

Abstract
A 57-year-old woman with pre-dialysis chronic kidney disease (CKD) was hospitalized because of fever and fatigue. On admission, increased inflammatory response and pyuria with bacteriuria were observed. Pyelonephritis was successfully treated with antibiotics, whereas her fatigue continued and she developed progressive hypercalcemia and hyponatremia; serum sodium level, 116 mEq/L and corrected serum calcium level, 13.4 mg/dL. Plasma concentrations of adrenocorticotropic hormone and cortisol and serum luteinizing hormone were under the detection level. Although the reaction of other anterior pituitary hormones and the serum antidiuretic hormone (ADH) was preserved, the response of serum luteinizing hormone to administration of luteinizing hormone releasing hormone was impaired. Magnetic resonance imaging showed no structural abnormality in the thalamus, hypothalamus, and pituitary gland. She was diagnosed with adrenal insufficiency caused by partial hypopituitarism in concomitant with pyelonephritis. After starting hydrocortisone replacement, serum levels of sodium and calcium were rapidly normalized. This case highlights the importance of adrenal insufficiency as a differential diagnosis of hypercalcemia in patients with pre-dialysis CKD, especially when hyponatremia was concomitantly observed. Besides, infection should be considered as an important trigger for the development of latent adrenal insufficiency since it could increase the physiological demand of corticosteroid in the body. Also, CKD may enhance the magnitude of hypercalcemia since CKD patients have decreased capacity to increase urinary calcium excretion.
Keywords: Adrenal insufficiency, Chronic kidney disease, Hypercalcemia, Hyponatremia, Partial hypopituitarism
Introduction
Hypercalcemia is a state of elevated serum ionized calcium (Ca) level that is occasionally lethal in both inpatients and outpatients [1]. Because serum ionized Ca level actively regulates a variety of physiological functions including muscle and heart contraction and also serves as a second messenger in various types of signal transduction, hypercalcemia leads to organ dysfunction, manifesting diverse signs and symptoms. The causes of hypercalcemia range widely, and malignancy and primary hyperparathyroidism are the two leading causes in the general population [2, 3]. As for pre-dialysis chronic kidney disease (CKD) patients, hypocalcemia is more common than hypercalcemia. Hypercalcemia in pre-dialysis CKD patients is often related to use of Ca-based phosphate binders in combination with or without oral vitamin D receptor activator use [4, 5]. However, pre-dialysis CKD patients occasionally develop hypercalcemia caused by other pathologies such as primary and secondary hyperparathyroidism, endogenous overproduction of calcitriol by granulomatous diseases, and endocrinological disorders [6–8].
We present here the case of a patient with familial adenomatous polyposis and pre-dialysis CKD who developed acute onset of hypercalcemia and hyponatremia as a manifestation of adrenal crisis.
Case report
A 57-year-old Japanese woman was hospitalized because of fever, general fatigue, and anorexia lasting for 2 weeks. She was diagnosed as familial adenomatous polyposis at age 29, and underwent subtotal colectomy, followed by total colectomy for the treatment of multiple colon cancers at age 40. At that time, she already had CKD with undetermined underlying kidney disease. She had been treated with montelukast, carbocysteine, chlorpheniramine, brotizolam, and loperamide before admission. Her kidney function and serum levels of mineral markers before admission are shown in Fig. 1.
Fig. 1.

Serial changes in surrogates of kidney function and serum mineral markers before admission. BUN blood urea nitrogen, Ca calcium, Cr creatinine, Na sodium, P phosphate
On admission, she was alert with a blood pressure of 80/48 mmHg, heart rate of 102 bpm, body temperature of 38.6 °C. Height was 151.7 cm, body weight was 42.7 kg, with a body mass index of 18.6 kg/m2. Physical examination revealed no abnormal pigmentation, and thyroid gland enlargement was not observed. However, knocking backache was prominent.
All laboratory data on admission are shown in Table 1. Briefly, hemoglobin level was 9.9 g/dL, leukocyte count was 13,260/µL with 3.9% of eosinophils (actual number 517/µL), and platelet count was 31.6 × 104/µL. Serum albumin level was 3.4 g/dL, serum creatinine was 1.96 mg/dL, serum sodium level was 134 mmol/L, serum potassium level was 5.1 mmol/L, serum chloride level was 100 mmol/L, serum Ca level was 10.7 mg/dl (albumin corrected Ca; 11.3 mg/dL), total cholesterol was 158 mg/dL, and serum C-reactive protein level was 16.8 mg/dL. Estimated glomerular filtration rate was 22 mL/min/1.73 m2. A dipstick test for urine showed 1+ for proteinuria and 3+ for bacteriuria, and 3+ for pyuria, but negative for hematuria. Urinary sediments were as follows: red blood cell: 1 > high power field (HPF), white blood cell: 50–100/HFP, and hyaline cast: 5–9/HFP. Intravenous administration of saline for hypotension and treatment with levofloxacin for pyelonephritis were immediately initiated. After treatment with levofloxacin, serum C-reactive protein and white blood cell count decreased over the next 5 days. However, she developed progressive hypercalcemia concomitant with hyponatremia: the highest corrected serum Ca level was 14.2 mg/dL and the lowest serum sodium level was 116 mEq/L, although apparent consciousness disturbance was not observed. At this point, further laboratory workup was performed.
Table 1.
Results of laboratory tests on admission
| Complete blood counts | ||
| Hemoglobin | 9.9 | g/dL |
| Hematocrit | 31.4 | % |
| White blood cells | 13,260 | /µL |
| Neutrophils | 10,873 | /µL |
| Lymphocytes | 1610 | /µL |
| Eosinophil | 517 | /µL |
| Platelets (× 104) | 31.6 | /µL |
| Serum biochemistries | ||
| Total protein | 6.6 | g/dL |
| Albumin | 3.4 | g/dL |
| Urea nitrogen | 14 | mg/dL |
| Creatinine | 1.96 | mg/dL |
| Uric acid | 5.5 | mg/dL |
| Total bilirubin | 0.2 | mg/dL |
| Glucose | 73 | mg/dL |
| C-reactive protein | 16.8 | mg/dL |
| Calcium | 10.7 | mg/dL |
| Phosphorus | 6.4 | mg/dL |
| Sodium | 134 | mmol/L |
| Potassium | 5.1 | mmol/L |
| Chloride | 100 | mmol/L |
| AST | 38 | U/L |
| ALT | 14 | U/L |
| Lactate dehydrogenase | 285 | U/L |
| Alkaline phosphatase | 280 | U/L |
| Amylase | 124 | U/L |
| Creatine kinase | 77 | U/L |
| Iron | 29 | µg/dL |
| UIBC | 217 | µg/dL |
| Ferritin | 86.6 | ng/mL |
| Total cholesterol | 158 | mg/dL |
| HDL cholesterol | 33 | mg/dL |
| LDL cholesterol | 47 | mg/dL |
| Triglyceride | 133 | mg/dL |
| Serum amyloid A | 7.1 | µg/mL |
| HbA1c | 5.3 | % |
| Immunological studies | ||
| Immunoglobulin G | 1341 | mg/dL |
| Immunoglobulin A | 286 | mg/dL |
| Immunoglobulin M | 148 | mg/dL |
| Rheumatoid factor | < 5 | U/mL |
| Anti-nuclear antibody | (–) | |
| MPO–ANCA | (–) | |
| PR3–ANCA | (–) | |
| Anti-SS-A antibody | (–) | |
| Anti-SS-B antibody | (–) | |
| Anti-HBs antigen | (–) | |
| Anti-HBs antibody | (–) | |
| Anti-HCV antibody | (–) | |
| Endocrinological studies | ||
| Plasma cortisol | < 0.4 | pg/mL |
| Plasma ACTH | < 2.0 | µg/mL |
| Serum TSH | 3.51 | µU/mL |
| Serum free T4 | 0.75 | ng/dL |
| Serum free T3 | 3.05 | pg/mL |
| Serum PTH | 26.3 | pg/mL |
| Serum calcitriol | 9.7 | pg/mL |
ACTH adrenocorticotropic hormone, ALT alanine aminotransferase, ANCA anti-neutrophilic antibody, AST aspartate aminotransferase, HBs hepatitis B surface, HCV hepatitis C virus, HDL low-density lipoprotein, LDL high-density lipoprotein, MPO myeloperoxidase, PR3 proteinase 3, PTH parathyroid hormone, SS Sjögren syndrome, TSH thyroid-stimulating hormone, UIBC unsaturated iron binding capacity
Serum calcitriol level was 9.7 pg/mL, serum parathyroid hormone (PTH) level measured by intact PTH assay was 3.5 pg/mL (normal range 15–65), and serum PTH-related peptide was under the detection level, showing that neither overproduction of calcitriol nor increased PTH or PTH-related protein was present. Urinary Ca–creatinine ratio was 0.44 (normal range 01–0.3), indicative of hypercalciuria. She was treated with neither thiazide diuretics, oxidized magnesium nor other drugs that induce hypercalcemia. Then endocrinological tests were performed.
Thyroid function was normal; thyroxin 1.20 ng/dL (normal range 0.97–1.72), tri-iodothyronine 3.71 pg/mL (normal range 2.1–4.3) and thyroid-stimulating hormone 3.84 µU/mL (normal range 0.5–3.5). Both anti-thyroglobulin antibody and anti-thyroid peroxidase antibody were negative. However, plasma levels of cortisol and adrenocorticotropic hormone (ACTH) were lower than the lowest detection limit; cortisol 0.8 µg/dL (normal range 4–18.3) and ACTH 3.5 pg/mL in the early morning (normal range 9–52). At this point, adrenal insufficiency was highly suspected. Hence, the following endocrinological tests were additionally conducted.
Circadian changes in serum cortisol were absent (data are not shown), and serum levels of ACTH and cortisol and urinary cortisol did not respond to administration of corticotropin-releasing hormone (CRH) on the 10th hospital day (Table 2). Provocation test on the 11th hospital day showed that basal levels of anterior pituitary hormone other than ACTH and luteinizing hormone (LH) were within normal ranges, and responded well to dynamic testing of anterior pituitary function (Table 3); response of LH was relatively small compared to normal individuals. Serum vasopressin level was 1.3 pg/mL (normal range 0.3–3.5), although severe hyponatremia was present. Plasma renin activity was 0.6 ng/mL/h (normal range 0.2–2.7) and plasma aldosterone concentration was 354 pg/mL (normal range 30–159). Serum and urinary osmolality were 271 mOsmol/kgH2O and 241 mOsmol/kgH2O, respectively. There was no space-occupying lesion in the pituitary gland and hypothalamus on magnetic resonance imaging. No abnormal lesions were detected in the bilateral adrenal glands on computed tomography. Neck ultrasonography did not show diffuse goiter. Although it was challenging to differentiate isolated ACTH deficiency from partial hypopituitarism (ACTH and LH dysfunction), we diagnosed her as partial hypopituitarism, because baseline serum LH level was low and LH reaction to LH-releasing hormone was also partially impaired.
Table 2.
Changes in plasma levels of ACTH and cortisol after CRH administration (CRH test)
| 0 (min) | 30 (min) | 60 (min) | 120 (min) | Normal range | |
|---|---|---|---|---|---|
| Plasma ACTH (pg/mL) | < 2.0 | < 2.0 | < 2.0 | < 2.0 | 7.2–63.3 |
| Plasma cortisol (µg/dL) | < 0.4 | < 0.4 | < 0.4 | < 0.4 | 4.0–18.3 |
Plasma levels of ACTH and cortisol did not respond to CRH stimulation
ACTH adrenocorticotropic hormone, CRH corticotropin-releasing hormone
Table 3.
Changes in serum anterior pituitary hormone levels after administration of THR, GHRP-2, and LHRP (provocation test)
| 0 (min) | 30 (min) | 60 (min) | 90 (min) | 120 (min) | |
|---|---|---|---|---|---|
| Serum LH (mU/mL) | < 0.2 | 0.8 | 1.4 | 1.4 | 1.3 |
| Serum FSH (mU/mL) | 1.2 | 5.3 | 11.6 | 14.2 | 15.8 |
| Serum PRL (ng/mL) | 13.4 | 57.7 | 59.3 | 51.1 | 46.3 |
| Serum GH (ng/mL) | 4.1 | 62.9 | 44.0 | 23.1 | 12.4 |
| Serum TSH (µU/mL) | 3.36 | 13.88 | 16.33 | 14.19 | 12.1 |
Provocation test included administration of TRH, GHRP-2, and LHRH
FSH follicle-stimulating hormone, GH growth hormone, GHRP-2 GH-releasing peptide 2, LH luteinizing hormone, LHRH LH-releasing hormone, PRL prolactin, TRH TSH-releasing hormone, TSH thyroid-stimulating hormone
Figure 2 shows the clinical course during her hospitalization. Immediately after the diagnosis of adrenal crisis, intravenous hydrocortisone administration (300 mg/day) was initiated from the 11th hospital day for 3 days, followed by 20 mg/day of oral hydrocortisone replacement, resulting in a marked improvement in fatigue within a few days. As shown in Fig. 2, corrected serum Ca level and serum sodium level returned to within normal ranges on the 17th hospital day. After she recovered from adrenal crisis and urinary tract infection, he was finally discharged after 45 days of hospitalization. At 8 years after the diagnosis of partial hypopituitarism, she was free of symptoms and signs related to adrenal insufficiency under daily replacement of hydrocortisone (20 mg/day). Her serum levels of Ca and sodium were drifted around 8.9–9.5 mg/dL and 136–141 mmol/L, respectively.
Fig. 2.
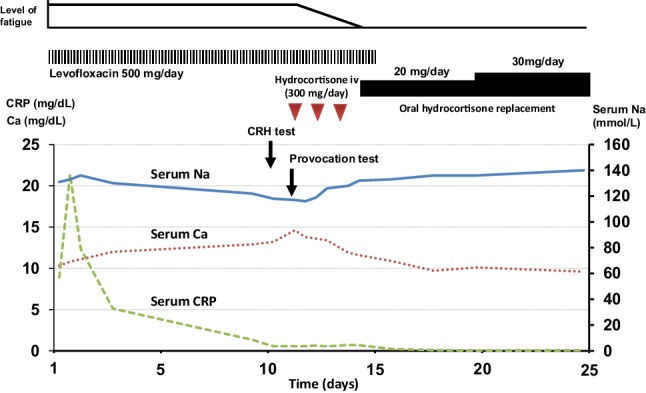
Clinical course during hospitalization. After initiation of levofloxacin treatment, serum CRP level decreased, whereas serum sodium level began to decrease and serum calcium level began to increase. CRH test and provocation test were then performed, because the presence of adrenal insufficiency was highly suspected. After the diagnosis of adrenal insufficiency, intravenous administration of hydrocortisone followed by oral hydrocortisone replacement was initiated. Soon after the replacement of hydrocortisone, the sensation of fatigue rapidly disappeared. Furthermore, hypercalcemia and hyponatremia were normalized. Ca calcium, CRH corticotropin-releasing hormone, CRP C-reactive protein, iv intravenous, Na sodium
Discussion
Hypercalcemia related to adrenal insufficiency was first described by Leeksma in 1957, and increasing number of cases have been reported ever since [9–12]. Acute or chronic cortisol deficiency is considered to be the main pathogenesis of hypercalcemia related to adrenal insufficiency, because ACTH has no known effects on Ca metabolism, and cortisol replacement can perfectly normalize hypercalcemia. Several possible mechanisms are proposed; increased bone resorption and decreased Ca excretion from the kidney [10]. However, the precise mechanisms of hypercalcemia related to adrenal insufficiency have not been fully investigated. As for patients with advanced CKD, because capacity to increase urinary Ca excretion is decreased, the magnitude of hypercalcemia may be greater than those without CKD or advanced CKD.
One of the important clues to identify the cause of hypercalcemia in the present case was concomitant hyponatremia. Hypercalcemia is not usually accompanied by disorders in serum sodium level. Among the previously reported causes of hypercalcemia, adrenal insufficiency is one of the major causes for hyponatremia. In this regard, measurement of serum sodium level can help us to make a correct diagnosis of hypercalcemia among various potential causes. As for the proposed mechanism responsible for hyponatremia in adrenal insufficiency, failed inhibition of ADH secretion by hypocortisolism is proposed, since cortisol is a physiological tonic inhibitor of ADH secretion [13].
Hypopituitarism is a rare clinical syndrome defined by a partial or complete defect in the secretion of one or more of the pituitary hormones [14]. In the present case, both serum cortisol and ACTH were not detected, serum ACTH level did not respond to CRH stimulation, baseline serum LH level was low, and LH did not respond to stimulation test sufficiently, leading to the diagnosis of hypopituitarism. Although hypopituitarism presents diverse signs and symptoms depending on the set of hormonal defects, our case presented adrenal crisis in the form of acute onset hypercalcemia and hyponatremia [5]. Because other anterior pituitary hormones and posterior pituitary hormones were normal and hypogonadism was not obvious, the patient was successfully treated with hydrocortisone replacement. Our case emphasizes the possibility of hypopituitarism as a differential diagnosis of hypercalcemia with hyponatremia.
The etiology of hypopituitarism is diverse. The cause of hypopituitarism is attributed to either pathology of the hypothalamus affecting the production of trophic hormones that act on the pituitary or direct pathology of the pituitary gland itself. The most common cause of hypopituitarism is the presence of pituitary tumors (both secretary and non-secretary), most of which are benign. Secondary metastases are less common. Another cause of hypopituitarism includes injury to the pituitary gland following traumatic brain injury or iatrogenically during surgery or cranial irradiation. Inflammatory conditions of the pituitary may also be responsible for the occurrence of hypopituitarism. The infectious agents are also causative. Lymphocytic hypophysitis usually presents in the post-partum period. Infiltrative diseases such as hemochromatosis, sarcoidosis, and histiocytosis may be associated with hypopituitarism. Pituitary apoplexy including acute ischemic infarction and hemorrhage can also cause hypopituitarism. Other causes include congenital absence of the pituitary gland, empty Sella syndrome, and Kallmann syndrome. In the present case, there was no space-occupying lesion in the thalamus and hypothalamus and her clinical course deny congenital or genetic mutation. Because she did not want further examination, we were unable to identify its etiology.
Differentiation between isolated ACTH deficiency and partial hypopituitarism is a subject of debate in the present case. Recent clinical studies have shown that isolated ACTH deficiency is more common than has been considered [6, 15]. In the present case, plasma ACTH was not detected, and did not respond to stimulation by CRH. In addition, serum LH was not detected, although plasma LH partially responded to stimulation test by LH-releasing hormone. Hence, we finally diagnosed the patient as partial hypopituitarism instead of isolated ACTH deficiency. However, it is also probable that incomplete response to stimulation test might be transient under stress state, leading to the possibility of isolated ACTH deficiency. Because the patient did not want to receive further evaluation after recovery from adrenal insufficiency, we could not determine whether LH function was finally normalized. Furthermore, the etiology of deficiency in the anterior pituitary hormone should be also considered when we diagnose the current case as isolated ACTH deficiency or partial hypopituitarism, although the etiology remains unclear in the present case.
Adrenal crisis is often triggered by physical stress including infection. In the present case, pyelonephritis might have been the trigger of manifestation of subclinical adrenal insufficiency secondary to hypopituitarism. Previous clinical studies have demonstrated that viral and bacterial infections are the most common cause of triggers that initiate adrenal crisis in patients with insidious adrenal insufficiency [16]. In the present case, serum levels of sodium and Ca were within the normal range at least 2 months prior to admission (data are not shown); hypercalcemia and hyponatremia manifested after hospitalization. Actually, hypercalcemia and hyponatremia were normalized immediately after hydrocortisone replacement and remission of bacterial infection, and did not relapse thereafter. Studies have also shown that subclinical hypopituitarism becomes overt under stressful conditions [17]. Taken together, it is reasonable to think that the patient potentially had been suffering from potential adrenal insufficiency secondary to subclinical partial hypopituitarism, which was manifested by acute pyelonephritis.
In summary, we described here the rare case of a patient with pre-dialysis CKD and familial adenomatous polyposis, who presented with hypercalcemia and hyponatremia as manifestation of adrenal crisis, which was secondary to partial hypopituitarism and triggered by urinary tract infection. This case emphasizes the importance of adrenal insufficiency as a differential diagnosis of hypercalcemia with hyponatremia in pre-dialysis CKD patients. Because adrenal crisis is fatal and rapidly reversed by the combination of fluid administration and hydrocortisone replacement, it is critical to make a quick diagnosis. Furthermore, CKD may predispose the magnitude of hypercalcemia because of the decreased capacity to increase urinary Ca excretion.
Conflict of interest
All the authors declare no competing interest.
Human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
A written informed consent was obtained from the patient included in this case study.
References
- 1.Assadi F. Hypercalcemia: an evidence-based approach to clinical cases. UJKD. 2009;3:71–79. [PubMed] [Google Scholar]
- 2.Carroll R, Matfin G. Endocrine and metabolic emergencies: hypercalcaemia. Ther Adv Endocrinol Metab. 2010;1:225–234. doi: 10.1177/2042018810390260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziegler R. Hypercalcemic crisis. J Am Soc Nephrol. 2001;12(Suppl 17):3–9. [PubMed] [Google Scholar]
- 4.Martin KJ, González EA. Prevention and control of phosphate retention/hyperphosphatemia in CKD-MBD: what is normal, when to start, and how to treat? Clin J Am Soc Nephrol. 2011;6:440–446. doi: 10.2215/CJN.05130610. [DOI] [PubMed] [Google Scholar]
- 5.Goldsmith DJ, Massy ZA, Brandenburg V. The uses and abuses of vitamin D compounds in chronic kidney disease-mineral bone disease (CKD-MBD) Semin Nephrol. 2014;34:660–668. doi: 10.1016/j.semnephrol.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Lunn MR, Muñoz Mendoza J, Pasche LJ, Norton JA, Ayco AL, Chertow GM. Hyperparathyroidism with hypercalcaemia in chronic kidney disease: primary or tertiary? NDT Plus. 2010;3:366–371. doi: 10.1093/ndtplus/sfq077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamada S, Tsujikawa H, Eriguchi M, Taniguchi M, Tsuruya K. Hypercalcemia and large abdominal mass. NDT Plus. 2011;4:213–214. doi: 10.1093/ndtplus/sfr016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemley KV. An unusual case of hypercalcemia in a patient with renal insufficiency. Pediatr Nephrol. 2014;29:1529–1533. doi: 10.1007/s00467-013-2611-8. [DOI] [PubMed] [Google Scholar]
- 9.Downie WW, Gunn A, Paterson CR, Howie GF. Hypercalcaemic crisis as presentation of Addison’s disease. Br Med J. 1977;1:145–146. doi: 10.1136/bmj.1.6054.145-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujikawa M, Kamihira K, Sato K, Okamura K, Kidota S, Lida M. Elevated bone resorption markers in a patient with hypercalcemia associated with post-partum thyrotoxicosis and hypoadrenocorticism due to pituitary failure. J Endocrinol Invest. 2004;27:782–787. doi: 10.1007/BF03347524. [DOI] [PubMed] [Google Scholar]
- 11.Kato A, Shinozaki S, Goga T, Hishida A. Isolated adrenocorticotropic hormone deficiency presenting with hypercalcemia in a patient on long-term hemodialysis. Am J Kidney Dis. 2003;42:E32-6. doi: 10.1016/S0272-6386(03)00672-3. [DOI] [PubMed] [Google Scholar]
- 12.Neary X, Neiman N. Adrenal insufficiency, etiology, diagnosis and treatment. Curr Opin Endocrionol Diabetes Obes. 2010;17:217–223. doi: 10.1097/MED.0b013e328338f608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuesta M, Garrahy A, Slattery D, Gupta S, Hannon AM, Forde H, McGurren K, Sherlock M, Tormey W, Thompson CJ. The contribution of undiagnosed adrenal insufficiency to euvolaemic hyponatraemia: results of a large prospective single-centre study. Clin Endocrinol (Oxf) 2016;85:836–844. doi: 10.1111/cen.13128. [DOI] [PubMed] [Google Scholar]
- 14.Ascoli P, Cavagnini F, Hypopituitarism Pituitary. 2006;9:335–342. doi: 10.1007/s11102-006-0416-5. [DOI] [PubMed] [Google Scholar]
- 15.Andrioli M, Pecori Giraldi F, Cavagnini F. Isolated corticotrophin deficiency. Pituitary. 2006;9:289–295. doi: 10.1007/s11102-006-0408-5. [DOI] [PubMed] [Google Scholar]
- 16.Rushworth RL, Torpy DJ. A descriptive study of adrenal crises in adults with adrenal insufficiency: increased risk with age and in those with bacterial infections. BMC Endocr Disord. 2014;14:79. doi: 10.1186/1472-6823-14-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Rodriguez E, Bernabeu I, Andujar-Plata P, Casanueva FF. Subclinical hypopituitarism. Best Pract Res Clin Endocrinol Metab. 2012;26:461–469. doi: 10.1016/j.beem.2011.10.007. [DOI] [PubMed] [Google Scholar]