253–5259. [PubMed] [Google Scholar]
- Arias-Palomo E, and Berger JM (2015). An Atypical AAA+ ATPase Assembly Controls Efficient Transposition through DNA Remodeling and Transposase Recruitment. Cell 162, 860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Palomo E, O’Shea VL, Hood IV, and Berger JM (2013). The bacterial DnaC helicase loader is a DnaB ring breaker. Cell 153, 438–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey S, Eliason WK, and Steitz TA (2007). Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 318, 459–463. [DOI] [PubMed] [Google Scholar]
- Bazin A, Cherrier MV, Gutsche I, Timmins J, and Terradot L (2015). Structure and primase-mediated activation of a bacterial dodecameric replicative helicase. Nucleic Acids Res 43, 8564–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleichert F, Botchan MR, and Berger JM (2017). Mechanisms for initiating cellular DNA replication. Science (80-. ) 355, eaah6317. [DOI] [PubMed] [Google Scholar]
- Bleichert F, Leitner A, Aebersold R, Botchan MR, and Berger JM (2018). Conformational control and DNA-binding mechanism of the metazoan origin recognition complex. Proc. Natl. Acad. Sci 115, E5906–E5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers JL, Randell JCW, Chen S, and Bell SP (2004). ATP hydrolysis by ORC catalyzes reiterative Mcm2–7 assembly at a defined origin of replication. Mol Cell 16, 967–978. [DOI] [PubMed] [Google Scholar]
- Brézellec P, Vallet-Gely I, Possoz C, Quevillon-Cheruel S, and Ferat J-L (2016). DciA is an ancestral replicative helicase operator essential for bacterial replication initiation. Nat. Commun 7, 13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodavarapu S, Jones AD, Feig M, and Kaguni JM (2016). DnaC traps DnaB as an open ring and remodels the domain that binds primase. Nucleic Acids Res 44, 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Ilves I, Tamberg N, Petojevic T, Nogales E, Botchan MR, and Berger JM (2011). The structural basis for MCM2–7 helicase activation by GINS and Cdc45. Nat Struct Mol Biol 18, 471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig N, Green R, Greider C, Storz G, Wolberger C, and Cohen-Fix O (2014). Molecular Biology. Principles of Genome Function (Oxford University Press)
- Davey MJ, and O’Donnell M (2003). Replicative helicase loaders: ring breakers and ring makers. Curr Biol 13, R594–6. [DOI] [PubMed] [Google Scholar]
- Davey MJ, Fang L, McInerney P, Georgescu RE, and O’Donnell M (2002). The DnaC helicase loader is a dual ATP/ADP switch protein. Embo J 21, 3148–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W (2002). The PyMOL Molecular Graphics System
- Duderstadt KE, Chuang K, and Berger JM (2011). DNA stretching by bacterial initiators promotes replication origin opening. Nature 478, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueber ELC, Corn JE, Bell SD, and Berger JM (2007). Replication origin recognition and deformation by a heterodimeric archaeal Orc1 complex. Science 317, 1210–1213. [DOI] [PubMed] [Google Scholar]
- Duzdevich D, Warner MD, Ticau S, Ivica NA, Bell SP, and Greene EC (2015). The dynamics of eukaryotic replication initiation: origin specificity, licensing, and firing at the single-molecule level. Mol. Cell 58, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Erzberger JP, and Berger JM (2006). Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct 35, 93–114. [DOI] [PubMed] [Google Scholar]
- Fang L, Davey MJ, and O’Donnell M (1999). Replisome assembly at oriC, the replication origin of E. coli, reveals an explanation for initiation sites outside an origin. Mol Cell 4, 541–553. [DOI] [PubMed] [Google Scholar]
- Frick DN, and Richardson CC (2001). DNA primases. Annu. Rev. Biochem 70, 39–80. [DOI] [PubMed] [Google Scholar]
- Frigola J, He J, Kinkelin K, Pye VE, Renault L, Douglas ME, Remus D, Cherepanov P, Costa A, and Diffley JFX (2017). Cdt1 stabilizes an open MCM ring for helicase loading. Nat. Commun 8, 15720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funnell BE, Baker T. a, and Kornberg a (1987). In vitro assembly of a prepriming complex at the origin of the Escherichia coli chromosome. J. Biol. Chem 262, 10327–10334. [PubMed] [Google Scholar]
- Galletto R, Rajendran S, and Bujalowski W (2000). Interactions of nucleotide cofactors with the Escherichia coli replication factor DnaC protein. Biochemistry 39, 12959–12969. [DOI] [PubMed] [Google Scholar]
- Galletto R, Jezewska MJ, and Bujalowski W (2003). Interactions of the Escherichia coli DnaB helicase hexamer with the replication factor the DnaC protein. Effect of nucleotide cofactors and the ssDNA on protein-protein interactions and the topology of the complex. J Mol Biol 329, 441–465. [DOI] [PubMed] [Google Scholar]
- Ioannou C, Schaeffer PM, Dixon NE, and Soultanas P (2006). Helicase binding to DnaI exposes a cryptic DNA-binding site during helicase loading in Bacillus subtilis. Nucleic Acids Res 34, 5247–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsathitphaisarn O, Wing R. a, Eliason WK, Wang J, and Steitz T. a (2012). The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell 151, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Leipe DD, Koonin EV, and Aravind L (2004). Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol 146, 11–31. [DOI] [PubMed] [Google Scholar]
- Katayama T, Ozaki S, Keyamura K, and Fujimitsu K (2010). Regulation of the replication cycle: conserved and diverse regulatory systems for DnaA and oriC. Nat Rev Microbiol 8, 163–170. [DOI] [PubMed] [Google Scholar]
- Kelch BA, Makino DL, O’Donnell M, and Kuriyan J (2012). Clamp loader ATPases and the evolution of DNA replication machinery. BMC Biol 10, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelch B. a, Makino DL, O’Donnell M, and Kuriyan J (2011). How a DNA polymerase clamp loader opens a sliding clamp. Science 334, 1675–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJE (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D, Forsberg BO, Scheres SH, and Lindahl E (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobori JA, and Kornberg A (1982a). The Escherichia coli dnaC gene product. II Purification, physical properties, and role in replication. J Biol Chem 257, 13763–13769. [PubMed] [Google Scholar]
- Kobori JA, and Kornberg A (1982b). The Escherichia colidnaC gene product. III. Properties of the dnaB-dnaC protein complex. J Biol Chem 257, 13770–13775. [PubMed] [Google Scholar]
- Koonin EV (1992). DnaC protein contains a modified ATP-binding motif and belongs to a novel family of ATPases including also DnaA. Nucleic Acids Res 20, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchta RD, and Stengel G (2010). Mechanism and evolution of DNA primases. Biochim. Biophys. Acta Proteins Proteomics 1804, 1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBowitz JH, and McMacken R (1986). The Escherichia colidnaB replication protein is a DNA helicase. J Biol Chem 261, 4738–4748. [PubMed] [Google Scholar]
- Li N, Lam WH, Zhai Y, Cheng J, Cheng E, Zhao Y, Gao N, and Tye B-K (2018). Structure of the origin recognition complex bound to DNA replication origin. Nature 559, 217–222. [DOI] [PubMed] [Google Scholar]
- Liu B, Eliason WK, and Steitz TA (2013). Structure of a helicase–helicase loader complex reveals insights into the mechanism of bacterial primosome assembly. Nat. Commun 4, 2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Y-H, Tsai K-L, Sun Y-J, Chen W-T, Huang C-Y, and Hsiao C-D (2009). The crystal structure of a replicative hexameric helicase DnaC and its complex with single-stranded DNA. Nucleic Acids Res 37, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscha KV, Jaudzems K, Ioannou C, Su XC, Hill FR, Otting G, Dixon NE, and Liepinsh E (2009). A novel zinc-binding fold in the helicase interaction domain of the Bacillus subtilis DnaI helicase loader. Nucleic Acids Res 37, 2395–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlam AV, McNatt MW, Carr KM, and Kaguni JM (2001). Essential amino acids of Escherichia coli DnaC protein in an N-terminal domain interact with DnaB helicase. J Biol Chem 276, 27345–27353. [DOI] [PubMed] [Google Scholar]
- Lyubimov AY, Costa A, Bleichert F, Botchan MR, and Berger JM (2012). ATP-dependent conformational dynamics underlie the functional asymmetry of the replicative helicase from a minimalist eukaryote. Proc Natl Acad Sci U S A 109, 11999–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeill SA (2011). Protein-protein interactions in the archaeal core replisome. Biochem Soc Trans 39, 163–168. [DOI] [PubMed] [Google Scholar]
- Makowska-Grzyska M, and Kaguni JM (2010). Primase directs the release of DnaC from DnaB. Mol. Cell 37, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar D, Trakselis MA, and Benkovic SJ (2004). On the Solution Structure of the T4 Sliding Clamp (gp45). Biochemistry 43, 12723–12727. [DOI] [PubMed] [Google Scholar]
- Mott ML, Erzberger JP, Coons MM, and Berger JM (2008). Structural synergy and molecular crosstalk between bacterial helicase loaders and replication initiators. Cell 135, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DL, and Cox MM (2012). Lehninger Principles of Biochemistry (Sixth Edition) (Freeman WH).
- Neuwald AF, Aravind L, Spouge JL, and Koonin EV (1999). AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9, 27–43. [PubMed] [Google Scholar]
- O’Donnell ME, and Li H (2018). The ring-shaped hexameric helicases that function at DNA replication forks. Nat. Struct. Mol. Biol 25, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea VL, and Berger JM (2014). Loading strategies of ring-shaped nucleic acid translocases and helicases. Curr. Opin. Struct. Biol 25, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T, and Wilkinson AJ (2001). AAA+ superfamily ATPases: common structure--diverse function. Genes Cells 6, 575–597. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pomerantz RT, and O’Donnell M (2007). Replisome mechanics: insights into a twin DNA polymerase machine. Trends Microbiol 15, 156–164. [DOI] [PubMed] [Google Scholar]
- Randell JCW, Bowers JL, Rodríguez HK, and Bell SP (2006). Sequential ATP Hydrolysis by Cdc6 and ORC Directs Loading of the Mcm2–7 Helicase. Mol. Cell 21, 29–39. [DOI] [PubMed] [Google Scholar]
- Reha-Krantz LJ, and Hurwitz J (1978). The dnaB gene product of Escherichia coli. II. Single stranded DNA-dependent ribonucleoside triphosphatase activity. J. Biol. Chem 253, 4051–4057. [PubMed] [Google Scholar]
- Rosenthal PB, and Henderson R (2003). Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol 333, 721–745. [DOI] [PubMed] [Google Scholar]
- Roychowdhury A, Szymanski MR, Jezewska MJ, and Bujalowski W (2009). Escherichia coli DnaB helicase-DnaC protein complex: allosteric effects of the nucleotides on the nucleic acid binding and the kinetic mechanism of NTP hydrolysis. 3. Biochemistry 48, 6747–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui K, and Stillman B (2007). ATP-dependent Assembly of the Human Origin Recognition Complex. J. Biol. Chem 282, 32370–32383. [DOI] [PubMed] [Google Scholar]
- Soultanas P (2002). A functional interaction between the putative primosomal protein DnaI and the main replicative DNA helicase DnaB in Bacillus. Nucleic Acids Res 30, 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultanas P (2012). Loading mechanisms of ring helicases at replication origins. Mol Microbiol 84, 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck C, and Stillman B (2007). Cdc6 ATPase Activity Regulates ORC—Cdc6 Stability and the Selection of Specific DNA Sequences as Origins of DNA Replication. J. Biol. Chem 282, 11705–11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck C, Chen Z, Li H, and Stillman B (2005). ATPase-dependent cooperative binding of ORC and Cdc6 to origin DNA. Nat. Struct. Mol. Biol 12, 965–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story RM, Weber IT, and Steitz TA (1992). The structure of the E. coli recA protein monomer and polymer. Nature 355, 318–325. [DOI] [PubMed] [Google Scholar]
- Strycharska MS, Arias-Palomo E, Lyubimov AY, Erzberger JP, O’Shea VL, Bustamante CJ, and Berger JM (2013). Nucleotide and Partner-Protein Control of Bacterial Replicative Helicase Structure and Function. Mol. Cell 52, 844–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen ND, and Berger JM (2009). Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell 139, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen ND, Lawson MR, Witkowsky LB, Qu S, and Berger JM (2016). Molecular mechanisms of substrate-controlled ring dynamics and substepping in a nucleic acid-dependent hexameric motor. Proc. Natl. Acad. Sci. U. S. A 113, E7691–E7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakselis MA, Alley SC, Abel-Santos E, and Benkovic SJ (2001). Creating a dynamic picture of the sliding clamp during T4 DNA polymerase holoenzyme assembly by using fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. U. S. A 98, 8368–8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakselis MA, Berdis AJ, and Benkovic SJ (2003). Examination of the role of the clamp-loader and ATP hydrolysis in the formation of the bacteriophage T4 polymerase holoenzyme. J. Mol. Biol 326, 435–451. [DOI] [PubMed] [Google Scholar]
- Tsai K-L, Lo Y-H, Sun Y-J, and Hsiao C-D (2009). Molecular interplay between the replicative helicase DnaC and its loader protein DnaI from Geobacillus kaustophilus. J. Mol. Biol 393, 1056–1069. [DOI] [PubMed] [Google Scholar]
- Velten M, McGovern S, Marsin S, Ehrlich SD, Noirot P, and Polard P (2003). A two-protein strategy for the functional loading of a cellular replicative DNA helicase. Mol Cell 11, 1009–1020. [DOI] [PubMed] [Google Scholar]
- Wahle E, Lasken RS, and Kornberg A (1989a). The dnaB-dnaC replication protein complex of Escherichia coli. II. Role of the complex in mobilizing dnaB functions. J Biol Chem 264, 2469–2475. [PubMed] [Google Scholar]
- Wahle E, Lasken RS, and Kornberg A (1989b). The dnaB-dnaC replication protein complex of Escherichia coli. I. Formation and properties. J Biol Chem 264, 2463–2468. [PubMed] [Google Scholar]
- Wang G, Klein MG, Tokonzaba E, Zhang Y, Holden LG, and Chen XS (2008). The structure of a DnaB-family replicative helicase and its interactions with primase. Nat. Struct. Mol. Biol 15, 94–100. [DOI] [PubMed] [Google Scholar]
- Wendler P, Ciniawsky S, Kock M, and Kube S (2012). Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta 1823, 2–14. [DOI] [PubMed] [Google Scholar]
- Wickner S, and Hurwitz J (1975). Interaction of Escherichia colidnaB and dnaC(D) gene products in vitro. Proc Natl Acad Sci U S A 72, 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao N, and O’Donnell M (2016). Bacterial and Eukaryotic Replisome Machines. JSM Biochem. Mol. Biol 3. [PMC free article] [PubMed] [Google Scholar]
- Yardimci H, and Walter JC (2014). Prereplication-complex formation: a molecular double take? Nat. Struct. Mol. Biol 21, 20–25. [DOI] [PubMed] [Google Scholar]
- Yuan Z, Riera A, Bai L, Sun J, Nandi S, Spanos C, Chen ZA, Barbon M, Rappsilber J, Stillman B, et al. (2017). Structural basis of Mcm2–7 replicative helicase loading by ORC-Cdc6 and Cdt1. Nat. Struct. Mol. Biol 24, 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y, Cheng E, Wu H, Li N, Yung PYK, Gao N, and Tye B-K (2017). Open-ringed structure of the Cdt1-Mcm2–7 complex as a precursor of the MCM double hexamer. Nat. Struct. Mol. Biol 24, 300–308. [DOI] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Linear morphing of a DnaB dimer from a closed-ring/constricted state to the open-ring/constricted configuration found in the apo DnaBC complex.
Linear morphing of DnaC ATPase domains from apo to the DNA-bound configuration.
Linear morphing of DnaC N-terminal domain from apo to the DNA-bound configuration.
Linear morphing of DnaB domains within DnaBC from the apo to the nucleic acid-bound state.
Linear morphing of DnaB domains within DnaBC from the apo to the nucleic acid-bound state.
Linear morphing of DnaBC movements accompanying helicase binding, opening, and ssDNA loading.