

Abstract
Background and Purpose
Myristoylated alanine‐rich C kinase substrate (MARCKS), a PKC substrate, facilitates mucus production and neutrophil migration. However, the effects of therapeutic procedures targeting the phosphorylation site of MARCKS on steroid‐resistant asthma and the mechanisms underlying such effects have not yet been investigated. We designed a peptide that targets the MARCKS phosphorylation site (MPS peptide) and assessed its therapeutic potential against steroid‐resistant asthma.
Experimental Approach
Mice were sensitized with ovalbumin (OVA), alum, and challenged with aerosolized OVA five times a week for 1 month. The mice were intratracheally administered MPS peptides three times a week, 1 hr before OVA challenge. Asthma symptoms and cell profiles in the bronchoalveolar lavage were assessed, and key proteins were analysed using Western blotting.
Key Results
Phosphorylated (p)‐MARCKS was highly expressed in inflammatory and bronchial epithelial cells in OVA‐immunized mice. MPS peptide reduced eosinophils, neutrophils, mucus production, collagen deposition, and airway hyper‐responsiveness. Dexamethasone (Dexa) did not alleviate steroid‐resistant asthma symptoms. MPS peptide caused a decrease in p‐MARCKS, nitrotyrosine and the expression of oxidative stress enzymes, NADPH oxidase dual oxidase 1 and inducible NOS, in lung tissues. Compared to Dexa, MPS peptides inhibited C5a production and attenuated IL‐17A and KC production in the airway more effectively, thus suppressing asthma symptoms.
Conclusions and Implications
Our findings indicate that targeting MARCKS phosphorylation through MPS treatment may inhibit neutrophilic inflammation and relieve asthma symptoms, thereby highlighting its potential as a therapeutic agent for steroid‐resistant asthma.
Abbreviations
- AHR
airway hyperresponsiveness
- BAL
bronchoalveolar lavage
- C5a
complement component 5a
- Dexa
dexamethasone
- Duox
dual oxidase
- FITC
fluorescein isothiocyanate
- GATA‐3
GATA binding protein 3
- HBE
human bronchial epithelial
- H&E
haematoxylin&eosin
- IHC
immunohistochemistry
- IL‐1RA
IL‐1 receptor antagonist
- iNOS
inducible NOS
- MARCKS
myristoylated alanine‐rich c‐kinase substrate
- MDR
multiple drug resistance
- MPS
MARCKS phosphorylation site
- OVA
ovalbumin
- PAS
periodic acid–Schiff
- PSD
phosphorylation site domain
- RORγt
RAR‐related orphan receptor γt
- T‐bet
T‐box transcription factor
- TH
T helper cell
- TIMP
tissue inhibitors of metalloproteinase
What is already known
Myristoylated alanine‐rich C kinase substrate (MARCKS), a PKC substrate, facilitates mucus production and neutrophil migration
What this study adds
MARCKS phosphorylation highly expressed in airway epithelial cells and inflammatory cells in a murine model of steroid‐resisitant‐like asthma. A peptide targeting MARCKS phosphorylation site domain showed inhibitory effects of airway dysfunction of allergic asthma.
What is the clinical significance
There is no effective drug for treatment of steroid‐resistance asthma. Targeting MARCKS phosphorylation through MPS treatment may inhibit neutrophilic inflammation and relieve asthma symptoms, thereby highlighting its potential as a therapeutic agent for steroid‐resistant asthma.
1. INTRODUCTION
Asthma, which is a chronic inflammatory disease, is considered a health problem worldwide. According to the World Health Organization, 235 million people suffer from asthma globally (http://www.who.int/respiratory/asthma/en/), and its prevalence has increased in the past two decades. Most manage their symptoms with drugs. However, 10–25% of patients require corticosteroids to control asthma symptoms. Even with high‐dosage steroid treatments, symptoms may recur frequently (Bel et al., 2011; Chan, 1998; Kupczyk & Wenzel, 2012; Nelson, Leung, & Bloom, 2003; Szefler et al., 2002), indicating the development of steroid‐resistant asthma (Chan, 1998; Kupczyk & Wenzel, 2012; Nelson et al., 2003). Therefore, there is a need for a novel therapeutic method to treat this form of asthma.
Myristoylated alanine‐rich C kinase substrate (MARCKS), an 87‐kDa protein, is the major substrate of PKC and is composed of three highly conserved regions: the N‐terminal myristoylated domain, the phosphorylation site domain (PSD), and the multiple homology 2 domain. MARCKS is anchored to the cell membrane through an amide bond formed between the amino group of the N‐terminal residue and the highly basic PSD of several positively charged lysine residues interacting with negatively charged acidic membrane phospholipids (Harlan, 1991; Sundaram, Cook, & Byers, 2004; Wu, Walaas, Nairn, & Greengard, 1982). The PSD comprises 25 amino acids containing four serine residues that may be phosphorylated by PKC. Phosphorylated MARCKS regulates secretion, migration, and invasion in various cells. Membrane‐bound MARCKS is bound to phosphatidylinositol 4,5‐bisphosphate (PIP2) and phosphatidylserine. Upon phosphorylation by PKC, MARCKS moves from the cell membrane into the cytosol and binds to actin filaments, inducing leukocyte migration (Eckert, Neuder, Park, Adler, & Jones, 2010; Green et al., 2012), lung cancer cell metastasis (Chen et al., 2014), and airway goblet cell mucus secretion (Singer et al., 2004). MARCKS protein is an important molecule that regulates mucin hyper‐production and secretion in asthma (Singer et al., 2004).
Mechanisms underlying severe asthma remain unclear. However, activation of CD4+ T helper cells (TH1, TH2, and TH17) and oxidative stress may contribute to severe asthma. MARCKS activation through phosphorylation regulates PIP2‐related pathways, important for lymphocyte activation and inflammatory cell migration. Targeting the phosphorylation site of MARCKS may be a potential therapeutic strategy for steroid‐resistant asthma. We designed a peptide targeting the MARCKS phosphorylation site (MPS) and investigated its effects on steroid‐resistant asthma in a mouse model and the mechanism underlying these effects. We investigated whether the MPS could be utilized as a novel therapeutic target for steroid‐resistant asthma.
2. METHODS
2.1. Animals
Six‐week‐old female BALB/cJ mice were purchased from the National Laboratory Animal Centre, National Research Laboratories, Taiwan (RRID:IMSR_JAX:000651). All mice were housed in a pathogen‐free environment at the animal centre of China Medical University and were provided with a standard chow diet (carbohydrate: 70%; protein: 20%; fat: 10%) and water ad libitum. The animals were housed in groups of seven per cage under a 12 hr light–dark cycle and fed for 8 weeks. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010) and with the recommendations made by the British Journal of Pharmacology. All animal experiments were approved by the Institutional Animal Care and Use Committee of China Medical University. Protocols for animal experiments were approved by the Animal Care and Use Committee of China Medical University (Protocol No. 103‐128‐N).
2.2. Animal experiments
On Day 0, 6‐week‐old female BALB/c mice were immunized with 50 μg of ovalbumin (OVA) and 2 mg of alum adjuvant dissolved in 200 μl of PBS through i.p. injection. Boosters composed of 25 μg OVA and 2 mg alum were administered on Days 4 and 7 (Reddy, Lakshmi, & Reddy, 2012). Sensitized mice were challenged with 4%, 5 ml aerosolized OVA in PBS using a nebulizer (AG‐AL1000, AeronebR Lab Nebulizer Unit, Standard VMD) once a day, 5 days per week, for 1 month as previously described (Lee et al., 2013; Figure 1a). On Days 12, 14, 16, 19, 21, 23, 26, 28, 30, 33, 35, and 37, the mice were administered 0.46 and 2.3 mg·kg−1 MPS, 2.3 mg·kg−1 A‐MPS, and 2.3 mg·kg−1 D‐MPS peptides (figures represent A‐MPS and D‐MPS as groups) or 1 mg·kg−1 dexamethasone (Dexa), intratracheal instillation 1 hr before OVA exposure. Each group was composed of five to seven mice. On Day 38, the mice were killed via CO2 exposure, and chronic inflammatory responses were noted (Figure 1b).
Figure 1.
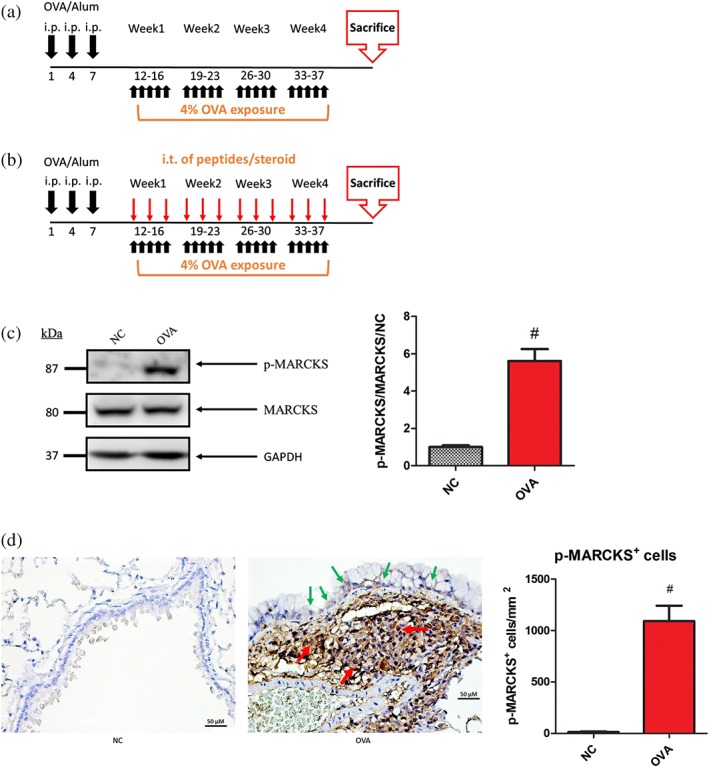
Elevation of phosphorylated (p)‐MARCKS levels in the airways of OVA‐immunized mice. (a) OVA‐induced steroid‐resistant‐like asthma model. Six‐week‐old mice were sensitized with OVA/alum, as described in 2. On Day 12, mice were exposed to PBS or OVA (4% weight per volume diluted in 0.9% saline; 5 ml per inhalation) 5 days per week for 4 weeks. On Day 38, mice were killed; the lungs were embedded in paraffin and analysed via Western blot and immunohistochemical (IHC) staining. (b) Brief scheme of the generation of the mouse steroid‐resistant model. Mice were sensitized with aerosolized OVA 5 times·week−1 for 1 month (black arrow). On Days 12, 14, 16, 19, 21, 23, 26, 28, 30, 33, 35, and 37, mice were administered various MPS peptides/steroids which were intratracheally instilled 1 hr before OVA challenge (red arrow). On Day 38, mice were killed, and the lungs were collected for further experimentation. (c) Proteins were extracted from homogenized lungs and subjected to Western blot to detect p‐MARCKS. p‐MARCKS was quantified using ImageJ. (d) Paraffin‐embedded sections of mouse airway tissues were de‐paraffinized, IHC stained with anti‐Ser152/164 p‐MARCKS antibody (green arrows represent epithelial cells and red arrows represent inflammatory cells). The scale bar is 50 μm. Data are expressed as means and SEM for NC and OVA groups (n > 11). # P < 0.05, as compared to the NC group
2.3. Bronchoalveolar lavage and lung histology
Bronchoalveolar lavage (BAL) was performed by injecting 1 ml of HBSS and gently aspirating 1 ml of BAL. The procedure was performed three times. BAL samples were centrifuged at 200× g and 4°C for 5 min. Supernatant from the first BAL sample was stored at −20°C for cytokine analysis by elisa. BAL cells were counted using a haematocytometer. Cytospin slides of BAL cells were stained with May–Giemsa stain. The cell profile of 300 cells in at least four fields of view was determined under a light microscope (Lee et al., 2010; Lee et al., 2013). The lungs were fixed with 10% formalin and sectioned for haematoxylin & eosin (H&E) staining, periodic acid–Schiff (PAS) staining, and Masson's trichrome staining, which were performed to quantify the number of infiltrating cells, mucus production, and collagen deposition. Quantification of inflammatory cells, PAS‐positive bronchial epithelial cells, and the collagen deposition area was performed according to a previous study (Lee et al., 2010).
2.4. Immunohistochemistry
Lung sections were de‐paraffinized and hydrated, and epitope retrieval was performed according to manufacturer's protocol (Thermo Quanto Detection system, CA). Briefly, the lung sections were stained with hydrogen peroxide and protein block and treated with rabbit polyclonal anti‐Ser152/156 phosphorylated MARCKS (cat#07‐1238, Millipore, Bedford, MA) at 1:100 dilution rate and PIP2 antibody (ab811039, Abcam, Cambridge, MA) afterwards at a 3 μg·ml−1 dilution rate. Sections were then stained with secondary HRP‐labelled polymer and DAB (Antibody amplifier + System‐HRP, Thermo) and counterstained with haematoxylin. Immunostained sections were viewed under a Zeiss microscope (Stuttgart, Germany). The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander, Roberts et al., 2018).
2.5. Cytokine assay
BAL supernatants were stored at −20°C prior to analysis via elisa, performed according to the manufacturer's instructions. Standards were prepared from recombinant mouse IFN‐γ, IL‐4, IL‐13, IL‐17, IL‐33, eotaxin (CCL11), and KC (CXCL1) (R&D systems, Minneapolis, MN).
2.6. Western blot analysis
Lung tissues were homogenized via a lysis buffer (25 mM Tris–HCl, 2 mM EDTA, 1 mM benzamidine, 1 mM PMSF; pH 7.4) containing protease and phosphatase inhibitors (Thermo Fisher Scientific). Appropriate homogenate volumes, containing 50 μg of proteins per well, were quantified using a Bradford Assay Kit (cat#500‐0006, Bio‐Rad, Hercules, CA) and separated via 10% SDS‐PAGE. The proteins were then transferred to PVDF membranes and probed with anti‐Ser159/163 phosphorylated MARCKS antibody (ab81295, RRID:AB_2773726; Abcam, Cambridge, MA) at a 1:2,000 dilution for Western blotting. Anti‐FOXP3 at a concentration of 4 μg·ml−1 (ab20034, RRID:AB_445284; Abcam), anti‐MARCKS at 1:500 dilution, anti‐DUOX‐1 (dual oxidase 1) at 1:500 dilution, anti‐DUOX‐2 at 1:2,000 dilution, anti‐total PKC‐βII at 1:500 dilution, anti‐GATA binding protein 3 (GATA‐3) at 1:500 dilution, anti‐RAR‐related orphan receptor γt (RORγt) at 1:500 dilution, anti‐T‐box transcription factor (T‐bet) at 1:500 dilution (SC‐100777, RRID:AB_1125958; SC‐48858, RRID:AB_2094184; SC‐49939, RRID:AB_2094195; SC‐210, RRID:AB_2252825; SC‐268, RRID:AB_2108591; SC‐28559, RRID:AB_2285218; SC‐21749, RRID:AB_2094195; Santa Cruz, CA, USA), anti‐3‐nitrotyrosine at 1:1500 dilution (ab61392, RRID:AB_942087; Abcam), anti‐complement component 5a (C5a; Genetax 11625, RRID:AB_1949789), anti‐Ser660 phosphorylated PKC‐βII at 1:1,000 dilution (Cell Signaling #9371, RRID:AB_2168219), anti‐inducible NOS (iNOS) at 1:500 dilution, and anti‐GAPDH at 1:1000 dilution (#NB300‐605, RRID:AB_10002794; #NB300‐221, RRID:AB_10077627, mouse monoclonal, Novus Biologicals, Littleton, CO; Chan, Lien, Lee, & Huang, 2016) were used. Following incubation with HRP‐conjugated secondary antibodies, signals were visualized using a Millipore ECL Kit (Millipore, Bedford, MA) according to the manufacturer's instructions.
2.7. Measurement of airway hyperresponsiveness by non‐invasive whole body plethysmography
Mice were placed in whole body plethysmography (BUXCO, USA) chambers for 15 min. Baseline readings of airway resistance were collected every 2 s for 3 min. The baseline for resistance index was determined using PBS as a control, and 500 μl each of varying methacholine concentrations (0, 3.125, 6.25, and 12.5 mg·ml−1) was used for repeat nebulization for 2 min and an additional 6‐min cycle. Airway resistance was determined by calculating the dimensionless parameter enhanced pause (Reddy et al., 2012).
2.8. In vivo fluorescence imaging with the IVIS spectrum system
Retention time of MPS in the lung tissues was determined via in vivo fluorescence imaging using the IVIS spectrum imaging system (Caliper Life Sciences) 0 min, 3 min, 6 hr, 24 hr, and 48 hr following intratracheal instillation of FITC‐conjugated MPS peptide (2.3 mg·kg−1) into BALB/c mice. At specified time periods following intratracheal instillation of MPS peptide, the mice were killed and the lungs isolated for fluorescence imaging. Fluorescence intensities of MPS peptides were analysed using accessory software (Living Image Version 4.0; Caliper Life Sciences). Ratios of fluorescence intensities in the MPS peptide‐treated group and those in the control group were calculated using ImageJ software. (ImageJ, RRID:SCR_003070).
2.9. Statistical analysis
Results are presented as mean ± SD for n ≥ 5. The programme Instat (GraphPad software) was used for statistical analysis. Group comparisons using one‐way ANOVA followed by Newman–Keuls post hoc test were conducted for cell counts in BAL fluid, H&E, PAS, Masson's trichrome pathology index, airway hyperresponsiveness (AHR), Western blotting, and cytokine levels. In Figure 1c,d, data were analysed using Student's paired t‐test. Statistical significance was set at P < 0.05. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology.
2.10. Reagents and chemicals
Ovalbumin (OVA; grade V), methacholine, and dexamethasone (Dexa) were purchased from Sigma Chemical Co. (St. Louis, MO). HBSS was obtained from Invitrogen (Carlsbad, CA). Alum adjuvant was purchased from Thermo Fisher Scientific (AlumImuject; Pierce Chemical, Rockford, IL). The MPS peptide (N‐KKKKRFSFKKSFKLSGFSFKKNKK‐C), A‐MPS peptide; (N‐KKKKRFAFKKAFKLAGFAFKKNKK‐C), D‐MPS peptide (N‐KKKKRFDFKKDFKLSGFSFKKNKK‐C), and MPS, A‐MPS, and D‐MPS peptides conjugated to fluorescein isothiocyanate (FITC) at the C‐terminal were purchased from KareBay™ Biochem's (Monmouth Junction, NJ).
2.11. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Cidlowski et al., 2017; Alexander, Fabbro et al., 2017).
3. RESULTS
3.1. Elevation of MARCKS phosphorylation in the lungs in a murine model of steroid‐resistant asthma
To create a murine steroid‐resistant asthma model, mice were sensitized with OVA combined with alum and exposed to OVA for 4 weeks (Figure 1a,b). MARCKS was highly expressed in the lungs of OVA‐immunized mice (Figure 1c). It was also expressed in bronchial epithelial cells (green arrows) and inflammatory cells (red arrows; Figure 1d). Steroid‐resistant‐like mice exhibited severe airway inflammation (Figures 2c,d), mucus production (Figure 2e), collagen deposition (Figure 2f), and AHR (Figure 2g). The mice were resistant to Dexa treatment.
Figure 2.
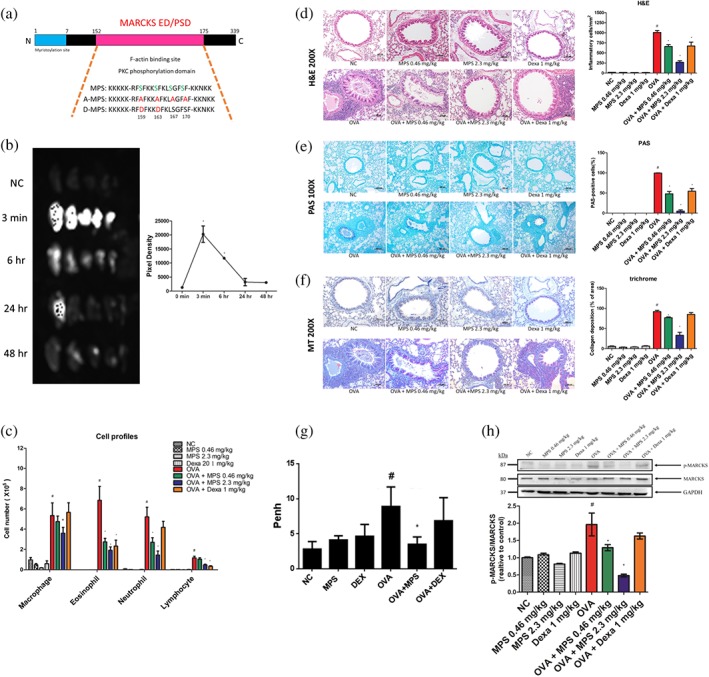
Asthma symptoms were alleviated by a peptide targeting the MARCKS phosphorylation site. (a) Amino acid sequences of MPS, A‐MPS, and D‐MPS peptides. (b) Retention time of MPS peptides in the airway. MPS‐FITC peptides at concentrations of 0.46 and 2.3 mg·kg−1 were intratracheally instilled in mice at different time points (0 min, 3 min, 24 hr, 48 hr, and 72 hr), and the lungs were dissected, and fluorescence was measured under an IVIS system. (c) As described in 2, mice were killed on Day 38, and differential cell counts of BAL were assessed via May–Giemsa staining. (d–f) The lungs were stained using H&E, periodic acid–Schiff (PAS), and Masson's trichrome (MT) staining. Bar represents 100 μm for H&E and MT; 200 μm for PAS. (g) Airway resistance was determined via whole body plethysmography. (h) Lung proteins were subjected to Western blot to assess Ser159/163 p‐MARCKS, MARCKS, and GAPDH expression. Proteins were quantified using ImageJ. Data are expressed as mean ± SD (n ≧ 5). # P < 0.05; *P < 0.05, compared to NC and OVA groups
3.2. MPS peptide inhibits asthma symptoms by interfering with MARCKS phosphorylation in a murine steroid‐resistant‐like asthma model
We designed a peptide that targets the MARCKS phosphorylation to investigate potential therapeutic effects of MPS peptides on steroid‐resistant asthma (Figure 2a). To determine the retention time of MPS peptides, MPS peptides conjugated to FITC (2.3 mg·kg−1) were intratracheally instilled in mice and MPS peptide levels measured at different time points. The fluorescence of MPS peptide reached its peak at 3 min (Figure 2b), declined sharply, sustained at 48 hr, and disappeared at 72 hr (Figure S4). The fluorescence of low dose MPS peptide (0.46 mg·kg−1) reached its peak at 3 min but disappeared at 24 hr (left panel; Figure S4). We investigated the ability of three designated MPS peptides in human bronchial epithelial (HBE) cell line to enter cells and localize intracellularly. HBE cells were treated with FITC‐labelled MPS, A‐MPS, and D‐MPS peptides. MPS, A‐MPS, and D‐MPS peptides anchor in the cell membrane and enter HBE cells as a complete cytosolic green fluorescence, extending into the nuclei (Figure S1). Peptide fluorescence was sustained for 48 hr in HBE cells (data not shown). Based on these results, the time interval between intratracheal instillation of MPS peptide and the relevant assays was set as 48 hr. Following intratracheal instillation of MPS peptide at 0.46 and 2.3 mg·kg−1, OVA‐induced airway inflammation, mucus production, collagen deposition, and AHR were inhibited in a dose‐dependent manner (Figure 2c–g). Analysis of inflammatory cell profiles of BAL showed that MPS peptide inhibited infiltration of neutrophils, eosinophils, and lymphocytes in steroid‐resistant‐like mice. Dexa was able to suppress the infiltration of eosinophils and lymphocytes but not that of neutrophils (Figure 2c). To determine whether suppression of asthma symptoms following MPS peptide treatment was associated with MARCKS phosphorylation, we assessed p‐MARCKS expression in the lungs. The expression of p‐MARCKS was higher in the lungs of OVA‐immunized mice. It decreased upon treatment with MPS peptide but was unaffected by Dexa treatment (Figure 2h). Thus, targeting p‐MARCKS with MPS peptides may block asthma symptoms.
3.3. MPS peptide reduces IL‐17A, C5a expression, and oxidative stress production and contributes to the inhibition of asthma symptoms in a murine steroid‐resistant asthma model
T helper (TH) cells, TH1, TH2, and TH17, are involved in airway inflammation in steroid‐resistant asthma (Hansbro et al., 2017; Linden, 2006; Zijlstra, Ten Hacken, Hoffmann, van Oosterhout, & Heijink, 2012). We observed a similar phenomenon. In steroid‐resistant mice, TH1‐specific cytokine IFN‐γ, TH2‐related cytokines, IL‐4, IL‐13, and eotaxin, and TH17‐related cytokines, IL‐17 and KC, were all increased in BAL or in the lungs (Figure 3a). Following treatment with 2.3 mg·kg−1 MPS peptide, all these cytokines were suppressed. In contrast, Dexa inhibited IFN‐γ, IL‐4, and eotaxin and slightly attenuated IL‐13 and KC production but did not inhibit IL‐17. MPS peptide also inhibited expression of TH‐specific transcription factors (T‐bet TH1, GATA‐3 TH2, and RORγt TH17 in lung tissues). By contrast, Dexa inhibited GATA‐3 expression and slightly suppressed T‐bet expression but not RORγt (Figure 3b). This trend was similar to that for TH‐specific cytokines. In a previous study it was found that mice subjected to prolonged 3% OVA exposure (three times a week for a total period of 6 weeks) displayed airway tolerance with a reduction in total BAL cells, eosinophils, neutrophils, and lymphocytes; however, exposure to different concentrations of OVA for 6 weeks caused airway inflammation (Sethi & Naura, 2018). To confirm effects of immune tolerance in our mouse model, we investigated regulatory T cell specific transcription factor, FOXP3, expression in the lungs. FOXP3 expression in lung tissue was not increased in our steroid‐resistant‐like asthma model (Figure 3b). Therefore, airways exhibiting severe allergic asthma symptoms did not induce naturally occurring Foxp3+CD4+CD25+ T cell (nTreg) activation in our mouse model.
Figure 3.
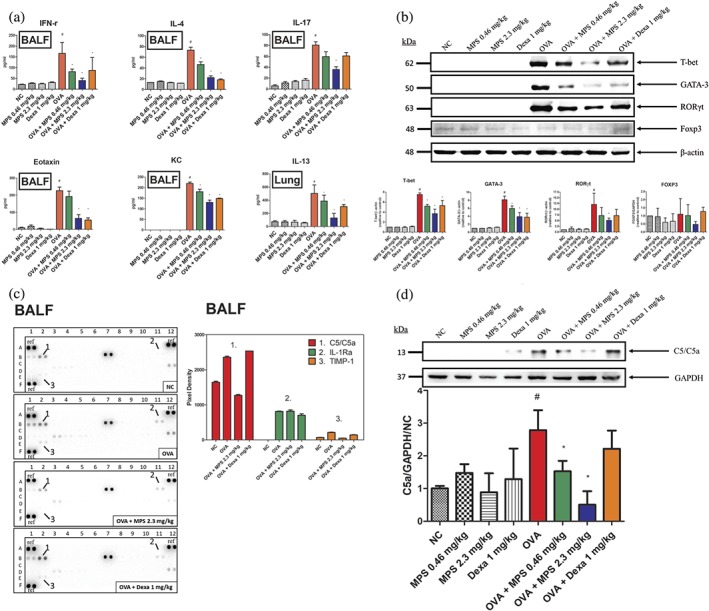
MPS peptides decreased TH1‐, TH2‐, and TH17‐related cytokines and specific‐transcriptional factors as well as C5a expression in the lungs. (a) On Day 38, mice were killed, BAL fluids collected, and lung proteins analysed via elisa. (b) Proteins were extracted from homogenized lungs and subjected to Western blotting to assess T‐bet, GATA‐3, RORγt, FOXP3, and β‐actin expressions. (c) BAL fluids were analysed using a mouse cytokine array to screen 40 cytokines, chemokines, and complement and acute‐phase proteins in the BAL. Pixel density plots of C5/C5a, IL‐1Ra, and TIMP‐1 were analysed using ImageJ. (d) Proteins were extracted from homogenized lungs and subjected to Western blotting to assess C5a and GAPDH expressions. These proteins were quantified using ImageJ. Data are expressed as mean ± SD (n ≧ 5). # P < 0.05, compared to the NC group; *P < 0.05, compared to the OVA group
Next, cytokine/chemokine arrays were utilized to characterize the cytokine/chemokine profiles following MPS peptide and Dexa treatment. Of the 40 cytokines, chemokines and acute‐phase proteins that were screened, C5a and tissue inhibitors of metalloproteinase (TIMP‐1), which were markedly overexpressed in steroid‐resistant mice, were down‐regulated by MPS peptide but not by Dexa (Figure 3c). C5a is important for the recruitment and activation of granulocytes (Ehrengruber, Geiser, & Deranleau, 1994; Zeck‐Kapp, Kroegel, Riede, & Kapp, 1995). In addition, IL‐1 receptor antagonist, IL‐1Ra (receptor antagonist for IL‐1β), levels in BAL were lower in Dexa‐treated steroid‐resistant mice than in OVA‐ or MPS peptide‐treated mice (Figure 3c). Other chemokines CXCL‐13, soluble intercellular adhesion molecule‐1 (sICAM‐1), KC and the stromal cell‐derived factor‐1 (SDF‐1), were also increased in the OVA treatment group (Figure S5). MPS peptide and Dexa both reduced CXCL‐13, KC, and SDF‐1 levels but not that of sICAM‐1.
In steroid‐resistant asthma, airway inflammatory cell activation causes oxidative stress in the lungs, suppressing lung function, increasing mucus production, and reducing corticosteroid responsiveness in asthma (Saleh, Ernst, Lim, Barnes, & Giaid, 1998). Thus, oxidative stress is a pathological mechanism, which induces steroid‐resistant responses in asthma. Therefore, we examined oxidative stress in lung tissues. We found that numerous proteins had been nitrosylated. Also, levels of the oxidative indicator, 3‐nitrotyrosine (3‐NT), and related enzymes including dual oxidase‐1 (Duox‐1), Duox‐2, and iNOS in the lung tissues of the OVA group were increased (Figure 4). The 2.3 mg·kg−1 MPS peptide treatment group, but not Dexa treatment, significantly reduced 3‐NT, Duox‐1, and iNOS expressions in the lungs.
Figure 4.
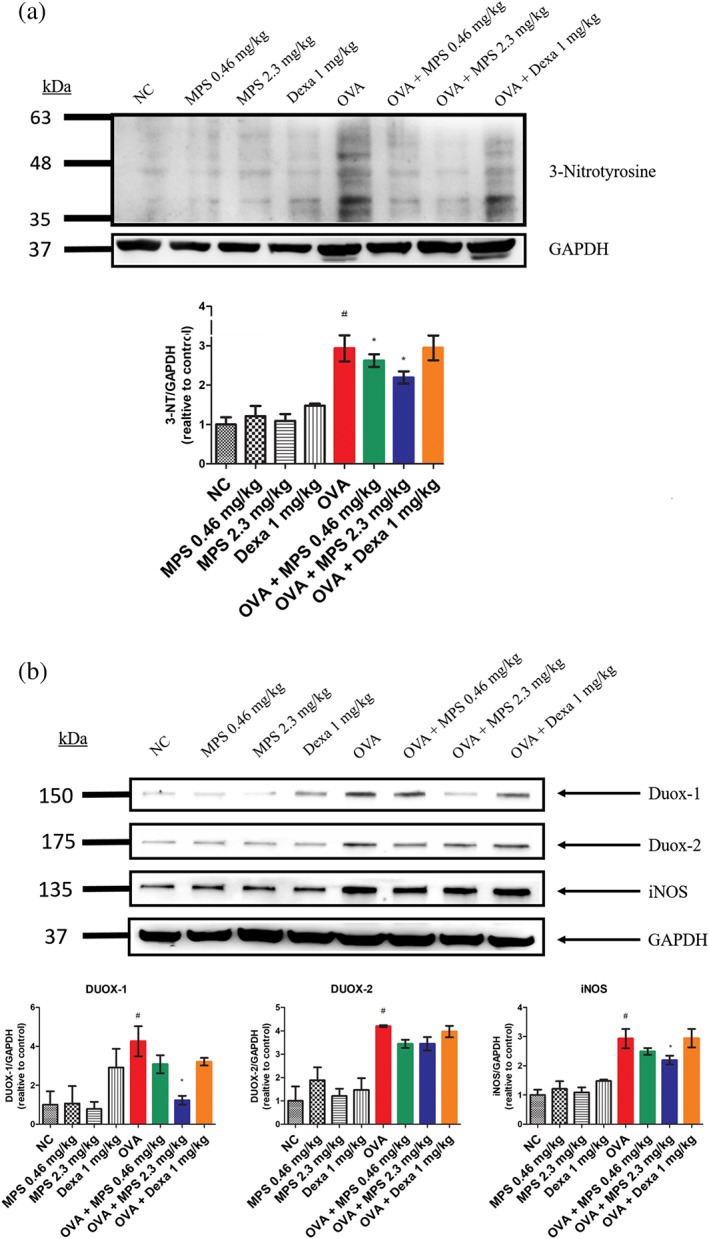
Suppression of oxidative stress in lung tissues from steroid‐resistant mice. Proteins were extracted from homogenized lungs and subjected to Western blotting to assess the expression of (a) 3‐nitrotyrosine (3‐NT) and (b) dual oxidase‐1 (Duox‐1) and Duox‐2, inducible NOS (iNOS), and GAPDH. These proteins were quantified using ImageJ. Data are expressed as mean ± SD (n = 5). # P < 0.05, as compared with the NC group; *P < 0.05, as compared with the OVA group
3.4. MPS peptide interferes with p65 phosphorylation but does not affect the reduction of HDAC‐2 in the lungs in a murine steroid‐resistant‐like asthma model
To clarify mechanisms underlying the effect of MPS peptide on steroid‐resistant signalling, histone deacetylase‐2 (HDAC‐2) protein expression and NF‐κB activation, involved in steroid‐resistant signalling (Barnes, 2011; Ito et al., 2008), were investigated in lung tissue. HDAC‐2 protein expression was significantly decreased in the OVA‐immunized group that showed steroid‐resistant features, whereas Dexa treatment did not prevent this HDAC‐2 reduction (Figure S2). However, MPS peptide was also unable to reverse HDAC decline in the OVA‐immunized group. As NF‐κB, which is a key transcription factor activating inflammatory cytokines, is induced by high oxidative stress in the OVA‐immunized group in the steroid‐resistant‐like model, we investigated whether MPS peptide inhibited asthma symptoms by activating NF‐κB. A high dose of MPS peptide treatment suppressed high pp65 expression in the OVA‐immunized group whereas Dexa treatment did not change pp65 expression (Figure S6).
3.5. Mutation of Ser residues in MPS to Asp or Ala blocks MPS peptide‐induced anti‐asthma effects in a murine steroid‐resistant asthma model
We investigated the molecular mechanisms underlying inhibition of asthma symptoms by MPS peptide treatment. First, two different mutant MPS peptides were designed (Figure 2a): A‐MPS (four Ser residues mutated to Ala which cannot be phosphorylated) and D‐MPS (two Ser residues mutated to Asp to mimic protein structure of phosphorylated serine residue). Intratracheal instillation of A‐MPS or D‐MPS (2.3 mg·kg−1) blocked MPS peptide‐induced inhibition of asthma symptoms, including airway inflammation (Figures 5a,b), mucus production (Figure 5c), collagen deposition (Figure 5d), and AHR (Figure 5e–g). Furthermore, A‐MPS and D‐MPS did not inhibit OVA‐induced MARCKS phosphorylation in the lungs (Figure 6). Finally, unlike MPS peptide, A‐MPS and D‐MPS peptide were unable to suppress an increase in phosphorylated PKC expression in the lungs of murine steroid resistant‐like‐asthma models (Figure 6).
Figure 5.
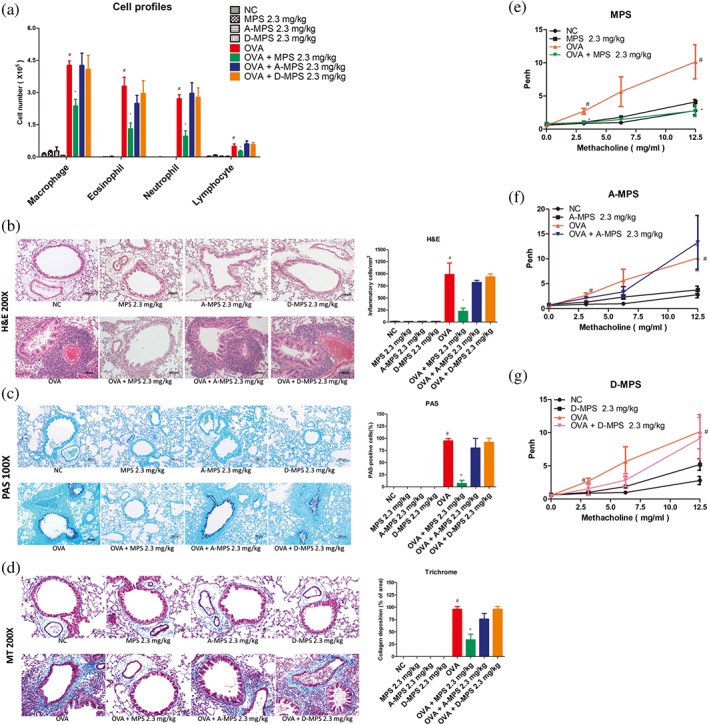
The effects of MPS peptide, A‐MPS, and D‐MPS on asthma symptoms in steroid‐resistant mice. (a) BAL differential cell counts were assessed via May–Giemsa staining. (b–d) Lung sections were stained using H&E, PAS, and Masson's trichrome (MT) to assess inflammatory cell infiltration, mucus production, and collagen deposition around the airways, respectively. High magnification bar represents 100 μm for H&E and MT and 200 μm for PAS. (e–g) Airway resistance was determined through whole body plethysmography. Data are expressed as mean ± SD of pulmonary resistance values (RL) (n ≧ 5). # P < 0.05, compared to the NC group; *P < 0.05, compared to the OVA group
Figure 6.
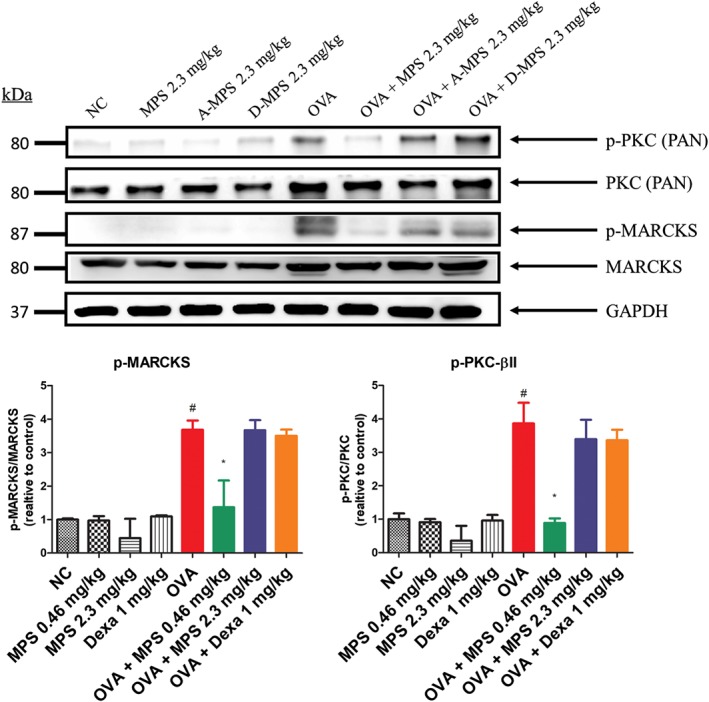
Suppression of p‐MARCKS expression by MPS peptides in steroid‐resistant mice. Lung proteins were subjected to Western blot to assess the expression of phospho‐PKC (p‐PKC), total PKC‐βII, p‐MARCKS, MARCKS, and GAPDH. Proteins were quantified using ImageJ. Data are expressed as mean ± SD (n = 5). # P < 0.05, compared to NC group; *P < 0.05, compared to the OVA group
4. DISCUSSION
The protein, MARCKS, induces vesicle release during exocytosis and cell migration via actin cytoskeleton remodelling. It plays an important role in the pathophysiology of several pulmonary diseases such as allergic asthma (Singer et al., 2004) and in the development of lung cancer (Chen et al., 2014), breast cancer (Browne et al., 2013; Chen et al., 2015), renal cell carcinoma (Chen et al., 2017), hepatocellular carcinoma (Naboulsi et al., 2016), and pancreatic cancer (Brandi et al., 2016). However, the role of MARCKS phosphorylation in airway inflammatory diseases such as severe, steroid‐resistant asthma remains unclear. We detected an elevation in p‐MARCKS expression in the lungs of murine steroid‐resistant asthma models. We showed that MPS peptide, which targets the MPS, alleviated asthma symptoms including airway inflammation, mucus production, airway remodelling, and airway hyper‐responsiveness in steroid‐resistant models. This suggested that MPS is a potential target for the treatment of steroid‐resistant asthma. These results suggested that MPS peptide inhibited asthma symptoms by interfering with MARCKS phosphorylation. The phosphorylation of MARCKS by PKC causes translocation of MARCKS into the cytosol and the release of PIP2 (Glaser & Wanaski, 1996; Morton et al., 2012). PIP2 is then activated by PLC‐γ1 and phosphatidylinositol‐4, 5‐bisphosphate 3‐kinase (PtdIns‐3‐OH kinase) allowing it to activate downstream signalling pathways involving immune cell activation. We also found that MPS peptide attenuated PKC phosphorylation which may reduce MARCKS phosphorylation. However, PKC is activated by calcium ions (Ca2+) and DAG, released following MARCKS phosphorylation. Therefore, inhibition of PKC phosphorylation may occur after MPS peptide interferes with MARCKS phosphorylation. This may require further investigation. We also investigated PIP2 expression in the lungs using immunohistochemical. PIP2, which was detected in both bronchial and alveolar epithelial cells of normal lungs, was highly expressed in epithelial cells and inflammatory cells of inflamed lungs (Figure S7). Following MPS peptide treatment, the PIP2 level was reduced through attenuation of inflammatory cell infiltration in the lungs. The effect of MPS peptide on PIP2 downstream signalling pathways needs further investigation.
To understand the mechanisms by which MPS peptide inhibits MARCKS phosphorylation, two MPS mutant peptides, A‐MPS and D‐MPS, were used. Both peptides did not alleviate asthma symptoms in a murine steroid‐resistant‐like asthma model. This indicates that Ser residues in MPS peptide are important for relieving asthma symptoms. D‐MPS did not inhibit MARCKS phosphorylation because D‐MPS mimics phosphorylated MPS in that it possesses excess negative charge, thereby reducing its electrostatic interaction with acidic lipids such as phosphatidylserine (McLaughlin & Aderem, 1995; Seykora, Myat, Allen, Ravetch, & Aderem, 1996). Therefore, D‐MPS was unable to bind to the cell membrane. However, A‐MPS did not suppress MARCKS phosphorylation and asthma symptoms, although no excess negative charge was introduced. Such inability of A‐MPS to reduce asthma symptoms and MARCKS phosphorylation will be investigated.
The cytokine profile in BAL was analysed. MPS peptide inhibited expression of T helper cell (TH) 1‐, TH2‐, TH17‐specific cytokines (IFN‐γ, IL‐4, and IL‐17) and transcription factors (T‐bet, GATA‐3, and RORγt) in the lungs. This may be due to inhibition of MARCKS phosphorylation, which attenuated activation of lymphocytes, including TH cells, by down‐regulating PIP2/PLC/inositol 1,4,5‐triphosphate, DAG/NF‐κB, activator protein 1, and nuclear factor of activated T‐cell signalling pathways (Foster, De Hoog, & Mann, 2003; Saito et al., 2003; Strzelecka‐Kiliszek, Korzeniowski, Kwiatkowska, Mrozinska, & Sobota, 2004). In contrast, Dexa inhibited the production of TH1‐ and TH2‐specific cytokines but not that of the TH17‐specific cytokine IL‐17. TH17 causes steroid resistance in murine asthma models (Ano et al., 2013), patients with inflammatory diseases, and bronchial epithelial cells (Ramesh et al., 2014; Schewitz‐Bowers et al., 2015; Vazquez‐Tello et al., 2010). The mechanisms underlying TH17‐induced steroid resistance are still unclear. Ramesh et al. (2014) demonstrated the expression of the protein multi‐drug resistance type 1 (MDR1) by TH17 cells, which is refractory to glucocorticoid‐mediated T cell suppression in Crohn's disease. The role of MDR1+ TH17 cells in the pathological mechanism of steroid‐resistant asthma and inhibition of TH17 cell activation by MPS peptide in our model still needs investigation. In addition to TH17 cells, neutrophils released IL‐17A and RORγt in mouse models and human in vitro studies (Taylor et al., 2014). Therefore, attenuation of IL‐17A‐dependent asthma symptoms by MPS peptide may contribute to inhibition of neutrophil infiltration in the airways.
The complement system is an evolutionarily component of the innate immune system. C5a, a powerful chemoattractant, regulates inflammatory responses and function and recruits granulocytes and monocytes (Guo & Ward, 2005). In our study, the C5a increase in the lungs, and BAL observed in a murine steroid‐resistant‐like asthma model was inhibited by MPS peptide. However, the levels of C5a, reportedly a steroid‐resistant inflammatory mediator, which were shown to be elevated in the BAL of asthmatic patients (Krug, Tschernig, Erpenbeck, Hohlfeld, & Köhl, 2001), were shown to be inhibited by steroid treatment in cardiopulmonary bypass patients (Engelman et al., 1995). Therefore, the mechanism underlying the loss of response to steroids in steroid‐resistant asthma needs further investigation.
In conclusion, the MARCKS phosphorylation site may serve as a potential therapeutic target in the treatment of steroid‐resistant asthma. Additionally, MPS peptide shows potential as a steroid‐resistant asthma drug.
AUTHOR CONTRIBUTIONS
This study was designed, directed, and coordinated by R.W., and C.‐C.L. C.‐C.L., as the corresponding author, provided conceptual and manuscript editing. C.‐N.W. performed the experiments, modulated the animal models, analysed the data, and wrote and edited the manuscript. Y.‐C.L. and B.‐C.C. assisted C.‐N.W. to carry out the experiment and data analysis.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1. Quantification of mouse cytokines, chemokines, and complement and acute‐phase proteins in bronchoalveolar lavage (BAL) fluids by cytokine arrays.
Figure S1. Intracellular localization of MPS, A‐MPS, and D‐MPS peptides in NHBE cells. NHBE cells were seeded in a chamber slide for 24 h and then incubated with 100 nmol MPS‐FITC, A‐MPS‐FITC, and D‐MPS‐FITC at 37°C for 5 min. The cells were washed thrice with 1 ml HBSS and then subjected to confocal microscopy. Nuclei were stained with DAPI which showed blue fluorescence. Green fluorescence indicates different MPS peptides. Scale bar: 25 μm, N = 5.
Figure S2. The effect of HDAC‐2 protein expression in a murine model of steroid‐resistant like asthma. The lung proteins were subjected to western blot analysis to assess the expression of HDAC‐2 and GAPDH. The proteins were quantified using Image J. Data are expressed as the mean ± S.D. (n = 5). # P < 0.05, as compared with the NC group.
Figure S3. Pulmonary inflammation and pathology at different times of OVA exposure. (A, D, G) Mice at 6 weeks of age were sensitized with OVA‐alum. On day 12, the mice were exposed to PBS or 5 mL 4 % OVA for five days per week for 1, 2, and 4 weeks, respectively. (B, E, H) Mice were intratracheally instilled with 1 mg/kg of dexamethasone twice, at 24 h and 48 h before dissection. On day 17, 24, and 38, the mice were sacrificed and differential cell counts in the BAL were assessed by May‐Giemsa staining. (C, F, I) Lungs were subjected to hematoxylin and eosin (H&E), periodic acid–Schiff (PAS), and Masson's trichrome (MT) staining. High magnification (bar represents 100 μm for H&E and MT, 200 μm for PAS). Data are expressed as mean ± S.D. (n ≧5). # P < 0.05, compared to the NC group.; * P < 0.05 compared to OVA group.
Figure S4. Retention time of MPS peptides in the airway. MPS‐FITC peptides (0.46 mg/kg and 2.3 mg/kg) were intratracheally instilled in mice at different time points (0 min, 3 min, 24 h, 48 h, and 72 h), and the lungs were then dissected and observed under an IVIS System to measure and quantify the fluorescence intensity. Data are expressed as mean ± S.D. (n ≧ 5). # P < 0.05, as compared with the NC.
Figure S5. Cytokine expression after MPS treatment using a cytokine array in a murine model of steroid‐resistant asthma. BAL fluid (BALF) was analyzed with a mouse cytokine array to screen for 40 cytokines, chemokines, and complement and acute‐phase proteins in the BAL. Pixel density plots of chemokine (C‐X‐C motif) ligand (CXCL)‐13 (A), soluble intercellular adhesion molecule‐1 (sICAM‐1) (B), KC (C) and stromal cell‐derived factor (SDF)‐1 (D) in BALF, and their quantification using Image J.
Figure S6. The effect of phospho‐p65 and p‐65 in mice with steroid‐resistant like asthma. Lung proteins were subjected to western blot analysis to assess the expression of p‐p65, p‐65, and GAPDH. The proteins were quantified using Image J. Data are expressed as the mean ± S.D. (n = 5). # P < 0.05, as compared with the NC group. * P < 0.05, as compared with the OVA group.
Figure S7. The effect of phosphatidylinositol 4,5‐biphosphate (PIP2) levels on the airways of OVA‐immunized mice. Mice at 6 weeks of age were sensitized with OVA/alum, as described in Figure 1B. Paraffin sections of mouse airway tissues were deparaffinized and IHC stained with PIP2 antibody (green arrows represent bronchial epithelial cells, black arrows represent alveolar epithelial cells, and red arrows represent for inflammatory cells). High magnification (bar represents 100 μm).
ACKNOWLEDGEMENTS
This work was financially supported by the “Drug Development Centre, China Medical University” from The Featured Areas Research Centre Programme within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. This investigation was supported by grants from the China Medical University of Taiwan (CMU102‐BC‐10; CMU105‐S‐08; CMU105‐BC‐1‐4; CMU‐S‐12; CMU106‐TC‐01) and Ministry of Science and Technology, ROC (MOST 103‐2320‐B‐039‐022‐MY3; MOST 106‐2320‐B‐039‐007). Experiments and data analysis were performed in part through the use of the Medical Research Core Facilities Centre, Office of Research & Development at China Medical University, Taichung, Taiwan.
Wang C‐N, Lin Y‐C, Chang B‐C, Chen C‐H, Wu R, Lee C‐C. Targeting the phosphorylation site of myristoylated alanine‐rich C kinase substrate alleviates symptoms in a murine model of steroid‐resistant asthma. Br J Pharmacol. 2019;176:1122–1134. 10.1111/bph.14596
Contributor Information
Reen Wu, Email: rwu@ucdavis.edu.
Chen‐Chen Lee, Email: leechenchen@mail.cmu.edu.tw.
REFERENCES
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … Collaborators, C. G. T. P. (2017). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. British Journal of Pharmacology, 174(S1), S208–S224. 10.1111/bph.13880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY. British Journal of Pharmacology, 174(Suppl 1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ano, S. , Morishima, Y. , Ishii, Y. , Yoh, K. , Yageta, Y. , Ohtsuka, S. , … Hizawa, N. (2013). Transcription factors GATA‐3 and RORγt are important for determining the phenotype of allergic airway inflammation in a murine model of asthma. The Journal of Immunology, 190, 1056–1065. 10.4049/jimmunol.1202386 [DOI] [PubMed] [Google Scholar]
- Barnes, P. J. (2011). Glucocorticosteroids: Current and future directions. British Journal of Pharmacology, 163, 29–43. 10.1111/j.1476-5381.2010.01199.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bel, E. H. , Sousa, A. , Fleming, L. , Bush, A. , Chung, K. F. , Versnel, J. , … on behalf of the members of the Unbiased Biomarkers for the Prediction of Respiratory Disease Outcome (U‐BIOPRED) Consortium, Consensus Generation (2011). Diagnosis and definition of severe refractory asthma: An international consensus statement from the Innovative Medicine Initiative (IMI). Thorax, 66, 910–917. 10.1136/thx.2010.153643 [DOI] [PubMed] [Google Scholar]
- Brandi, J. , Dalla Pozza, E. , Dando, I. , Biondani, G. , Robotti, E. , Jenkins, R. , … Cecconi, D. (2016). Secretome protein signature of human pancreatic cancer stem‐like cells. Journal of Proteomics, 136, 1–12. 10.1016/j.jprot.2016.01.017 [DOI] [PubMed] [Google Scholar]
- Browne, B. C. , Hochgrafe, F. , Wu, J. , Millar, E. K. , Barraclough, J. , Stone, A. , … Daly, R. J. (2013). Global characterization of signalling networks associated with tamoxifen resistance in breast cancer. The FEBS Journal, 280, 5237–5257. 10.1111/febs.12441 [DOI] [PubMed] [Google Scholar]
- Chan, C. Y. , Lien, C. H. , Lee, M. F. , & Huang, C. Y. (2016). Quercetin suppresses cellular migration and invasion in human head and neck squamous cell carcinoma (HNSCC). Biomedicine (Taipei), 6, 15 10.7603/s40681-016-0015-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, M. T. S. (1998). Difficult‐to‐control asthma: Clinical characteristics of steroid‐insensitive asthma. The Journal of Allergy and Clinical Immunology, 101, 594–601. 10.1016/S0091-6749(98)70165-4 [DOI] [PubMed] [Google Scholar]
- Chen, C. H. , Fong, L. W. R. , Yu, E. , Wu, R. , Trott, J. F. , & Weiss, R. H. (2017). Upregulation of MARCKS in kidney cancer and its potential as a therapeutic target. Oncogene, 36, 3588–3598. 10.1038/onc.2016.510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. H. , Statt, S. , Chiu, C. L. , Thai, P. , Arif, M. , Adler, K. B. , & Wu, R. (2014). Targeting myristoylated alanine‐rich C kinase substrate phosphorylation site domain in lung cancer. Mechanisms and therapeutic implications. American Journal of Respiratory and Critical Care Medicine, 190, 1127–1138. 10.1164/rccm.201408-1505OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. H. , Thai, P. , Yoneda, K. , Adler, K. B. , Yang, P. C. , & Wu, R. (2014). A peptide that inhibits function of myristoylated alanine‐rich C kinase substrate (MARCKS) reduces lung cancer metastasis. Oncogene, 33, 3696–3706. 10.1038/onc.2013.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.‐H. , Cheng, C. T. , Yuan, Y. , Zhai, J. , Arif, M. , Fong, L. W. R. , … Ann, D. K. (2015). Elevated MARCKS phosphorylation contributes to unresponsiveness of breast cancer to paclitaxel treatme. Oncotarget, 6, 15194–15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, R. E. , Neuder, L. E. , Park, J. , Adler, K. B. , & Jones, S. L. (2010). Myristoylated alanine‐rich C‐kinase substrate (MARCKS) protein regulation of human neutrophil migration. American Journal of Respiratory Cell and Molecular Biology, 42, 586–594. 10.1165/rcmb.2008-0394OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrengruber, M. U. , Geiser, T. , & Deranleau, D. A. (1994). Activation of human neutrophils by C3a and C5A. Comparison of the effects on shape changes, chemotaxis, secretion, and respiratory burst. FEBS Letters, 346, 181–184. [DOI] [PubMed] [Google Scholar]
- Engelman, R. M. , Rousou, J. A. , Flack, J. E. 3rd , Deaton, D. W. , Kalfin, R. , & Das, D. K. (1995). Influence of steroids on complement and cytokine generation after cardiopulmonary bypass. The Annals of Thoracic Surgery, 60, 801–804. 10.1016/0003-4975(95)00211-3 [DOI] [PubMed] [Google Scholar]
- Foster, L. J. , De Hoog, C. L. , & Mann, M. (2003). Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proceedings of the National Academy of Sciences of the United States of America, 100, 5813–5818. 10.1073/pnas.0631608100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser, M. , & Wanaski, S. (1996). Myristoylated alanine‐rich C kinase substrate (MARCKS) produces reversible inhibition of phospholipase C by sequestering phosphatidylinositol 4,5‐bisphosphate in lateral domains*. The Journal of Biological Chemistry, 27, 26187–26193. [DOI] [PubMed] [Google Scholar]
- Green, T. D. , Park, J. , Yin, Q. , Fang, S. , Crews, A. L. , Jones, S. L. , & Adler, K. B. (2012). Directed migration of mouse macrophages in vitro involves myristoylated alanine‐rich C‐kinase substrate (MARCKS) protein. Journal of Leukocyte Biology, 92, 633–639. 10.1189/jlb.1211604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, R.‐F. , & Ward, P. A. (2005). Role OF C5A in inflammatory responses. Annual Review of Immunology, 23, 821–852. 10.1146/annurev.immunol.23.021704.115835 [DOI] [PubMed] [Google Scholar]
- Hansbro, P. M. , Kim, R. Y. , Starkey, M. R. , Donovan, C. , Dua, K. , Mayall, J. R. , … Horvat, J. C. (2017). Mechanisms and treatments for severe, steroid‐resistant allergic airway disease and asthma. Immunological Reviews, 278, 41–62. 10.1111/imr.12543 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlan, D. M. (1991). The human myristoylated alanine‐rich C kinase substrate (MARCKS) gene (MACS). The Journal of Biological Chemistry, 266, 14399–14405. [PubMed] [Google Scholar]
- Ito, K. , Herbert, C. , Siegle, J. S. , Vuppusetty, C. , Hansbro, N. , Thomas, P. S. , … Kumar, R. K. (2008). Steroid‐resistant neutrophilic inflammation in a mouse model of an acute exacerbation of asthma. American Journal of Respiratory Cell and Molecular Biology, 39, 543–550. 10.1165/rcmb.2008-0028OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug, N. , Tschernig, T. , Erpenbeck, V. J. , Hohlfeld, J. M. , & Köhl, J. (2001). Complement factors C3a and C5a are increased in bronchoalveolar lavage fluid after segmental allergen provocation in subjects with asthma. American Journal of Respiratory and Critical Care Medicine, 164, 1841–1843. 10.1164/ajrccm.164.10.2010096 [DOI] [PubMed] [Google Scholar]
- Kupczyk, M. , & Wenzel, S. (2012). U.S. and European severe asthma cohorts: What can they teach us about severe asthma? Journal of Internal Medicine, 272, 121–132. 10.1111/j.1365-2796.2012.02558.x [DOI] [PubMed] [Google Scholar]
- Lee, C. C. , Lai, Y. T. , Chang, H. T. , Liao, J. W. , Shyu, W. C. , Li, C. Y. , & Wang, C. N. (2013). Inhibition of high‐mobility group box 1 in lung reduced airway inflammation and remodeling in a mouse model of chronic asthma. Biochemical Pharmacology, 86, 940–949. 10.1016/j.bcp.2013.08.003 [DOI] [PubMed] [Google Scholar]
- Lee, C. C. , Wang, C. N. , Lai, Y. T. , Kang, J. J. , Liao, J. W. , Chiang, B. L. , … Cheng, Y. W. (2010). Shikonin inhibits maturation of bone marrow‐derived dendritic cells and suppresses allergic airway inflammation in a murine model of asthma. British Journal of Pharmacology, 161, 1496–1511. 10.1111/j.1476-5381.2010.00972.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden, A. (2006). Interleukin‐17 and airway remodelling. Pulmonary Pharmacology & Therapeutics, 19, 47–50. 10.1016/j.pupt.2005.02.004 [DOI] [PubMed] [Google Scholar]
- McLaughlin, S. , & Aderem, A. (1995). The myristoyl‐electrostatic switch: A modulator of reversible protein‐membrane interactions. Trends in Biochemical Sciences, 20, 272–276. 10.1016/S0968-0004(00)89042-8 [DOI] [PubMed] [Google Scholar]
- Morton, L. A. , Yang, H. , Saludes, J. P. , Fiorini, Z. , Beninson, L. , Chapman, E. R. , … Yin, H. (2012). MARCKS‐ED peptide as a curvature and lipid sensor. ACS Chemical Biology, 8, 218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naboulsi, W. , Megger, D. A. , Bracht, T. , Kohl, M. , Turewicz, M. , Eisenacher, M. , … Sitek, B. (2016). Quantitative tissue proteomics analysis reveals versican as potential biomarker for early‐stage hepatocellular carcinoma. Journal of Proteome Research, 15, 38–47. 10.1021/acs.jproteome.5b00420 [DOI] [PubMed] [Google Scholar]
- Nelson, H. S. , Leung, D. Y. M. , & Bloom, J. W. (2003). Update on glucocorticoid action and resistance. Journal of Allergy and Clinical Immunology, 111, 3–22. 10.1067/mai.2003.97 [DOI] [PubMed] [Google Scholar]
- Ramesh, R. , Kozhaya, L. , McKevitt, K. , Djuretic, I. M. , Carlson, T. J. , Quintero, M. A. , … Sundrud, M. S. (2014). Pro‐inflammatory human Th17 cells selectively express P‐glycoprotein and are refractory to glucocorticoids. The Journal of Experimental Medicine, 211, 89–104. 10.1084/jem.20130301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, A. T. , Lakshmi, S. P. , & Reddy, R. C. (2012). Murine model of allergen induced asthma. Journal of Visualized Experiments, 63, e3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, K. , Tolias, K. F. , Saci, A. , Koon, H. B. , Humphries, L. A. , Scharenberg, A. , … Carpenter, C. L. (2003). BTK regulates PtdIns‐4,5‐P2 synthesis: Importance for calcium signaling and PI3K activity. Immunity, 19, 669–678. 10.1016/S1074-7613(03)00297-8 [DOI] [PubMed] [Google Scholar]
- Saleh, D. , Ernst, P. , Lim, S. , Barnes, P. J. , & Giaid, A. (1998). Increased formation of the potent oxidant peroxynitrite in the airways of asthmatic patients is associated with induction of nitric oxide synthase: Effect of inhaled glucocorticoid. The FASEB Journal, 12, 929–937. 10.1096/fasebj.12.11.929 [DOI] [PubMed] [Google Scholar]
- Schewitz‐Bowers, L. P. , Lait, P. J. P. , Copland, D. A. , Chen, P. , Wu, W. , Dhanda, A. D. , … Lee, R. W. J. (2015). Glucocorticoid‐resistant Th17 cells are selectively attenuated by cyclosporine A. Proceedings of the National Academy of Sciences, 112, 4080–4085. 10.1073/pnas.1418316112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi, G. S. , & Naura, A. S. (2018). Progressive increase in allergen concentration abrogates immune tolerance in ovalbumin‐induced murine model of chronic asthma. International Immunopharmacology, 60, 121–131. 10.1016/j.intimp.2018.04.047 [DOI] [PubMed] [Google Scholar]
- Seykora, J. T. , Myat, M. M. , Allen, L. A. , Ravetch, J. V. , & Aderem, A. (1996). Molecular determinants of the myristoyl‐electrostatic switch of MARCKS. The Journal of Biological Chemistry, 271, 18797–18802. 10.1074/jbc.271.31.18797 [DOI] [PubMed] [Google Scholar]
- Singer, M. , Martin, L. D. , Vargaftig, B. B. , Park, J. , Gruber, A. D. , Li, Y. , & Adler, K. B. (2004). A MARCKS‐related peptide blocks mucus hypersecretion in a mouse model of asthma. Nature Medicine, 10, 193–196. 10.1038/nm983 [DOI] [PubMed] [Google Scholar]
- Strzelecka‐Kiliszek, A. , Korzeniowski, M. , Kwiatkowska, K. , Mrozinska, K. , & Sobota, A. (2004). Activated FccRII and signalling molecules revealed in rafts by ultra‐structural observations of plasma‐membrane sheets. Molecular Membrane Biology, 21, 101–108. 10.1080/09687680310001639094 [DOI] [PubMed] [Google Scholar]
- Sundaram, M. , Cook, H. W. , & Byers, D. M. (2004). The MARCKS family of phospholipid binding proteins: Regulation of phospholipase D and other cellular components. Biochemistry and Cell Biology, 82, 191–200. 10.1139/o03-087 [DOI] [PubMed] [Google Scholar]
- Szefler, S. J. , Martin, R. J. , King, T. S. , Boushey, H. A. , Cherniack, R. M. , Chinchilli, V. M. , … Sorkness, C. A. (2002). Significant variability in response to inhaled corticosteroids for persistent asthma. Journal of Allergy and Clinical Immunology, 109, 410–418. 10.1067/mai.2002.122635 [DOI] [PubMed] [Google Scholar]
- Taylor, P. R. , Roy, S. , Leal, S. M. Jr. , Sun, Y. , Howell, S. J. , Cobb, B. A. , … Pearlman, E. (2014). Activation of neutrophils by autocrine IL‐17A‐IL‐17RC interactions during fungal infection is regulated by IL‐6, IL‐23, RORγt and dectin‐2. Nature Immunology, 15, 143–151. 10.1038/ni.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez‐Tello, A. , Semlali, A. , Chakir, J. , Martin, J. G. , Leung, D. Y. , Eidelman, D. H. , & Hamid, Q. (2010). Induction of glucocorticoid receptor‐β expression in epithelial cells of asthmatic airways by T‐helper type 17 cytokines. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology, 40, 1312–1322. 10.1111/j.1365-2222.2010.03544.x [DOI] [PubMed] [Google Scholar]
- Wu, W. C. , Walaas, S. I. , Nairn, A. C. , & Greengard, P. (1982). Calcium/phospholipid regulates phosphorylation of a Mr “87k” substrate protein in brain synaptosomes. Proceedings of the National Academy of Sciences, 79, 5249–5253. 10.1073/pnas.79.17.5249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeck‐Kapp, G. , Kroegel, C. , Riede, U. N. , & Kapp, A. (1995). Mechanisms of human eosinophil activation by complement protein C5a and platelet‐activating factor: Similar functional responses are accompanied by different morphologic alterations. Allergy, 50, 34–47. 10.1111/j.1398-9995.1995.tb02481.x [DOI] [PubMed] [Google Scholar]
- Zijlstra, G. J. , Ten Hacken, N. H. , Hoffmann, R. F. , van Oosterhout, A. J. , & Heijink, I. H. (2012). Interleukin‐17A induces glucocorticoid insensitivity in human bronchial epithelial cells. The European Respiratory Journal, 39, 439–445. 10.1183/09031936.00017911 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Quantification of mouse cytokines, chemokines, and complement and acute‐phase proteins in bronchoalveolar lavage (BAL) fluids by cytokine arrays.
Figure S1. Intracellular localization of MPS, A‐MPS, and D‐MPS peptides in NHBE cells. NHBE cells were seeded in a chamber slide for 24 h and then incubated with 100 nmol MPS‐FITC, A‐MPS‐FITC, and D‐MPS‐FITC at 37°C for 5 min. The cells were washed thrice with 1 ml HBSS and then subjected to confocal microscopy. Nuclei were stained with DAPI which showed blue fluorescence. Green fluorescence indicates different MPS peptides. Scale bar: 25 μm, N = 5.
Figure S2. The effect of HDAC‐2 protein expression in a murine model of steroid‐resistant like asthma. The lung proteins were subjected to western blot analysis to assess the expression of HDAC‐2 and GAPDH. The proteins were quantified using Image J. Data are expressed as the mean ± S.D. (n = 5). # P < 0.05, as compared with the NC group.
Figure S3. Pulmonary inflammation and pathology at different times of OVA exposure. (A, D, G) Mice at 6 weeks of age were sensitized with OVA‐alum. On day 12, the mice were exposed to PBS or 5 mL 4 % OVA for five days per week for 1, 2, and 4 weeks, respectively. (B, E, H) Mice were intratracheally instilled with 1 mg/kg of dexamethasone twice, at 24 h and 48 h before dissection. On day 17, 24, and 38, the mice were sacrificed and differential cell counts in the BAL were assessed by May‐Giemsa staining. (C, F, I) Lungs were subjected to hematoxylin and eosin (H&E), periodic acid–Schiff (PAS), and Masson's trichrome (MT) staining. High magnification (bar represents 100 μm for H&E and MT, 200 μm for PAS). Data are expressed as mean ± S.D. (n ≧5). # P < 0.05, compared to the NC group.; * P < 0.05 compared to OVA group.
Figure S4. Retention time of MPS peptides in the airway. MPS‐FITC peptides (0.46 mg/kg and 2.3 mg/kg) were intratracheally instilled in mice at different time points (0 min, 3 min, 24 h, 48 h, and 72 h), and the lungs were then dissected and observed under an IVIS System to measure and quantify the fluorescence intensity. Data are expressed as mean ± S.D. (n ≧ 5). # P < 0.05, as compared with the NC.
Figure S5. Cytokine expression after MPS treatment using a cytokine array in a murine model of steroid‐resistant asthma. BAL fluid (BALF) was analyzed with a mouse cytokine array to screen for 40 cytokines, chemokines, and complement and acute‐phase proteins in the BAL. Pixel density plots of chemokine (C‐X‐C motif) ligand (CXCL)‐13 (A), soluble intercellular adhesion molecule‐1 (sICAM‐1) (B), KC (C) and stromal cell‐derived factor (SDF)‐1 (D) in BALF, and their quantification using Image J.
Figure S6. The effect of phospho‐p65 and p‐65 in mice with steroid‐resistant like asthma. Lung proteins were subjected to western blot analysis to assess the expression of p‐p65, p‐65, and GAPDH. The proteins were quantified using Image J. Data are expressed as the mean ± S.D. (n = 5). # P < 0.05, as compared with the NC group. * P < 0.05, as compared with the OVA group.
Figure S7. The effect of phosphatidylinositol 4,5‐biphosphate (PIP2) levels on the airways of OVA‐immunized mice. Mice at 6 weeks of age were sensitized with OVA/alum, as described in Figure 1B. Paraffin sections of mouse airway tissues were deparaffinized and IHC stained with PIP2 antibody (green arrows represent bronchial epithelial cells, black arrows represent alveolar epithelial cells, and red arrows represent for inflammatory cells). High magnification (bar represents 100 μm).