

Abstract
The use of Drosophila cell cultures has positively impacted both fundamental and biomedical research. The most widely used cell lines: Schneider, Kc, the CNS and imaginal disc lines continue to be the choice for many applications. Drosophila cell lines provide a homogenous source of cells suitable for biochemical experimentations, transcriptomics, functional genomics and biomedical applications. They are amenable to RNAi and serve as a platform for high-throughput screens to identify relevant candidate genes or drugs for any biological process. Currently, CRISPR-based functional genomics are also being developed for Drosophila cell lines. Even though many uniquely derived cell lines exist, cell genetic techniques such the transgenic UAS-GAL4 based RasV12 oncogene expression, CRISPR-Cas9 editing and Recombination Mediated Cassette Exchange (RMCE) are likely to drive the establishment of many more lines from specific tissues, cells or genotypes. However, the pace of creating new lines is hindered by several factors inherent to working with Drosophila cell cultures: single cell cloning, optimal media formulations and culture conditions capable of supporting lines from novel tissue sources or genotypes. Moreover, even though many Drosophila cell lines are morphologically and transcriptionally distinct it may be necessary to implement a standard for Drosophila cell line authentication, ensuring the identity and purity of each cell line. Altogether, recent advances and a standardized authentication effort should improve the utility of Drosophila cell cultures as a relevant model for fundamental and biomedical research.
Graphical Abstract

Introduction
Ever since the first immortalized Drosophila cell lines were created (G. Echalier & Ohanessian, 1969; Gvozdev, Birshtein, Kakpakov, & Polukarova, 1971; Kakpakov, Polukarova, & Gvozdev, 1971), Drosophila cell lines have become a valuable tool to not only complement in vivo work but also as a primary tool for the discovery of basic and conserved biological questions. Like mammalian cell lines, these cell lines can be passaged many rounds without the effects of senescence; they can be cryopreserved and then revived to be cultured again.
The use of Drosophila cell culture provides: (1) The rapid testing of potential in vivo transgenic constructs for proper biochemical and cellular functions before creating transgenic flies; (2) The ability to follow the immediate responses to a stimulus in a population of homogenous cells; (3) A vast set of molecular reagents available for genetic manipulation, including CRISPR-Cas9 editing tools and many expression-ready tagged ORF cDNA collections; (4) The amenability to high-throughput functional genomics, including the well-developed protocols for chemical screens and RNA interference (RNAi); (6) Transcriptomics for at least 41 different cell lines (L. Cherbas et al., 2011; Stoiber, Celniker, Cherbas, Brown, & Cherbas, 2016; Wen et al., 2014).
Currently, there are over 150 unique cell lines available at the Drosophila Genomics Resource Center (DGRC). These cell lines were mostly derived from Drosophila melanogaster, including a few lines from other Drosophila species. The five most widely distributed Drosophila melanogaster cell lines are the embryonic-derived S2R+, S2-DGRC, S2-DRSC, Kc167 and the larval CNS-derived ML-BG3-c2 cell lines (DGRC, unpublished). Other cell lines used in the model organism Encyclopedia of DNA Elements (modENCODE) project are popular as well.
Recent advances in Drosophila somatic cell genetics, including the use of RasV12 transgene, CRISPR-Cas9 genome editing and RMCE, are likely to increase the number of new cell lines. Here, we begin by covering the major applications of Drosophila cell lines in both fundamental and biomedical research, emphasizing their suitability for high-throughput screens. We then review the various methods used to generate the main collection of existing Drosophila cell lines and discuss best practices for working with them. Finally, we present current challenges associated with working with Drosophila cell lines and the opportunities to improve the utility and relevance of the system for both fundamental and biomedical research.
Drosophila cell cultures: impact on diverse biological research areas
How researchers have used Drosophila cell lines is too broad to cover in this review. Here, we highlight the major applications for the most commonly used lines and how they have been utilized successfully to address various biological questions.
Extensive transcriptional and genome data are available for many of the lines, which is often an entry point to choosing the right line to study the biological process of interest (L. Cherbas et al., 2011; H. Lee et al., 2014a; Stoiber et al., 2016; Wen et al., 2014). Drosophila cell lines have been used to study various topics in biology ranging from biomedical applications, signal transduction, metal biology, metabolism, cell morphology, innate immunity and many others. Notably, Drosophila cell cultures are suited for high-throughput functional genomic studies due to its amenability to RNAi. We highlight below research topics and applications that have benefited from using Drosophila cell culture system.
Biomedical applications: Schneider lines
Drosophila cell lines offer several advantages over mammalian cell cultures for biomedical applications. The system is easier to work with, relatively simpler and less expensive. An important attribute for studying many evolutionarily conserved processes, Drosophila has a well-annotated genome that retains a high proportion of non-redundant genes with confirmed mammalian orthologs. Its smaller genome allows for the design of sequence-specific RNAi/CRISPR reagents with lower risks for inadvertently targeting similar sequences elsewhere. Furthermore, the high-throughput scalability of using Drosophila cell lines is relevant for biomedical applications such as chemical screening and drug discovery.
Drosophila cell lines are relatively easier for transgenesis.S2R+ and Kc167 cells are also capable of processing T2A-sequence polypeptide self-cleavage and express multicistronic vectors (Gonzalez et al., 2011). These attributes are relevant to the expression of therapeutic recombinant proteins. While mammalian cells are the main system for commercial production of therapeutic proteins, it remains an expensive model. There have been efforts to optimize the S2 cells to produce therapeutic vaccines. Adhering to current Good Manufacturing Practice (cGMP) standards, S2 cells were recently used to produce a full-length Plasmodium falciparum reticulocyte-binding protein homolog 5 (PfRH5) vaccine against malaria (Manoff et al., 2015). The immunogenic recombinant protein vaccine, determined to be suitable for human testing, has made it to phase I/IIa clinical trial. Other S2-derived vaccine products against Dengue virus also made it to clinical trials (Manoff et al., 2015). Therefore, the prospect of using Drosophila cell cultures to produce other recombinant protein vaccines is promising (Jin et al., 2018). Furthermore, the ease for stably transforming Drosophila cells is also relevant for the development of biosensor-based diagnostic for disease markers (H. C. Lau et al., 2015).
Drosophila immune response genes have homologs in mammals.The most widely used Drosophila lines are of hemocyte origins that exhibit intact innate immune response genes. Studies using these lines helped to elucidate host-pathogen requirements for infection and identify new targets for drug development. In fact, Drosophila cell cultures can support the complete in vitro development of the malaria parasite, Plasmodium falciparum (Warburg & Schneider, 1993) and Plasmodium berghei (Hurd, Al-Olayan, & Butcher, 2003). Drosophila cell lines have also been used to study other human pathogens including the infection processes of Chlamydia trachomatis, Listeria monocytogenes, Mycobacteria marinum, and Ehrlichia chaffeensis (Luce-Fedrow, Von Ohlen, Boyle, Ganta, & Chapes, 2008).
Receptor-ligand interaction: Schneider lines
Signal transduction underlies cell-to-cell communication. Cell communication is critical for many biological processes that occur during organismal development: cell fate determination, division, movement and apoptosis, among many others.
The cellular interaction between the Notch receptor and its Delta ligand was demonstrated using S2 cells (Fehon et al., 1990; Rebay et al., 1991). S2 cells normally do not express these proteins, but when transiently expressed in separate populations, Delta-Notch interactions and trans-endocytosis were observed via cell aggregation assays. More recently, Schneider lines were used as a platform to probe receptor-ligand interaction in a high throughput manner, in what was termed the “Extracellular Interactome Assay”(Ozkan et al., 2013). The group curated and expressed 202 clones for extracellular domains (ECD) of Immunoglobulin Superfamily (IgSF) and Leucine-rich repeat (LRR)-containing family of proteins, in bait and prey format to assay for receptor ligand interaction in up to 42,000 pairwise experiment. Overall, the screen identified pairwise interacting candidates that may be relevant in vivo (Ozkan et al., 2013).
Signal transduction pathways: Schneider lines, Kc, Cl.8
Drosophila cell lines provide a suitable model for studying cell signaling pathways (Ammeux, Housden, Georgiadis, Hu, & Perrimon, 2016; L. Cherbas et al., 2011). The evolutionarily conserved signaling pathways in Drosophila cell cultures (Notch, Insulin, EGFR, Wnt, JAK/STAT, Hippo, JNK, TGF-beta/BMP, Hedgehog, TNF-a, and PVR) have low activity in many cell lines and they can be further induced by exogenous ligands (Ammeux et al., 2016; Franz, Shlyueva, Brunner, Stark, & Basler, 2017; Lum et al., 2003; Stoiber et al., 2016; C. Zhang, Williams, Guo, Lum, & Beachy, 2004).
The ability to track immediate responses to a ligand, in a model that provides a homogenous cell population, is ideal in distinguishing direct or indirect secondary responses in signaling pathways. This type of cell-based analysis allows for systems-level analysis of signaling pathways, in which variables such as signal strength or duration may be systematically tested to determine the signaling output that would elicit a meaningful biological outcome. Detailed assay methods and relevant reagents for the various signaling methods are available in the following research and review articles (Albert & Bokel, 2017; J. Li, Housden, & Bray, 2014; Shaposhnikov et al., 2013; Vincent, 2014).
Circadian rhythm biology: Schneider lines
S2 cells have been successfully used to recapitulate certain aspects of the circadian clock. S2 cells do not express circadian proteins and this simplified system continues to be ideal for addressing the regulatory mechanisms of the CLOCK proteins. For example, using S2 cell and luciferase-based transcription assay, the transcription of period (per) and timeless (tim) were found to be directly regulated the activity of the CLOCK (CLK) and Cryptochrome C (CYC) dimer. Furthermore, the heterodimer of PER and TIM were found to have inhibitory control on CLOCK/CYC activity (Darlington et al., 1998). In addition, post-translational regulation and degradation of PER, TIM and CRY were determined using S2-based assays (Ko, Jiang, & Edery, 2002; Koh, Zheng, & Sehgal, 2006; F. J. Lin, Song, Meyer-Bernstein, Naidoo, & Sehgal, 2001).
Metal metabolism or homeostasis: Schneider lines
Metal ion homeostasis is important for healthy cellular functions. Drosophila cell culture has proved to be a highly suitable system to study the impact of metal imbalance. For example, the role of iron in cell cycle progression has been tested in mbn2 cells (Metzendorf & Lind, 2010). Recently, S2R+ cells have been utilized as a genomic cell-based screening platform to identify factors that are responsible for zinc homeostasis (S. E. Mohr et al., 2018).
Cellular stress response: Kc167 and Schneider lines
Stress granules are cytoplasmic ribonucleoprotein particles that form when cells experience various stresses. Kc167 and S2 cells are commonly used to biochemically assay and visualize the regulation and formation of cytoplasmic stress granules after various chemical treatments or genetic modifications (Aguilera-Gomez et al., 2017; Jevtov et al., 2015; Luhur, Buddika, Ariyapala, Chen, & Sokol, 2017).
Neuronal morphology, cell migration and communication: Miyake laboratory lines
Nineteen CNS derived lines from the Miyake laboratory are available from the DGRC (Ui, Ueda, & Miyake, 1987), and we briefly profile a few lines for their utility.
ML-BG2-c2 line is useful for studying mitochondrial functions in dendritic arborizations and neurite extensions (T. Y. Lin et al., 2009; Minin et al., 2006; Tsuyama, Tsubouchi, Usui, Imamura, & Uemura, 2017). Larval CNS derived ML-BG3-c2 line displays neuronal characteristics (Ui et al., 1994). It has been used as a neuronal milieu to express microRNA-processing factors to the analysis of small RNA processing kinetics (Chawla & Sokol, 2014; Luhur, Chawla, Wu, Li, & Sokol, 2014). ML-DmD17-c3 (D17) cells, derived from third instar larval haltere disc are robustly motile, amenable to RNAi and has been used to study cell migration and communication (J. D. Currie & Rogers, 2011; D. Zhang et al., 2011).
Innate immunity: mbn2, tumorous blood cell line
The lethal (2) malignant blood cell line, commonly referred to as the mbn2 cell line, was derived from tumorous larval blood cells (Gateff et al., 1980). This line displays phagocytic activity, immuno-competency and has been used to study the various aspects of Drosophila innate immunity (B. Liu et al., 2016; Nonaka et al., 2017; Samakovlis, Asling, Boman, Gateff, & Hultmark, 1992).
Functional genomic: RNAi in Drosophila cultured cells
Drosophila cell lines are amenable to RNAi (Clemens et al., 2000; Ulvila et al., 2006). Most cells take up dsRNA easily from the culture media without the use of lipid transfection agent (Clemens et al., 2000). For increased efficiency and prolonged knockdown duration, dsRNA/shRNA expression constructs can be transfected (Baum & Cherbas, 2008; Zhou, Mohr, Hannon, & Perrimon, 2013).
The use of Drosophila cell cultures is highly suited for RNAi-based high throughput functional genomic analysis (S. E. Mohr, 2014; S. E. Mohr, Smith, Shamu, Neumuller, & Perrimon, 2014). Well-designed RNAi screens have contributed to understanding the regulation of many basic biological processes (Boutros & Ahringer, 2008; S. Mohr, Bakal, & Perrimon, 2010). Screen design, reagent libraries, assay development and analysis have been extensively covered elsewhere (Billmann & Boutros, 2016; Boutros & Ahringer, 2008; S. Mohr et al., 2010; S. E. Mohr, 2014). RNAi screening centers such as the Drosophila RNAi Screening Center (DRSC)/ Transgenic RNAi Project (TRiP) Functional Genomics Resources, the New York University (NYU) RNAi Core, the Division of Signaling and Functional Genomics (Boutros laboratory, DKFZ, Heidelberg, Germany) and the Sheffield RNAi Screening Facility (SRSF) offer cell-based RNAi reagents and assist researchers in conducting and analyzing high-throughput RNAi screens. Information on databases for RNAi screen results, online resources for screen design and analysis have been reviewed comprehensively elsewhere (Echeverri & Perrimon, 2006; Hu et al., 2017; Hu et al., 2013; S. E. Mohr, Hu, Kim, Housden, & Perrimon, 2014; S. E. Mohr, Smith, et al., 2014).
The Schneider lines, Kc167, ML-BG2 and Cl.8 have been used for functional genomic screens. The ease of culture, RNAi amenability, semi-adherent properties, distinct morphologies and the availability of transcriptomic data for these lines are major draws. With genome engineering and the oncogene RasV12 method for generating novel cell lines, it is conceivable that cell lines of more diverse origins or cell lines with known genotypes will be utilized for RNAi screens in the future (Housden et al., 2015; S. E. Mohr, Smith, et al., 2014).
Using multiple cell lines in a functional genomics screen to query the regulation of a cellular process can facilitate a comparative analysis and identify functionally similar or related genes (Boutros et al., 2004; T. Liu, Sims, & Baum, 2009; Rohn et al., 2011). This may be important because cell lines have very distinct gene expression profiles, and will likely help identify common or cell-specific regulators of cellular processes.
There are many strategies that complement high-throughput RNAi screens in Drosophila cell lines. Genetic modifier screens that use double RNAi (Horn et al., 2011; Nir, Bakal, Perrimon, & Berger, 2010), genetically sensitized cells (Housden et al., 2017; Housden et al., 2015) or small molecules (Albert & Bokel, 2017; Castoreno et al., 2010; Eggert et al., 2004; Serbus et al., 2012; Shah et al., 2012; Smurnyy, Toms, Hickson, Eck, & Eggert, 2010) have successfully identified new genetic interactions and drug targets. Transcriptome analysis can also complement high-throughput RNAi screens. The hits from the Drosophila cell-based zinc toxicity RNAi screen, along with transcriptomics data from wildtype and genetically Zn-sensitized mutant cells stressed by mild Zinc supplementation, is a comprehensive list of candidates genes relevant for furthering metal homeostasis research in vivo (S. E. Mohr et al., 2018).
Data analysis and validation are key steps of cell-based RNAi screen. For detailed discussion on the topic, including assay readouts, bioinformatics, molecular verification, validation using genome-editing approaches and cross-species genomic rescue strategy, see (Kondo, Booker, & Perrimon, 2009; S. Mohr et al., 2010; S. E. Mohr, 2014; S. E. Mohr, Hu, et al., 2014; S. E. Mohr & Perrimon, 2012; S. E. Mohr, Smith, et al., 2014).
CRISPR-Cas9 mediated transcriptional inactivation (CRISPRi) and activation (CRISPRa)
The complete loss of essential genes leads to lethality. CRISPRi can facilitate the analysis of essential gene functions by reducing its expression levels in a controlled manner.
Ghosh et al. demonstrated CRISPRi using Cl.8 cells, in which the transcriptional repression of roX locus is achieved by expressing catalytically inactive dCas9 (dead Cas9) together with specific guide RNAs (gRNA) (Ghosh, Tibbit, & Liu, 2016). Depending on the location where dCas9 is targeted, it can repress gene expression by blocking transcription initiation at the transcriptional start site (TSS), or by blocking transcriptional elongation (Qi et al., 2013).
The strength of repression by CRISPRi can be tuned. The use of multiple gRNAs with dCas9 increased the level of suppression, suggesting that the increased numbers of dCas9 binding near the target TSS enhanced the repression (Ghosh et al., 2016). An alternative method that fused various repressor domains to dCas9, which has been shown to be functional in mammalian cell cultures (Gilbert et al., 2013), has not been tested in Drosophila lines.
CRISPR can also mediate transcriptional activation (CRISPRa) of specific genes in Drosophila cell lines. The fusions of various individual activator domains (e.g. VP64, p65, Rta) or a chimera of VP64-p65-Rta (VPR) to dCas9 are generally effective for activating the transcription for multiple genes in S2R+ cells (Chavez et al., 2015; Chavez et al., 2016; S. Lin, Ewen-Campen, Ni, Housden, & Perrimon, 2015). The VPR chimera performed well for many tested genes. Other promising strong activators tested include the SAM and Suntag systems (Chavez et al., 2016).
CRISPR-based screen in Drosophila cell lines
Unlike RNAi knockdowns, mutations induced by CRISPR-mediated knockouts generally result in the complete loss of gene function. This feature allows the identification of essential genes; however, the function of essential genes cannot be easily probed with CRISPR knockouts.
Importantly, CRISPR screen in Drosophila cell lines can carried out in a pooled format (Bassett, Kong, & Liu, 2015; Okamoto et al., 2018; Viswanatha, Li, Hu, & Perrimon, 2018). CRISPRi and CRISPRa may also be used in a pooled format, and the strength of the repression or activation may be tuned. CRISPRa may offer a highly scalable approach to perform screens based on the overexpression from endogenous loci. In contrast, similar ORF overexpression screens would involve challenges such as the cloning of multiple isoforms and different transfection efficiencies due to construct size variations. There are limitations associated with gRNA design and potential off-target effects (OTE) in CRISPR-based functional genomics. The guidelines for gRNA design and discussion on the underexplored gRNA OTE in Drosophila cell lines have been covered elsewhere (S. E. Mohr et al., 2016). These guidelines may evolve, as more CRISPR-based screens are carried out and data pertaining to OTE becomes available.
GENERATING DROSOPHILA CELL LINES
The criteria for cultured Drosophila cells to be considered a permanent cell line has been proposed rather arbitrarily, ranging from 40 subcultures to 120 mitoses (Schneider & Blumenthal, 1978; A. A. Simcox, Sobeih, & Shearn, 1985). However, in most cases, Drosophila cell line immortality seems to be achieved after 10 passages, or an average of 6 months (Table 1).
Table 1.
A survey of the starting materials, approaches and results from the various laboratories in which Drosophila cell lines had been established.
| Echalier & Ohanessian, 1970 | Schneider, 1972 | Debec, 1978 | Milner Lab, 1988 | Miyake Lab 1987, 1994 | Simcox et al., 2008 | Lecland et al., 2013 | |
|---|---|---|---|---|---|---|---|
| Sample | e/se | Ore-R | 1182ts | Ore-R | y v f mal | Act-GAL4, UAS-GFP Act-GAL4, UAS-RasV12 | DSas-4 |
| Tissue source | 6–12h embryo | 20–24h embryo | Embryo | Late L3 discs | 4–8h embryo, Larval discs and CNS | Embryo | Embryo |
| Surface treatment & sterilization (min) | 2.5% NaOCl (10) | 2.5% NaOCl, 70% EtOH (20) 0.05% HgCl2 70% EtOH (~ 5) | 2.5% NaOCl, (10) | 50% NaOCl (5) | 2.5% NaOCl (2) 10% saponated cresol (10) | 50% bleach (3–5) 0.02% Triton-X | 40% bleach (15) 0.025% Triton-X |
| Homogenizer (Volume) | Conical glass homogenizer (N/A) | Chopped 0.2% trypsin 20–45′ (1.25 ml) | Conical glass homogenizer (N/A) | Cut, PBS, 0.1% Trypsin, 2ng/ml dispase, 45′ (0.8 ml) | 5ml Potter homogenizer Teflon pestle, 0.2mm clearance (5 ml) | 5ml Teflon pestle (3 ml) | 2ml Tenbroeck grinder (2 ml) |
| Media | D-22 10–20% FCS | Schneider’s 15% FCS Bactopeptone Vitamins | M3:D-22 (1:1) 20% FCS | M3* 2 % FCS Insulin Fly Extract | Modified M3 10 % FCS Insulin | Schneider’s 10% FCS | M3 20% FCS |
| Flask (# of strokes) | - (N/A) | 9 cm2 (N/A) | - (N/A) | 96 well (2–3) | 35mm plates (several) | 25cm2 (3) | 12.5 cm2 (5–7) |
| Antibiotic | - | - | - | - | - | + | + |
| Duration PC to confluence (Vessel size) | 5–6 mo (<25cm2) | 3 wk – 8 mo (9 cm2) | 4 wk (NA) | 4 wk (96 well) | Disc: 4– 12 wk CNS: 4– 8 wk (35mm plates) | Control: 16–29 wk RasV12: 3–6 wk (25cm2) | 3–8 wk (12.5 cm2) |
| Duration to passage 10 (temp) | > 5–6 mo (25–27°C) | 3– 8 mo (22°C) | 5–6 mo (19°C) | 5–6 mo (25°C) | 5–8 mo (25°C) | Control: 12–18 mo RasV12: 5–8 mo (22°C) | up to 6 mo (23°C) |
| % Immortalized (n) | NA | NA | NA | 44.1% (15/34) | Disc: 21% (4/19) CNS: 44% (8/18) | Control: 11.1% (3/27) RasV12: 96.4% (27/28) | 8.4% (11/131) |
Supplemented with 0.125 i.u/ml insulin, 1ng/ml 20-HE and 5% fly extract
Drosophila cell lines originate from primary cell cultures, in which some cells spontaneously achieve immortality likely through mutations that perturb tumor suppressor genes and/or activate oncogenes. Many Drosophila cell lines were established from tissues of wildtype (Debec, 1978; G. Echalier & Ohanessian, 1970; Schneider & Blumenthal, 1978) or mutant animals displaying phenotypes such as temperature sensitivity (Debec, 1978; A. A. Simcox et al., 1985; Woods & Poodry, 1984), lethality (Gateff et al., 1980) or female sterility (Niki, Yamaguchi, & Mahowald, 2006). Other cell lines were derived from larval tissues such as the CNS and the imaginal discs (D. A. Currie, Milner, & Evans, 1988; Ui et al., 1994; Ui et al., 1987) (Table 1).
Generating Drosophila cell lines de novo from tissues
Many of the cell lines are derived from the different stages of embryonic development. At the DGRC, 62% (111/180) of the currently distributed cell lines have embryonic origins, 37% (67/180) are derived from larval tissues, and the remaining 1% (2/180) are from adult organs.
Developmentally staged embryos (between 3–24 hours after egg laying) are composed of many cell types and from these embryonic primary cultures, some fraction of cells become immortalized. Though it can be difficult to determine the origin or lineage of the line, the method for deriving these cell lines is straightforward (Debec, Megraw, & Guichet, 2016). Briefly, embryos from flies of the desired genotype were collected and aged. The embryos were surface sterilized and then gently homogenized. The resulting primary cultures were maintained between 23–25°C with routine media changing. When the primary cultures had become confluent, they were subcultured until spontaneous immortalization was achieved.
Generating Drosophila cell lines from primary cultures is both an art and science. Many factors can influence the efficiency as well as the effectiveness of establishing a cell line (Table 1). We refer readers to a detailed protocol by (Debec et al., 2016), from which a number of interesting cell lines have been established recently: haploid 1182–4H cell line (Debec, 1978), GFP-Jupiter (Karpova, Bobinnec, Fouix, Huitorel, & Debec, 2006) for studies on microtubule dynamics during mitosis, JW18 and LDW1 lines (Serbus et al., 2012; White et al., 2017) for the identification of compounds affecting Wolbachia, and acentriolar Jup-GFP-DSas-4 lines for studies of mitotic spindle apparatus assembly (Lecland et al., 2013).
It is important to start with many primary cultures because most will eventually undergo arrested growth, differentiation or senescence. Typically, only 8–10% of primary cultures will become immortalized (Lecland et al., 2013). The efficiency of immortalization is also dependent on the technique and the genotype of the animal (Table 1).
The genotype of the animal influences the success rate of establishing cell lines. For reasons unknown other than the relative efficiency (21–44%), the Miyake laboratory consistently used the fly strain bearing X-linked mutations: yellow, vermilion, forked, maroon-like (y v f mal) as a tissue source for generating imaginal disc and CNS lines (Ui et al., 1994; Ui et al., 1987). Similar attempts to establish CNS cell lines derived from animals with a different genetic background known to be efficient for establishing embryonic cell lines produced only one immortalized CNS line out of 25 primary cultures (Ui et al., 1994).
The loss of tumor suppressor function can increase the efficiency of establishing cell lines. Two notable lines were created based on deregulated cellular proliferation in the animal. The mbn2 cell line originated from neoplastic primary cell cultures derived from larval blood cells mutant for l(2)mbn (Gateff et al., 1980). l(2)mbn larvae were punctured to collect the larval hemocytes, and the tumorous blood cells were directly cultured a dish. The cell line was “immortalized” within six weeks as compared to many months needed for other classical embryonic lines (Table 1). Similarly, the co-culture of female Germline Stem cell/ Ovarian Somatic Sheath (fGS/OSS) cell line was established from bag of marbles (bam) mutant ovaries. The tumor suppressor mutant germ tissues had abnormal cysts that contain excess number of tumorigenic and undifferentiated cells (Niki et al., 2006).
Transgenic overexpression of constitutively active oncogene RasV12
The expression of oncogenic RasV12 is an effective genetic method to promote proliferation in Drosophila primary culture cells. Primary cultures expressing RasV12 were immortalized or reached the tenth passage within 5–8 months, while control cultures expressing UAS-GFP transgene took 12–18 months (Table 1). Notably, the duration to immortalization in RasV12 cell lines was comparable to other existing cell lines (Table 1). Most importantly, expressing RasV12 increased the efficiency of establishing cell lines dramatically (Table 1). The expression of RasV12 promotes cellular proliferation via the increased activity of MAPK and the insulin signaling pathway (A. Simcox et al., 2008). One caveat of using the RasV12 cell line is the extent to which endogenous signaling pathways have been altered. Therefore, proper controls are important when using these cells for signal transduction studies.
The RasV12 lines were derived from embryos ubiquitously expressing RasV12 and their cellular origins were not clear initially (A. Simcox et al., 2008). Expression profiling revealed that these cells have characteristic signatures that most resemble ML-DmD8 (Ui et al., 1987), a wing disc cell line with adult muscle precursors (AMPs) characteristics (L. Cherbas et al., 2011; Dequeant et al., 2015). RasV12 cells retain the ability to differentiate into muscle cells when treated with ecdysone (Dequeant et al., 2015). The basis for the proliferative and undifferentiated state of RasV12 lines may be attributed to the increased activity of genes in the E2 promoter binding factor/Retinoblastoma (E2F/Rb) and the Polycomb Group (PcG) pathways, respectively (Dequeant et al., 2015). The RasV12 approach has been successfully used to create several cell lines from animals with many specific genotypes (Table 2).
Table 2.
Drosophila cell lines derived from using the RasV12 strategy.
| Cell line | Biological relevance | Ref |
|---|---|---|
| RasV12; wtsRNAi | piRNA pathway biogenesis, transposon, proliferation | a |
| rumi26, RasV12 | Notch signaling pathway | a, b |
| Df(2R)Su(z) 2–1.b8, RasV12 | Deletion removing Psc and Su(z)2 genes. Epigenetic | c |
| Su(z)124, RasV12 | Epigenetic studies | d |
| Pc3, RasV12 | Epigenetic studies | d |
| EZ2–2: E(z)61, RasV12 | Epigenetic studies | d |
| LoqsKO, RasV12 | miRNA gene silencing | e |
| RasV12 attP-L1 | RMCE de novo | [ |
Targeted loss of tumor suppressors
The loss of tumor suppressors, either alone or in conjunction with activating oncogenic mutations, can impart immortality to mammalian cells (Payne & Kemp, 2005). A list of mutant tumor suppressor genes was systematically tested for their effectiveness and efficiency to immortalize Drosophila primary cultures. Notably, the loss of neoplastic tumor suppressors such as scrib, dlg1, lgl and brat did not promote cell line establishment, while mutations in the hyperplastic class of tumor suppressors from the members of the Hippo pathway (hpo and wts) and the insulin pathway (pten) were likely to accelerate immortalization (Justiniano, Mathew, Mitra, Manivannan, & Simcox, 2012).
The loss of a specific tumor suppressor such as pten offers an important alternative strategy to establish cell lines. PTEN (Phosphatase and tensin homolog) functions as a tumor suppressor in the insulin signaling pathway. Pten117 primary cultures displayed accelerated growth, as fast as primary cultures from transgenic RasV12 lines and yet combining the two did not result in additional improvements (Justiniano et al., 2012). There are characteristic differences between the established Pten and RasV12 lines. Unlike RasV12 lines, Pten lines generally have longer doubling time and a lower confluent density. Interestingly, RasV12 expressing lines fail to reach high confluent density in a Pten mutant background. The targeted loss of pten offers an alternative approach to establish a cell population with a signaling state different from RasV12 lines, facilitating a different context for the genetic analysis of other mutations. Other cell lines that have been derived based on the loss of tumor suppressors include the fGS/OSS from bam adult ovaries (Niki et al., 2006) and the mbn2 cell line (Gateff et al., 1980).
Opportunities and potential improvements
With UAS-GAL4 and transgenic RasV12 genetic approach, there is immense potential for generating cell-type or stage-specific cell lines. There are specific GAL4 drivers that direct expression in for example the neuronal, glial cell lineages or the adult intestine epithelia. With genome engineering, it may be possible to establish such lines from wildtype or animals carrying known mutations. The main challenge lies with finding the optimal media formulations or culture conditions to establish and maintain these lines.
Cell lines derived from specific tissues appear to have different media requirements. Most cell lines have been derived with M3 and Schneider’s media. Systematically testing other media such D-22, CCM3 (Hyclone) or EX-CELL 420 (Sigma) may help discover media that support the immortalization process of a distinct cell types. Furthermore, media supplementations such as fly extract and insulin have promoted the establishment of CNS, imaginal disc, ovarian cell lines (D. A. Currie et al., 1988; Niki et al., 2006; Saito et al., 2009; Ui et al., 1994; Ui et al., 1987) and may be useful in establishing new lines.
The components of fly extract are largely unknown, and there has yet to be a systematic assessment of whether there are relevant differences between extracts derived from various developmental stages or genotypes. For instance, using fly extracts derived from animals over-expressing DILP2 may circumvent the need of supplementing with xenogenic insulin sources.
It has been suggested that the endosymbiont Wolbachia can increase the efficiency of establishing cell lines (Debec et al., 2016). The maternally transmitted intracellular bacteria are commonly found in many Drosophila stocks. Although the mechanism is unknown, primary cultures from animals infected with Wolbachia exhibited significantly higher rates of immortalization ranging from 25% (Serbus et al., 2012; White et al., 2017) to 70% (Debec et al., 2016). The presence of Wolbachia in cell lines alters endoplasmic reticulum (ER) morphology (White et al., 2017) but how the presence of Wolbachia is affecting the growth of these cell lines, compared to treated lines, is not very well studied.
Editing the genome of existing cells to create novel lines
Stable transformation of existing cell lines
Transfection protocols in Drosophila cell lines are well established and have been used successfully to generate many different stable cell lines. Commercial liposome-based transfection agents can deliver exogenous DNA into most Drosophila cell lines, although with varying efficiencies among different lines (Baum & Cherbas, 2008). Transfected cells can take up many DNA plasmid molecules, such that multiple different plasmids can be introduced concurrently (Baum & Cherbas, 2008; Lucy Cherbas & Cherbas, 2000; L. Cherbas & Cherbas, 2007; L. Cherbas, Moss, & Cherbas, 1994).
To generate stably transformed cell lines, antibiotic resistance or fluorescence markers can be used to enrich and select for the cells that bear random transgene insertions into the genome. The stably transformed cells can then be further propagated. One major drawback of this approach is the inability to control the strength of transgene expression due to the heterogenous transfected cell population. Often the inserted gene forms unstable tandem arrays with the propensity to expand or be deleted due to homologous recombination over time (L. Cherbas & Cherbas, 1997). As a result, the copy number of the integrated DNA, and therefore the strength of transgene expression cannot be easily controlled, even after single cell cloning. To address this issue, there were significant efforts to create new and engineer existing cell lines such that it has become possible to stably insert single copy transgenes at known genomic loci in cell lines (L. Cherbas, Hackney, Gong, Salzer, Mauser, et al., 2015; Manivannan et al., 2015). This is important especially for experiments that assay for the effects of different transgenes, so that transgenes can be expressed at comparable levels.
Using P-element transformation, Cherbas et al. transformed existing Kc167 and Sg4 lines to carry a single attP flanked GFP cassette at multiple random loci (L. Cherbas, Hackney, Gong, Salzer, Mauser, et al., 2015). In contrast, Manivannan et al. generated de novo Ras-attP1 and Ras-attP2, two act-GAL4 driven RasV12 cell lines derived from flies that harbor existing attP-flanked cassettes at known genomic loci (Manivannan et al., 2015). Both approaches demonstrated successful Recombination Mediated Cassette Exchange (RMCE) facilitated by φC31 integrase (Bateman, Lee, & Wu, 2006) via the use of targeting constructs containing attB sites. The resulting stable cell lines carry single copy transgenes of inducible mCherry, Tubulin, Jupiter or Cas9 at defined genomic loci (L. Cherbas, Hackney, Gong, Salzer, Mauser, et al., 2015).
CRISPR mediated gene editing
CRISPR-Cas9 has been used to edit the genome of at least three Drosophila cell lines (Bassett, Tibbit, Ponting, & Liu, 2014; Bottcher et al., 2014; Franz, Shlyueva, et al., 2017; Ishizu, Sumiyoshi, & Siomi, 2017; Kunzelmann, Bottcher, Schmidts, & Forstemann, 2016; H. Lee et al., 2014a; S. E. Mohr et al., 2018). The list of genome-edited Drosophila cell lines is likely to grow rapidly. CRISPR-Cas9 has been successfully used to edit the genome of pseudo-tetraploid cells such as S2R+ and Kc167 cells. Recent loss of function mutants generated in S2R+ lines include Stat92E, TSC1, TSC2, ZnT63C, IA2, loquacious and r2d2 (Bassett et al., 2014; Housden et al., 2015; S. E. Mohr et al., 2018; Tants et al., 2017). Genes knocked out in another pseudo-tetraploid Kc167 lines include armadillo and pangolin (Franz, Shlyueva, et al., 2017). This suggests the possibility of CRISPR-based genome modifications for other Drosophila cell lines with lower ploidy. In fact, the mainly diploid adult ovary derived OSC cells has successfully been edited to carry a null mutation of the tumor suppressor gene lethal (3) malignant brain tumor, creating Δmbt-OSC line (Sumiyoshi et al., 2016). Detailed protocols from recent publications are available for generating CRISPR-modified cell lines (Franz, Brunner, & Basler, 2017; Housden et al., 2015; Ishizu et al., 2017; Kunzelmann et al., 2016).
The growth characteristics of a stable continuous cell line can be modified by knocking out specific tumor suppressor genes. For example, when S2R+ cells carry with TSC1/2 mutations, the cell lines are bigger and require no serum no proliferate (Housden et al., 2015). Not surprisingly, the molecular characteristics of Δmbt-OSC cells have been altered because of the modification. The cells phenocopied undifferentiated germ cells that accumulate in the mutant ovaries and ectopically express germline markers Aub, Vasa and AGO3 (Coux, Teixeira, & Lehmann, 2018; Ishizu et al., 2017; Saito et al., 2009; Sumiyoshi et al., 2016).
Genes such as ago1, pgk, tub56D, or act5C loci have been epitope-tagged in S2 cells (S2R+) (Bassett et al., 2014; Bottcher et al., 2014; Kunzelmann et al., 2016). In addition, the zucchini locus from the mainly diploid OSC line has been successfully FLAG-tagged at its 3′ end (Ishizu et al., 2017). Notably, more than half of the edited OSC clones represent bi-allelic modifications.
CRISPR-Cas9 /Recombination Mediated Cassette Exchange (RMCE)
In order to engineer cell lines capable of RMCE, CRISPR-Cas9 can be used direct the placement of attP cassette docked at defined genomic locations, with the option of concurrently inactivating a gene of interest. This has has been successfully done in flies (X. Zhang, Koolhaas, & Schnorrer, 2014). In addition, CRISPR-Cas9 may facilitate the generation of a cell line that carries a known copy number of transgenes at desired genomic loci. Together with the UAS-RasV12 approach and the vast collection of GAL4 drivers, the potential exists to create cell lines with known genotype and lineage that are also amenable to RMCE.
WORKING WITH DROSOPHILA CELL LINES
Culturing Drosophila cell lines
Maintenance
The facility requirements of growing Drosophila cell lines are minimal compared to mammalian cells. Drosophila cell lines are generally maintained between 22°C to 25°C in a standard incubator without CO2 exchange. Cell culture work is ideally performed in a laminar flow hood to minimize contamination (see aseptic techniques).
Cell passaging or subculturing is accompanied by routine observation of cellular phenotypes such as morphology, doubling time and the presence of microorganismal contaminants. Typically, the health of a culture can be determined simply by looking at cellular morphology (Figure 1). Under light microscopy, the presence of bright cytoplasmic vacuoles and irregularity of the cell membrane often indicate an unhealthy, deteriorating culture.
Figure 1.
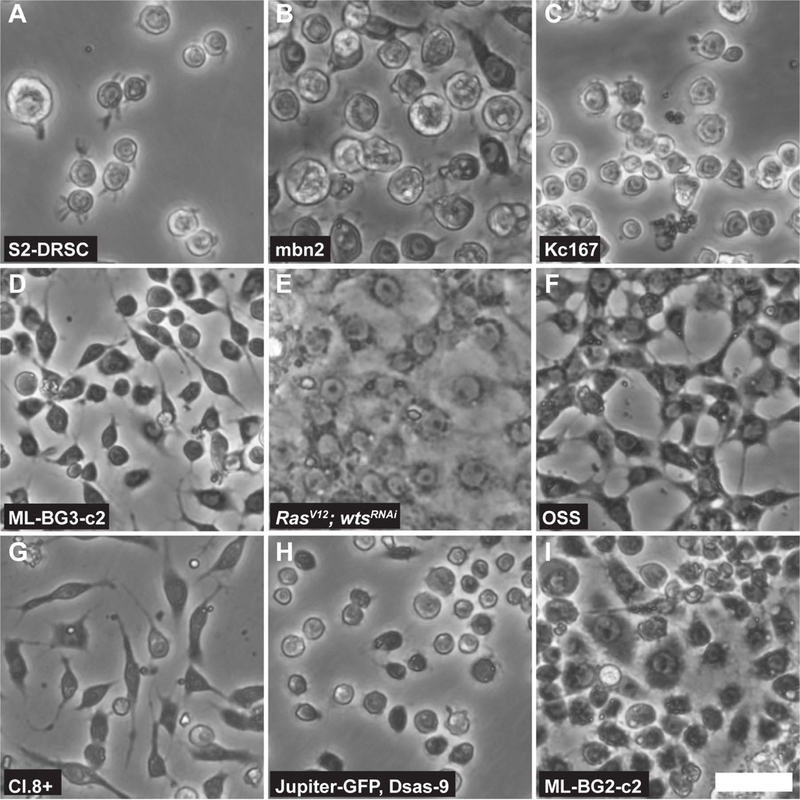
Multiple Drosophila cell lines with different morphologies. Round cells of S2-DRSC (A), mbn2 (B), Kc167 (C) and Jupiter-GFP, Dsas-9 cells (H). CNS-derived cell lines ML-BG3-c2 (D) and ML-BG2-c2 (I) show very distinct morphologies. RasV12; wtsRNAi (WRR1) cell line with epithelial appearance (E) and ovaryderived OSS (F) cell line. Larval wing disc Cl.8+ cells (G) with fibroblast-like morphology. Scale bar = 25m (Images courtesy of DGRC, Johnny Roberts).
Drosophila cell lines can be grown in tissue-culture grade flask and plates. The weakly adherent cells can be dislodged from the surface by dispensing medium over the vessel surface using a serological or Pasteur pipettes. Alternatively, for more surface adherent cell lines such as the RasV12 cell lines, trypsin-EDTA can be used to aid in resuspending cells. Strongly adherent cells can be mechanically dislodged using a cell scraper.
Drosophila cell lines do not exhibit contact inhibition and passaging them prevents overcrowding. Subculturing feeds the cells and ensures that metabolite waste products are diluted from the media. A routine for subculturing depends on the proliferation rate and the density of the culture. Cells should be subcultured when they are in the middle exponential (log) phase of growth, and never during the lag period. The ratio of subculturing is such that if cell lines take too long or too early to reach confluence, they need to be seeded at a higher and lower density, respectively.
Most Drosophila cells typically grow well at a density between 106 to 107 cells/ml. Cells to be passaged are seeded at a density that allows the cells to enter the log phase of growth with minimal or no lag time to allow them to proliferate until they reach confluence. How often one subcultures will depend on the doubling time distinct for each cell line and the passage dilution factor, which ranges from 1:2 to 1:10, after confluence is reached. When handling a new cell line in which the user has little experience with, it is advisable to passage the cell line with a low split ratio (1:2 or 1:4) during the first few passages. Once the user gains more experience with handling the cell line, or when then cell line is more established in the laboratory, it is possible to subculture at a higher split ratio (for example 1:10), while retaining at least one flask with a lower ratio as a back-up.
A hemocytometer or an automated particle counter facilitates cell counting. It is recommended to calculate cell density when dealing with individual cell lines at the time of subculturing and to determine the split ratio accordingly. The cell morphology of different cell lines can vary (Figure 1). For example, S2 cells are generally smaller than mbn2 cells, such that a confluent S2 culture will have more cells than a confluent mbn2 culture. There are also cell lines that tend to clump together (e.g. fGS/OSS) and form epithelial-like sheets (e.g. WRR-1) (A. Simcox et al., 2008) (Figure 1). For such lines, trypsin can be used to aid with dissociating the cell aggregates to facilitate accurate cell counts.
Cell freezing
Drosophila cell lines can be cryopreserved. There are several reasons for freezing them. First, cell lines are dynamic. Their gene copy numbers are under constant selection by the culture conditions and can change over time (H. Lee et al., 2014a; Y. Zhang et al., 2010). In addition, despite aseptic guidelines, cell lines may be contaminated by exogenous microorganisms such as bacteria, fungi, viruses and by other cell lines. Second, cell lines not in use can be frozen to minimize maintenance time and costs. Third, storing frozen cell lines in multiple locations insures against catastrophic events affecting either the incubators or the cryopreservation tanks. Finally, frozen cell lines can be easily distributed to other researchers.
Prior to freezing, most lines are grown to mid-log phase reaching a concentration of at least 5 X 106 cells/ml. The cells are collected by centrifugation, and the cell pellet resuspended to a high cell density (5 X 107 cells/ml) in the freezing media that contains 20% FCS and 10% DMSO. Slow cooling is essential in cryopreserving Drosophila cell lines. After the cells are aliquoted into cryopreservation vials, they are placed in a cooler flask filled with isopropanol before being transferred into a −80°C freezer for at least 24 hours for slow controlled freezing at a rate of −1°C/minute. For prolonged storage, the ampoules of cells are then transferred into liquid nitrogen.
Thawing
Frozen cells are to be thawed rapidly and seeded at a high cell density. A frozen ampoule containing 2.5 X 107 cells in a 0.5ml volume can be resuspended to 5ml volume in a T25 flask, with a resulting cell density of 2.5 X 106 cells/ml. Resuspending the cells in the growth media and changing the media after cell adherence serve to dilute the toxic DMSO degradation byproducts from the freezing media.
Cell culture media
The uniqueness of each cell line suggests that each line requires a different media for growth and proliferation. While each cell line is historically cultured in the media that it was originally established in, the DGRC has adapted most cell lines to proliferate in M3 basal media (Table 1), supplemented with various additions such as yeast extract, bactopeptone, and fetal bovine serum. Certain lines require media additions such as insulin and adult fly extract.
Transformation
Drosophila cell lines can be transfected to transiently express the exogenous DNA of interest. With a corresponding selection drug, or a traceable phenotype, stably transformed cell lines can be recovered. Various transfection procedures have been covered in details in other reviews (Lucy Cherbas & Cherbas, 2000; L. Cherbas & Gong, 2014; L. Cherbas et al., 1994). Briefly, the various transformation methods are: (1) calcium phosphate-DNA coprecipitation (Bourouis & Jarry, 1983), (2) electroporation (Swevers, Cherbas, Cherbas, & Iatrou, 1996), (3) baculovirus infection (Bryson, Weber, & Henikoff, 2010; D. F. Lee, Chen, Hsu, & Juang, 2000), and the more commonly used lipid-based delivery methods (Felgner et al., 1987).
Selection
Drug sensitivity can enrich for the desired cells after transfection in order to establish stably transformed lines. Depending on the selection marker, drugs such as puromycin, blasticidin, methotrexate (MTX) or hygromycin have been used to select desired cells, while Thymidine Kinase-conferred sensitivity to Ganciclovir (GCV) has been used to remove cells with illegitimate insertions (L. Cherbas, Hackney, Gong, Salzer, Mauser, et al., 2015; Franz, Brunner, et al., 2017; Franz, Shlyueva, et al., 2017; Kunzelmann et al., 2016; Manivannan et al., 2015). Alternatively, cells carrying fluorescence markers can be selected by flow cytometry (Neumuller et al., 2012).
For CRISPR-mediated tagging of genes, the selection cassette inserted near the gene of interest can have unintended consequences. It has been reported that functional small interfering RNAs (siRNAs) were generated at and near the site where CRISPR-directed integration had occurred (Kunzelmann & Forstemann, 2017). These siRNAs were found to repress the expression of the tagged target gene, Act5C-GFP. The phenomenon has been attributed to the selection cassette. The bidirectional copia promoter in the selection cassette may generate long dsRNAs, which were subsequently processed by the endogenous RNAi machinery. This produces functional siRNAs with sequences capable of dicing the transcripts arising from the tagged gene. Removing selection cassette has been shown to abolish siRNA activation (Kunzelmann & Forstemann, 2017). Therefore, it is recommended that selection cassettes be removed after the establishment of CRISPR-tagged lines.
Single cell cloning
Most cell lines are likely heterogenous at the time of establishment. Cellular homogeneity can be achieved by deriving sublines originating from a single cell, a process termed “single cell cloning”.
Single cell cloning of Drosophila cell lines remains a challenging process. A single Drosophila cell typically does proliferate when isolated, likely due to the less than optimal media formulations. Current protocols for single cell cloning are designed to provide the solitary cell a media environment that will promote proliferation. Several cloning methods for Drosophila cell cultures are available, including the use of feeder cells, conditioned media, limited dilutions or combinations of the methods. We briefly discuss single cell cloning methods used in recent publications below and refer readers to other sources for a comprehensive review of single cell cloning methods (L. Cherbas & Cherbas, 2007; L. Cherbas et al., 1994; G. Echalier, 1997).
The feeder layer method (Puck & Marcus, 1955) uses metabolically active feeder cells of the parental origin, which have been rendered incapable of cellular division. Parental cell lines of Sg4 and Kc167 cell lines can be treated with up to 60 kRad of ionizing radiation to generate Kc167 and Sg4 feeder cells, respectively. These cells were distributed into 96 well plates at 1.5 X105 cells per well to support single cell clones isolated by flow cytometry (L. Cherbas, Hackney, Gong, Salzer, Zhang, et al., 2015). Notably, the culture media influence single cell cloning efficiency. The cloning efficiency for Kc167 cells ranged between 10–20% when M3+BPYE media supplemented with 5% FCS was used. In contrast, attempts to clone single Kc167 cells supported by Kc feeder cells cultured in CCM3 media failed to induce any clone growth (L. Cherbas, Hackney, Gong, Salzer, Zhang, et al., 2015). As an alternative to using irradiated cells, it is possible to limit the proliferation of feeder cells by culturing them in a selective media that only allow the transformed and drug-resistant single cells to proliferate (Neumuller et al., 2012).
Cells secrete metabolic and signaling molecules that promote proliferation into the culture media. S2R+ -conditioned media can support single cell clones of S2R+ cell lines with a reported average cloning efficiency of 16% (Housden et al., 2015). In contrast, the cloning efficiency was 0% with using non-conditioned media. Franz et al. cloned single Kc167 cells by using conditioned media and performing limiting dilutions from pre-selected cell aggregates (Franz, Brunner, et al., 2017). The reported cloning efficiency was 24%. Attempts to use S2-DRSC conditioned media to clone single S2-DRSC cells resulted in only 2.8% (30/1056) cloning efficiency (DGRC, unpublished).
It remains underexplored whether using conditioned media from other cell lines would achieve higher cloning efficiencies. Notably, the cloning efficiency might vary between different cell lines using the same conditioned media (Nakajima and Miyake, 1976)
Adherent single cells can be seeded sparsely on a culture plate and allowed to form colonies (Ishizu et al., 2017; Sumiyoshi et al., 2016). This method could be used together with the semi-solid agar overlay in order to minimize the chances of clonal contamination, which could affect loosely adherent cells (G. Echalier, 1971). Visible colonies can be gradually transferred into larger culture flasks for clonal expansion.
Drosophila cell culture contaminants
Cell lines are prone to contamination by exogenous bacteria, fungi, and viruses. In addition, some cell types intrinsically harbor bacterial endosymbionts of flies such as Wolbachia or Spiroplasma.
Bacteria
The use of antibiotics to prevent bacterial contamination is not necessary, as long as sterile techniques are practiced. If antibiotics need to be included, Penicillin-Streptomycin (60 – 100ug/ml) can be added to the media.
Mycoplasmas
Mycoplasma contaminants in cell culture are mainly introduced from human or bovine sources (Echalier, 1997). Mycoplasma infections are not cytopathic; however, they might alter the growth, and the molecular properties of the cells. PCR-based Mycoplasma detection kits with species-specific primers are available commercially and they can detect and identify Mycoplasma infections. Alternatively, fluorescent microscopy using DNA dyes such as DAPI and Hoechst 33258 can reveal potential Mycoplasma contamination (Echalier, 1997). Mycoplasma will appear as specks of fluorescent signal in the spaces surrounding the cells. Tetracycline antibiotic is recommended for treating Mycoplasma infections. However, it is still advisable to discard the infected culture in order to minimize the risk of spreading the contamination (Echalier, 1997).
Fungi
Drosophila cell lines do not tolerate anti-fungal drugs well. Once contaminated with yeast, mold or fungus, they will have to be discarded immediately to prevent spreading.
Bacterial endosymbionts
Certain cell lines were derived from fly strains that harbor bacterial endosymbionts such as Wolbachia and Spiroplasma. The presence of Wolbachia can be assayed molecularly by PCR detection or visually by a fluorescent DNA stain. Wolbachia contaminants be treated by culturing in tetracyclin (50μM) supplemented media for up to five weeks (Debec et al., 2016).
Endogenous Viruses
Virus sequences are readily detected from the modENCODE Drosophila cell culture data set (Webster et al., 2015), corroborating the widely known fact that Drosophila cell cultures are infected with viruses. Another potential source of viral infection comes from the calf serum. There are protocols described to ensure the establishment and maintenance of virus-free cell lines (Echalier, 1997) but there is currently no method for eliminating viral infections from Drosophila cell cultures.
Best practices: aseptic techniques working with Drosophila cell cultures
The objective of aseptic techniques in cell culture is to prevent microorganismal contaminations. Adventitious contaminants may find their way into the cultures through the user, environment and many other sources. It is essential to adhere to a set of strict guidelines, especially if multiple users share the same workplace. Aseptic techniques are a combination of precautions that can drastically minimize the risk of contamination. Working with Drosophila cell lines, the aseptic techniques prescribed for cell cultures in general are applicable and have been thoroughly covered elsewhere (Phelan & May, 2017a, 2017b). Table 3 briefly summarizes the do and don’ts for aseptic techniques.
Table 3.
Important guidelines for aseptic technique for handling Drosophila cell cultures
| Do | Do not | |
|---|---|---|
| Laminar hood | Use appropriate safety level laminar hood.+ Swab with 70% alcohol before and after use. Sterilize hood and allow 5 minutes between working with different cell lines. Wipe spills immediately. Leave laminar flow hoods to run continuously. |
Clutter the hood. (Figure 2) Create mess. Work behind an open vessel (horizontal laminar flow). Work above an open vessel (vertical laminar flow). |
| Glassware | Separate glassware for cell cultures. Sterilize by oven-baking (up to 200°C) Double autoclaving.* |
Use glassware for chemicals. |
| Media | Swab bottle with 70% alcohol before placing in hood. | Share media among users. |
| Label media bottles. | Pour from one sterile container to another. | |
| Contamination | Check cultures routinely. Quarantine and dispose contaminated cultures properly. |
Open contaminated cultures in the hood or in the facility. |
| Handling | Work with one cell line at a time. Label plates and flasks carefully. Store culture vessels in a plastic box. |
Store multiple different cell lines in the same plastic box. |
Use vertical laminar flow hood when working with baculovirus to infect Drosophila cells.
Fungal spores can be fully killed by double-autoclaving a bottle or flask containing sterile water. This is important for growing suspension cultures of Drosophila cells.
Best practices: nomenclature
Cell lines should have a standard nomenclature. It is very important that when a cell line nomenclature has been adopted, to adhere strictly to it. The Drosophila S2 cell line served as an example of how poor reporting in the literature made it impossible to determine the isolate or source of the S2 cells. The DGRC assigns each individual cell line with an accession number. In addition, each cell line has its own RRID number, maintained by the Cellosaurus database (Bairoch, 2018). When reporting the use of Drosophila cell lines, it is imperative to indicate these identifying numbers.
Best practices: maintenance of records
It is important to keep detailed records of subculturing. Table 4 illustrates a set of parameters to be recorded. If establishing a new cell line from primary cultures, a similar record for feeding the cells is recommended. The inclusion of the passage number is recommended whenever applicable.
Table 4.
A sample of data recording for routine maintenance of Drosophila cell culture.
| Date | X-X-2018 | |
|---|---|---|
| Cell line | Name | S2-DGRC |
| Passage number | N/A | |
| Visual Status | Cell Morphology | Round |
| Density | 90% confluent | |
| Color of medium | Clear yellow | |
| Dissociation method | Dislodge by pipetting media over cells | X |
| Trypsin-EDTA | ||
| Cell scraper | ||
| Cell count | Cell density before subculture | 8.0 X 106 cells/ml |
| Volume (ml) | 10 ml | |
| Total cell counts before subculture | 8.0 X 107 cell | |
| Seeding | Number/Type of flask | 4 / 100mm plate |
| Final cell density (cell/ml) | 1.0 X 106 cells/ml | |
| Volume (ml) | 10 | |
| Split ratio | 1:2 | |
| Medium | Medium name | M3 + BPYE |
| Serum (batch and %) | SH30070 (AXH47535) 10% | |
| Adult fly extract | - | |
| Insulin | - | |
| Others | Media changes | - |
Best practices: cryopreservation
An outline of the stages of cryopreservation in the accession process is depicted in Figure 3. Briefly, as soon as a cell line is established and determined to be free of contamination, a seed stock acts as the primary cell bank, from which the working or distribution stock is derived from.
Figure 3.
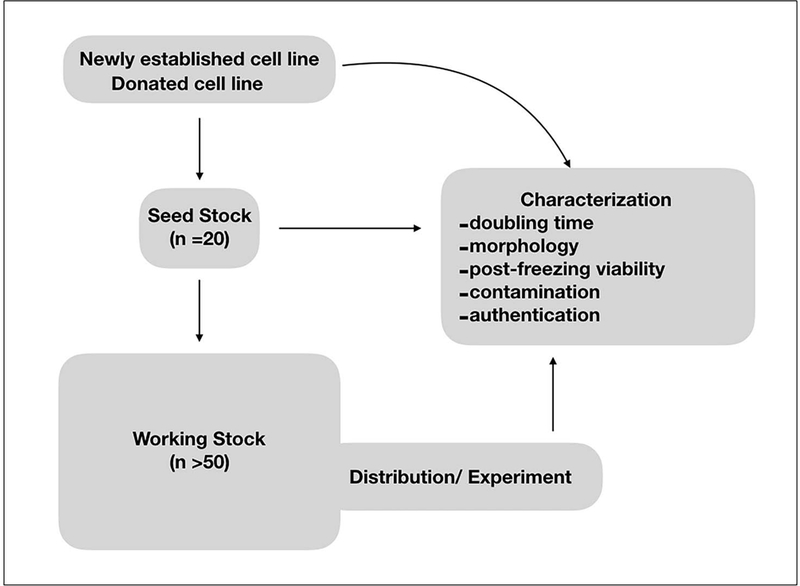
The accession process and cryopreservation of a Drosophila cell line.
Best practices: handling contamination
If cultures are contaminated by microorganisms, discard contents into a disinfectant bucket inside a fume hood, away from the cell culture room. Alternatively, the contents must be autoclaved to ensure safe and proper disposal.
CHALLENGES AND OPPORTUNITIES FOR DROSOPHILA CELL CULTURES
The challenges of working with Drosophila cultured cells may be progressively overcome if opportunities are taken to improve media formulations that support the proliferation of many lines, such that the efficiency of single cell cloning may be higher, or media that readily support the establishment of epithelial cell lines. Other challenges are associated with the nature of somatic cell genetics, for example the validation of gene functions from both low- and high-throughput genome-editing experiments. There are also opportunities to elevate the standard for using Drosophila cell cultures through cell line authentication, to prevent the use of misidentified or cross-contaminated lines and ensure the continued relevance of Drosophila cell lines for biomedical applications.
Media formulations for culturing Drosophila cell lines
Many formulations for Drosophila melanogaster cell culture media have been devised in the last five decades. The media were originally developed according to the properties (osmolarity, pH, ion balance) and components (amino acids, organic acids of the TCA cycle, vitamins, sugars) of the larval hemolymph. Depending on the line, supplements for the culture media have since included insulin, ecdysone, Juvenile Hormone, the largely undefined adult fly extract, exogenous bactopeptone, yeast extract, fetal bovine serum, or a combination of these additives.
The DGRC has adapted most of its cell line collection to grow in M3 media (Cross & Sang, 1978; Shields, Dubendorfer, & Sang, 1975). M3 contains a lower concentration of chloride salts compared to Schneider’s medium and uses glutamate salts as the source for Na+ and K+ ions (Na+/K+ = 1.8). The defined individual amino acid supplementation, relative lack of organic acids, and the use Bis-tris as the media buffering agent characterize the M3 media. At the DGRC, M3 is often supplemented with bactopeptone and yeast extract to satisfy the requirement for vitamins, and other complex constituents.
With regard to Kc cell lines, the late Guy Echalier Ph.D., a pioneer in Drosophila cell cultures had used the relatively simpler Drosophila 20 (D20, later D22) media, often supplemented with up to 10% fetal calf serum. Using this formulation, the Kc lines were continuously subcultured for decades (Guy Echalier, Perrimon, & Mohr, 2017). Kc167 and a few other robust cell lines can grow in commercial serum-free HyClone-CCM3 or cGMP Hyclone protein-free SFX-Insect media (DGRC, unpublished). SFX-Insect media was developed based on the analysis of cellular metabolic profiles during culture conditions (Metabolic Pathway Design, GE Healthcare), and optimized to sustain the proliferation and viability for several insect cell lines. The reduced cost, increased consistency, and cGMP compliance are the main advantages of using serum-free or protein-free media for Drosophila cells. In addition, there is no need for serum screening. However, the composition of these media is proprietary and therefor it cannot be considered chemically defined.
With S2 isolates, the DGRC uses M3 as the base media, while many others have used Schneider’s media (Schneider & Blumenthal, 1978) to culture S2 cells. There are noted differences in doubling time, for instance, S2-DRSC lines cultured in M3+BPYE have a doubling time of 46 hours, while the same lines cultured in Schneider’s medium have a doubling time of 93 hours (DGRC, unpublished). Both media are supplemented with 10% FBS.
The opportunities for improving and designing holidic or chemically defined culture media are there for the taking. Until recently, few groups have undertaken the systematic effort to identify bioactive components and formulate chemically defined media capable of supporting the growth and proliferation of cultured cells. These efforts have interestingly come from attempts to optimize ex vivo organ cultures. The lessons learnt from media formulation in organ culture might be applicable to cell lines.
In searching for a media capable of sustaining in vitro culture of larval wing discs, Zartman et al. tested, blended various media and formulated wing disc medium 1 (WM1) that maintained the proliferation of Clone.8 (Cl.8) cells and S2 cells (Zartman, Restrepo, & Basler, 2013). WM1 uses Schneider’s media as a base (which contains a twice as much Yeastolate as M3 medium), and is supplemented with 5% fly extract, and 6.2 μg/ml of insulin. Notably, WM1 does not contain xenogenic FBS, and instead is replaced by the adult fly extract supplementation.
What then is in the adult fly extract? Earlier work points to the mitogenic role of adenosine deaminase-related growth factors that deplete extracellular adenosine and that various cell lines react differently to extracellular adenosine in the culture media (Fleischmannova et al., 2012; Zurovec, Dolezal, Gazi, Pavlova, & Bryant, 2002). Fractionation of fly extracts identified the enzyme adenosine deaminase (ADA) and it functions to promote the proliferation and maintenance of wing disc cultures in M3-based media (Strassburger et al., 2017) (Table 6). The iron-carrier ferritin was also identified as an active ingredient in the fly extract and it promotes Cl.8 cell proliferation in a dose-dependent manner (S. Li, 2010).
Table 6.
Comparison of the composition of various organ culture media: WM1, ZB media and media used by Strassburger et al.
| WM1 Media | ZB media | Strassburger |
|---|---|---|
| Basal media | ||
| Schneider’s | ZO (Wyss | M3 |
| Supplements | ||
| Human insulin (6.2 mg/ml) | Human insulin (5.0 mg/ml) | Bovine insulin (10.0 mg/ml) |
| Fly extract (5%) | ||
| Spermidine trihydrochloride (0.001mM) | ||
| 20-HE (5.0 nM) | ||
| JH (1.6 nM) | ||
| ADA (0.0083 mg/ml) | ||
Insulin signaling plays major role in cell growth and proliferation. Drosophila melanogaster genome encodes for eight insulin-like peptides (ILP). Bovine or human insulin are often added to the media to promote Drosophila cell line proliferation. Bovine or human insulin is likely not the perfect agonist for Drosophila Insulin Receptor (dInR). Handke et al. showed that 0.5 μM of DILP2 could elicit mitogenic effects as strong as using 35 μM of bovine insulin (Handke, Szabad, Lidsky, Hafen, & Lehner, 2014).
Burnette et al. (2014) screened for bioactive compounds that support growth and proliferation of Cl.8, S2-DRSC and Kc167 cells (Burnette, Brito-Robinson, Li, & Zartman, 2014). Semi-chemically defined ZO media (Wyss, 1982a) was selected as the base media and was fortified with insulin, trehalose, ala-gln and A2P. This medium can facilitate Cl.8 cell attachment to the growth surface and supported limited proliferation. Importantly, the screen identified spermidine, which significantly increased cell proliferation when added to the ZO media.
ZB media (ZO media + Spermidine) can support long-term Cl.8 and Kc167 cell proliferation (Table 5 and Table 6). In addition, Cl.8 and Kc167 were able to recover from cryopreservation using ZB-based freezing media containing 0.2M trehalose and 10% DMSO (Burnette et al., 2014). However, ZB media failed to support S2-DRSC cells beyond the fourth passage. Therefore, opportunities exist for optimizing media for S2-DRSC cells.
Table 5.
Comparison of the media composition of Schneider’s media (SM), Shield and Sang’s M3 media and ZB media. The formulation of Schneider’s, M3 and ZB media were reproduced based on a comparison of the table compiled by Echalier et al. (2017) with components described by Schneider’s media (Sigma) and Shields and Sang M3 media (Sigma).
| SM | M3 | ZB | ||||||
|---|---|---|---|---|---|---|---|---|
| mg/L | mM | mg/L | mM | mg/L | mM | |||
| Salts | ||||||||
| CaCl2 | 600 | 5.40 | CaCl2. 6H2O | 1500 | 6.8 | CaCl2 | 111 | 1.0 |
| MgSO4. 7H2O | 3700 | 15.00 | MgSO4. 7H2O | 4400 | 17.9 | MgSO4 | 600.6 | 5.0 |
| KCl | 1600 | 21.50 | KHCO3 | 500 | 5.0 | KCl | 2000 | 26.8 |
| KH2PO4 | 450 | 3.30 | C5H8KNO4.H2O | 788 | 38.8 | |||
| Na2HPO4 | 700 | 4.90 | CH3COONa.3H2O | 25 | 0.18 | |||
| NaHCO3 | 400 | 4.70 | Na2PO4.2H2O | 880 | 5.6 | NaH2PO4.H2O | 420 | 3.0 |
| NaCl | 2100 | 35.90 | C5H8NaNO4 | 653 | 38.6 | NaCl | 3200 | 54.7 |
| Amino acids | ||||||||
| L-⍺-Alanine | - | - | L-⍺-Alanine | 250 | 2.8 | L-⍺-Alanine | 45 | 0.50 |
| L-β-Alanine | 500 | 5.60 | L-β-Alanine | 500 | 2.9 | L-β-Alanine | 45 | 0.50 |
| L-Arginine | 400 | 2.30 | L-Arginine | 300 | 2.25 | L-Arginine | 177.3 | 1.00 |
| L-Asparagine.H2O | 171.1 | 1.14 | ||||||
| L-Aspartic acid | 400 | 3.00 | L-Aspartic acid | 300 | 2.27 | L-Aspartic acid | 133 | 1.00 |
| L-Cysteine HCl | 60 | 0.50 | L-Cysteine HCl | 200 | 1.27 | L-Cysteine HCl.H2O | 351 | 2.00 |
| L-Cystine | 100 | 0.40 | ||||||
| Alanyl-glutamine (Aln-gln) | 2600 | 11.96 | ||||||
| L-Glutamic acid | 800 | 5.40 | L-Glutamic acid | 406.5 | 2.76 | |||
| L-Glutamine | 1800 | 12.30 | L-Glutamine | 600 | 4.1 | L-Glutamine | 680 | 4.64 |
| Glycine | 250 | 3.30 | Glycine | 500 | 6.6 | Glycine | 750 | 10.00 |
| L-Histidine | 400 | 2.60 | L-Histidine | 550 | 3.5 | L-Histidine | 155 | 1.00 |
| L-Hydroxyproline | 65 | 0.50 | ||||||
| L-Isoleucine | 150 | 1.10 | L-Isoleucine | 250 | 2.0 | L-Isoleucine | 131 | 1.00 |
| L-Leucine | 150 | 1.10 | L-Leucine | 400 | 3.00 | L-Leucine | 131 | 1.00 |
| L-Lysine HCl | 1650 | 9.0 | L-Lysine HCl | 850 | 4.60 | L-Lysine HCl | 182 | 1.00 |
| L-Methionine | 800 | 5.40 | L-Methionine | 250 | 1.60 | L-Methionine | 149 | 1.00 |
| L-Ornithine-HCl | 84 | 0.50 | ||||||
| L-Phenylalanine | 150 | 0.90 | L-Phenylalanine | 250 | 1.50 | L-Phenylalanine | 165 | 1.00 |
| L-Proline | 1700 | 14.8 | L-Proline | 400 | 3.50 | L-Proline | 115 | 1.00 |
| L-Serine | 250 | 2.40 | L-Serine | 350 | 3.30 | L-Serine | 525 | 5.00 |
| L-Threonine | 350 | 2.00 | L-Threonine | 500 | 4.20 | L-Threonine | 119 | 1.00 |
| L-Tryptophan | 100 | 0.50 | L-Tryptophan | 100 | 0.50 | L-Tryptophan | 102 | 0.50 |
| L-Tyrosine | 500 | 2.70 | L-Tyrosine | 250 | 1.40 | L-Tyrosine 2Na. 2H2O | 112.6 | |
| L-Valine | 300 | 2.50 | L-Valine | 400 | 3.60 | L-Valine | 117 | 1.00 |
| Sugars | ||||||||
| Glucose | 2000 | 11.00 | Glucose | 10000 | 55.50 | D-Glucose | 2000 | 11.00 |
| Trehalose | 2000 | 5.50 | Trehalose | 10000 | 26.43 | |||
| Organic acids | ||||||||
| ⍺-Ketoglutaric acid | 200 | 1.40 | ||||||
| Fumaric acid | 100 | 0.80 | ||||||
| Malic acid | 100 | 0.75 | Malic acid | 670 | 5.00 | |||
| Succinic acid | 100 | 0.80 | ||||||
| Oxaloacetic acid | 250 | 1.90 | Oxaloacetic acid | 250 | 1.90 | |||
| Sodium pyruvate | 110 | 1.00 | ||||||
| Succinic acid | 60 | 0.50 | ||||||
| Supplements | ||||||||
| TC Yeastolate | 2000 | TC Yeastolate | 1000 | |||||
| Peptone | 5000 | |||||||
| Bis-Tris (Sigma) | 1050 | 5.0 | ||||||
| Glutathione | 0.005 | |||||||
| 1-Thioglycerol (β-ME) | 5.4 | 0.05 | ||||||
| Inosine | 2.7 | 0.01 | ||||||
| Thymidine | 2.4 | 0.01 | ||||||
| Uridine | 2.4 | 0.01 | ||||||
| DL-Carnitine-HCl | 1000 | 5.05 | ||||||
| Human insulin | 5.0 | |||||||
| Spermidine trihydrochloride | 0.001 | |||||||
| Vitamins | mg/L | μM | ||||||
| Vitamin B12 (cobalamin) | 0.10 | 0.07 | ||||||
| Biotin (B7) | 0.10 | 0.41 | ||||||
| Folic acid (B9) | 0.10 | 0.23 | ||||||
| Niacinamide (B3) | 0.10 | 0.82 | ||||||
| D-Calcium panthothenate (B5) | 10.00 | 40.00 | ||||||
| Pyridoxal hydrochloride (B6) | 30.00 | 147.33 | ||||||
| Riboflavin (B2) | 0.01 | 0.03 | ||||||
| Thiamin hydrochloride (B1) | 10.00 | 29.65 | ||||||
| Nicotinic acid (B3) | 10.00 | 81.3 | ||||||
| Pyridoxine (B6) | 0.10 | 0.49 | ||||||
| Choline chloride | 20.00 | 0.14 | ||||||
| m-Inositol | 20.0 | 111.0 | ||||||
| L-⍺-Lecithin (distearoyl) | 0.01 | 0.013 | ||||||
| L-⍺-Lecithin (dimyristoyl) | 0.01 | 0.015 | ||||||
| L-⍺-Lecithin (dipalmityl) | 0.005 | 0.007 | ||||||
| Sodium-⍺-glycerophosphate | 0.16 | 0.74 | ||||||
| Calcium phosphoryl choline | 0.13 | 0.39 | ||||||
| Phosphoethanolamine | 0.07 | 0.50 | ||||||
| Cholestrol | 0.002 | 0.005 | ||||||
| Oleic acid | 0.05 | 0.18 | ||||||
| Linoleic acid | 0.05 | 0.18 | ||||||
| Ferrous sulphate. 7H2O | 0.95 | 3.42 | ||||||
| Zinc sulphate. 7H2O | 0.99 | 3.44 | ||||||
| Copper Sulphate. 5H2O | 0.002 | 0.008 | ||||||
| Manganese Chloride . 4H2O | 0.69 | 3.49 | ||||||
| L-Ascorbic acid 2 -phosphate sesquimagnesium salt hydrate | 23.13 | 78.89 | ||||||
The most commonly used Drosophila cell culture media contain natural additions such as bovine serum, yeast extracts, bactopeptone or insulin. Some cell lines require adult fly extracts. Notably, batch variations are common for natural media supplements. A few Drosophila cell lines can grow in commercial serum-free/protein-free media and this suggests the potential path towards chemically defined media. The goal of formulating a chemically defined media is driven with the aim of achieving consistency for Drosophila cell culture performance. Towards this goal, the workflow adopted by Burnette et al. may be expanded to systematically identify synergistic active components that will support the maintenance of most cultured Drosophila cell lines (Burnette et al., 2014). This step-wise approach to media design and evolution, unfortunately, can be laborious. For discussion on media optimization strategies, we refer readers to a review on the recent progress of mammalian cell culture media development (Yao & Asayama, 2017)
Searching for Drosophila epithelial cell line
To our current knowledge, there is arguably no true Drosophila epithelial cell line (Baum, 2011; Baum & Cherbas, 2008). wtsRNAi and RasV12, wtsRNAi (WRR-1) cell lines have epithelial morphology and strongly express an epithelial cell junction marker such as E-Cadherin (A. Simcox, 2013; A. Simcox et al., 2008). However, it remains unknown whether these cells exhibit cell polarity, a hallmark feature for epithelial cells.
The first insect epithelial cell line derived from the primary cultures of the midge (Chironomus tentans) were responsive to ecdysone and were capable of continuous proliferation (Wyss, 1982a). Notably, the epithelial cells could alternate between forming organized spherical vesicles made of many cells and cell sheets attached to the vessel surface. Intermediate luminal structures that were observed include tubes, bulges, and protrusions.
TcA, a continuous cell line derived from the primary co-cultures of pupal and adult tissue from the red flour beetle (Tribolium Castaneum) displays epithelial morphology (Goodman et al., 2012). TcA cells can form sheets of cells, as well as multicellular macro spherical vesicles. Transcriptome data for the cell line suggest an epidermal origin (Silver et al., 2014). The commercial EX-CELL420 (Sigma) media supplemented with bovine serum was used to establish the TcA line. Notably, attempts to establish the beetle continuous cell line were not successful when other media such as M3 or Schneider’s media were used (Goodman et al., 2012).
Several factors may influence the success of deriving epithelial Drosophila cell lines. The use of UAS/GAL4 system driving expression of RasV12 in specifically in epithelial cells could favor the establishment of epithelial cell lines. The choice of developmental stage for the starting tissues may be crucial as well. Media formulations such as the use of ZW media (Wyss, 1982a, 1982b) or EX-CELL420 (Sigma) may be effective for establishing epithelial cell lines. In addition, culture conditions may need to be optimized to promote the formation multicellular luminal structures, a characteristic of many epithelial cell lines (A. Simcox, 2013).
Functional genomics
sgRNA design, off target effects and validation
One of the challenges associated with CRISPR-based gene disruption for functional genomics pertain to both sgRNA design and the underexplored off-target effects (OTE) of the gRNAs (Fu et al., 2013; Pattanayak et al., 2013). Despite limitations on variations in gRNA efficiency and gRNA design for a target gene at a genome level, gRNA libraries with multiple gRNAs/target gene are available (Guy Echalier et al., 2017; S. E. Mohr et al., 2016; Viswanatha et al., 2018).
Analyzing and validating phenotypes from CRISPR knockout cell lines
CRISPR OTEs are likely a more challenging issue in cell culture than in the whole fly, because in vivo transheterozygotes or hemizygotes in the whole animal can minimize the risks of analyzing off-target phenotypes.
It is best practice in somatic cell genetics to create multiple distinct mutant cell lines using different gRNAs (S. E. Mohr et al., 2016). It was estimated that 97% of genes in the Drosophila genome could be targeted with specific gRNAs (Housden et al., 2015). Therefore, this specificity may mitigate the risks of OTE and may circumvent the need for establishing mutant cell lines derived from different gRNAs, which can be time-consuming and laborious. In addition, it is also important to analyze sublines from a single gRNA modification to ensure phenotype reproducibility between sublines.
A validation strategy that uses gene rescue remains the gold standard for assigning gene function. For CRISPR-KO lines, cross-species genomic DNA rescue reagents may be used by subjecting cells stably transfected with D.pseudoobsura fosmids to the CRISPR gene-targeting workflow, a strategy similar to what has been demonstrated for RNAi rescue in Drosophila cell cultures (Kondo et al., 2009). In addition, validation using a standard wildtype transgene rescue can be a practical approach, as long as the wild type transgene has been modified to be impervious to the gRNA (S. E. Mohr et al., 2016).
Genome editing: different cell lines have different genome/gene copy number
Drosophila cells have varying and evolving genome copy numbers (H. Lee et al., 2014b). We hypothesize that homozygous editing could be achieved more readily in cell lines that are “minimally diploid” such as BG3-c2, Cl.8, D20-c2, D20-c5, D4-c1, L1, S1, W2, and D8, OSS, and more challenging in “minimally tetraploid” cell lines such as S2-DRSC, S2R+, S3, Sg4, Kc167, D16-c3, and D17-c3. The success of CRISPR-based editing in the pseudo tetraploid Schneider and Kc lines is promising for other minimally diploid Drosophila cell lines.
Cell line authentication
Currently, there is no published report of misidentified Drosophila cell lines. The use of misidentified cell lines is a problem, compromising data reproducibility and accuracy in biomedical research (Almeida, Cole, & Plant, 2016). This problem is endemic in mammalian cell culture, in which the reported cell lines are no longer what they originally were. The cell lines might have been cross-contaminated by other cell lines, either from intraspecies or interspecies sources. In addition, cell lines can also be contaminated with non-cytopathic adventitious agents that could affect their phenotypes. The International Cell Line Authentication Committee (ICLAC), as well as the Cellosaurus database, maintains a list of misidentified cell lines (Bairoch, 2018).
Cell lines evolve and can change their phenotypes as they continue to be cultured by multiple independent laboratories for prolonged periods. The most widely used S2 line suffered in identity when it became obvious from the scientific literature that there were no careful tracking and annotation of their origin in each laboratory. Multiple S2 isolates are now available: S2-DGRC, S2-DRSC, S2R+, Sg4, and S2-ATCC. These S2 isolates, cultured independently, have diverged substantially, based on their transcriptional profiles (L. Cherbas et al., 2011; Wen et al., 2014). The S2R+ line, because of its history, is likely the most similar to the original cell line established by Schneider (Guy Echalier et al., 2017; Schneider, 1972). Researchers using S2 lines must be careful to accurately identify the different isolates.
To address the problem of cell line misidentification, the National Institutes of Health (NIH) has guidelines (NIH Notice number: NOT-OD-15–103) for funding applications and journal reporting to authenticate key biological resources, including cell lines used in research. The goal of authentication is to help establish confidence regarding the cell line identity and the validity of the biological results from using cell line resources.
Transcriptional profiling of more than 40 Drosophila cell lines distributed from the DGRC indicates that the lines are largely distinct (L. Cherbas et al., 2011; N. C. Lau et al., 2009; Stoiber et al., 2016; Wen et al., 2014). Most of the cell lines available at the DGRC were obtained from the laboratory in which the cell lines were created. Assuming best practices for culturing cells including minimal passaging, these lines are likely similar to their original lines.
The advances in somatic cell genetic techniques will likely create more Drosophila cell lines with known modifications. It is important for us as a community to begin setting standards and guidelines for researchers and repositories to authenticate existing, widely used and new Drosophila cell lines.
Several methods may be considered to form the standards for assessing Drosophila cell lines (Table 7):
Table 7.
Various proposed methods for Drosophila cell line authentication and their associated authentication metrics.
| Method |
STR/S NP |
Microscopy Karyotyping |
CO1 barcoding/ species specific primers |
Whole genome sequencing/Targeted resequencing |
Transcriptome Analysis |
|---|---|---|---|---|---|
| Authentication metric | |||||
|
Interspecies identification |
✓ |
✓ |
✓ |
✓ |
|
| Intraspecies identification | ✓ | ✓ | ✓ | ✓ | |
| Contamination with adventitious agents | ✓* | ✓ | ✓ | ✓ | |
| Specific markers | ✓* | ✓ | ✓ | ||
| Morphology, doubling time | ✓* | ||||
| Chromosomal aberration | ✓* | ✓ | |||
Immunostaining with specific antibodies and/or the use of DNA stains can help detect Mycoplasma contamination.
Karyotyping
A panel of Drosophila cell lines has been systematically karyotyped by DNA-sequencing and mitotic chromosomal spreads (H. Lee et al., 2014a). DNA-sequencing ratiometry presents minimal ploidy, not absolute ploidy, and is relatively high-throughput. The method requires expertise for data processing and analysis. In contrast, chromosomal spread karyotyping, which has been used to detect interspecies misidentified cell lines (Nelson-Rees, Daniels, & Flandermeyer, 1981a, 1981b), provides absolute ploidy. However, the method is low-throughput, time-consuming and requires cytological expertise.
Results from DNA-sequencing ratiometry suggest that the karyotype of a Drosophila cell line is arguably stable at the population level. In contrast, mitotic chromosome spreads indicate that the karyotype of the individual cells in the same population can be variable (H. Lee et al., 2014a). However, cellular ploidy can change over time, as exemplified by the originally haploid cell line 1182–4H (Debec, 1978). The cell line has evolved into a minimally diploid state (H. Lee et al., 2014a).
Short Tandem Repeat (STR) profiling
STR profiling is a molecular technique applied routinely to authenticate mammalian cell cultures (Barallon et al., 2010; Masters et al., 2001). The current standards already set by the authentication of human cell lines, make STR profiling a potential choice for authenticating Drosophila cell lines. Potential polymorphic STR markers from sequencing the Drosophila Genetic Reference Panel (DGRP) could be used (Fondon, Martin, Richards, Gibbs, & Mittelman, 2012). However, whether the STR markers can discriminate between the Drosophila cell lines remains to be tested.
DNA barcoding with cytochrome c oxidase subunit I (COI)
There is species to species sequence variability in the COI gene and probing the sequences of CO1 (CO1 barcoding) can facilitate Drosophila species identification (Yassin, Markow, Narechania, O’Grady, & DeSalle, 2010). However, the method has not been tested on the cell lines derived from Drosophila melanogaster and other Drosophila species.
Gene expression profile
The modENCODE project produced detailed information on the transcriptome of the cell lines and indicated that the tested cell lines are distinct. Furthermore, their transcriptional responses to hormonal stimulations are largely different. However, cellular gene expression profiles are likely to vary depending on culture conditions and therefore cellular identity authentication by transcriptome analyses may not be ideal.
Assaying known characteristics (doubling time, expression of specific markers)
It is important to routinely monitor the cell morphology, doubling time and the expression of specific markers when working with cell lines during cell banking and distribution stages. The cell shapes and sizes can vary between cell lines derived from different tissues (Figure 1). In addition, cell lines that are known to carry specific markers (GFP, mCherry) can be confirmed for marker retention during cell banking.
6. Next Generation or Nanopore DNA-sequencing
Theoretically, NGS technology provides a high-throughput platform for authenticating cell lines. Bioinformatics processing of DNA sequencing data should be capable of distinguishing between species, genotype, and report the presence of adventitious agents including Mycoplasma, Wolbachia, and viral sources. In addition, DNA-sequencing can provide cellular ploidy information (H. Lee et al., 2014a). As long as the cost associated with DNA sequencing continues to drop (Miller, Staber, Zeitlinger, & Hawley, 2018), this approach is practical in that it satisfies all the requirements of cell line authentication, including the identity of the cell line and the test for the presence of contamination.
Ultimately, it is important to note that authentic cell lines that have been distributed and maintained in multiple different laboratories are likely to evolve independently with long periods of subculturing. There is no clear consensus on how many passages a cell line has to undergo before it is considered changed. The problem could potentially be exacerbated in the absence of best practices in cell culture. It is recommended that users thaw new frozen lines every few months in order to minimize the effects arising from genetic drift (Baum & Cherbas, 2008).
Conclusion
Drosophila cell lines have taken on a dual role as a complementary tool for in vivo genetic approaches to address some of the most basic biological questions as well as a robust tool for high throughput functional genomics. With the growing utilization of CRISPR-based gene editing and the RasV12 approach for making cell lines, we anticipate increased numbers of novel cell lines.
Towards this goal, systematic efforts to develop defined media formulations and optimize culture conditions are necessary.
As the use Drosophila cell culture continues to be embraced for many applications, best practices for working with Drosophila cell lines must be observed. A standard for the authentication of Drosophila cell lines is necessary as routine authentication will identify potential problems early and are likely to reduce the risks of using misidentified or contaminated cell lines. These efforts should increase the utility and relevance of the Drosophila cell culture system for both fundamental and biomedical applications.
Figure 2.

(Top) Ideal arrangement of the workspace in the laminar hood and handling one cell line at a time. (Bottom) A cluttered and overcrowded work area in the laminar hood compromises the airflow and thus the sterility in the laminar hood.
Acknowledgments
We thank Drs. Nicholas Sokol and Jason Tennessen for comments, and the National Institutes of Health (Award NIH P40OD010949) for continued financial support of the Drosophila Genomics Resource Center.
Contributor Information
Arthur Luhur, Department of Biology, Drosophila Genomics Resource Center, Indiana University Bloomington, aluhur@indiana.edu.
Kristin M Klueg, Department of Biology, Drosophila Genomics Resource Center, Indiana University Bloomington, kklueg@indiana.edu.
Andrew C Zelhof, Department of Biology, Drosophila Genomics Resource Center, Indiana University Bloomington, azelhof@indiana.edu.
References
- Aguilera-Gomez A, Zacharogianni M, van Oorschot MM, Genau H, Grond R, Veenendaal T, … Rabouille C (2017). Phospho-Rasputin Stabilization by Sec16 Is Required for Stress Granule Formation upon Amino Acid Starvation. Cell Rep, 20(9), 2277. doi: 10.1016/j.celrep.2017.08.054 [DOI] [PubMed] [Google Scholar]
- Albert EA, & Bokel C (2017). A cell based, high throughput assay for quantitative analysis of Hedgehog pathway activation using a Smoothened activation sensor. Sci Rep, 7(1), 14341. doi: 10.1038/s41598-017-14767-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida JL, Cole KD, & Plant AL (2016). Standards for Cell Line Authentication and Beyond. PLoS Biol, 14(6), e1002476. doi: 10.1371/journal.pbio.1002476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammeux N, Housden BE, Georgiadis A, Hu Y, & Perrimon N (2016). Mapping signaling pathway cross-talk in Drosophila cells. Proc Natl Acad Sci U S A, 113(35), 9940–9945. doi: 10.1073/pnas.1610432113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A (2018). The Cellosaurus, a Cell-Line Knowledge Resource. J Biomol Tech. doi: 10.7171/jbt.18-2902-002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barallon R, Bauer SR, Butler J, Capes-Davis A, Dirks WG, Elmore E, … Kerrigan L (2010). Recommendation of short tandem repeat profiling for authenticating human cell lines, stem cells, and tissues. In Vitro Cell Dev Biol Anim, 46(9), 727–732. doi: 10.1007/s11626-010-9333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AR, Kong L, & Liu JL (2015). A genome-wide CRISPR library for high-throughput genetic screening in Drosophila cells. J Genet Genomics, 42(6), 301–309. doi: 10.1016/j.jgg.2015.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AR, Tibbit C, Ponting CP, & Liu JL (2014). Mutagenesis and homologous recombination in Drosophila cell lines using CRISPR/Cas9. Biol Open, 3(1), 42–49. doi: 10.1242/bio.20137120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman JR, Lee AM, & Wu CT (2006). Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics, 173(2), 769–777. doi: 10.1534/genetics.106.056945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum B (2011). Finding gold in yellowing papers. Nat Rev Mol Cell Biol, 12(4), 205. doi: 10.1038/nrm3092 [DOI] [PubMed] [Google Scholar]
- Baum B, & Cherbas L (2008). Drosophila cell lines as model systems and as an experimental tool In Dahmann C (Ed.), Drosophila: Methods and Protocols (Vol. 420, pp. 391–424): Humana Press. [DOI] [PubMed] [Google Scholar]
- Billmann M, & Boutros M (2016). Methods for High-Throughput RNAi Screening in Drosophila Cells. Methods Mol Biol, 1478, 95–116. doi: 10.1007/978-1-4939-6371-3_5 [DOI] [PubMed] [Google Scholar]
- Bottcher R, Hollmann M, Merk K, Nitschko V, Obermaier C, Philippou-Massier J, … Forstemann K (2014). Efficient chromosomal gene modification with CRISPR/cas9 and PCR-based homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res, 42(11), e89. doi: 10.1093/nar/gku289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourouis M, & Jarry B (1983). Vectors containing a prokaryotic dihydrofolate reductase gene transform Drosophila cells to methotrexate-resistance. EMBO J, 2(7), 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, & Ahringer J (2008). The art and design of genetic screens: RNA interference. Nat Rev Genet, 9(7), 554–566. doi: 10.1038/nrg2364 [DOI] [PubMed] [Google Scholar]
- Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, … Heidelberg Fly Array C (2004). Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science, 303(5659), 832–835. doi: 10.1126/science.1091266 [DOI] [PubMed] [Google Scholar]
- Bryson TD, Weber CM, & Henikoff S (2010). Baculovirus-encoded protein expression for epigenomic profiling in Drosophila cells. Fly (Austin), 4(3), 258–265. doi: 10.4161/fly.4.3.12177 [DOI] [PubMed] [Google Scholar]
- Burnette M, Brito-Robinson T, Li J, & Zartman J (2014). An inverse small molecule screen to design a chemically defined medium supporting long-term growth of Drosophila cell lines. Mol Biosyst, 10(10), 2713–2723. doi: 10.1039/c4mb00155a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoreno AB, Smurnyy Y, Torres AD, Vokes MS, Jones TR, Carpenter AE, & Eggert US (2010). Small molecules discovered in a pathway screen target the Rho pathway in cytokinesis. Nat Chem Biol, 6(6), 457–463. doi: 10.1038/nchembio.363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, E PRI, … Church GM (2015). Highly efficient Cas9-mediated transcriptional programming. Nat Methods, 12(4), 326–328. doi: 10.1038/nmeth.3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Tuttle M, Pruitt BW, Ewen-Campen B, Chari R, Ter-Ovanesyan D, … Church G (2016). Comparison of Cas9 activators in multiple species. Nat Methods, 13(7), 563–567. doi: 10.1038/nmeth.3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla G, & Sokol NS (2014). ADAR mediates differential expression of polycistronic microRNAs. Nucleic Acids Res, 42(8), 5245–5255. doi: 10.1093/nar/gku145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbas L, & Cherbas P (1997). “Parahomologous” gene targeting in Drosophila cells: an efficient, homology-dependent pathway of illegitimate recombination near a target site. Genetics, 145(2), 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbas L, & Cherbas P (2000). Drosophila cell culture and transformation In Sullivan W, Ashburner M, & Hawley RS (Eds.), Drosophila: A Laboratory Manual, 2nd ed. (2 ed.). Cold Spring Harbor, NY: Cold Spring Harbor Press. [Google Scholar]
- Cherbas L, & Cherbas P (2007). Transformation of Drosophila cell lines: an alternative approach to exogenous protein expression In Murhammer DW (Ed.), Baculovirus Expression Protocols (2 ed., Vol. 388, pp. 317–340): Humana Press. [DOI] [PubMed] [Google Scholar]
- Cherbas L, & Gong L (2014). Cell lines. Methods, 68(1), 74–81. doi: 10.1016/j.ymeth.2014.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbas L, Hackney J, Gong L, Salzer C, Mauser E, Zhang D, & Cherbas P (2015). Tools for Targeted Genome Engineering of Established Drosophila Cell Lines. Genetics, 201(4), 1307–1318. doi: 10.1534/genetics.115.181610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbas L, Hackney JF, Gong L, Salzer C, Zhang D, & Cherbas P (2015). Tools for targeted genome engineering of established Drosophila cell lines. Genetics, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbas L, Moss R, & Cherbas P (1994). Transformation techniques for Drosophila cell lines. Methods Cell Biol, 44, 161–179. [DOI] [PubMed] [Google Scholar]
- Cherbas L, Willingham A, Zhang D, Yang L, Zou Y, Eads BD, … Cherbas P (2011). The transcriptional diversity of 25 Drosophila cell lines. Genome Research, 21(2), 301–314. doi: 10.1101/gr.112961.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens JC, Worby CA, Simonson-Leff N, Muda M, Maehama T, Hemmings BA, & Dixon JE (2000). Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc Natl Acad Sci U S A, 97(12), 6499–6503. doi: 10.1073/pnas.110149597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coux RX, Teixeira FK, & Lehmann R (2018). L(3)mbt and the LINT complex safeguard cellular identity in the Drosophila ovary. Development, 145(7). doi: 10.1242/dev.160721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DP, & Sang JH (1978). Cell culture of individual Drosophila embryos. I. Development of wild-type cultures. J Embryol Exp Morphol, 45, 161–172. [PubMed] [Google Scholar]
- Currie DA, Milner MJ, & Evans CW (1988). The growth and differentiation in vitro of leg and wing imaginal disc cells from Drosophila melanogaster. Development, 102, 805–814. [Google Scholar]
- Currie JD, & Rogers SL (2011). Using the Drosophila melanogaster D17-c3 cell culture system to study cell motility. Nature Protocols, 6(10), 1632–1641. doi: 10.1038/Nprot.2011.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlington TK, Wager-Smith K, Ceriani MF, Staknis D, Gekakis N, Steeves TD, … Kay SA (1998). Closing the circadian loop: CLOCK-induced transcription of its own inhibitors per and tim. Science, 280(5369), 1599–1603. [DOI] [PubMed] [Google Scholar]
- Debec A (1978). Haploid cell cultures of Drosophila melanogaster. Nature, 274(5668), 255–256. [DOI] [PubMed] [Google Scholar]
- Debec A, Megraw TL, & Guichet A (2016). Methods to Establish Drosophila Cell Lines. Methods Mol Biol, 1478, 333–351. doi: 10.1007/978-1-4939-6371-3_21 [DOI] [PubMed] [Google Scholar]
- Dequeant ML, Fagegaltier D, Hu Y, Spirohn K, Simcox A, Hannon GJ, & Perrimon N (2015). Discovery of progenitor cell signatures by time-series synexpression analysis during Drosophila embryonic cell immortalization. Proc Natl Acad Sci U S A, 112(42), 12974–12979. doi: 10.1073/pnas.1517729112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echalier G (1971). II. Established diploid cell lines of Drosophila melanogaster as potential material for the study of genetics of somatic cells. Curr Top Microbiol Immunol, 55, 220–227. [DOI] [PubMed] [Google Scholar]
- Echalier G (1997). Drosophila Cells in Culture. San Diego: Academic Press. [Google Scholar]
- Echalier G, & Ohanessian A (1969). [Isolation, in tissue culture, of Drosophila melangaster cell lines]. C R Acad Sci Hebd Seances Acad Sci D, 268(13), 1771–1773. [PubMed] [Google Scholar]
- Echalier G, & Ohanessian A (1970). In vitro culture of Drosophila melanogaster embryonic cells. In Vitro, 6(3), 162–172. [DOI] [PubMed] [Google Scholar]
- Echalier G, Perrimon N, & Mohr S (2017). Drosophila cells in culture (2nd edition ed.). Waltham, MA: Elsevier. [Google Scholar]
- Echeverri CJ, & Perrimon N (2006). High-throughput RNAi screening in cultured cells: a user’s guide. Nat Rev Genet, 7(5), 373–384. doi: 10.1038/nrg1836 [DOI] [PubMed] [Google Scholar]
- Eggert US, Kiger AA, Richter C, Perlman ZE, Perrimon N, Mitchison TJ, & Field CM (2004). Parallel chemical genetic and genome-wide RNAi screens identify cytokinesis inhibitors and targets. PLoS Biol, 2(12), e379. doi: 10.1371/journal.pbio.0020379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehon RG, Kooh PJ, Rebay I, Regan CL, Xu T, Muskavitch MA, & Artavanis-Tsakonas S (1990). Molecular interactions between the protein products of the neurogenic loci Notch and Delta, two EGF-homologous genes in Drosophila. Cell, 61(3), 523–534. [DOI] [PubMed] [Google Scholar]
- Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, … Danielsen M (1987). Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci U S A, 84(21), 7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmannova J, Kucerova L, Sandova K, Steinbauerova V, Broz V, Simek P, & Zurovec M (2012). Differential response of Drosophila cell lines to extracellular adenosine. Insect Biochem Mol Biol, 42(5), 321–331. doi: 10.1016/j.ibmb.2012.01.002 [DOI] [PubMed] [Google Scholar]
- Fondon JW 3rd, Martin A, Richards S, Gibbs RA, & Mittelman D (2012). Analysis of microsatellite variation in Drosophila melanogaster with population-scale genome sequencing. PLoS One, 7(3), e33036. doi: 10.1371/journal.pone.0033036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz A, Brunner E, & Basler K (2017). Generation of genome-modified Drosophila cell lines using SwAP. Fly (Austin), 11(4), 303–311. doi: 10.1080/19336934.2017.1372068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz A, Shlyueva D, Brunner E, Stark A, & Basler K (2017). Probing the canonicity of the Wnt/Wingless signaling pathway. PLoS Genet, 13(4), e1006700. doi: 10.1371/journal.pgen.1006700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, & Sander JD (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol, 31(9), 822–826. doi: 10.1038/nbt.2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gateff E, Gissmann L, Shrestha R, Plus N, Pfister H, Schroder J, & Hausen HZ (1980). Characterization of two tumorous blood cell lines of Drosophila melanogaster and the viruses they contain In Kurstak E, Maramorosch K, & Dubendorfer A (Eds.), Invertebrate Systems in Vitro. Fifth International Conference on Invertebrate Tissue Culture, Rigi-Kaltbad, Switzerland, 1979 (pp. 517–533). Amsterdam: Elsevier. [Google Scholar]
- Ghosh S, Tibbit C, & Liu JL (2016). Effective knockdown of Drosophila long non-coding RNAs by CRISPR interference. Nucleic Acids Res, 44(9), e84. doi: 10.1093/nar/gkw063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, … Qi LS (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell, 154(2), 442–451. doi: 10.1016/j.cell.2013.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, Martin-Ruiz I, Jimenez S, Pirone L, Barrio R, & Sutherland JD (2011). Generation of stable Drosophila cell lines using multicistronic vectors. Sci Rep, 1, 75. doi: 10.1038/srep00075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CL, Stanley D, Ringbauer JA Jr., Beeman RW, Silver K, & Park Y (2012). A cell line derived from the red flour beetle Tribolium castaneum (Coleoptera: Tenebrionidae). In Vitro Cell Dev Biol Anim, 48(7), 426–433. doi: 10.1007/s11626-012-9524-x [DOI] [PubMed] [Google Scholar]
- Gvozdev VA, Birshtein VY, Kakpakov VT, & Polukarova LG (1971). Activity of a sex-linked gene in Drosophila melanogaster embryonic cell sublines. The Soviet journal of developmental biology, 2(3), 243–248. [PubMed] [Google Scholar]
- Handke B, Szabad J, Lidsky PV, Hafen E, & Lehner CF (2014). Towards long term cultivation of Drosophila wing imaginal discs in vitro. PLoS One, 9(9), e107333. doi: 10.1371/journal.pone.0107333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn T, Sandmann T, Fischer B, Axelsson E, Huber W, & Boutros M (2011). Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat Methods, 8(4), 341–346. doi: 10.1038/nmeth.1581 [DOI] [PubMed] [Google Scholar]
- Housden BE, Li Z, Kelley C, Wang Y, Hu Y, Valvezan AJ, … Perrimon N (2017). Improved detection of synthetic lethal interactions in Drosophila cells using variable dose analysis (VDA). Proc Natl Acad Sci U S A, 114(50), E10755–E10762. doi: 10.1073/pnas.1713362114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housden BE, Valvezan AJ, Kelley C, Sopko R, Hu Y, Roesel C, … Perrimon N (2015). Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR-based knockouts with RNAi. Sci Signal, 8(393), rs9. doi: 10.1126/scisignal.aab3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Comjean A, Roesel C, Vinayagam A, Flockhart I, Zirin J, … Mohr SE (2017). FlyRNAi.org-the database of the Drosophila RNAi screening center and transgenic RNAi project: 2017 update. Nucleic Acids Res, 45(D1), D672–D678. doi: 10.1093/nar/gkw977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Roesel C, Flockhart I, Perkins L, Perrimon N, & Mohr SE (2013). UP-TORR: online tool for accurate and Up-to-Date annotation of RNAi Reagents. Genetics, 195(1), 37–45. doi: 10.1534/genetics.113.151340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd H, Al-Olayan E, & Butcher GA (2003). In vitro methods for culturing vertebrate and mosquito stages of Plasmodium. Microbes Infect, 5(4), 321–327. [DOI] [PubMed] [Google Scholar]
- Ishizu H, Sumiyoshi T, & Siomi MC (2017). Use of the CRISPR-Cas9 system for genome editing in cultured Drosophila ovarian somatic cells. Methods, 126, 186–192. doi: 10.1016/j.ymeth.2017.05.021 [DOI] [PubMed] [Google Scholar]
- Jevtov I, Zacharogianni M, van Oorschot MM, van Zadelhoff G, Aguilera-Gomez A, Vuillez I, … Rabouille C (2015). TORC2 mediates the heat stress response in Drosophila by promoting the formation of stress granules. J Cell Sci, 128(14), 2497–2508. doi: 10.1242/jcs.168724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Tarrant RD, Bolam EJ, Angell-Manning P, Soegaard M, Pattinson DJ, … Draper SJ (2018). Production, quality control, stability, and potency of cGMP-produced Plasmodium falciparum RH5.1 protein vaccine expressed in Drosophila S2 cells. NPJ Vaccines, 3, 32. doi: 10.1038/s41541-018-0071-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justiniano SE, Mathew A, Mitra S, Manivannan SN, & Simcox A (2012). Loss of the tumor suppressor Pten promotes proliferation of Drosophila melanogaster cells in vitro and gives rise to continuous cell lines. PLoS One, 7(2), e31417. doi: 10.1371/journal.pone.0031417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn TG, Stenberg P, Pirrotta V, & Schwartz YB (2014). Combinatorial interactions are required for the efficient recruitment of pho repressive complex (PhoRC) to polycomb response elements. PLoS Genet, 10(7), e1004495. doi: 10.1371/journal.pgen.1004495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakpakov VT, Polukarova LG, & Gvozdev VA (1971). Stability and variability of karyotype in continually cultivated sublines of Drosophila melanogaster embryonic cells. The Soviet journal of developmental biology, 2(3), 236–242. [PubMed] [Google Scholar]
- Karpova N, Bobinnec Y, Fouix S, Huitorel P, & Debec A (2006). Jupiter, a new Drosophila protein associated with microtubules. Cell Motil Cytoskeleton, 63(5), 301–312. doi: 10.1002/cm.20124 [DOI] [PubMed] [Google Scholar]
- Ko HW, Jiang J, & Edery I (2002). Role for Slimb in the degradation of Drosophila Period protein phosphorylated by Doubletime. Nature, 420(6916), 673–678. doi: 10.1038/nature01272 [DOI] [PubMed] [Google Scholar]
- Koh K, Zheng X, & Sehgal A (2006). JETLAG resets the Drosophila circadian clock by promoting light-induced degradation of TIMELESS. Science, 312(5781), 1809–1812. doi: 10.1126/science.1124951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Booker M, & Perrimon N (2009). Cross-species RNAi rescue platform in Drosophila melanogaster. Genetics, 183(3), 1165–1173. doi: 10.1534/genetics.109.106567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzelmann S, Bottcher R, Schmidts I, & Forstemann K (2016). A Comprehensive Toolbox for Genome Editing in Cultured Drosophila melanogaster Cells. G3 (Bethesda), 6(6), 1777–1785. doi: 10.1534/g3.116.028241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzelmann S, & Forstemann K (2017). Reversible perturbations of gene regulation after genome editing in Drosophila cells. PLoS One, 12(6), e0180135. doi: 10.1371/journal.pone.0180135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau HC, Lee IK, Ko PW, Lee HW, Huh JS, Cho WJ, & Lim JO (2015). Non-invasive screening for Alzheimer’s disease by sensing salivary sugar using Drosophila cells expressing gustatory receptor (Gr5a) immobilized on an extended gate ion-sensitive field-effect transistor (EG-ISFET) biosensor. PLoS One, 10(2), e0117810. doi: 10.1371/journal.pone.0117810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Robine N, Martin R, Chung WJ, Niki Y, Berezikov E, & Lai EC (2009). Abundant primary piRNAs, endo-siRNAs, and microRNAs in a Drosophila ovary cell line. Genome Res, 19(10), 1776–1785. doi: 10.1101/gr.094896.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecland N, Debec A, Delmas A, Moutinho-Pereira S, Malmanche N, Bouissou A, … Guichet A (2013). Establishment and mitotic characterization of new Drosophila acentriolar cell lines from DSas-4 mutant. Biol Open, 2(3), 314–323. doi: 10.1242/bio.20133327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DF, Chen CC, Hsu TA, & Juang JL (2000). A baculovirus superinfection system: efficient vehicle for gene transfer into Drosophila S2 cells. J Virol, 74(24), 11873–11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, McManus C, Cho DY, Eaton M, Renda F, Somma M, … MacAlpine D (2014a). DNA copy number evolution in Drosophila cell lines. Genome Biol, 15(8), R70. doi: 10.1186/gb-2014-15-8-r70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HG, Kahn TG, Simcox A, Schwartz YB, & Pirrotta V (2015). Genome-wide activities of Polycomb complexes control pervasive transcription. Genome Res, 25(8), 1170–1181. doi: 10.1101/gr.188920.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi J, Fernandez-Valdivia R, Li YD, Simcox AA, & Jafar-Nejad H (2011). Multiple O-glucosylation sites on Notch function as a buffer against temperature-dependent loss of signaling. Development, 138(16), 3569–3578. doi: 10.1242/dev.068361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Housden BE, & Bray SJ (2014). Notch signaling assays in Drosophila cultured cell lines. Methods Mol Biol, 1187, 131–141. doi: 10.1007/978-1-4939-1139-4_10 [DOI] [PubMed] [Google Scholar]
- Li S (2010). Identification of iron-loaded ferritin as an essential mitogen for cell proliferation and postembryonic development in Drosophila. Cell Res, 20(10), 1148–1157. doi: 10.1038/cr.2010.102 [DOI] [PubMed] [Google Scholar]
- Lim MY, Ng AW, Chou Y, Lim TP, Simcox A, Tucker-Kellogg G, & Okamura K (2016). The Drosophila Dicer-1 Partner Loquacious Enhances miRNA Processing from Hairpins with Unstable Structures at the Dicing Site. Cell Rep, 15(8), 1795–1808. doi: 10.1016/j.celrep.2016.04.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FJ, Song W, Meyer-Bernstein E, Naidoo N, & Sehgal A (2001). Photic signaling by cryptochrome in the Drosophila circadian system. Mol Cell Biol, 21(21), 7287–7294. doi: 10.1128/MCB.21.21.7287-7294.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Ewen-Campen B, Ni X, Housden BE, & Perrimon N (2015). In Vivo Transcriptional Activation Using CRISPR/Cas9 in Drosophila. Genetics, 201(2), 433–442. doi: 10.1534/genetics.115.181065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TY, Huang CH, Kao HH, Liou GG, Yeh SR, Cheng CM, … Juang JL (2009). Abi plays an opposing role to Abl in Drosophila axonogenesis and synaptogenesis. Development, 136(18), 3099–3107. doi: 10.1242/dev.033324 [DOI] [PubMed] [Google Scholar]
- Liu B, Zheng Y, Yin F, Yu J, Silverman N, & Pan D (2016). Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila. Cell, 164(3), 406–419. doi: 10.1016/j.cell.2015.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Sims D, & Baum B (2009). Parallel RNAi screens across different cell lines identify generic and cell type-specific regulators of actin organization and cell morphology. Genome Biol, 10(3), R26. doi: 10.1186/gb-2009-10-3-r26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luce-Fedrow A, Von Ohlen T, Boyle D, Ganta RR, & Chapes SK (2008). Use of Drosophila S2 cells as a model for studying Ehrlichia chaffeensis infections. Applied and Environmental Microbiology, 74(6), 1886–1891. doi:Doi 10.1128/Aem.02467-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhur A, Buddika K, Ariyapala IS, Chen S, & Sokol NS (2017). Opposing Post-transcriptional Control of InR by FMRP and LIN-28 Adjusts Stem Cell-Based Tissue Growth. Cell Rep, 21(10), 2671–2677. doi: 10.1016/j.celrep.2017.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhur A, Chawla G, Wu YC, Li J, & Sokol NS (2014). Drosha-independent DGCR8/Pasha pathway regulates neuronal morphogenesis. Proc Natl Acad Sci U S A, 111(4), 1421–1426. doi: 10.1073/pnas.1318445111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum L, Zhang C, Oh S, Mann RK, von Kessler DP, Taipale J, … Beachy PA (2003). Hedgehog signal transduction via Smoothened association with a cytoplasmic complex scaffolded by the atypical kinesin, Costal-2. Mol Cell, 12(5), 1261–1274. [DOI] [PubMed] [Google Scholar]
- Manivannan SN, Jacobsen TL, Lyon P, Selvaraj B, Halpin P, & Simcox A (2015). Targeted integration of single-copy transgenes in Drosophila melanogaster tissue culture cells using recombination-mediated cassette exchange. Genetics, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoff SB, George SL, Bett AJ, Yelmene ML, Dhanasekaran G, Eggemeyer L, … Coller BA (2015). Preclinical and clinical development of a dengue recombinant subunit vaccine. Vaccine, 33(50), 7126–7134. doi: 10.1016/j.vaccine.2015.09.101 [DOI] [PubMed] [Google Scholar]
- Masters JR, Thomson JA, Daly-Burns B, Reid YA, Dirks WG, Packer P, … Debenham PG (2001). Short tandem repeat profiling provides an international reference standard for human cell lines. Proc Natl Acad Sci U S A, 98(14), 8012–8017. doi: 10.1073/pnas.121616198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzendorf C, & Lind MI (2010). The role of iron in the proliferation of Drosophila l(2) mbn cells. Biochem Biophys Res Commun, 400(3), 442–446. doi: 10.1016/j.bbrc.2010.08.100 [DOI] [PubMed] [Google Scholar]
- Miller DE, Staber C, Zeitlinger J, & Hawley RS (2018). Highly Contiguous Genome Assemblies of 15 Drosophila Species Generated Using Nanopore Sequencing. G3 (Bethesda), 8(10), 3131–3141. doi: 10.1534/g3.118.200160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin AA, Kulik AV, Gyoeva FK, Li Y, Goshima G, & Gelfand VI (2006). Regulation of mitochondria distribution by RhoA and formins. J Cell Sci, 119(Pt 4), 659–670. doi: 10.1242/jcs.02762 [DOI] [PubMed] [Google Scholar]
- Mohr S, Bakal C, & Perrimon N (2010). Genomic screening with RNAi: results and challenges. Annu Rev Biochem, 79, 37–64. doi: 10.1146/annurev-biochem-060408-092949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE (2014). RNAi screening in Drosophila cells and in vivo. Methods, 68(1), 82–88. doi: 10.1016/j.ymeth.2014.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Hu Y, Ewen-Campen B, Housden BE, Viswanatha R, & Perrimon N (2016). CRISPR guide RNA design for research applications. FEBS J, 283(17), 3232–3238. doi: 10.1111/febs.13777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Hu Y, Kim K, Housden BE, & Perrimon N (2014). Resources for functional genomics studies in Drosophila melanogaster. Genetics, 197(1), 1–18. doi: 10.1534/genetics.113.154344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, & Perrimon N (2012). RNAi screening: new approaches, understandings, and organisms. Wiley Interdiscip Rev RNA, 3(2), 145–158. doi: 10.1002/wrna.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Rudd K, Hu Y, Song WR, Gilly Q, Buckner M, … Perrimon N (2018). Zinc Detoxification: A Functional Genomics and Transcriptomics Analysis in Drosophila melanogaster Cultured Cells. G3 (Bethesda), 8(2), 631–641. doi: 10.1534/g3.117.300447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Smith JA, Shamu CE, Neumuller RA, & Perrimon N (2014). RNAi screening comes of age: improved techniques and complementary approaches. Nat Rev Mol Cell Biol, 15(9), 591–600. doi: 10.1038/nrm3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson-Rees WA, Daniels DW, & Flandermeyer RR (1981a). Cross-contamination of cells in culture. Science, 212(4493), 446–452. [DOI] [PubMed] [Google Scholar]
- Nelson-Rees WA, Daniels DW, & Flandermeyer RR (1981b). Human embryonic lung cells (HEL-R66) are of monkey origin. Arch Virol, 67(1), 101–104. [DOI] [PubMed] [Google Scholar]
- Neumuller RA, Wirtz-Peitz F, Lee S, Kwon Y, Buckner M, Hoskins RA, … Perrimon N (2012). Stringent analysis of gene function and protein-protein interactions using fluorescently tagged genes. Genetics, 190(3), 931–940. doi: 10.1534/genetics.111.136465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki Y, Yamaguchi T, & Mahowald AP (2006). Establishment of stable cell lines of Drosophila germ-line stem cells. Proc Natl Acad Sci U S A, 103(44), 16325–16330. doi: 10.1073/pnas.0607435103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nir O, Bakal C, Perrimon N, & Berger B (2010). Inference of RhoGAP/GTPase regulation using single-cell morphological data from a combinatorial RNAi screen. Genome Res, 20(3), 372–380. doi: 10.1101/gr.100248.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Ando Y, Kanetani T, Hoshi C, Nakai Y, Nainu F, … Nakanishi Y (2017). Signaling pathway for phagocyte priming upon encounter with apoptotic cells. J Biol Chem, 292(19), 8059–8072. doi: 10.1074/jbc.M116.769745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto N, Viswanatha R, Bittar R, Li Z, Haga-Yamanaka S, Perrimon N, & Yamanaka N (2018). A Membrane Transporter Is Required for Steroid Hormone Uptake in Drosophila. Dev Cell. doi: 10.1016/j.devcel.2018.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkan E, Carrillo RA, Eastman CL, Weiszmann R, Waghray D, Johnson KG, … Garcia KC (2013). An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell, 154(1), 228–239. doi: 10.1016/j.cell.2013.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, & Liu DR (2013). High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol, 31(9), 839–843. doi: 10.1038/nbt.2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne SR, & Kemp CJ (2005). Tumor suppressor genetics. Carcinogenesis, 26(12), 2031–2045. doi: 10.1093/carcin/bgi223 [DOI] [PubMed] [Google Scholar]
- Phelan K, & May KM (2017a). Mammalian Cell Tissue Culture. Curr Protoc Hum Genet, 94, A 3G 1-A 3G 22. doi: 10.1002/cphg.41 [DOI] [PubMed] [Google Scholar]
- Phelan K, & May KM (2017b). Mammalian Cell Tissue Culture Techniques. Curr Protoc Mol Biol, 117, A 3F 1-A 3F 23. doi: 10.1002/cpmb.31 [DOI] [PubMed] [Google Scholar]
- Puck TT, & Marcus PI (1955). A Rapid Method for Viable Cell Titration and Clone Production with Hela Cells in Tissue Culture: The Use of X-Irradiated Cells to Supply Conditioning Factors. Proc Natl Acad Sci U S A, 41(7), 432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, & Lim WA (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell, 152(5), 1173–1183. doi: 10.1016/j.cell.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebay I, Fleming RJ, Fehon RG, Cherbas L, Cherbas P, & Artavanis-Tsakonas S (1991). Specific EGF repeats of Notch mediate interactions with Delta and Serrate: implications for Notch as a multifunctional receptor. Cell, 67(4), 687–699. [DOI] [PubMed] [Google Scholar]
- Rohn JL, Sims D, Liu T, Fedorova M, Schock F, Dopie J, … Baum B (2011). Comparative RNAi screening identifies a conserved core metazoan actinome by phenotype. J Cell Biol, 194(5), 789–805. doi: 10.1083/jcb.201103168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Inagaki S, Mituyama T, Kawamura Y, Ono Y, Sakota E, … Siomi MC (2009). A regulatory circuit for piwi by the large Maf gene traffic jam in Drosophila. Nature, 461(7268), 1296–1299. doi: 10.1038/nature08501 [DOI] [PubMed] [Google Scholar]
- Samakovlis C, Asling B, Boman HG, Gateff E, & Hultmark D (1992). In vitro induction of cecropin genes--an immune response in a Drosophila blood cell line. Biochem Biophys Res Commun, 188(3), 1169–1175. [DOI] [PubMed] [Google Scholar]
- Schneider I (1972). Cell lines derived from late embryonic stages of Drosophila melanogaster. J Embryol Exp Morphol, 27(2), 353–365. [PubMed] [Google Scholar]
- Schneider I, & Blumenthal AB (1978). Drosophila cell and tissue culture In Ashburner M & Wright TRF (Eds.), The Genetics and Biology of Drosophila (Vol. 2A, pp. 265–315): Academic Press. [Google Scholar]
- Serbus LR, Landmann F, Bray WM, White PM, Ruybal J, Lokey RS, … Sullivan W (2012). A cell-based screen reveals that the albendazole metabolite, albendazole sulfone, targets Wolbachia. PLoS Pathog, 8(9), e1002922. doi: 10.1371/journal.ppat.1002922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Yarrow C, Dunning R, Cheek B, Vass S, Windass J, & Hadfield S (2012). Insecticide detoxification indicator strains as tools for enhancing chemical discovery screens. Pest Manag Sci, 68(1), 38–48. doi: 10.1002/ps.2218 [DOI] [PubMed] [Google Scholar]
- Shaposhnikov AV, Kryndushkin AS, Nikolenko Iu V., Panov VV, Nabirochkina EN, Lebedeva LA, & Shidlovskii Iu V. (2013). [Activation of JAK/STAT signaling pathway in S2 Drosophila melanogaster cell culture]. Mol Biol (Mosk), 47(3), 486–491. [DOI] [PubMed] [Google Scholar]
- Shields G, Dubendorfer A, & Sang JH (1975). Differentiation in vitro of larval cell types from early embryonic cells of Drosophila melanogaster. J Embryol Exp Morphol, 33(1), 159–175. [PubMed] [Google Scholar]
- Silver K, Jiang H, Fu J, Phillips TW, Beeman RW, & Park Y (2014). The Tribolium castaneum cell line TcA: a new tool kit for cell biology. Sci Rep, 4, 6840. doi: 10.1038/srep06840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcox A (2013). Progress towards Drosophila epithelial cell culture. Methods Mol Biol, 945, 1–11. doi: 10.1007/978-1-62703-125-7_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcox A, Mitra S, Truesdell S, Paul L, Chen T, Butchar JP, & Justiniano S (2008). Efficient genetic method for establishing Drosophila cell lines unlocks the potential to create lines of specific genotypes. PLoS Genet, 4(8), e1000142. doi: 10.1371/journal.pgen.1000142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcox AA, Sobeih MM, & Shearn A (1985). Establishment and characterization of continuous cell lines derived from temperature-sensitive mutants of Drosophila melanogaster. Somat Cell Mol Genet, 11(1), 63–70. [DOI] [PubMed] [Google Scholar]
- Smurnyy Y, Toms AV, Hickson GR, Eck MJ, & Eggert US (2010). Binucleine 2, an isoform-specific inhibitor of Drosophila Aurora B kinase, provides insights into the mechanism of cytokinesis. ACS Chem Biol, 5(11), 1015–1020. doi: 10.1021/cb1001685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoiber M, Celniker S, Cherbas L, Brown B, & Cherbas P (2016). Diverse Hormone Response Networks in 41 Independent Drosophila Cell Lines. G3 (Bethesda), 6(3), 683–694. doi: 10.1534/g3.115.023366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassburger K, Lorbeer FK, Lutz M, Graf F, Boutros M, & Teleman AA (2017). Oxygenation and adenosine deaminase support growth and proliferation of ex vivo cultured Drosophila wing imaginal discs. Development, 144(13), 2529–2538. doi: 10.1242/dev.147538 [DOI] [PubMed] [Google Scholar]
- Sumiyoshi T, Sato K, Yamamoto H, Iwasaki YW, Siomi H, & Siomi MC (2016). Loss of l(3)mbt leads to acquisition of the ping-pong cycle in Drosophila ovarian somatic cells. Genes Dev, 30(14), 1617–1622. doi: 10.1101/gad.283929.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swevers L, Cherbas L, Cherbas P, & Iatrou K (1996). Bombyx EcR (BmEcR) and Bombyx USP (BmCF1) combine to form a functional ecdysone receptor. Insect Biochem Mol Biol, 26(3), 217–221. [DOI] [PubMed] [Google Scholar]
- Tants JN, Fesser S, Kern T, Stehle R, Geerlof A, Wunderlich C, … Sattler M (2017). Molecular basis for asymmetry sensing of siRNAs by the Drosophila Loqs-PD/Dcr-2 complex in RNA interference. Nucleic Acids Res, 45(21), 12536–12550. doi: 10.1093/nar/gkx886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuyama T, Tsubouchi A, Usui T, Imamura H, & Uemura T (2017). Mitochondrial dysfunction induces dendritic loss via eIF2alpha phosphorylation. J Cell Biol, 216(3), 815–834. doi: 10.1083/jcb.201604065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ui K, Nishihara S, Sakuma M, Togashi S, Ueda R, Miyata Y, & Miyake T (1994). Newly established cell lines from Drosophila larval CNS express neural specific characteristics. In Vitro Cell Dev Biol Anim, 30A(4), 209–216. [DOI] [PubMed] [Google Scholar]
- Ui K, Ueda R, & Miyake T (1987). Cell lines from imaginal discs of Drosophila melanogaster. In Vitro Cell Dev Biol, 23(10), 707–711. [DOI] [PubMed] [Google Scholar]
- Ulvila J, Parikka M, Kleino A, Sormunen R, Ezekowitz RA, Kocks C, & Ramet M (2006). Double-stranded RNA is internalized by scavenger receptor-mediated endocytosis in Drosophila S2 cells. J Biol Chem, 281(20), 14370–14375. doi: 10.1074/jbc.M513868200 [DOI] [PubMed] [Google Scholar]
- Vincent JP (2014). Modulating and measuring Wingless signalling. Methods, 68(1), 194–198. doi: 10.1016/j.ymeth.2014.03.015 [DOI] [PubMed] [Google Scholar]
- Viswanatha R, Li Z, Hu Y, & Perrimon N (2018). Pooled genome-wide CRISPR screening for basal and context-specific fitness gene essentiality in Drosophila cells. Elife, 7. doi: 10.7554/eLife.36333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg A, & Schneider I (1993). In vitro culture of the mosquito stages of Plasmodium falciparum. Exp Parasitol, 76(2), 121–126. doi: 10.1006/expr.1993.1014 [DOI] [PubMed] [Google Scholar]
- Wen J, Mohammed J, Bortolamiol-Becet D, Tsai H, Robine N, Westholm JO, … Lai EC (2014). Diversity of miRNAs, siRNAs, and piRNAs across 25 Drosophila cell lines. Genome Research, 24(7), 1236–1250. doi: 10.1101/gr.161554.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White PM, Serbus LR, Debec A, Codina A, Bray W, Guichet A, … Sullivan W (2017). Reliance of Wolbachia on High Rates of Host Proteolysis Revealed by a Genome-Wide RNAi Screen of Drosophila Cells. Genetics, 205(4), 1473–1488. doi: 10.1534/genetics.116.198903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods DF, & Poodry CA (1984). Cell surface proteins of Drosophila: I. Changes induced by 20-hydroxyecdysone. Develomental Biology, 96(1), 23–31. [DOI] [PubMed] [Google Scholar]
- Wyss C (1982a). Chironomus tentans epithelial cell lines sensitive to ecdysteroids, juvenile hormone, insulin and heat shock. Exp Cell Res, 139(2), 309–319. [DOI] [PubMed] [Google Scholar]
- Wyss C (1982b). Ecdysterone, insulin and fly extract needed for the proliferation of normal Drosophila cells in defined medium. Exp Cell Res, 139(2), 297–307. [DOI] [PubMed] [Google Scholar]
- Yao T, & Asayama Y (2017). Animal-cell culture media: History, characteristics, and current issues. Reprod Med Biol, 16(2), 99–117. doi: 10.1002/rmb2.12024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassin A, Markow TA, Narechania A, O’Grady PM, & DeSalle R (2010). The genus Drosophila as a model for testing tree- and character-based methods of species identification using DNA barcoding. Mol Phylogenet Evol, 57(2), 509–517. doi: 10.1016/j.ympev.2010.08.020 [DOI] [PubMed] [Google Scholar]
- Zartman J, Restrepo S, & Basler K (2013). A high-throughput template for optimizing Drosophila organ culture with response-surface methods. Development, 140(3), 667–674. doi: 10.1242/dev.088872 [DOI] [PubMed] [Google Scholar]
- Zhang C, Williams EH, Guo Y, Lum L, & Beachy PA (2004). Extensive phosphorylation of Smoothened in Hedgehog pathway activation. Proc Natl Acad Sci U S A, 101(52), 17900–17907. doi: 10.1073/pnas.0408093101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Grode KD, Stewman SF, Diaz-Valencia JD, Liebling E, Rath U, … Sharp DJ (2011). Drosophila katanin is a microtubule depolymerase that regulates cortical-microtubule plus-end interactions and cell migration. Nat Cell Biol, 13(4), 361–370. doi: 10.1038/ncb2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Koolhaas WH, & Schnorrer F (2014). A versatile two-step CRISPR- and RMCE-based strategy for efficient genome engineering in Drosophila. G3 (Bethesda), 4(12), 2409–2418. doi: 10.1534/g3.114.013979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Malone JH, Powell SK, Periwal V, Spana E, Macalpine DM, & Oliver B (2010). Expression in aneuploid Drosophila S2 cells. PLoS Biol, 8(2), e1000320. doi: 10.1371/journal.pbio.1000320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Mohr S, Hannon GJ, & Perrimon N (2013). Inducing RNAi in Drosophila cells by transfection with dsRNA. Cold Spring Harb Protoc, 2013(5), 461–463. doi: 10.1101/pdb.prot074351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurovec M, Dolezal T, Gazi M, Pavlova E, & Bryant PJ (2002). Adenosine deaminase-related growth factors stimulate cell proliferation in Drosophila by depleting extracellular adenosine. Proc Natl Acad Sci U S A, 99(7), 4403–4408. doi: 10.1073/pnas.062059699 [DOI] [PMC free article] [PubMed] [Google Scholar]