

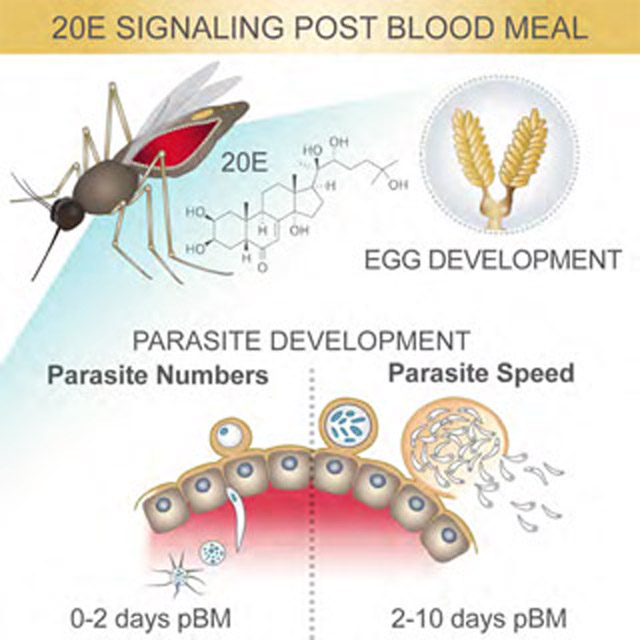
Summary
Transmission of malaria parasites occurs when a female Anopheles mosquito feeds on an infected host to acquire nutrients for egg development. How parasites are affected by oogenetic processes, principally orchestrated by the steroid hormone 20-hydroxyecdysone (20E), remains largely unknown. Here we show that Plasmodium falciparum development is intimately but not competitively linked to processes shaping Anopheles gambiae reproduction. We unveil a 20E-mediated positive correlation between egg and oocyst numbers, such that impairing oogenesis by multiple 20E manipulations decreases parasite intensities. These manipulations, however, accelerate Plasmodium growth rates, allowing sporozoites to become infectious sooner. Parasites exploit mosquito lipids for faster growth, but they do so without further impacting egg development. These results suggest that P. falciparum has adopted a non-competitive evolutionary strategy of resource exploitation to optimize transmission while minimizing fitness costs to its mosquito vector. Our findings have profound implications for currently proposed control strategies aimed at suppressing mosquito populations.
Graphical Abstract
eTOC blurb:
The development of the malaria-causing parasite Plasmodium falciparum depends on its ability to exploit the sexual cycle of its mosquito host in a non-competitive manner.
Introduction
Blood feeding behavior has independently evolved in multiple insect taxa as a means to acquire the necessary nutrient resources for successful reproduction. While increasing insect reproductive fitness, the evolution of this behavior has inadvertently provided a new and effective transmission route for a multitude of human pathogens, including the mosquito-borne dengue and Zika viruses as well as Plasmodium parasites, the causative agents of malaria. Unquestionably, the most lethal of these pathogens is Plasmodium falciparum, yearly killing over 420,000 people, mostly young African children, via the bite of highly effective anopheline species (WHO, 2018). The blood meal-triggered events that lead to egg development have been comprehensively characterized in the arboviral mosquito vector Aedes aegypti. In this species, blood feeding initiates the release of ovarian ecdysteroidogenic hormone (OEH) (Brown et al., 1998) and insulin-like peptides (ILPs) (Wu and Brown, 2006; Brown et al., 2008) from the brain. In turn, these factors stimulate the production of the steroid hormone ecdysone (E) from the ovarian epithelium (Brown et al., 1998; Riehle and Brown, 1999), followed by E hydroxylation to the active hormone 20E in the fat body, the mosquito fat storage organ. Upon binding to its nuclear receptor, the heterodimer ecdysone receptor (EcR)/ultraspiracle (USP), 20E triggers a vast transcriptional program that promotes the accumulation of lipids and other nutrients into the ovaries via the function of the lipid transporter lipophorin (Lp) and the yolk protein precursor vitellogenin (Vg), leading to egg development (Raikhel, 1992; Raikhel and Dhadialla, 1992; van Heusden et al., 1997; van Heusden et al., 1998; Attardo et al., 2005). Pathways important for oogenesis appear to be largely conserved in other mosquito species including the major malaria vector Anopheles gambiae (Noriega et al., 2006; Bai et al., 2010).
In An. gambiae, the 2–3 day period needed for egg development is also critical for the successful establishment of Plasmodium parasite infection in the midgut (Mitchell and Catteruccia, 2017). Shortly after ingestion within the blood meal, male and female gametes fuse to form a diploid zygote that then transforms during the next 21–24 h into a motile ookinete, which traverses the midgut epithelium before encysting beneath the midgut basal lamina and differentiating into an oocyst. During this transition, the parasite is subject to attack by the mosquito innate immune system, including lysis triggered by the complement-like thioester-containing protein TEP1 (Blandin et al., 2004; Frolet et al., 2006). After an additional 7–10 days, surviving Plasmodium oocysts rupture to release thousands of sporozoites that invade the salivary glands and are injected into another human host at the mosquito’s next blood meal. The time from ingestion to infectivity in the mosquito is known as the parasite extrinsic incubation period (EIP), and it is a key component of malaria transmission: faster parasites have a higher chance of being transmitted given the short lifespan of Anopheles females (Macdonald, 1956; Shapiro et al., 2016; Shapiro et al., 2017).
During the initial phases of infection, Plasmodium parasites are exposed to rapidly changing physiological conditions largely regulated by the function of ecdysteroid hormones and may be substantially affected by this blood meal-induced physiological ‘storm’. However, whether they utilize available nutritional resources or compete for them with their mosquito hosts remains unclear. Previous work has suggested that parasites can accumulate and are affected by mosquito lipids (Atella et al., 2009; Rono et al., 2010; Costa et al., 2018), and numerous studies, albeit using unnatural vector-parasite associations, emphasize that parasites exert a fitness cost to mosquito fecundity (Hogg and Hurd, 1995; Jahan and Hurd, 1997; Ahmed et al., 1999; Ferguson et al., 2003). Yet, the relative fitness of parasites and mosquitoes during the course of a natural infection remains largely undetermined or difficult to interpret due to confounding factors associated with field infections (Hogg and Hurd, 1997; Sangare et al., 2014).
Here, we show that development of P. falciparum parasites in the An. gambiae female is intimately linked to physiological processes modulated by 20E during egg development. By analyzing both egg and parasite numbers in individual females, we unveil an unexpected positive correlation between mosquito and parasite fitness that is dependent on 20E signaling. While impairing oogenesis by multiple manipulations of 20E function decreases oocyst numbers, parasites respond to these metabolic changes by increasing their speed of development, becoming infectious sooner. Although faster growth depends on mosquito lipid resources via the lipid transporter Lp, it does not impair the mosquito oogenesis cycle, indicating that parasites use surplus nutrients that are not required for host reproduction. Thus, our data suggest that P. falciparum has adopted an evolutionary strategy that minimizes reproductive costs to its natural mosquito vector, partly explaining the formidable spread of these parasites in sub-Saharan Africa. These findings have important implications for malaria control strategies targeting mosquito reproduction.
Results:
P. falciparum infections are linked to egg development in An. gambiae
To determine whether Plasmodium infections are linked to the reproductive fitness of the mosquito vector, we infected An. gambiae (G3 strain) females with a P. falciparum culture (NF54 strain), and then counted both the number of eggs developed in the ovaries and the number of oocysts present in the midgut of each individual female 8 days (d) post infectious blood meal (pIBM). Surprisingly, when we plotted the paired egg-parasite data and performed a correlation analysis, we found that oogenesis and intensity of infection were positively correlated (Figure 1A), demonstrating a positive link between these two processes in contrast to the theory of vector-parasite competition predicting costs of infection on mosquito reproduction (Schwenke et al., 2016). This correlation was observed independent of mosquito size, approximated by measuring wing lengths (Spearman’s r = 0.3224, N = 106, p = 0.0007), and therefore independent of blood meal volume.
Figure 1: Parasite and egg development are linked.

(A) There is a positive correlation between the number of An. gambiae eggs and P. falciparum oocysts developing in females (Spearman’s correlation. Line shows data trend). (B) A transgenic vasa2-Cas9 mosquito line was crossed with a U657-Zpg gRNA transgenic line to generate Δzpg mosquitoes. (C–D) Blood-fed Δzpg females (C) fail to produce eggs (scale bar = 500 μm) and (D) support fewer parasites compared to Zpg/+ controls (Cntrl) (Mann-Whitney), while there is no effect on infection prevalence (P) (Chi-Squared). (E) Δzpg females produce lower levels of ecdysteroids at 26 h and 36 h pBM (Kruskal-Wallis, Dunn’s correction). N = sample size.
To understand the nature of the egg-parasite correlation, we next explored whether successful establishment of P. falciparum infection depends on the presence of fully functional ovaries. To this end, we generated transgenic An. gambiae females (Δzpg) in which zero population growth (zpg), a gene essential for germ cell development (Tazuke et al., 2002; Thailayil et al., 2011), was mutated by CRISPR/Cas9 mutagenesis (Figure 1B). As expected, Δzpg females had ablated or severely underdeveloped ovaries, and largely failed to produce eggs after a blood meal (Figure 1C). When fed on P. falciparum-infected blood, Δzpg mutant females that failed to develop eggs showed significantly fewer oocysts compared to control siblings (Figure 1D), and again, this effect was independent of body size (Mann Whitney: oocysts ÷ wing length3, U = 257.5, p = 0.0241). There was no difference in the prevalence of infection between the two groups (Figure 1D).
The steroid hormone 20E regulates parasite development via its nuclear receptor
The reduced oocyst load observed in Δzpg mutants suggests that parasite development relies on processes that are regulated by the correct function of ovarian tissues. Given the key role of 20E in oogenesis, we went on to assess whether the function of this steroid hormone affects parasite numbers. As a first step, we determined ecdysteroid levels in Δzpg mutant females at 26 h and 36 h post-blood meal (pBM), around the peak of 20E synthesis (Redfern, 1982), and found severely reduced titers, as we had hypothesized given that multiple steps of 20E synthesis occur in the ovaries (Figure 1E). To provide a direct link between 20E levels and Plasmodium infection, we then experimentally lowered ecdysteroid levels in An. gambiae by injecting wild type females with ecdysone 22-oxidase (E22O), which oxidizes 20E (and its precursor E) in the C22 position thereby reducing its activity (Kamimura et al., 2012). Females injected with E22O showed a 4-fold decrease in ecdysteroid levels 26 h pBM (Figure 2A). Moreover, E22O injections prior to P. falciparum infection negatively affected egg development (Figure 2B), and also significantly reduced oocyst intensities, while the prevalence of infection remained unaffected (Figure 2C). These data provide additional evidence that 20E may regulate mosquito processes important for P. falciparum development.
Figure 2: The steroid hormone 20E regulates both egg and parasite numbers.

(A) Injecting wild type females with the ecdysteroid oxidase E22O reduces ecdysteroid levels in the female 26 h pBM compared to BSA-injected controls (Cntrl) (unpaired t-test). (B–C) E22O-injected females (B) produce fewer eggs (Mann-Whitney) and (C) have fewer oocysts at 8 d pIBM (GLM, Poisson distribution). There is no effect on infection prevalence (P) (Chi-Squared). (D) Egg and oocyst numbers are positively correlated in control and E22O-injected females (Spearman’s correlations. Lines show data trends). N = sample size.
Although E22O-injected females had limited reproductive fitness and reduced parasite loads, these females maintained a positive correlation between eggs and oocysts (Spearman’s ρ = 0.2989, N = 109, p = 0.0016, Figure 2D). However, when we disrupted steroid hormone signaling—rather than 20E levels—by RNAi silencing of the heterodimeric nuclear receptor EcR/USP, this correlation was lost. While still observing decreased egg and parasite loads in females injected with double-stranded RNA targeting EcR (dsEcR) compared to dsGFP-injected controls (dsCntrl) (Figure 3A, 3B) (see STAR Methods), the number of oocysts in the midgut became unlinked from the number of eggs developed in the ovaries (Spearman’s ρ = 0.1737, N = 107, p > 0.05, Figure 3C). We verified that 20E signaling was successfully suppressed in dsEcR females by detecting reduced transcripts of Vg (Figure 3D), which is normally induced by 20E through its nuclear receptor after blood feeding (Raikhel, 1992). Furthermore, the correlation between eggs and oocysts was also lost after silencing the 20E co-receptor USP (Spearman’s ρ = 0.1556, N = 95, p > 0.05, Figure 3E), confirming that the positive interaction between mosquito and parasite fitness depends on signaling pathways controlled by 20E via its nuclear receptor.
Figure 3: 20E controls the egg-parasite correlation via its nuclear receptor.

(A–B) Females injected with dsEcR (EcR) and fed on a P. falciparum infected blood meal (A) produce fewer eggs (Mann-Whitney) and (B) develop fewer oocysts at 7 d pIBM (GLM, Poisson distribution) compared to dsGFP-injected females (Cntrl). There is no effect on infection prevalence (P) (Chi-Squared). (C) The positive correlation between egg and oocyst numbers is lost in dsEcR-injected females (Spearman’s correlation). (D) dsEcR females have lower Vg expression at 24 h pBM compared to controls (means ± SEM, unpaired t-test, FDR corrected). NBF = non-blood fed. (E) The positive egg-oocyst correlation is also lost in dsUSP-injected females (USP) (Spearman’s correlation). N = sample size. Lines show data trends.
20E signaling affects early oocyst development in a TEP1-independent manner
In order to establish when P. falciparum parasite numbers are reduced following disrupted 20E signaling, we analyzed earlier developmental stages in dsEcR and control females by microscopy. We dissected midguts at 24 h and 48 h pIBM and, after eliminating the blood bolus, performed immunofluorescence assays using an antibody targeting the P. falciparum Pfs25 surface protein to determine the number of ookinetes crossing the midgut and the number of early oocysts, respectively. While ookinete numbers were unaffected (Figure 4A), we detected a significant reduction in early oocysts in dsEcR females (Figure 4B, 4C). Consistent with the lack of an observed effect on ookinete numbers, co-silencing of TEP1, the major immune effector that kills ookinetes during midgut invasion, did not rescue the number of oocysts in dsEcR/dsTEP1 co-injected females (Figure 4D). 20E signaling therefore affects parasites during their transition into oocysts via TEP1-independent mechanisms.
Figure 4: 20E signaling affects parasites during the ookinete-oocyst transition.

(A) dsEcR injections (EcR) have no effect on the number (GLM, Poisson distribution) or prevalence (P) (Fisher’s Exact) of ookinetes traversing the midgut at 24 h pIBM, detected by immunofluorescent labeling of Pfs25 after blood bolus removal. Control = dsGFP-injected (Cntrl). (B–C) By 2 d pIBM, dsEcR females have fewer oocysts compared to controls (Log(y) transformed, GLM, normal distribution), as detected by Pfs25 (green) labeling. Red = actin (phalloidin) (scale bar = 50 μm). There remains no effect on infection prevalence (P) (Fisher’s Exact). (D) TEP1 silencing does not rescue oocyst numbers in dsEcR females. Females injected with dsEcR or co-injected with dsEcR and dsTEP1 (EcR/TEP1) have the same number of oocysts at 7 d pIBM, and both groups have reduced oocyst numbers compared to controls. There is no effect of dsTEP1 (TEP1) alone on oocyst numbers (Kruskal-Wallis, Dunn’s correction). Infection prevalence (P) is unaffected (Fisher’s Exact). N = sample size.
Disrupting 20E signaling accelerates P. falciparum growth rates
When analyzing Plasmodium infections in midguts 7–8 d pIBM, we noticed that dsEcR females had larger oocysts than controls, which was confirmed by measuring the cross-sectional area of individual oocysts (Figure 5A, 5B). This effect was observed throughout oocyst development, as early as 5 d pIBM, the first time point when we could reliably measure parasite size. By 12 d pIBM, when some oocysts have burst to release sporozoites, there was no longer a difference between the two groups (Figure 5B). Increased size was not the result of reduced competition between parasites at lower intensities, as we observed no correlation between oocyst size and numbers (dsEcR: Spearman’s ρ = 0.1103, N = 77, p > 0.05; dsGFP: Spearman’s ρ = 0.0444, N = 75, p > 0.05).
Figure 5: Disrupting 20E signaling promotes faster parasite development.

(A) Oocysts are larger in dsEcR females (EcR) compared to dsGFP controls (Cntrl) at 8 d pIBM (scale bar = 50 μm). (B) The mean oocyst area per midgut is higher in dsEcR females from 5–10 d pIBM. By 12 d pIBM, as oocysts burst, there is no difference between dsEcR and controls (Least Squares model). (C–D) Salivary glands of dsEcR females show (C) a higher prevalence of sporozoites at 10 d pIBM (Chi-Squared) and (D) significantly more sporozoites at 12 d pIBM (Mann-Whitney). No difference is observed at later time points (Mann-Whitney). (E) Salivary gland sporozoites collected from dsEcR females at 12 d pIBM are as infectious as controls when applied to primary human hepatocytes in vitro (medians ± IQR, unpaired t-test) (EEFs = exoerythrocytic forms). (F) The EIP50 (time to 50% infectious) of dsEcR females is significantly reduced compared to controls, as determined from sporozoite prevalence data shown in (C) and (D) (means ± 95% CI, unpaired t-test). N = sample size with dsGFP listed first. See Figures S1–S2 and Table S1.
Larger oocysts may represent faster developing parasites or may be the consequence of an aberrant developmental program. To address this question, we analyzed sporozoite prevalence and intensity in the female salivary glands over time. Consistent with accelerated oocyst growth, sporozoites were detectable earlier and in greater numbers after perturbing 20E signaling. EcR-silenced females were 2.67 times more likely to have sporozoites in the glands than controls at 10 d pIBM (odds ratio: 95% CI = 1.13–6.25 and Figure 5C), an early time point for sporozoite invasion of salivary glands. Infection intensities were also elevated in dsEcR females at 12 d pIBM, while by 14 d pIBM the number of sporozoites was equal in both groups (Figure 5D). No difference was observed in the number of sporozoites per oocyst (Table S1), showing that the higher prevalence and intensity of infection observed in dsEcR females were due to accelerated growth rather than increased oocyst productivity.
Faster parasites are infectious to the vertebrate host
Faster parasite growth could potentially occur at the expense of sporozoite fitness, causing reduced infectivity to the human host. We therefore assessed whether sporozoites collected from the salivary glands of dsEcR females were infectious to primary human hepatocytes, a well-established in vitro method to measure P. falciparum infectivity to humans (Mazier et al., 1985; March et al., 2013). Sporozoites dissected from dsEcR females at 12 d pIBM were as able to invade and infect primary human hepatocytes as controls, as determined by the quantification of exoerythrocytic forms (EEFs) (Figure 5E). Consistently, we did not find any differences in sporozoite gliding motility or cell invasion, demonstrating the equivalent fitness of these faster-developing sporozoites (Figure S1A, S1B). Sporozoites from EcR-silenced females remained as infective as controls at later time points (18 d pIBM) (Figure S1C–E).
We used our prevalence data to extrapolate the length of sporogonic development in the two groups and found that the EIP50 (median time when 50% of mosquitoes are infectious) of EcR-silenced females was significantly reduced compared to controls (Figure 5F). Importantly, increased parasite growth was also detected in E22O-injected and Δzpg mutant females (Figure S2A, S2B). Moreover, E22O-injected females were 2.09 times more likely than controls to have sporozoites in their salivary glands at 11–12 d pIBM, similar to our observations after EcR silencing (odds ratio: 95%CI = 1.04–4.23 and Figure S2C). Altogether these data demonstrate that reducing 20E function shortens the EIP of P. falciparum in An. gambiae—a trait critical for parasite transmission by the mosquito vector.
Lipid accumulation accelerates P. falciparum growth via the lipid transporter Lp
What factors could be inducing faster oocyst development after 20E manipulations? Previous work determined that silencing the lipid transporter Lp affects oocyst size (Rono et al., 2010; Costa et al., 2018), while avian Plasmodium oocysts were found to incorporate lipids and lipid transporters in the midgut (Atella et al., 2009). We therefore examined the Lp expression profile of EcR-silenced and control females and found that whereas in controls Lp transcripts peaked at 12 h pBM before declining (similar to findings in Ae. aegypti (Sun et al., 2000)), in EcR-silenced females levels of Lp remained high up to 48 h pBM (Figure 6A). These data were confirmed by western blot using a specific antibody targeting the ApoI subunit of Lp (Figure S3A).
Figure 6: Faster parasite development is mediated by Lp-transported lipids with no additional cost to fecundity.

(A) Lp expression is elevated in dsEcR females (EcR) after blood feeding relative to dsGFP controls (Cntrl) (means ± SEM, unpaired t-test, FDR corrected). NBF = non-blood fed. (B) Total lipids, TAGs, and PCs are significantly higher in dsEcR midguts at 48 h pIBM while DAGs are increased independent of time (means ± SEM, Least Squares model). (C) Co-injecting females with both dsEcR and dsLp (EcR/Lp) largely returns mean oocyst size to control levels at 8 d pIBM (Kruskal-Wallis, Dunn’s correction). (D) In dsGFP controls, there is a negative correlation between the number of eggs and the mean size of oocysts in individual females at 8 d pIBM. This correlation is lost in dsLp females (Spearman’s correlations. Lines show data trends). (E) dsEcR females develop the same number of eggs after either an infectious or inactivated P. falciparum blood meal (Mann-Whitney). N = sample size with dsGFP listed first. See Figures S3–S4.
Since mis-regulated Lp implies altered lipid trafficking, we next determined the lipid profile of midgut tissues at 24 h and 48 h pBM. dsEcR midguts had a significantly higher lipid content at 48 h pBM (Figure 6B)—a time point when early oocysts are beginning development. Specifically, four lipid species were enriched after EcR silencing: triacylglycerides (TAGs), diacylglycerides (DAGs), phosphatidylcholines (PCs), and lysophosphatidylethanolamines (LPEs) (Figure 6B, Figure S4A). Impairing 20E signaling thus results in a significantly altered midgut lipid profile following a blood meal.
Given the parallel increases in lipid levels and Lp expression in EcR-depleted females, we next assessed whether lipid trafficking by this lipid transporter was responsible for faster oocyst growth. We silenced Lp alone and in combination with EcR, and analyzed egg and parasite numbers as well as oocyst size. Strikingly, co-silencing of Lp and EcR largely suppressed the increase in oocyst size observed in dsEcR-injected females (Figure 6C), implicating lipid species transported by this lipoprotein in parasite development. In addition, Lp silencing reduced egg and oocyst numbers (Figure S3B, S3C) as previously shown (Vlachou et al., 2005; Rono et al., 2010).
We next determined whether 20E levels affect the speed of parasite growth in an Lp-dependent manner also in physiological conditions. To this end, we examined if there was a relationship between egg numbers (a proxy for 20E levels) and oocyst size (a proxy for EIP) in our control females and found a negative correlation between these parameters (Spearman’s ρ = −0.2057, N = 153, p = 0.0107, Figure 6D). This negative correlation was however lost in Lp-silenced females, where mean oocyst size per midgut—although in our experiments not significantly reduced compared to controls as in Costa et al., 2018—was independent of egg numbers (Spearman’s ρ = 0.0404, N = 114, p > 0.05, Figure 6D).
Altogether these data point to 20E-regulated lipids trafficked by Lp as important factors in shaping the speed of P. falciparum development—the first demonstration to our knowledge of mosquito factors impacting the parasite EIP in the Anopheles female.
Faster P. falciparum growth does not induce a reproductive fitness cost in An. gambiae
To rule out the possibility that during oocyst growth, parasites deplete resources destined for egg development, we plotted the total oocyst area in individual midguts (mean oocyst area × total oocyst number) against the number of eggs in the same female and found a positive correlation between these parameters (Spearman’s ρ = 0.2837, N = 153, p = 0.0004). This suggests parasites are not competing for resources critical for oogenesis, as we would otherwise expect their cumulative area in the midgut to be negatively correlated to the number of eggs developed.
Furthermore, we determined whether the accelerated parasite growth observed in the absence of EcR signaling occurs at the expense of egg development. To this end, we compared the number of eggs developed by EcR-depleted females fed either an infectious or heat-inactivated P. falciparum blood meal. Heat exposure renders gametocytes non-infectious while allowing females to feed on a blood meal with otherwise identical nutrition. Consistent with a lack of competition, females from both groups developed comparable numbers of eggs regardless of the presence of infectious parasites (Figure 6E).
Combined, these results point to a non-competitive strategy adopted by P. falciparum for optimal transmission whereby parasites exploit excess nutritional resources that the Anopheles female does not invest towards reproduction.
Silencing the TAG lipase TL2 induces faster parasite growth
To establish a more direct link between lipid levels and parasite growth, we experimentally increased neutral lipids by impairing lipolysis through silencing TAG Lipase 2 (TL2, AGAP000211). TL2 possesses the catalytic serine residue found in the lipase active site motif (Pagni et al., 2007) and is enriched in blood-fed females (Marinotti et al., 2006). As expected, dsTL2 injections caused an accumulation of midgut lipids including TAGs, DAGs, and PCs (Figure 7A) as also observed by an abundance of lipid droplets in dsTL2 midguts (Figure 7B). Three additional lipids classes were also enriched (Figure S4B). Besides raising levels of Lp transcripts (Figure 7C), TL2 silencing induced a remarkable increase in oocyst size throughout development (Figure 7D), closely phenocopying the effects observed in EcR-silenced females. There was no effect on the prevalence or intensity of P. falciparum infection (Figure S5A), and once again, we observed no correlation between oocyst size and numbers, confirming a lack of competition between parasites (Spearman’s ρ = 0.1554, N = 37, p > 0.05). Together with the EcR/Lp co-silencing results, these data further support a role for Lp-transported neutral lipids in determining parasite growth.
Figure 7: Silencing the TAG lipase TL2 induces faster parasite growth.

(A) Total lipids, TAGs, DAGs, and PCs are higher in dsTL2 midguts (TL2) at 24 h and 48 h pBM compared to dsGFP controls (Cntrl) (means ± SEM, Least Squares model). (B) Midguts of dsTL2 females stained with the neutral lipid dye LD540 (red) show an accumulation of lipid droplets at 48 h pBM (scale bar = 20 μm). Blue = DAPI. (C) Lipophorin expression is elevated in dsTL2 fat bodies at 24 h pBM (means ± SEM, Least Squares model). (D) dsTL2 females develop larger oocysts from 5–10 d pIBM (Mann-Whitney). N = sample size with dsGFP listed first. See Figures S4–S5.
Discussion
The parasite EIP is a key parameter of malaria transmission dynamics. Given the relatively short Anopheles lifespan, estimated to be around 2–3 weeks depending on species and environmental conditions (Clements and Paterson, 1981; Costantini et al., 1996), parasites with faster sporogonic development are more likely to be transmitted to the next human host (Macdonald, 1956; Shapiro et al., 2016; Shapiro et al., 2017). Despite its relevance, however, little is known about factors regulating the length of the P. falciparum EIP (Ohm et al., 2018). Notable exceptions are increased temperature (Paaijmans et al., 2010; Shapiro et al., 2017) and plentiful mosquito nourishment during larval stages (Shapiro et al., 2016), both associated with a shorter parasite EIP. Here we demonstrate that P. falciparum has the ability to respond to metabolic changes within the mosquito vector, taking advantage of resources that become available to achieve faster development without compromising sporozoite infectivity. Specifically, our data points to the 20E-dependent accumulation of glycerolipids (TAGs, DAGs) and phospholipids (PCs) in the midgut as the driver of faster, Lp-mediated parasite growth. Interestingly, in Ae. aegypti, which transmits several arboviruses including dengue and Zika, inhibition of EcR also leads to increased TAG storage after a blood meal, possibly by down-regulation of hepatocyte nuclear factor 4 (HNF4), which controls the expression of several TAG lipolytic genes (Wang et al., 2017). In our experiments however, EcR silencing did not affect expression of TL2 or other lipolytic enzymes (Figure S5B–D), while its effects were significantly reduced by co-silencing the lipid transporter Lp, suggesting that lipid trafficking rather than lipolysis is predominantly affected by 20E signaling. How might P. falciparum parasites exploit additional lipid resources to decrease EIP? The substantial growth and subdivision needed to generate thousands of sporozoites during oocyst development requires significant biosynthesis of phospholipid-containing cellular membranes. Increased availability of lipids may accelerate this process, in a manner similar to host lipid scavenging that occurs during human stages of infection (Mi-Ichi et al., 2007; Itoe et al., 2014; Brancucci et al., 2017).
Disrupting egg development through zpg mutation, 20E inactivation, or EcR silencing all induced faster parasite growth, likely through the same metabolic mechanism (see Figure S6 for a scheme). These findings are an important warning for the deployment of mosquito population suppression methods such as gene drive systems aimed to favor the spread of factors that decrease female reproductive fitness (Burt, 2003; Hammond et al., 2016). Indeed, methods that induce changes to blood meal metabolism could lead to fewer mosquitoes that nevertheless allow faster parasite development, potentially causing an increase in Plasmodium transmission over a period of time. Careful assessment of interactions between pathogen development and mosquito reproductive processes is therefore essential when generating any genetically modified strain intended for field release.
Faster oocyst growth after EcR silencing, albeit mediated by lipid accumulation, did not further affect egg numbers. Moreover, we found no negative relationship between egg numbers and total parasite area. Based on these observations, an intriguing hypothesis is that P. falciparum may have followed a non-competitive evolutionary strategy based on exploiting available mosquito resources that are not needed by the female for egg development, as previously postulated (Costa et al., 2018). Reproductive costs inflicted by infection with Plasmodium parasites have been documented in mosquitoes, but it is worth noting that those studies used animal infection models such as P. yoelii nigeriensis and P. chabaudi (Hogg and Hurd, 1995; Jahan and Hurd, 1997; Ahmed et al., 1999; Ferguson et al., 2003) that have not coevolved with anthropophilic vectors such as An. gambiae. Indeed, these murine models also stimulate strong complement-mediated immune reactions (Blandin et al., 2004; Dong et al., 2006; Frolet et al., 2006) that P. falciparum has evolved to avoid via the surface expression of Pfs47 (Molina-Cruz et al., 2013). One caveat of this interpretation is that we analyzed egg development after a single blood meal rather than across the entire female reproductive lifespan. Theory however predicts that the evolutionary costs of impairing the development of the first egg batch would be substantially larger than costs imposed during subsequent gonotrophic cycles (Koella et al., 2009). Overall, it seems plausible that a strategy of non-competitive resource exploitation during oocyst development may have favored adaptation of P. falciparum to An. gambiae, partly explaining why this mosquito-pathogen combination is so formidable at ensuring malaria transmission in sub-Saharan Africa.
The unexpected positive correlation between egg and oocyst numbers that we found across a number of experiments also reinforces the notion of a non-competitive host-parasite interaction. Although we could not establish the specific mechanisms behind the reduction in parasite numbers observed after decreasing 20E titers or impairing 20E signaling, our results suggest that 20E may regulate the function of TEP1-independent immune processes targeting the early oocyst. Established theory predicts organisms balance their energetic investment into multiple life processes at any given time due to a limited pool of available resources, often leading to reciprocal fitness trade-offs between survival, immunity, and reproduction (Schwenke et al., 2016). Our findings point to a possible regulation of these trade-offs by steroid hormones. When high 20E levels increase investment in oogenesis, a concomitant general repression of somatic or immune functions may occur, explaining how more parasites develop in females producing more eggs. In contrast, with low 20E levels, resources may be allocated preferentially towards immune or somatic processes that negatively affect parasite survival, resulting in reduced Plasmodium numbers as observed in our experiments. Notably, parasite numbers were reduced during the ookinete-to-oocyst transition, which broadly corresponds to the peak of 20E synthesis. Steroid hormone levels after blood feeding vary largely between individuals of a mosquito population (i.e. Figure 1E), likely depending on diverse factors including larval nutritional status, microbial load and environmental conditions as well as genetic determinants (Mamai et al., 2014). Other interpretations not involving a reproduction-immunity tradeoff cannot be excluded, for instance steroid hormone function may affect the expression of midgut components that are required for the ookinete-oocyst transition or for oocyst attachment to the basal lamina (Arrighi et al., 2005). Regardless of the mechanism, it appears that P. falciparum parasites have evolved their developmental strategy to respond to the mosquito reproductive investment: they either grow faster (when the female’s reproductive investment is reduced) or they are more abundant (when the female invests more significantly in egg development). Moreover, they appear not to compete with each other, as we see no interaction between parasite numbers and oocyst size.
Our work sheds new light on the parasite’s journey within the female mosquito. By unraveling the intricate interplay between An. gambiae reproductive physiology and P. falciparum survival and growth, these studies identify the steroid hormone 20E as a major factor regulating the development of the deadliest human parasites in their most effective mosquito vectors.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Flaminia Catteruccia (fcatter@hsph.harvard.edu).
Experimental Model and Subject Details
Rearing of Anopheles gambiae mosquitoes
Anopheles gambiae mosquitoes (wild-type G3 and transgenic strains) were reared in cages at 27°C, 70–80% humidity on a 12 h light:12 h dark cycle. Adults in colony cages were fed on 10% glucose solution ad libitum and weekly on donated human blood (Research Blood Components, Boston, MA). Males and females were separated as pupae to ensure the virgin status of females. The wild-type G3 has been verified as An. gambiae by PCR and DNA sequencing of the amplified products (Santolamazza et al., 2008).
Culturing of Plasmodium falciparum NF54 parasites
P. falciparum (NF54 strain) was cultured as asexual stages between 0.2 and 2% parasitemia at 37°C in human erythrocytes at 5% hematocrit (Interstate Blood Bank, Memphis TN) using RPMI medium 1640 supplemented with 25mM HEPES, 10mg/l hypoxanthine, 0.2% sodium bicarbonate, and 10% heat-inactivated human serum (Interstate Blood Bank) under a gas mixture of 5% O2, 5% CO2, balanced N2 for up to 8 weeks according to established protocols (Trager and Jensen, 1976; Ifediba and Vanderberg, 1981). Gametocytogenesis was induced by raising parasitemia >4% and incubating cultures for 14–20 d to accumulate stage IV and V male and female gametocytes, with media replaced daily. To inactivate gametocytes and render cultures non-infectious, infectious cultures were heat-exposed at 42°C for 15 min immediately prior to feeding of mosquitoes. This strain has been confirmed to be P. falciparum by PCR followed by DNA sequencing of the amplified products (Snounou et al., 1993). The Plasmodium falciparum NF54 strain is used under the permissions of a material transfer agreement from the laboratory of Carolina Barillas-Mury, National Institutes of Health, Bethesda, MD, USA.
Culturing of primary human hepatocytes
Cryopreserved male human primary hepatocytes (BioIVT, Westbury, NY) were cultured according to the protocols extensively laid out in (March et al., 2015). Briefly, thawed cells were seeded onto micropatterned collagen islands in well plates and cultured at 37°C, 5% CO2 using DMEM with L-glutamine medium supplemented with 1% penicillin-streptomycin. These cells were validated by the distributor. After 24 h hepatocyte islands were surrounded by supporting cells from the male mouse fibroblast cell line 3T3–J2 (Rheinwald and Green, 1975) and then co-cultured as above. Fibroblasts were cultured at 37°C, 5% CO2 using DMEM medium supplemented with 10% bovine serum and 1% penicillin-streptomycin.
Method Details
P. falciparum infections of An. gambiae mosquitoes
Cages of female mosquitoes aged 4–7 d were blood fed on ~320 μl P. falciparum culture for 30–60 min via heated membrane feeders and introduced into a custom-built glove box (Inert Technology, Amesbury MA). Mosquitoes not fully engorged were removed, and blood-fed mosquitoes were fed 10% glucose solution ad libitum until dissection. At dissection time points, mosquitoes were aspirated into 80% ethanol and transferred to 1× phosphate-buffered saline (PBS) (ookinete, oocyst stages) or aspirated into ice-cold PBS or RPMI (sporozoites stages). At least 3 biological replicates of each infection were performed.
Ookinete and early oocyst counts:
Mosquitoes were dissected at 24 h (ookinetes) or 48 h (early oocysts) pIBM and any remaining blood meal was removed. Midguts were fixed in 4% formaldehyde for 30–40 min, permeabilized and blocked for 1 h in PBS, 0.1% Triton (PBS-T), 1% bovine serum albumin (BSA) at 22°C, and stained with a mouse αPfs25 monoclonal primary antibody 4B7 (B.E.I Resources, Atlanta, GA) (1/200), followed by an Alexa 488 goat amouse secondary antibody (Thermo Fisher Scientific, Waltham, MA) (1/400). Immunofluorescently-labelled midguts were mounted in Vectashield with DAPI (Vector Laboratories, Burlingame, CA), imaged at 100× or 200× on a Zeiss Inverted Observer Z1 and parasites counted.
Late oocyst counts and measurements:
From 5 d pIBM onwards, midguts were stained directly in 2 mM mercurochrome (Sigma-Aldrich, St. Louis, MO) for 12 min, while ovarian development was also scored. The mosquito carcass including ovaries was preserved at 22°C in >70% ethanol until eggs could be counted later. Mercurochrome-stained midguts were imaged at 100× or 200× on an Olympus Inverted CKX41 microscope, and oocysts were counted and measured using scaled images in FIJI (Schindelin et al., 2012). Burst oocysts were excluded from oocyst size analysis. Mean oocyst size was calculated for each midgut to avoid pseudoreplication.
Sporozoite counts:
Mosquitoes were decapitated and the salivary glands of individual females were collected in a small volume of RPMI and disrupted using a handheld disposable pestle. Released sporozoites were spun at 8000 g for 10 min at 4°C and resuspended in a known volume of RPMI. Sporozoites in 0.1 μl were counted using a disposable hemocytometer at 200× or 400× magnification on an Olympus Inverted CKX41 microscope with phase-contrast microscopy. Sporozoite totals for each mosquito were calculated. Median sporozoite totals per oocyst were estimated at the population level using mosquitoes collected on different days following an infectious blood meal on d 1: median sporozoite intensity at d 18 was divided by median oocyst intensity at d 7–8. Data from infections presented in Figure 3 and 5 were pooled for this calculation.
Generation of transgenic mosquito lines
gRNA design:
The zpg locus (AGAP006241) was sequenced to identify SNPs present within our An. gambiae strains. gRNA design tools available at the Broad Institute (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design (Doench et al., 2016)) and the Zhang laboratory, MIT (http://crispr.mit.edu (Hsu et al., 2013)) were used to design three gRNAs with minimal off-target sites and mathematically determined optimal cutting. Multiple gRNA sequence targets were chosen to maximize the chances of generating gene knockouts (gRNAa: 5’-GCGGCTTCACTGTCGTGTGA–3’; gRNAb: 5’-CCAAGTGTTTGCATTCCTGG-3’; gRNAc: 5’-GATCCGATCACGCAGTCGAT-3’). gRNAs were synthetized as gBlocks (Integrated DNA Technologies, Skokie, IL) under the control of the U657 promoter (322bp upstream of AGAP013557) (Hammond et al., 2016) and followed by a polyT sequence terminator. gRNAa and gRNAb were synthesized within a single gBlock as a tethered pair connected by a 21bp sequence (5’-TTCACTGTGCGCATTATATAT-3’) predicted not to interfere with gRNA secondary structure (RNAfold) (Hofacker et al., 1994). gRNAc was synthesized as a second gBlock.
Plasmid construction:
A 2.3 kb fragment of the vasa2 promoter (Papathanos et al., 2009) was amplified from genomic DNA with primers 5’-CAGGTCTCAATCCCGATGTAGAACGCGAG-3’ and 5’-CGGTCTCACATATTGTTTCCTTTCTTTATTCACCGG-3’. SpCas9 was amplified from plasmid PX165 (Addgene #48137) (Ran et al., 2013) with primers 5’-CAGGTCTCATATGGACTATAAGGACCACGACGGAG-3’ and 5’-CAGGTCTCAAAGCTTACTTTTTCTTTTTTGCCTGGCC03’. The fragments were cloned via Golden Gate ligation into pDSAR16, a transgenesis plasmid that provides an SV40 terminator, an attB site for ϕC31 transgenesis into attP-containing docking lines, and a 3xPax3-RFP selectable marker (Volohonsky et al., 2015). The two gRNA-containing gBlocks were cloned into the pDSAY transgenesis plasmid (Volohonsky et al., 2015). To enable in vivo fluorescence detection of the adult germline, an EYFP reporter under the control of the vasa2 promoter was included on this transgene. The vasa2 promoter region was amplified from genomic DNA with primers (5’-CGGTCTCACGCGCGATGTAGAACGCGAGCAAA-3’) and (5’-CAGGTCTCACCATATTGTTTCCTTTCTTTATTCACCGG3’). The EYFP-SV40 terminator cassette was amplified from pDSAY using primers (5’–CAGGTCTCAATGGTGAGCAAGGGCG-3’) and (5’-CAGGTCTCACGCGGCTTTAAGATACATTGATGAGTTTGGAC–3’). The two fragments were cloned into the AscI site in the pDSAY plasmid via Golden Gate ligation. Completed plasmids were sequenced to verify correct assembly.
Transgenesis:
The zpg gRNA construct (400 ng/μl) and Cas9 construct (400 ng/μl) were injected into the posterior of freshly-laid embryos from An. gambiae X13 (N = 1663) and X1 (N = 2585) attP-containing lines (Volohonsky et al., 2015), respectively, in conjunction with 80 ng/μl of a helper plasmid expressing ϕC31 integrase. Adults surviving injection were crossed to the wild type G3 strain to identify and isolate transgenic lines gZPG(Zpg) and VZC(Cas9), which were both verified as single insertions by DNA sequencing around the attP insertion sites. These lines were subsequently backcrossed four times to wild type. Transgenes were made homozygous using the fluorescence intensity of the neuronal Pax marker and true breeding was verified in subsequent generations by the absence of non-fluorescent progeny.
Generation of Δzpg females
Zpg/+ control and Zpg/Cas9 mutant females, hereafter Δzpg mutants, were generated as the F1 progeny of Zpg transgene homozygotes and Cas9/+ heterozygotes. Rare Zpg/+ F1 females with ovarian phenotypes due to the maternal contribution of Cas9 to the egg were excluded from the analysis. To eliminate any bias, rearing and feeding effects in experiments, controls and Δzpg mutants were not separated during larval development or in adulthood by the DsRed fluorescence of the Cas9 transgene until after blood feeding (and, if applicable, P. falciparum development) had taken place. Controls and Δzpg mutant females did not differ in wing length size or in their ability to take a blood meal. The ovarian phenotype was scored after dissection or through the cuticle via the vasa2-EYFP marker in the Zpg transgene, present in all individuals, under a Leica M80 fluorescence dissecting scope.
Ecdysteroid inactivation using the oxidase E22O
The fungal ecdysteroid-22-oxidase (E22O) from Metarhizium rileyi (Kamimura et al., 2012) catalyzes the inactivation of 20E through the oxidation of the hydroxyl group at the carbon backbone position 22. The gene encoding E22O was codon-optimized for mammalian expression and synthesized by BioBasic (Markham, ON, Canada). The E22O construct was engineered to contain the mature protein and a 6×-His tag before the stop codon, subcloned into the VR2001 TOPO vector (a modified version of the VR1020 vector used in Calvo et al., 2006; Vical Incorporated, San Diego CA), and transformed in One Shot TOP10 Chemically Competent E. coli (Thermo Fisher Scientific). A total of 1 mg of plasmid DNA (VR2001-E22O construct) was obtained using GeneElute HP endotoxin-free plasmid MEGA prep kit (Sigma-Aldrich). Plasmid DNA was sterilized through a 0.22 μm filter, and recombinant protein was produced by transfecting human embryonic kidney cells (HEK293E; American Type Culture Collection, Manassas, VA). The cell supernatant was collected 72 h after transfection and loaded onto Nickel-charged HiTrap Chelating HP column (5 ml bed volume; GE Healthcare Life Sciences, Marlborough MA) following the manufacturer’s directions. Fractions were step-eluted with 5, 20, 300 and 1000 mM imidazole (in 50 mM Tris, 500 mM NaCl, pH 8.0) and then loaded onto a size-exclusion column (Superdex 200 HR10/30; GE Healthcare Life Sciences) using the ÄKTA purifier system (GE Healthcare Life Sciences). Proteins were eluted isocratically at a flow rate of 0.5 ml/min in 50 mM Tris, 150 mM NaCl, pH 8.0. Purified E22O was separated in a 4–12% Bis-Tris NuPAGE Gel (Thermo Fisher Scientific) and visualized by Coomassie stain. Protein identity was verified by automated Edman degradation at the Research Technologies Branch, NIAID, NIH. The catalytic activity of recombinant E22O was determined as described in Kamimura et al., 2012. A total of 69 nl (Figure S2C) or 138 nl (Figure 2, Figure S2B) of purified protein (0.5 mg/ml) was injected into wild-type females 4–5 d after eclosion, and 3–6 h prior to feeding on a P. falciparum-infected blood meal. Control females were injected with an equal volume and concentration of BSA (66 kDa), which has a comparable molecular mass to E22O (76 kDa). Females were randomly assigned to injection groups and surviving females were used in experiments.
Measurement of ecdysteroid levels
Control or Δzpg mutant females and E22O- or BSA-injected females were blood fed on human blood and then anaesthetized on ice at 26 h or 36 h pBM as required. Egg development was scored in Δzpg mutant females through the cuticle by the expression of a vasa2-EYFP transgene to preserve circulating ecdysteroids in the hemolymph within the body. Heads were removed from females and carcasses were stored in pairs at −80°C in 80 μl of 100% methanol. Ecdysteroid levels were determined using the 20E enzyme immunoassay (EIA) kit (Cayman Chemical, Ann Arbor, MI), following the recommended protocol. Each sample was measured in duplicate. This assay detects 20E but according to the manufacturer, also cross reacts with ecdysone, 2-deoxy-20-hydroxyecdysone, and 2-deoxy-edysone, hence we assayed ecdysteroid levels. EIA plates were read on a spectrophotometer at 412 nm after 90 min.
Gene expression knockdown using dsRNA
Creation of templates for dsRNA production:
PCR fragments of EcR (AGAP028634; 435 bp) and the eGFP control (495 bp) were amplified from plasmids pCR2.1-EcR and pCR2.1-eGFP described previously (Baldini et al., 2013). A 481 bp fragment of USP (AGAP002095) was amplified from cDNA prepared from female whole bodies minus head using USP-FWD: 5’-GGAAGCAATGGAGGTGGAG-3’ and USP-REV: 5’-CATAGAATTCTGGCCAACGC-3’, and then cloned into the pCR2.1 construct (Thermo Fisher Scientific). A 600 bp fragment of Lp (AGAP001826) corresponding to bases 9452–10051 of the Lp cDNA was amplified from plasmid pLL10-Lp, a gift from Miranda Whitten and Elena A. Levashina (Max Planck Institute for Infection Biology, Berlin), using a primer matching the inverted T7 promoters: 5’–TAATACGACTCACTATAGGG–3’. Purified constructs were recovered from NEB Turbo chemically-competent E. coli (New England Biolabs, Ipswich, MA) and verified by DNA sequencing prior to use. Amplifications from pCR2.1-based vectors used universal primers against the vector backbone to add the T7 promoter (lowercase) to both ends: pCR2.1-T7F: 5’–taatacgactcactatagggCCGCCAGTGTGCTGGAA–3’ pCR2.1-T7R: 5’–taatacgactcactatagggGGATATCTGCAGAATTCGCCC–3’. PCR fragments of TEP1 (AGAP010815; 395 bp) and TL2 (AGAP000211; 406 bp) were amplified directly from cDNA prepared from female whole bodies minus head using the following primers: TEP1-FWD: 5’–taatacgactcactatagggTTTGTGGGGCTGAAAGCGCTG–3’, TEP1-REV: 5’–taatacgactcactatagggACCACGTAACCGCTCGGTAAG–3’, based on Jaramillo-Gutierrez et al., 2009; TL2-FWD: 5’-taatacgactcactatagggCTTTAGTCGGGTGGAGGACA-3’, TL2–REV: 5’-taatacgactcactatagggGATGATCGGTGTTCGGCTT-3’. PCR product sizes and yield were confirmed by gel electrophoresis. dsRNA was transcribed and purified from the PCR templates using the Megascript T7 transcription kit (Thermo Fisher Scientific) as described previously (Blandin et al., 2004; Rogers et al., 2009).
dsRNA injections:
For GFP, EcR, USP, Lp and TL2 single knockdown experiments, 690 ng of dsRNA (dsGFP, dsEcR, dsUSP, dsLp, dsTL2) was injected (Nanoject II, Drummond) at a concentration of 10 ng/nl into adult females within 1 d of eclosion. In double knockdown groups, dsRNAs were mixed 1:1 and injected in twice the volume (1.38 μg). Females were randomly assigned to injection groups and surviving females were used in subsequent experiments. Gene knockdown levels were determined in at least 3 biological replicates by quantitative real-time PCR at 2 d post injection (Figure S7).
RNA extraction, cDNA synthesis and qRT-PCR
Female mosquitoes (N = 5–10) were decapitated in PBS and transferred to TRI reagent (Thermo Fisher Scientific). Samples were stored at −80°C and RNA was extracted according to the manufacturer’s instructions, with a modification to wash the pelleted RNA with 70% ethanol in RNAse-free water three times. RNA was DNAse treated using Turbo DNAse (Thermo Fisher Scientific) and quantified using a NanoDrop Spectrophotometer 2000c (Thermo Fisher Scientific). cDNA synthesis was performed as in Rogers et al., 2008. Approximately 2 μg of RNA was used in 100 μl cDNA reactions. Primers (Integrated DNA Technologies) used for quantitative real-time PCR (qRT-PCR) (Table S2) were previously published or designed using Primer-BLAST (Ye et al., 2012) where possible over exon-exon junctions or in separate exons and checked for potential off-target binding. cDNA samples from 3 biological replicates were diluted 10-fold for qRT-PCR. Quantification was performed in duplicate 15 μl reactions, containing 1× Fast SYBR Green Master mix for qRT-PCR (Thermo Fisher Scientific), primers and 5 μl of diluted cDNA. Reactions were run on a StepOnePlus qRT-PCR thermocycler (Thermo Fisher Scientific). Relative quantities were determined using a standard curve and normalized against the ribosomal gene RpL19 (AGAP004422), which does not change significantly with blood feeding (Marinotti et al., 2006). To determine the expression of additional TAG lipases, we assayed transcripts of AGAP000210, AGAP001764 and AGAP007672. Amplifications using 4 different primers sets designed against transcripts of AGAP001764 were attempted but were not successful, and no RNA sequencing reads were detected in mixed sex adults from this gene model (Gibbons et al., 2009). We concluded that this gene is a pseudogene.
Sporozoite gliding and infectivity assays
P. falciparum sporozoites from salivary glands of dsGFP and dsEcR females at 12 d and 18 d pIBM were assessed for their ability to glide on coated plates, invade, and infect micropatterned human primary hepatocytes in 96-well plate format assays according to protocols described in March et al., 2015. To assay sporozoite gliding ability, 7000 sporozoites were added per well of a 96 well plate coated with 5 μg/ml of a mouse αPfCSP monoclonal primary antibody 2A10 (B.E.I Resources). Plates were spun down at 3000 rpm for 5 min, and were incubated at 37°C for 45 min. Following incubation, plates were fixed and CSP trails were stained as in March et al., 2015.
For invasion and infection assays, 10000 and 100000 sporozoites, respectively, from each group, were added per well of a 96-well plate containing micropatterned human primary hepatocytes. Plates were spun down at 3000 rpm for 5 min, then incubated at 37°C. For the invasion assay, plates were fixed in 4% paraformaldehyde (PFA) at 3 h post-invasion. Cells were double-stained with αPfCSP to label both intracellular and extracellular parasites, and 3 wells were counted per condition, with a minimum of 100 events scored per well. For the infection assays, plates were fixed in 100% MeOH at 2 d post-invasion. Cells were stained with mouse αPfHSP70 monoclonal primary antibody 4C9 (Tsuji et al., 1994), following the protocol described in March et al., 2015.
Western blotting
Abdominal fat body (N = 8) was dissected from 4-d-old blood-fed females at 24 h and 48 h pBM in cold PBS and frozen at −80°C in modified breaking buffer (Hansen et al., 2005) (50 mM Tris, pH 8.0; 1% NP40; 0.25% sodium deoxycholate; 150 mM NaCl; 1 mM EDTA; 1× protease inhibitor mixture (Roche, Basel, Switzerland). Samples were homogenized, and the insoluble material removed by centrifugation at 20000 g at 4°C. Protein was quantified using a Quick Start Bradford assay (Bio-Rad, Hercules, CA). 30 μg of soluble protein was denatured and run in parallel between 3–8% Tris-Acetate NuPAGE and 4–12% Bis-Tris NuPAGE gels (Thermo Fisher Scientific) due to the large size difference between ApoI (288 kDa) and the actin loading control (42 kDa). Protein was transferred to PVDF membranes using an iBLOT2 transfer system (Thermo Fisher Scientific), and membranes were washed twice in 1× PBS-T, then blocked for 30 min at 22°C in Odyssey Blocking Buffer (Li-cor, Lincoln, NE). Membranes from 3–8% gels were incubated with rabbit aApoI (1:4000) polyclonal primary antibody and those from 4–12% gels were incubated with rat aActin monoclonal primary antibody MAC237 (Abcam, Cambridge, UK) (1:5000), both shaking overnight at 4°C. Affinity-purified antibodies targeting the large ApoI subunit of Lp were generated by Genscript (Piscataway, NJ) using the peptide FQRDASTKDEKRSGC conjugated to a carrier protein. Following the primary antibody, membranes were washed with PBS-T, and incubated with secondary antibodies (donkey arabbit 800CW and goat arat 680LT (Li-cor), both at 1:20,000) in blocking buffer with 0.01% SDS for 1 h at 22°C. Membranes were washed thoroughly with PBS-T before imaging with the Odyssey CLx scanner. Images and band intensities were analyzed in StudioImageLite (Li-cor).
Staining of neutral lipids with LD540
Midguts (N = 10) were dissected from dsGFP- or dsTL2-injected females at 48 h pBM and fixed with 4% PFA for 45 min at 22°C, and then neutral lipids were stained with LD540 in PBS (0.5 μg/ml) for 5 min at 22°C according to Spandl et al., 2009. Nuclei were stained with DAPI for 5 min before mounting with Vectashield (Vector Laboratories). Images were captured at 400× on a Zeiss Inverted Observer Z1.
Lipidomics of mosquito tissues
Midguts from dsGFP, dsEcR, or dsTL2 females were collected in pools of 4–5 into 100 μl of isopropanol (HPLC Grade; Honeywell) at 24 h and 48 h pBM, in 3 biological replicates for dsEcR or 4 replicates in dsTL2 analyses. Analyses of polar and non-polar lipids were conducted using an LC-MS system comprised of a Shimadzu Nexera X2 U-HPLC (Shimadzu Corp, Marlborough, MA) coupled to an Exactive Plus orbitrap mass spectrometer (Thermo Fisher Scientific). Tissues were homogenized in a TissueLyser (Qiagen, Hilden, Germany) with 2, 3mm tungsten beads for 4 min at 20 Hz. Lipids were extracted from tissue homogenates (10 μL) using 190 μL of isopropanol (HPLC Grade; Honeywell) containing 1,2-didodecanoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids, Alabaster, AL). Lipid extracts were then injected onto an ACQUITY BEH C8 column (100 × 2.1 mm, 1.7 μm; Waters, Milford, MA) and the column was eluted isocratically with 80% mobile phase A (95:5:0.1 vol/vol/vol 10 mM ammonium acetate/methanol/formic acid) for 1 min followed by a linear gradient to 80% mobile-phase B (99.9:0.1 vol/vol methanol/formic acid) over 2 min, a linear gradient to 100% mobile phase B over 7 min, then 3 min at 100% mobile-phase B. Mass spectrometry data was acquired using electrospray ionization in the positive ion mode over 200–1100 m/z and at 70,000 resolution. Raw data were processed using TraceFinder 3.3 (Thermo Fisher Scientific) and Progenesis QI (Nonlinear Dynamics, Newcastle upon Tyne, UK) software for detection and integration of LC-MS peaks. Lipid identities were determined based on comparison to reference standards and reference plasma extracts and denoted by total number of carbons in the lipid acyl chain(s) and total number of double bonds in the lipid acyl chain(s).
Quantification and Statistical Analysis
See figure legends for details of the statistical methods used in each experiment. Data was analyzed in GraphPad Prism 7 and JMP Pro 13. Normality was determined using the D’Agostino-Pearson omnibus normality test, using a significance cut-off value of p = 0.05. Transformations to generate normally distributed data were attempted prior to statistical testing using parametric methods, and, if unsuccessful, non-parametric statistical methods were used. A significance cut-off value of p = 0.05 was used for statistical tests and, where multiple tests were performed, the significance was corrected using a 5% false discovery rate (FDR), or Dunn’s multiple comparison correction. Where P. falciparum infection intensity was variable between biological replicates, or data were collected over time, Generalized Linear models (GLM) or Standard Least Squares models incorporating replicate, treatment, and time as factors were constructed, with interactions between factors excluded if they were not significant (see Table S3 for full details). Unless indicated, horizontal bars on graphs represent medians. Trendlines illustrate the overall shape of correlations using linear regression but non-parametric statistical methods were used to analyze the data. Proportional data for infection prevalence was compared using a Chi-squared test where N > 5 in all groups, or else using Fisher’s Exact test. Wing length, measured in millimeters from the proximal wing notch to the distal tip of the third cross vein, was cubed and used to normalize oocyst and egg count data for mosquito size.
Supplementary Material
Supplementary Figure 1: Sporozoite gliding and infectivity to hepatocytes. Related to Figure 5. (A–B) Sporozoites collected from the salivary glands of dsEcR (EcR) and dsGFP (Cntrl) females at 12 d pIBM display no significant difference in (A) gliding activity (means, Fisher’s Exact) or (B) hepatocyte invasion (medians ± IQR, Mann-Whitney), as determined through microscopy by identification of circumsporozoite surface protein (CSP) trails and percent sporozoites inside vs outside hepatocyte cells, respectively. (C–E) Similarly, sporozoites collected from the same groups at 18 d pIBM show no difference in their (C) gliding activity (means, Fisher’s Exact), (D) hepatocyte invasion (medians ± IQR, Mann-Whitney) and (E) development into exoerythrocytic forms (EEFs) (medians ± IQR, unpaired t-test).
Supplementary Figure 2: Parasites grow faster in Δzpg mutant and E22O-injected females. Related to Figure 5. (A–B) At 8 d pIBM, mean oocyst area is significantly greater in (A) Δzpg females (unpaired t-test, Welch’s correction) and (B) E22O-injected females (Mann-Whitney) compared to their respective controls (Cntrl). (C) Salivary glands of E22O-injected females show a higher prevalence of sporozoites at 11–12 d pIBM (Nominal Logistic Regression). There was no difference in intensity (Mann-Whitney). In all panels, N corresponds to the sample size. P = prevalence of infection.
Supplementary Figure 3: Lp accumulates in EcR-depleted females and its silencing reduces both egg and parasite numbers. Related to Figure 6. (A) ApoI protein (αApoI) accumulates in the fat bodies of dsEcR females (EcR) pIBM, with a more pronounced effect at 48 h pIBM. Actin (αActin) = loading control. (B–C) dsEcR-, dsLp- (Lp), and dsEcR/Lp- (EcR/Lp) injected females (B) produce fewer eggs (Kruskal-Wallis & Dunn’s correction) and (C) have fewer oocysts at 8 d pIBM compared to dsGFP-injected controls (Cntrl) (Kruskal-Wallis & Dunn’s correction). There is no effect on infection prevalence (Fisher’s Exact) (P = prevalence of infection). In all panels where applicable, N corresponds to the sample size.
Supplementary Figure 4: Minor lipid species enriched in dsEcR and dsTL2 midguts. Related to Figure 6 and Figure 7. (A) dsEcR-injected midguts (EcR) have elevated levels of LPEs compared to dsGFP controls (Cntrl) (means ± SEM, Standard Least Squares model) at 48 h pBM. (B) dsTL2-injected midguts (TL2) are enriched for three additional lipid species: phosphatidylserines (PSs), phosphatidylinositols (PIs), and phosphatidylethanolamines (PEs) (means ± SEM, Standard Least Squares model). See also Table S3.
Supplementary Figure 5: TL2 does not affect oocyst numbers and EcR does not transcriptionally regulate TAG lipases. Related to Figure 7. (A) dsTL2-injected females (TL2) infected with P. falciparum develop the same number of midgut oocysts by 7–8 d pIBM compared to dsGFP controls (Cntrl) (Mann-Whitney). There is also no difference in infection prevalence (Chi-Squared) (N corresponds to the sample size; P corresponds to prevalence of infection). (B–D) Expression of neither (B) TL2, (C) AGAP007672, nor (D) AGAP000210 was significantly different in dsEcR females (EcR) at any time point pBM (means ± SEM, unpaired t-tests, FDR corrected). NBF = non-blood fed.
Supplementary Figure 6: Summary scheme of experiments and findings. Related to all Figures. Egg development (top, 0–48 h) and parasite development (bottom, 2–10 d) are represented schematically in physiological conditions (left) and following different manipulations of 20E signaling (right). These manipulations lower 20E levels (Δzpg, E22O) or impair 20E signaling (dsEcR) after blood feeding, reduce egg and parasite numbers, and cause a lipid-mediated increase in parasite growth.
Supplementary Figure 7: Silencing efficiencies for dsRNA injections. Related to Figure 3–7. Using qRT-PCR, target gene expression was normalized to expression of the housekeeping ribosomal gene Rpl19, and then silencing efficiency was calculated as expression in the treatment group relative to expression in control, dsGFP, females (means ± SEM). dsEcR: 0.67 EcR expression relative to controls (33% knock down). dsUSP: 0.32 USP expression relative to controls (68% knock down). dsTEP1: 0.29 TEP1 expression relative to controls (71% knock down). dsLp: 0.07 Lp expression relative to controls (93% knock down). dsTL2: 0.24 TL2 expression relative to controls (76% knock down). See also Table S2.
Highlights:
Human malaria parasites interact non-competitively with their mosquito vectors
Mosquito hormone signaling co-regulates egg and parasite development
Parasites use host lipids for their growth via a mosquito lipid transporter
Parasites respond to mosquito metabolism, with consequences for vector control
Acknowledgements
We thank Emily Lund, Casey Hill, Kate Thornburg, Sean Scott, Jamaica Siwak and Ping Lui for help in mosquito rearing and P. falciparum parasite culture. We thank Evdoxia Kakani for sharing protocols, Eric Marois for the An. gambiae X1 and X13 lines and transgenesis plasmids, Christoph Thiele for the LD540 neutral lipid dye, Elena A. Levashina for the pLL10-Lp plasmid, Lucia Ricci for graphics support, and members of the Catteruccia laboratory for comments on the manuscript. F.C. is funded by a Faculty Research Scholar Award by the Howard Hughes Medical Institute (HHMI) and the Bill & Melinda Gates Foundation (BMGF) (Grant ID: OPP1158190), and by the National Institutes of Health (NIH) (R01 AI124165, R01 AI104956). A.L.S was funded by the NIH (F31 AI120480) and P.M was funded by a Merit/Term-time Research Fellowship at Harvard T.H. Chan School of Public Health. Lipidomics and infection work were supported by two Broad Institute BN10 grants, awarded to M.I., S.N.B. and F.C. The findings and conclusions within this publication are those of the authors and do not necessarily reflect positions or policies of the HHMI, the BMGF, or the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests
Data and Software Availability
The lipidomics data have been deposited in the EMBL-EBI Metabolights database under the ID code MTBLS827 and can be accessed at https://www.ebi.ac.uk/metabolights/MTBLS827.
References
- Ahmed AM, Maingon RD, Taylor PJ, and Hurd H (1999). The effects of infection with Plasmodium yoelii nigeriensis on the reproductive fitness of the mosquito Anopheles gambiae. Invertebr Repr Dev 36, 217–222. [Google Scholar]
- Arrighi RB, Lycett G, Mahairaki V, Siden-Kiamos I, and Louis C (2005). Laminin and the malaria parasite’s journey through the mosquito midgut. J Exp Biol 208, 2497–2502. [DOI] [PubMed] [Google Scholar]
- Atella GC, Bittencourt-Cunha PR, Nunes RD, Shahabuddin M, and Silva-Neto MA (2009). The major insect lipoprotein is a lipid source to mosquito stages of malaria parasite. Acta Trop 109, 159–162. [DOI] [PubMed] [Google Scholar]
- Attardo GM, Hansen IA, and Raikhel AS (2005). Nutritional regulation of vitellogenesis in mosquitoes: Implications for anautogeny. Insect Biochem Mol Biol 35, 661–675. [DOI] [PubMed] [Google Scholar]
- Bai H, Gelman DB, and Palli SR (2010). Mode of action of methoprene in affecting female reproduction in the African malaria mosquito, Anopheles gambiae. Pest Manag Sci 66, 936–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldini F, Gabrieli P, South A, Valim C, Mancini F, and Catteruccia F (2013). The Interaction between a Sexually Transferred Steroid Hormone and a Female Protein Regulates Oogenesis in the Malaria Mosquito Anopheles gambiae. PLoS Biol 11, e1001695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandin S, Shiao SH, Moita LF, Janse CJ, Waters AP, Kafatos FC, and Levashina EA (2004). Complement-like protein TEP1 is a determinant of vectorial capacity in the malaria vector Anopheles gambiae. Cell 116, 661–670. [DOI] [PubMed] [Google Scholar]
- Brancucci NMB, Gerdt JP, Wang C, De Niz M, Philip N, Adapa SR, Zhang M, Hitz E, Niederwieser I, Boltryk SD, et al. (2017). Lysophosphatidylcholine Regulates Sexual Stage Differentiation in the Human Malaria Parasite Plasmodium falciparum. Cell 171, 1532–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MR, Clark KD, Gulia M, Zhao Z, Garczynski SF, Crim JW, Suderman RJ, and Strand MR (2008). An insulin-like peptide regulates egg maturation and metabolism in the mosquito Aedes aegypti. Proc Natl Acad Sci U S A 105, 5716–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MR, Graf R, Swiderek KM, Fendley D, Stracker TH, Champagne DE, and Lea AO (1998). Identification of a steroidogenic neurohormone in female mosquitoes. J Biol Chem 273, 3967–3971. [DOI] [PubMed] [Google Scholar]
- Burt A (2003). Site-specific selfish genes as tools for the control and genetic engineering of natural populations. Proc Biol Sci 270, 921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo E, Mans BJ, Andersen JF, and Ribeiro JM (2006). Function and evolution of a mosquito salivary protein family. J Biol Chem 281, 1935–1942. [DOI] [PubMed] [Google Scholar]
- Clements AN, and Paterson GD (1981). The analysis of mortality and survival rates in wild populations of mosquitos. J Appl Ecol 18, 373–399. [Google Scholar]
- Costa G, Gildenhard M, Eldering M, Lindquist RL, Hauser AE, Sauerwein R, Goosmann C, Brinkmann V, Carrillo-Bustamante P, and Levashina EA (2018). Non-competitive resource exploitation within mosquito shapes within-host malaria infectivity and virulence. Nat Commun 9,3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini C, Li SG, Della Torre A, Sagnon N, Coluzzi M, and Taylor CE (1996). Density, survival and dispersal of Anopheles gambiae complex mosquitoes in a west African Sudan savanna village. Med Vet Entomol 10, 203–219. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Aguilar R, Xi Z, Warr E, Mongin E, and Dimopoulos G (2006). Anopheles gambiae immune responses to human and rodent Plasmodium parasite species. PLoS Pathog 2, e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson HM, Rivero A, and Read AF (2003). The influence of malaria parasite genetic diversity and anaemia on mosquito feeding and fecundity. Parasitology 127, 9–19. [DOI] [PubMed] [Google Scholar]
- Frolet C, Thoma M, Blandin S, Hoffmann JA, and Levashina EA (2006). Boosting NF-kappaB-dependent basal immunity of Anopheles gambiae aborts development of Plasmodium berghei. Immunity 25, 677–685. [DOI] [PubMed] [Google Scholar]
- Gibbons JG, Janson EM, Hittinger CT, Johnston M, Abbot P, and Rokas A (2009). Benchmarking next-generation transcriptome sequencing for functional and evolutionary genomics. Mol Biol Evol 26, 2731–2744. [DOI] [PubMed] [Google Scholar]
- Hammond A, Galizi R, Kyrou K, Simoni A, Siniscalchi C, Katsanos D, Gribble M, Baker D, Marois E, Russell S, et al. (2016). A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat Biotechnol 34, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen IA, Attardo GM, Roy SG, and Raikhel AS (2005). Target of rapamycin-dependent activation of S6 kinase is a central step in the transduction of nutritional signals during egg development in a mosquito. J Biol Chem 280, 20565–20572. [DOI] [PubMed] [Google Scholar]
- Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, and Schuster P (1994). Fast folding and comparison of RNA secondary structures. Monatshefte für Chemie 125, 167–188. [Google Scholar]
- Hogg JC, and Hurd H (1995). Plasmodium yoelii nigeriensis: the effect of high and low intensity of infection upon the egg production and bloodmeal size of Anopheles stephensi during three gonotrophic cycles. Parasitology 111, 555–562. [DOI] [PubMed] [Google Scholar]
- Hogg JC, and Hurd H (1997). The effects of natural Plasmodium falciparum infection on the fecundity and mortality of Anopheles gambiae s. l. in north east Tanzania. Parasitology 114, 325–331. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ,Wu X, Shalem O, et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifediba T, and Vanderberg JP (1981). Complete in vitro maturation of Plasmodium falciparum gametocytes. Nature 294, 364–366. [DOI] [PubMed] [Google Scholar]
- Itoe MA, Sampaio JL, Cabal GG, Real E, Zuzarte-Luis V, March S, Bhatia SN, Frischknecht F, Thiele C, Shevchenko A, et al. (2014). Host cell phosphatidylcholine is a key mediator of malaria parasite survival during liver stage infection. Cell Host Microbe 16, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahan N, and Hurd H (1997). The effects of infection with Plasmodium yoelii nigeriensis on the reproductive fitness of Anopheles stephensi. Ann Trop Med Parasitol 91, 365–369. [DOI] [PubMed] [Google Scholar]
- Jaramillo-Gutierrez G, Rodrigues J, Ndikuyeze G, Povelones M, Molina-Cruz A, and Barillas-Mury C (2009). Mosquito immune responses and compatibility between Plasmodium parasites and anopheline mosquitoes. BMC Microbiol 9, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura M, Saito H, Niwa R, Niimi T, Toyoda K, Ueno C, Kanamori Y, Shimura S, and Kiuchi M (2012). Fungal Ecdysteroid-22-oxidase, a New Tool for Manipulating Ecdysteroid Signaling and Insect Development. J Biol Chem 287, 16488–16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koella JC, Lynch PA, Thomas MB, and Read AF (2009). Towards evolution-proof malaria control with insecticides. Evol Appl 2, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald G (1956). Epidemiological basis of malaria control. Bull World Health Organ 15, 613–626. [PMC free article] [PubMed] [Google Scholar]
- Mamai W, Mouline K, Blais C, Larvor V, Dabire KR, Ouedraogo GA, Simard F, and Renault D (2014). Metabolomic and ecdysteroid variations in Anopheles gambiae s.l. mosquitoes exposed to the stressful conditions of the dry season in Burkina Faso, West Africa. Physiol Biochem Zool 87, 486–497. [DOI] [PubMed] [Google Scholar]
- March S, Ng S, Velmurugan S, Galstian A, Shan J, Logan DJ, Carpenter AE, Thomas D, Sim BK, Mota MM, et al. (2013). A microscale human liver platform that supports the hepatic stages of Plasmodium falciparum and vivax. Cell Host Microbe 14, 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March S, Ramanan V, Trehan K, Ng S, Galstian A, Gural N, Scull MA, Shlomai A, Mota MM, Fleming HE, et al. (2015). Micropatterned coculture of primary human hepatocytes and supportive cells for the study of hepatotropic pathogens. Nat Protoc 10, 2027–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinotti O, Calvo E, Nguyen QK, Dissanayake S, Ribeiro JM, and James AA (2006). Genome-wide analysis of gene expression in adult Anopheles gambiae. Insect Mol Biol 15, 1–12. [DOI] [PubMed] [Google Scholar]
- Mazier D, Beaudoin RL, Mellouk S, Druilhe P, Texier B, Trosper J, Miltgen F, Landau I, Paul C, Brandicourt O, et al. (1985). Complete development of hepatic stages of Plasmodium falciparum in vitro. Science 227, 440–442. [DOI] [PubMed] [Google Scholar]
- Mi-Ichi F, Kano S, and Mitamura T (2007). Oleic acid is indispensable for intraerythrocytic proliferation of Plasmodium falciparum. Parasitology 134, 1671–1677. [DOI] [PubMed] [Google Scholar]
- Mitchell SN, and Catteruccia F (2017). Anopheline Reproductive Biology: Impacts on Vectorial Capacity and Potential Avenues for Malaria Control. Cold Spring Harb Perspect Med 7, a025593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Cruz A, Garver LS, Alabaster A, Bangiolo L, Haile A, Winikor J, Ortega C, van Schaijk BC, Sauerwein RW, Taylor-Salmon E, et al. (2013). The human malaria parasite Pfs47 gene mediates evasion of the mosquito immune system. Science 340, 984–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noriega FG, Ribeiro JM, Koener JF, Valenzuela JG, Hernandez-Martinez S, Pham VM, and Feyereisen R (2006). Comparative genomics of insect juvenile hormone biosynthesis. Insect Biochem Mol Biol 36, 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohm JR, Baldini F, Barreaux P, Lefevre T, Lynch PA, Suh E, Whitehead SA, and Thomas MB (2018). Rethinking the extrinsic incubation period of malaria parasites. Parasit Vectors 11, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paaijmans KP, Blanford S, Bell AS, Blanford JI, Read AF, and Thomas MB (2010). Influence of climate on malaria transmission depends on daily temperature variation. Proc Natl Acad Sci U S A 107, 15135–15139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagni M, Ioannidis V, Cerutti L, Zahn-Zabal M, Jongeneel CV, Hau J, Martin O, Kuznetsov D, and Falquet L (2007). MyHits: improvements to an interactive resource for analyzing protein sequences. Nucleic Acids Res 35, W433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papathanos PA, Windbichler N, Menichelli M, Burt A, and Crisanti A (2009). The vasa regulatory region mediates germline expression and maternal transmission of proteins in the malaria mosquito Anopheles gambiae: a versatile tool for genetic control strategies. BMC Mol Biol 10, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raikhel AS (1992). Vitellogenesis in mosquitoes In Advances in Disease Vector Research (Springer; ), pp. 1–39. [Google Scholar]
- Raikhel AS, and Dhadialla TS (1992). Accumulation of yolk proteins in insect oocytes. Annu Rev Entomol 37, 217–251. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern CPF (1982). 20-Hydroxy-ecdysone and ovarian development in Anopheles stephensi. J Insect Physiol 28, 97–109. [Google Scholar]
- Rheinwald JG, and Green H (1975). Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell 6, 331–343. [DOI] [PubMed] [Google Scholar]
- Riehle MA, and Brown MR (1999). Insulin stimulates ecdysteroid production through a conserved signaling cascade in the mosquito Aedes aegypti. Insect Biochem Mol Biol 29, 855–860. [DOI] [PubMed] [Google Scholar]
- Rogers DW, Baldini F, Battaglia F, Panico M, Dell A, Morris HR, and Catteruccia F (2009). Transglutaminase-Mediated Semen Coagulation Controls Sperm Storage in the Malaria Mosquito. PLoS Biol 7, e1000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DW, Whitten MMA, Thailayil J, Soichot J, Levashina EA, and Catteruccia F (2008). Molecular and cellular components of the mating machinery in Anopheles gambiae females. Proc Natl Acad Sci U S A 105, 19390–19395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rono MK, Whitten MM, Oulad-Abdelghani M, Levashina EA, and Marois E (2010). The major yolk protein vitellogenin interferes with the anti-plasmodium response in the malaria mosquito Anopheles gambiae. PLoS Biol 8, e1000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangare I, Dabire R, Yameogo B, Da DF, Michalakis Y, and Cohuet A (2014). Stress dependent infection cost of the human malaria agent Plasmodium falciparum on its natural vector Anopheles coluzzii. Infect Genet Evol 25, 57–65. [DOI] [PubMed] [Google Scholar]
- Santolamazza F, Mancini E, Simard F, Qi Y, Tu Z, and della Torre A (2008). Insertion polymorphisms of SINE200 retrotransposons within speciation islands of Anopheles gambiae molecular forms. Malar J 7, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenke RA, Lazzaro BP, and Wolfner MF (2016). Reproduction-Immunity Trade-Offs in Insects. Annu Rev Entomol 61, 239–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro LL, Murdock CC, Jacobs GR, Thomas RJ, and Thomas MB (2016). Larval food quantity affects the capacity of adult mosquitoes to transmit human malaria. Proc Biol Sci 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro LLM, Whitehead SA, and Thomas MB (2017). Quantifying the effects of temperature on mosquito and parasite traits that determine the transmission potential of human malaria. PLoS Biol 15, e2003489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snounou G, Viriyakosol S, Zhu XP, Jarra W, Pinheiro L, do Rosario VE, Thaithong S, and Brown KN (1993). High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol Biochem Parasitol 61, 315–320. [DOI] [PubMed] [Google Scholar]
- Spandl J, White DJ, Peychl J, and Thiele C (2009). Live cell multicolor imaging of lipid droplets with a new dye, LD540. Traffic 10, 1579–1584. [DOI] [PubMed] [Google Scholar]
- Sun J, Hiraoka T, Dittmer NT, Cho K-H, and Raikhel AS (2000). Lipophorin as a yolk protein precursor in the mosquito, Aedes aegypti. Insect Biochem Mol Biol 30, 1161–1171. [DOI] [PubMed] [Google Scholar]
- Tazuke SI, Schulz C, Gilboa L, Fogarty M, Mahowald AP, Guichet A, Ephrussi A, Wood CG, Lehmann R, and Fuller MT (2002). A germline-specific gap junction protein required for survival of differentiating early germ cells. Development 129, 2529–2539. [DOI] [PubMed] [Google Scholar]
- Thailayil J, Magnusson K, Godfray HCJ, Crisanti A, and Catteruccia F (2011). Spermless males elicit large-scale female responses to mating in the malaria mosquito Anopheles gambiae. Proc Natl Acad Sci USA 108, 13677–13681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W, and Jensen JB (1976). Human malaria parasites in continuous culture. Science 193, 673–675. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Mattei D, Nussenzweig RS, Eichinger D, and Zavala F (1994). Demonstration of heat-shock protein 70 in the sporozoite stage of malaria parasites. Parasitol Res 80, 16–21. [DOI] [PubMed] [Google Scholar]
- van Heusden MC, Erickson BA, and Pennington JE (1997). Lipophorin levels in the yellow fever mosquito, Aedes aegypti, and the effect of feeding. Arch Insect Biochem Physiol 34, 301–312. [DOI] [PubMed] [Google Scholar]
- van Heusden MC, Thompson F, and Dennis J (1998). Biosynthesis of Aedes aegypti lipophorin and gene expression of its apolipoproteins. Insect Biochem Mol Biol 28, 733–738. [DOI] [PubMed] [Google Scholar]
- Vlachou D, Schlegelmilch T, Christophides GK, and Kafatos FC (2005). Functional genomic analysis of midgut epithelial responses in Anopheles during Plasmodium invasion. Curr Biol 15, 1185–1195. [DOI] [PubMed] [Google Scholar]
- Volohonsky G, Terenzi O, Soichot J, Naujoks DA, Nolan T, Windbichler N, Kapps D,Smidler AL, Vittu A, Costa G, et al. (2015). Tools for Anopheles gambiae Transgenesis. G3 (Bethesda) 5, 1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hou Y, Saha TT, Pei G, Raikhel AS, and Zou Z (2017). Hormone and receptor interplay in the regulation of mosquito lipid metabolism. Proc Natl Acad Sci U S A 114, E2709–E2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2018). World Malaria Report (World Health Organization).
- Wu Q, and Brown MR (2006). Signaling and function of insulin-like peptides in insects. Annu Rev Entomol 51, 1–24. [DOI] [PubMed] [Google Scholar]
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, and Madden TL (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Sporozoite gliding and infectivity to hepatocytes. Related to Figure 5. (A–B) Sporozoites collected from the salivary glands of dsEcR (EcR) and dsGFP (Cntrl) females at 12 d pIBM display no significant difference in (A) gliding activity (means, Fisher’s Exact) or (B) hepatocyte invasion (medians ± IQR, Mann-Whitney), as determined through microscopy by identification of circumsporozoite surface protein (CSP) trails and percent sporozoites inside vs outside hepatocyte cells, respectively. (C–E) Similarly, sporozoites collected from the same groups at 18 d pIBM show no difference in their (C) gliding activity (means, Fisher’s Exact), (D) hepatocyte invasion (medians ± IQR, Mann-Whitney) and (E) development into exoerythrocytic forms (EEFs) (medians ± IQR, unpaired t-test).
Supplementary Figure 2: Parasites grow faster in Δzpg mutant and E22O-injected females. Related to Figure 5. (A–B) At 8 d pIBM, mean oocyst area is significantly greater in (A) Δzpg females (unpaired t-test, Welch’s correction) and (B) E22O-injected females (Mann-Whitney) compared to their respective controls (Cntrl). (C) Salivary glands of E22O-injected females show a higher prevalence of sporozoites at 11–12 d pIBM (Nominal Logistic Regression). There was no difference in intensity (Mann-Whitney). In all panels, N corresponds to the sample size. P = prevalence of infection.
Supplementary Figure 3: Lp accumulates in EcR-depleted females and its silencing reduces both egg and parasite numbers. Related to Figure 6. (A) ApoI protein (αApoI) accumulates in the fat bodies of dsEcR females (EcR) pIBM, with a more pronounced effect at 48 h pIBM. Actin (αActin) = loading control. (B–C) dsEcR-, dsLp- (Lp), and dsEcR/Lp- (EcR/Lp) injected females (B) produce fewer eggs (Kruskal-Wallis & Dunn’s correction) and (C) have fewer oocysts at 8 d pIBM compared to dsGFP-injected controls (Cntrl) (Kruskal-Wallis & Dunn’s correction). There is no effect on infection prevalence (Fisher’s Exact) (P = prevalence of infection). In all panels where applicable, N corresponds to the sample size.
Supplementary Figure 4: Minor lipid species enriched in dsEcR and dsTL2 midguts. Related to Figure 6 and Figure 7. (A) dsEcR-injected midguts (EcR) have elevated levels of LPEs compared to dsGFP controls (Cntrl) (means ± SEM, Standard Least Squares model) at 48 h pBM. (B) dsTL2-injected midguts (TL2) are enriched for three additional lipid species: phosphatidylserines (PSs), phosphatidylinositols (PIs), and phosphatidylethanolamines (PEs) (means ± SEM, Standard Least Squares model). See also Table S3.
Supplementary Figure 5: TL2 does not affect oocyst numbers and EcR does not transcriptionally regulate TAG lipases. Related to Figure 7. (A) dsTL2-injected females (TL2) infected with P. falciparum develop the same number of midgut oocysts by 7–8 d pIBM compared to dsGFP controls (Cntrl) (Mann-Whitney). There is also no difference in infection prevalence (Chi-Squared) (N corresponds to the sample size; P corresponds to prevalence of infection). (B–D) Expression of neither (B) TL2, (C) AGAP007672, nor (D) AGAP000210 was significantly different in dsEcR females (EcR) at any time point pBM (means ± SEM, unpaired t-tests, FDR corrected). NBF = non-blood fed.
Supplementary Figure 6: Summary scheme of experiments and findings. Related to all Figures. Egg development (top, 0–48 h) and parasite development (bottom, 2–10 d) are represented schematically in physiological conditions (left) and following different manipulations of 20E signaling (right). These manipulations lower 20E levels (Δzpg, E22O) or impair 20E signaling (dsEcR) after blood feeding, reduce egg and parasite numbers, and cause a lipid-mediated increase in parasite growth.
Supplementary Figure 7: Silencing efficiencies for dsRNA injections. Related to Figure 3–7. Using qRT-PCR, target gene expression was normalized to expression of the housekeeping ribosomal gene Rpl19, and then silencing efficiency was calculated as expression in the treatment group relative to expression in control, dsGFP, females (means ± SEM). dsEcR: 0.67 EcR expression relative to controls (33% knock down). dsUSP: 0.32 USP expression relative to controls (68% knock down). dsTEP1: 0.29 TEP1 expression relative to controls (71% knock down). dsLp: 0.07 Lp expression relative to controls (93% knock down). dsTL2: 0.24 TL2 expression relative to controls (76% knock down). See also Table S2.