

Abstract
Eicosanoids represent a diverse family of lipid mediators with fundamental roles in physiology and disease. Within the eicosanoid superfamily are prostanoids, which are specifically derived from arachidonic acid by the enzyme cyclooxygenase (COX). COX has two isoforms; COX‐1 and COX‐2. COX‐2 is the therapeutic target for the nonsteroidal anti‐inflammatory drug (NSAID) class of pain medications. Of the prostanoids, prostacyclin, first discovered by Sir John Vane in 1976, remains amongst the best studied and retains an impressive pedigree as one of the fundamental cardiovascular protective pathways. Since this time, we have learnt much about how eicosanoids, COX enzymes and prostacyclin function in the cardiovascular system, knowledge that has allowed us, for example, to harness the power of prostacyclin as therapy to treat pulmonary arterial hypertension and peripheral vascular disease. However, there remain many unanswered questions in our basic understanding of the pathways, and how they can be used to improve human health. Perhaps, the most important and controversial outstanding question in the field remains; ‘how do NSAIDs produce their much publicized cardiovascular side‐effects?’ This review summarizes the history, biology and cardiovascular function of key eicosanoids with particular focus on prostacyclin and other COX products and discusses how our knowledge of these pathways can applied in future drug discovery and be used to explain the cardiovascular side‐effects of NSAIDs.
Linked Articles
This article is part of a themed section on Eicosanoids 35 years from the 1982 Nobel: where are we now? To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.8/issuetoc
Abbreviations
- ADMA
asymmetric dimethyl arginine
- BCL‐6
B‐cell lymphoma 6 protein
- cPLA2
cytosolic phospholipase A2
- CREB
cAMP response element‐binding protein
- EPAC
exchange protein directly activated by cAMP
- iPLA2
calcium‐independent phospholipase A2
- NFAT
nuclear factor of activated T cells
- NSAID
non‐steroidal anti‐inflammatory drug
- PGX
prostaglandin X (prostacyclin)
- sPLA2
secretory phospholipase A2
Introduction
Eicosanoids and the COX enzyme pathway feature in almost all aspects of health and disease. Indeed, it is difficult to find an organ system, homeostatic process or disease in which COX is not involved. COX has two isoforms; COX‐1 is constitutively expressed in many cell types whereas COX‐2 is constitutively expressed only in certain regions but is readily induced in inflammation and cancer. Arguably the most studied area of COX biology concerns inflammation and pain and this is not surprising as COX‐2 is the therapeutic target for nonsteroidal anti‐inflammatory drugs (NSAIDs), which are amongst the most commonly taken medications worldwide. Indeed, ibuprofen and aspirin, two members of the NSAID family, along with paracetamol, which is not an NSAID but exerts its analgesic effects by inhibiting COX, are members of the WHO Model List of Essential Medicines. However, the importance of COX to the cardiovascular system, particularly within blood vessels, cannot be overstated. This is illustrated by a PubMed search of the term ‘COX’ that delivers 56 082 returns, of these publications 9006 return with the terms ‘COX’ and ‘vascular’. This compares well with the 11 384 publications returning with the terms ‘COX’ and ‘inflammation’. The importance of prostanoids within the cardiovascular system was realized in the remarkable discovery by Sir John Vane in 1976 of a new member of the family made specifically by blood vessels, named at the time, PGX (Moncada et al., 1976). PGX, renamed prostacyclin, is a potent inhibitor of platelet aggregation and a vasodilator; both features of a cardioprotective hormone. The joint discoveries of prostacyclin and the mechanism of action of NSAIDs earned John Vane a share in the Nobel Prize for Physiology and Medicine in 1982. Since its discovery, major advances in our understanding of the importance of prostacyclin have been made. Its structure has been harnessed to produce a class of drugs to treat peripheral vascular disease and pulmonary arterial hypertension and the role its deficiency plays in cardiovascular disease appreciated. Nevertheless, there remains much that we do not know and the full potential of prostacyclin as a biomarker and therapeutic tool has yet to be realized. However, the most pressing issue relating to our understanding of cardiovascular prostacyclin biology is to solve the problem of the much reported cardiovascular side‐effects caused by inhibiting cardio‐protective COX‐2 with NSAIDs. This review focuses on the role of the eicosanoid sub‐group, prostanoids and COX within the cardiovascular system with particular emphasis on prostacyclin and the issue of cardiovascular side‐effects caused by NSAIDs.
Vascular eicosanoid biology
Eicosanoids
Eicosanoids are biologically active molecules made by oxidation of either ω‐3 or ω‐6 20‐carbon fatty acids. Eicosanoids are grouped into subcategories including leukotrienes, lipoxins, hydroxy‐eicosatetraenoic acids, hydroxy‐eicosapentaenoic acids, eoxins, isoprostanes, resolvins and prostanoids (Figure 1). Although other members of the eicosanoid family may have vascular effects (Pratico and Dogne, 2009), such as the isoprostane, 8‐isoPGF2α, which contracts vessels by acting on the thromboxane prostanoid TP receptor (Jourdan et al., 1997) or leukotrienes that stimulate vascular leak, contract vessels and propagates inflammation (Colazzo et al., 2017), in blood vessels the best characterized eicosanoids are the prostanoid family with prostacyclin being the predominant prostanoid released by both healthy and diseased blood vessels (Kent et al., 1981; Cannon, 1984; Kirkby et al., 2013c; Kirkby et al., 2014).
Figure 1.
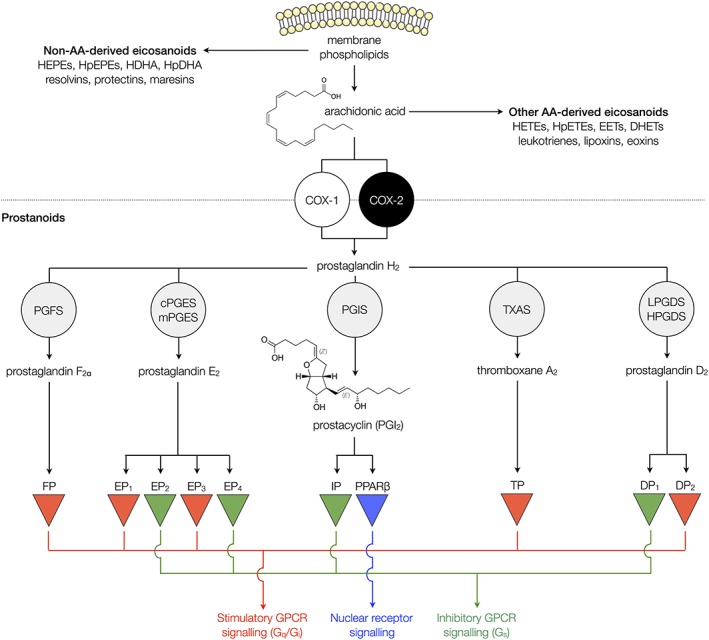
Eicosanoid and prostanoid synthesis. Arachidonic acid (AA) is liberated from membrane phospholipids by the action of PLA2. This can be converted to a range of eicosanoid mediators including prostanoids, hydroxyeicosatetraenoic acids (HETEs), hydroperoxyeicosatetraenoic acids (HpETEs), epoxyeicosatrienoic acids (EETs), dihydroxyeicosatrienoic acids (DHETs), leukotrienes, lipoxins and eoxins. Other eicosanoids are formed from other 20 carbon fatty acid substrates, including hydroxyeicosapentaenoic acids (HEPEs), hydroperoxyeicosapentaenoic acids (HpEPEs), hydroxydocosahexaenoic acids (HDHAs), hydroperoxydocosahexaenoic acids (HpDHAs), resolvins, protectins and maresins. For prostanoid synthesis, AA is converted to PGH2 by COX‐1 or COX‐2 enzymes. PGH2 is then further metabolized to individual prostanoid mediators by specific synthase enzymes including PGF synthase (PGFS), microsomal PGE synthases (mPGES)‐1 and ‐2, cytosolic PGE synthase (cPGES), prostacyclin synthase (PGIS), Tx synthase (TXAS), lipocalin‐type PGD synthase (LPGDS) and haematopoietic‐type PGD synthase (HPGDS). Each prostanoid acts preferentially on its corresponding receptors to activate inhibitory or stimulatory GPCRs or the nuclear receptor, PPARβ.
PLA2
Prostanoids are synthesized from the 20 carbon ω‐6 fatty acid arachidonic acid by the action of COX. Prostanoid release within most tissues, including blood vessels, is highly regulated and generally requires a strong stimulation before functionally relevant levels are made. COX is expressed in excess in most cells and thereby is generally not the limiting step. Instead, production is controlled by the availability of arachidonic acid. Due to its highly reactive nature and its sensitivity to oxidation, arachidonic acid is not normally free in cells and is instead stored in membrane phospholipids particularly phosphatidylethanolamine, phosphatidylcholine and phosphatidylinositol. The first step in prostanoid synthesis is therefore liberation of arachidonic acid from the membrane by phospholipases (Figure 1). This is mostly carried out by PLA2 enzymes, although some arachidonic acid can be formed by the action of PLC acting on arachidonic acid‐containing diacyl‐glycerols. PLA2 biology is complex with more than 30 different isoforms loosely classified as (i) cytosolic PLA2 (cPLA2), activated by μM concentrations of calcium, (ii) calcium independent (iPLA2) or (iii) secreted (sPLA2), activated by mM concentrations of calcium (Burke and Dennis, 2009; Murakami et al., 2011; Murakami et al., 2014).
Although there is evidence that sPLA2 isoforms and iPLA2β (Sharma et al., 2010; Majed and Khalil, 2012) can function in endothelial cells under some conditions, most evidence suggests that cPLA2ɑ (also referred to as group IVA cPLA2; encoded by the PLA2G4A gene) is the major isoform responsible for the release of arachidonic acid to produce prostanoids, in blood vessels.
This essential role of cPLA2ɑ in prostanoid production by cells of the cardiovascular system was confirmed in recent work from our group (Kirkby et al., 2015). We obtained cells from a patient with an extremely rare homozygous loss of function mutation of cPLA2ɑ and measured a full array of eicosanoids. Endothelial cells, leucocytes and platelets from this patient generated low or undetectable levels of prostanoids including prostacyclin, TxA2 and PGE2. The results from this patient are in agreement with an earlier study showing reduced production of eicosanoids by platelets from a patient carrying a compound heterozygous mutation of the same isoform of PLA2 (Adler et al., 2008).
COX
The second step in prostanoid synthesis, oxidation of the free arachidonic acid, is catalysed by the COX enzyme (Figure 1). COX is present in two isoforms, COX‐1 and COX‐2. There are structural differences between COX‐1 and COX‐2 within the active site, but for the conversion of arachidonic acid to prostanoids, the enzymic steps, oxidation forming PGG2 followed by peroxidation to yield PGH2, are identical for both isoforms. PGH2 is subsequently converted by downstream synthase/isomerase enzymes to the full range of prostanoids (Figure 1).
The first COX enzyme (what we now know as COX‐1) was purified in 1977 from sheep vesicular glands and found to be a haem protein of 70 kDa (Hemler and Lands, 1977). Just over 10 years later, this form of COX was sequenced (DeWitt and Smith, 1988) and the full‐length cDNA corresponding to a 2.8 kb mRNA was found to encode a protein of 600 amino acids. The authors had the foresight to comment ‘the availability of a full‐length cDNA clone coding for prostaglandin G/H synthase (COX) should facilitate studies of the regulation of expression of this enzyme and the structural features important for catalysis and for interaction with anti‐inflammatory drugs’ (DeWitt and Smith, 1988). They could not have known at the time that within a year, their work would be used to identify a completely new form of COX. Between 1989 and 1991, three separate groups using different experimental approaches identified a new 4.0 kb mRNA species encoding a similar ‘COX’ protein, also of ≈600 amino acids, which we now know as COX‐2 (Rosen et al., 1989; Kujubu et al., 1991; Xie et al., 1991). The first demonstration of human COX‐2, with full enzymic activity, was performed in 1992 using human endothelial cells stimulated with IL‐1 (Hla and Neilson, 1992).
A common feature in early studies of COX‐2 was that cells (Rosen et al., 1989; Kujubu et al., 1991; Xie et al., 1991; Hla and Neilson, 1992), blood vessels (Bishop‐Bailey et al., 1997) or laboratory animals (Vane et al., 1994) required an inflammatory insult for the enzyme to be ‘induced’ driven by transiently active transcription pathways including NF‐κB, nuclear factor of activated T cells (NFAT) and cAMP response element‐binding protein (CREB) (Figure 2). Indeed, it was this particular fact that led to the general idea that COX‐1 is constitutively expressed within tissues acting as a protective enzyme whereas COX‐2 is an inducible form present only in inflammation (Figure 2). We now know that this was a simplistic view and that COX‐2 is constitutively expressed in key regions of the body including the brain, lung, thymus, gut and kidney (Kirkby et al., 2013c; Figure 2). In the gut, COX‐2 together with COX‐1 protects the mucosa from injury (Wallace et al., 2000) while, in the kidney, constitutive COX‐2, driven by NFAT, regulates renal function and blood flow. The relevance of renal COX‐2 to cardio‐protection is discussed in detail later. The role of constitutive COX‐2 in the brain, lung and thymus is not completely understood and the transcriptional drivers have not yet been identified.
Figure 2.
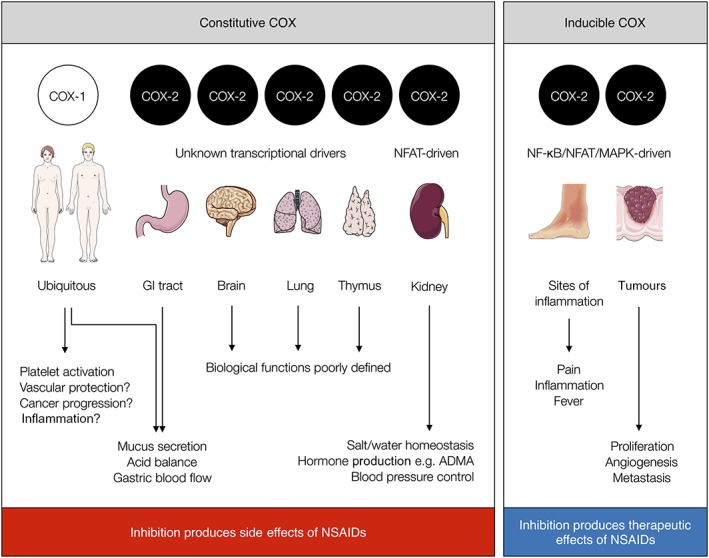
Constitutive and inducible COX enzymes. COX‐1 is a ubiquitous isoform expressed constitutively in all tissues. COX‐2 is constitutively expressed in specific tissue locations within the kidney, gastrointestinal tract, brain, lungs and thymus as well as other regions of the urogenital tract (not shown). Constitutive COX‐2 transcription pathways are known only for the kidney (NFAT). Inducible COX‐2 is expressed at the site of inflammation and in cancer. Inducible COX‐2 transcription pathways include NF‐κB, NFAT and CREB. Inducible COX‐2 is the therapeutic target of the NSAID class of drugs, but their inhibition of constitutive COX‐1 and COX‐2 results in their side‐effects.
Early work from our group (Mitchell et al., 1993) and others (Meade et al., 1993) showed that both COX‐1 and COX‐2 were inhibited by NSAIDs but with different potencies, leading us to suggest that COX‐2 is the therapeutic target of NSAIDs and that there was a therapeutic window for the development of COX‐2 selective drugs (Mitchell et al., 1993). Subsequently, COX‐2 selective NSAIDs such as Celebrex® (celecoxib), Vioxx® (rofecoxib) and Arcoxia® (etoricoxib) were introduced for the treatment of inflammation and pain, designed to diminish the gastric side‐effects associated with older style drugs such as diclofenac and ibuprofen. However, because neither the selective COX‐2 inhibitors nor the older NSAIDs discriminate between blocking the COX‐2 induced at the site of inflammation and the constitutively expressed COX‐2 that protects the cardiovascular system, they cause cardiovascular side‐effects. Indeed, understanding and solving the problem of cardiovascular side‐effects caused by NSAIDs remains amongst the most important unresolved questions in the vascular biology of eicosanoids. As such, the remainder of this review will focus on mechanisms of cardiovascular protection by COX‐2 and the involvement of specific eicosanoid and other mechanistic pathways in this aspect of COX‐2 biology.
Role of COX‐2 in cardiovascular protection and the cardiovascular side‐effects of NSAIDs
Scale of the problem
Cardiovascular side‐effects, which include hypertension, heart attacks and strokes, were initially thought to be limited to drugs that selectively targeted COX‐2, such as rofecoxib and celecoxib, introduced in the early 2000s. However, as a result of subsequent epidemiology analyses (Warner and Mitchell, 2008; McGettigan and Henry, 2011) and, most recently, the publication of two large clinical cardiovascular outcome studies, SCOT (MacDonald et al., 2017) and PRECISION (Nissen et al., 2016), it is clear that traditional NSAIDs, including ibuprofen and naproxen, carry at least a great cardiovascular risk as the COX‐2 selective drug celecoxib, with ibuprofen emerging as significantly more toxic to the kidney at therapeutic doses (Nissen et al., 2016). Aspirin is a special case in the NSAID family of drugs since it prevents rather than precipitates cardiovascular events. The mechanisms by which aspirin protects the cardiovascular system have been reviewed elsewhere (Crescente et al., 2019) but involve the unique pharmacology of aspirin as an irreversible inhibitor of COX which allows this NSAID to selectively target TXA2 release from the anucleate platelet. Thus, all NSAIDs, except aspirin, increase personal risk of a cardiovascular event by as much as 30% (McGettigan and Henry, 2011; Trelle et al., 2011; Coxib et al., 2013; Bally et al., 2017). Importantly, increased risk of cardiovascular events while on NSAIDs can be seen after as little as 2 weeks of regular use (Bally et al., 2017). To put this into context, there are ≈460 000 myocardial infarcts and strokes in the UK each year and an estimated 20–30% of the population regularly use NSAIDs. On this basis, the predicted increased risk of a cardiovascular adverse reaction while taking NSAIDs translates to a conservative estimate of around 30 000–50 000 NSAID‐attributable cardiovascular events per year or 10% of all myocardial infarcts and strokes.
The fear of adverse cardiovascular events caused by NSAIDs has led to a series of changes in drug regulation, including (i) the withdrawal of rofecoxib in 2004, (ii) the introduction of ‘black box’ warnings in 2005 and (iii) the reclassification of the over‐the‐counter medication diclofenac as prescription only in 2015 (UK).
Concern regarding cardiovascular events caused by NSAIDs has also resulted in the cautious prescribing of COX‐2 selective drugs (Scarpignato et al., 2015) in favour of the older NSAIDs that are more toxic to the gut or opioids, which carry their own different problems of tolerance and abuse, creating additional health and social burdens. This situation defies logic as we now know that ibuprofen, which is the most consumed NSAID and is still widely available without prescription, causes both cardiovascular side‐effects and is damaging to the gut.
Finally, it is important to remember that NSAIDs are effective chemo‐preventative therapies for cancer, with population‐based studies showing their long‐term use was associated with a 30–60% decrease in the risk of developing major cancers (Matos and Jordan, 2015), with colon cancer being the most extensively studied. Indeed, it should be noted that for colon cancer, evidence is not restricted to epidemiological data but backed up with randomized, placebo‐controlled clinical trials showing that rofecoxib (Baron et al., 2006) and celecoxib (Bertagnolli et al., 2006) were effective in preventing around 50% of tumour occurrence in some patient groups. Taking just this form of cancer as an example, with 43 000 new cases of colon cancer diagnosed in the UK each year, the use of COX‐2 inhibitors has the potential to prevent cancer in 22 000 of these cases annually. Clearly, the potential for NSAIDs to prevent other cancers adds additional importance to assessment of the role of these drugs in population health. However, since the withdrawal of Onsenal (celecoxib) for the prevention of cancer in 2011 because of safety concerns, including cardiovascular side‐effects, NSAIDs are no longer used to prevent cancer.
Prostacyclin as the major cardioprotective eicosanoid
Of the many eicosanoids produced by the vasculature, prostacyclin is the one most associated with cardiovascular protection and is centrally involved in the mechanisms by which NSAIDs cause cardiovascular side‐effects. As such, its production and signalling are discussed below.
The reason why blood vessels release predominantly prostacyclin is because they are enriched with both COX and prostacyclin synthase (CYP8A1; Figure 3). Prostacyclin synthase was first purified from bovine aorta (DeWitt and Smith, 1983). Although prostacyclin synthase knockout mice have been developed, surprisingly, they have not been extensively used in cardiovascular models. A single study describes prostacyclin synthase knockout mice as having altered renal developmental and renal dysfunction, increased BP and, consequently, vascular hypertrophy (Yokoyama et al., 2002). Perhaps, more informative, human mutations and polymorphisms in prostacyclin synthase are linked with essential hypertension, myocardial and cerebral infarction (Nakayama, 2010).
Figure 3.
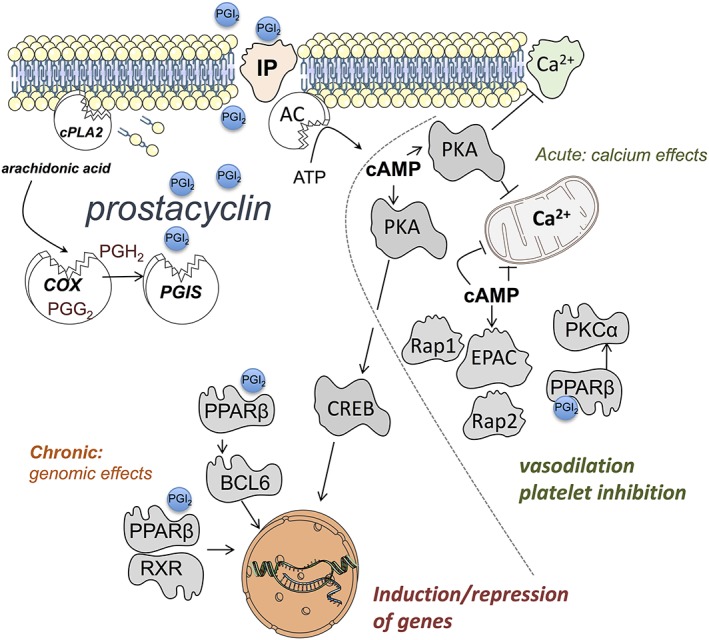
Synthesis and signalling pathways for prostacyclin (PGI2). Arachidonic acid is released by the actions of cytosolic PLA2 (cPLA2) then metabolized to prostacyclin by the concerted actions of COX and prostacyclin synthase (PGIS). Prostacyclin acts on cell surface IP receptors linked to action of adenylate cyclase (AC) resulting in the conversion of ATP to cAMP. cAMP then activates (i) PKA which phosphorylates substrate proteins leading to blockade of calcium channels and of release from intracellular stores. cAMP activated PKA also activates the transcription factor cAMP response element binding protein (CREB) leading to gene induction. cAMP also activates exchange protein directly activated by cAMP (EPAC), a Ras activate protein (Rap)1 and Rap2 guanine‐nucleotide‐exchange factor. Activated EPAC also prevents calcium elevation within cells (by unknown mechanisms). Prostacyclin also activates cytosolic PPARβ receptors that function in two ways; (i) via classical PPAR‐retinoid X receptor complex signalling to modulate gene expression and (ii) by binding and repressing the transcription factor BCL6 (inactive PPARβ) and PKCα, (active PPARβ). These pathways orchestrate acute signalling events characterized by calcium reduction leading to (for example) vasodilation and inhibition of platelet activation and chronic events mediated by gene induction.
Although the cardioprotective pedigree of prostacyclin is well established, apart from its effects on platelets, which are (i) inhibitory, (ii) highly reproducible across models and (iii) independent of platelet activation pathways, the effects of prostacyclin can be subtle and influenced by the resting state of the target cell or organ. This is because prostacyclin signalling utilizes different types of receptors and transduction pathways (Figures 1 and 3). For example, prostacyclin can exert acute effects, such as inhibition of platelet activation and relaxation of blood vessels, which are evident within seconds or minutes of adding prostacyclin to the system. Prostacyclin can also exert longer term genomic effects by directing gene transcription. Prostacyclin acts primarily on two receptors; the cell surface GPCR IP receptor and the cytosolic nuclear receptor PPARβ (Mitchell et al., 2014). Additionally, in some situations, prostacyclin can act on other prostanoid receptors to produce common and/or contradictory effects. This aspect of prostacyclin signalling has been reviewed in detail elsewhere (Luo et al., 2016).
IP receptors are linked to activation of membrane‐bound adenylate cyclase, which converts ATP to the intracellular signalling molecule cAMP. cAMP then activates PKA, engaging the ‘PKA pathway’ and/or exchange protein directly activated by cAMP (EPAC), engaging the ‘EPAC pathway’. These pathways, reviewed in detail elsewhere (Torres‐Quesada et al., 2017; Wang et al., 2017), are complex and incompletely understood. For vessels to contract or platelets to aggregate, intracellular calcium must be increased. Activation of PKA and/or engagement of EPAC in platelets or vascular smooth muscle cells results in immediate and profound reduction in levels of intracellular calcium through incompletely understood pathways (Yan et al., 2016).
However, for PKA, at a very basic level, activation results in phosphorylation of its substrate proteins (of which there are many) (Shabb, 2001); including those that regulate intracellular calcium handling and mediate contraction or aggregation. This leads to (i) depletion and/or blockade of release from intracellular calcium stores, (ii) inhibition of subsequent store‐operated calcium entry (Cuinas et al., 2016), (iii) blockade of calcium entry from voltage‐gated (and other) calcium channels (Shabb, 2001) and activation of MaxiK and ATP sensitive potassium channels and (iv) reduced sensitivity of effector proteins such as myosin light chain kinase that mediate contraction/aggregation.
Less is known about the EPAC pathway. There are two EPAC isoforms, EPAC1 and EPAC2 (Lezoualc'h et al., 2016), both of which are guanine‐nucleotide exchange factors for the Ras‐like GTPases, Rap1 and Rap2. EPAC mediates vasodilation by indirectly regulating calcium‐sensitive and ATP‐sensitive potassium channels and by lowering contractility by inhibition of small GTPase RhoA activity (Lezoualc'h et al., 2016). Although Rap1 and Rap2 signalling is known to be involved in platelet adhesion (Stefanini and Bergmeier, 2016), the role of EPAC in prostacyclin signalling in platelets has not been addressed. The chronic effects of IP receptor activation can be explained in part by PKA‐dependent activation of the transcription factor CREB. CREB binds to the CRE sequences in DNA to orchestrate transcriptional changes of ≈4000 target genes in the human genome (Zhang et al., 2005), although how this relates to specific responses associated with IP receptor activation within cardiovascular structures remains to be fully explored.
Work from our group and of others has shown that activation of PPARβ can also explain some of the acute effects of prostacyclin in platelets and blood vessels (Ali et al., 2009a; Baron et al., 2006; Harrington et al., 2010; Li et al., 2012; Kirkby et al., 2018). Initially, this was a surprising discovery as PPARβ was thought to be a pathway that regulates gene induction rather than acute signalling events; however, now we know that PPARβ activation leads to the engagement of both genomic and non‐genomic pathways (Mitchell et al., 2014). First, activation of PPARβ can act via classical PPAR‐retinoid X receptor signalling to regulate target gene induction. Target genes of PPARβ include ANGPTL4 and a range of lipid metabolism and angiogenic genes (Kojonazarov et al., 2013). The angiogenic profile of PPARβ explains in part why selective agonists of this receptor can induce cancer and provides a mechanism by which prostacyclin drives proliferation in some cells including endothelial progenitor cells (He et al., 2008). Second, in its resting state, PPARβ binds and represses the transcription factor BCL‐6 (Lee et al., 2003) and, in its active state, binds the protein kinase PKCα (Ali et al., 2009b; Mitchell et al., 2014). Consequently, when prostacyclin binds PPARβ, BCL‐6 is released, and PKCα is repressed to regulate inflammatory gene transcription and the acute effects seen in platelet activation respectively. It remains unclear precisely how PPARβ agonists induced acute vasodilation (Toral et al., 2017), although this may involve the RhoA pathway (Harrington et al., 2010).
While there is much we do not yet know about prostacyclin and its signalling pathways, drugs acting on prostacyclin receptors have been developed and used, for example, in the treatment of pulmonary arterial hypertension (see Mitchell et al., 2014). Considering the powerful effects of prostacyclin in the cardiovascular system, it is likely that prostacyclin has further potential as a therapy for more common forms of cardiovascular diseases, and this may come from a full understanding of its receptor and signalling pathways. However, to solve the problem of cardiovascular side‐effects caused by NSAIDs, we also need to understand how endogenous prostacyclin works to protect the cardiovascular system.
Mechanisms of NSAID cardiovascular side‐effects
When COX‐2 was discovered as an isoform of COX induced at the site of inflammation, it was known that COX‐1 and prostacyclin synthase were both constitutively expressed in endothelium. Thus, based on what was known at the time, studies showing that COX‐2 induced in vessels produced prostacyclin (Mitchell and Evans, 1998) and that rofecoxib or celecoxib selectively reduced urinary prostacyclin metabolites (Catella‐Lawson et al., 1999), raised concerns about the cardiovascular safety of COX‐2 inhibitors before clinical data were available. In this regard, the field was ‘primed’ and alerted to the possibility that COX‐2 selective NSAIDs might cause cardiovascular side‐effects. In 2000, when results of the VIGOR study (Bombardier et al., 2000) showed higher numbers of heart attacks in people taking rofecoxib than the comparator drug, naproxen, serious debate about cardiovascular side‐effects began. However, it was only after the publication of the APPROVe trial (Bresalier et al., 2005) where rofecoxib was tested against placebo and showed increased cardiovascular events that this became a widely accepted side‐effect and resulted in the withdrawal of the drug worldwide.
Despite almost 20 years of research in the area, we still do not fully understand how COX‐2 protects the cardiovascular system. It is generally agreed that prostacyclin is the major cardio‐protective prostanoid, but an important limitation in the field has been separating the respective roles of COX‐1 and COX‐2, in prostacyclin production and, furthermore, to determine the location of cardio‐protective COX‐2. It is now believed that cardio‐protective COX‐2 is located in the kidney and/or the systemic vascular endothelium. Although there is debate regarding the importance of homeostatic COX‐2 between these locations, it is known that constitutively expressed COX‐2 (Cheng et al., 2006; Ahmetaj‐Shala et al., 2015; Kirkby et al., 2016), rather than an inflammation‐driven isoform protects the cardiovascular system. This understanding is based on the findings that (i) NSAID‐associated cardiovascular events occur in patients, without overt cardiovascular disease or systemic inflammatory disease and (ii) blocking COX‐2 in healthy people or mice impairs renal function (Whelton and Hamilton, 1991; Ahmetaj‐Shala et al., 2015) and increases BP (Ahmetaj‐Shala et al., 2015) and thrombosis (Yu et al., 2012) in otherwise healthy laboratory animals.
Role of COX‐1 and COX‐2 in vascular prostacyclin production
The case for COX‐2 expressed in the systemic vascular endothelium producing prostacyclin is based on two lines of indirect research. The first is that a metabolite of prostacyclin, PGI‐M, which serves as a urinary marker of this prostanoid, is decreased in subjects taking COX‐2 selective NSAIDs (McAdam et al., 1999). However, the idea that urinary markers of prostacyclin reflect levels in the systemic circulation is not universally accepted (Mitchell and Warner, 2006; Flavahan, 2007) and current evidence suggests that urinary PGI‐M is, in fact, a renal metabolite, i.e. it reflects more renal levels, rather than systemic levels of prostacyclin (Mitchell, 2016; Kirkby et al., 2017a; Mitchell et al., 2017). The second line comes from studies showing that endothelial COX‐2 gene deletion in mice causes thrombosis (Yu et al., 2012). However, these data and reported phenotypes may also be explained by the loss of COX‐2 locally expressed in the renal rather than in systemic vascular endothelium. Further, the idea that COX‐2 is the major determinant of prostacyclin levels in the systemic circulation does not take account of research showing the importance of COX‐1 in prostacyclin production. COX‐1 and prostacyclin synthase are both constitutively expressed in endothelial cells (Figure 4). Consequently, blood vessels from COX‐1 knockout mice, stimulated with calcium ionophore to activate PLA2, release negligible levels of prostacyclin (Kirkby et al., 2012; Liu et al., 2012; Kirkby et al., 2013c; Luo et al., 2016) whereas prostacyclin release is essentially unchanged in freshly prepared arteries from COX‐2 knockout mice (Kirkby et al., 2012; Kirkby et al., 2013c). Similarly, plasma levels of prostacyclin are reduced in COX‐1 knockout mice but unchanged in mice where COX‐2 is deleted (Kirkby et al., 2012; Kirkby et al., 2013c). Most recently, our data using novel, tissue‐specific, COX knockout mice have confirmed that in systemic blood vessels, COX‐1 in endothelial cells accounts entirely for prostacyclin release from endothelium (Mitchell et al., 2017). This is also the case for vessels from atherosclerotic mice where although COX‐2 is expressed locally in lesions, its role in prostacyclin release in comparison with COX‐1 is negligible (Kirkby et al., 2014). In fact, to model a situation experimentally in mice where COX‐2 takes over from COX‐1 as the major source of prostacyclin production, severe systemic inflammation needs to be induced (Kirkby et al., 2013a). Clearly then, in vivo, and in defined blood vessels ex vivo, COX‐1 and not COX‐2 determines prostacyclin production. While this conclusion is not universally accepted, wherever investigated at the protein, gene or bioactivity level, COX‐2 is sparse, compared with COX‐1, in the majority of endothelial sites. To explain this, it has been suggested that COX‐2 in endothelial cells is induced by shear stress and is very unstable outside the body (Funk and FitzGerald, 2007). This suggestion has been addressed in experiments using fresh vessels and by showing that in plasma, basal as well as bradykinin‐induced prostacyclin release in vivo is COX‐1, and not COX‐2 dependent (Kirkby et al., 2012). Most recently, it has been suggested that low levels of COX‐2 activity can still contribute importantly to prostacyclin production, because this isoform requires lower levels of arachidonic acid for activation compared with those required by COX‐1 (Grosser et al., 2017). However, any small differences in the enzyme kinetics do not adequately explain the overwhelmingly dominant role of COX‐1 over COX‐2 in the control of basal plasma prostacyclin levels (Kirkby et al., 2012; Kirkby et al., 2013c) and ex vivo vascular prostacyclin production over a range of stimuli or substrate levels (Kirkby et al., 2013c).
Figure 4.
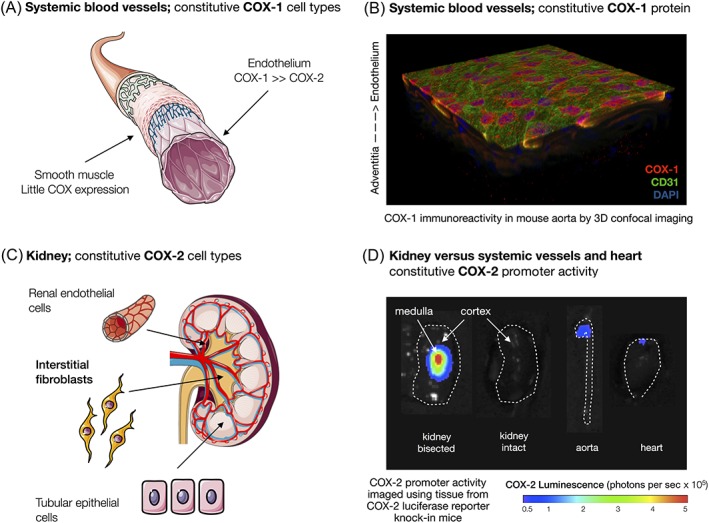
Expression of COX‐1 and COX‐2 at systemic vascular and renal sites. COX‐1 expression predominates over COX‐2 in the endothelium of systemic vessels (A) shown as red staining, with nuclei stained blue and CD31 stained green (B). COX‐2 is expressed in high levels within particular cells within the kidney including the specialized fibroblasts within the interstia of the renal medulla, renal tubule cells and the endothelium of renal blood vessels (C). COX‐2 gene expression is visualized as an intense area of activity within the bisected kidney that maps to the inner renal medulla where levels are very much higher than in systemic blood vessels or the heart (D).
Role of the kidney in cardiovascular protection by COX‐2
By contrast to the lack of clear, direct evidence for a role of COX‐2 in vascular prostacyclin production, the alternative case for a role for renal COX‐2 in cardiovascular protection is supported by a wealth of data. This includes studies which have shown COX‐2 as being constitutively expressed in interstitial fibroblasts in the renal medulla, tubular epithelial cells and renal endothelial cells (Harris, 2006) (Figure 4). Our recent work (Kirkby et al., 2016) confirms the medullary fibroblast region as a major site for COX‐2 and shows that expression is driven by the NFAT transcription factor, in the absence of inflammation. At a functional level, constitutive COX‐2 regulates fundamental aspects of renal homeostasis, including renin release, sodium excretion, renal blood flow and BP (see Harris, 2006). For example, whereas COX‐2 knockout mice exhibit negligible changes in the transcriptome of the aorta, heart or blood, such deletion of COX‐2 produces a profound effect on the renal transcriptome (>1000 genes) directly demonstrating the importance of COX‐2 in the kidney, relative to other cardiovascular structures (Ahmetaj‐Shala et al., 2015). Consequently, the effects of NSAIDs in the kidney may explain the cardiovascular side‐effects caused by inhibitors of COX‐2.
Role of eNOS in COX‐2 cardiovascular protection
Until recently, precisely how inhibition of COX‐2 derived prostacyclin resulted in hypertension, thrombosis and atherosclerosis was unclear and not adequately explained by the simple removal of prostacyclin as the sole event. A significant advancement in our understanding of how COX‐2 mediates cardiovascular protection was made in recent work from our group (Ahmetaj‐Shala et al., 2015) and others (Yu et al., 2012) describing a link between inhibition of COX‐2 and the endothelial NOS (eNOS) pathway (Figure 5). Our work (Ahmetaj‐Shala et al., 2015) showed that when COX‐2 is deleted in mice or blocked with NSAIDs in mice or healthy human volunteers, an inhibition of the methylarginine pathway is removed that results in increased levels of the endogenous eNOS inhibitor, asymmetric dimethylarginine (ADMA). We have now repeated, validated and extended these observations to show that the effect of COX‐2 inhibition on ADMA levels is reproduced in mice lacking prostacyclin synthase but not in PGE2 synthase knockout mice (Raouf et al., 2016), firmly establishing prostacyclin as the protective COX‐2 end product that limits ADMA levels (Figure 5).
Figure 5.
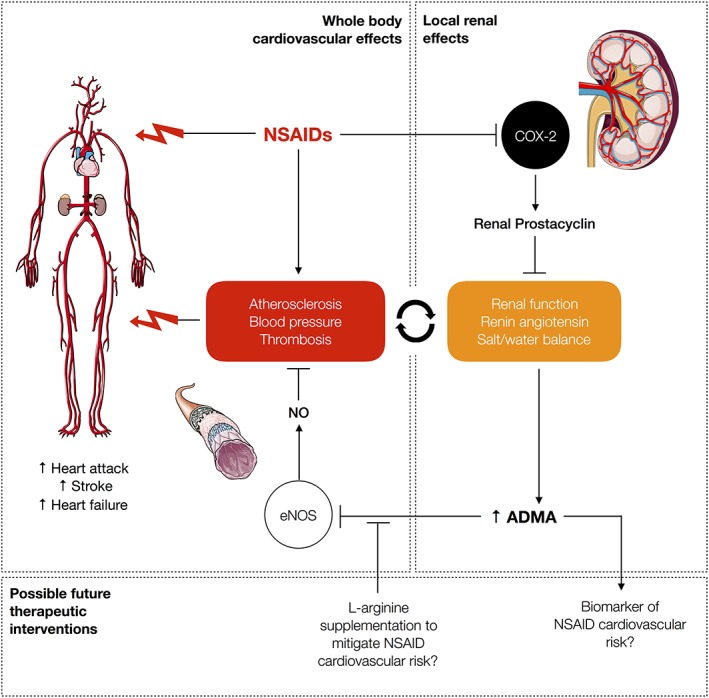
Hypothesis: pathways that link inhibition of renal COX‐2 prostacyclin with endothelial NOS (eNOS) inhibition and increased risk of cardiovascular events. Blocking COX‐2 with NSAIDs in the kidney results in increased levels of ADMA in the systemic circulation, where it inhibits eNOS. Inhibition of eNOS increases atherosclerosis, hypertension and thrombosis. Blocking COX‐2 in the kidney also decreases renal function and increases the renin‐angiotensin and sodium retention pathways. The eNOS‐dependent systemic effects and local effects within the kidney, work together to amplify the cardiovascular dysfunction and work in concert to predispose people taking NSAIDs to cardiovascular side‐effects.
The constitutive isoform, eNOS, is expressed throughout the vasculature where it protects the cardiovascular system against atherosclerosis, thrombosis and hypertension. The effects of eNOS and COX‐2 together in protecting the cardiovascular system is biologically enhanced as NO and prostacyclin work additively to maintain vascular function (Lidbury et al., 1989) and in a synergistic manner against thrombosis (Levin et al., 1982; Macdonald et al., 1988; Lidbury et al., 1989; Kirkby et al., 2013b). This pathway, which links COX‐2 and eNOS, via prostacyclin, provides a plausible mechanism for how NSAIDs could precipitate cardiovascular side‐effects and highlights the circulating levels of ADMA as a potential biomarker of such side‐effects. Finally, because the inhibitory effects of ADMA on eNOS can be prevented by L‐arginine, a simple, safe, nutritional supplement available without prescription, this mechanism also provides a possible rescue therapy for those people at risk who need to take NSAIDs to go about their daily lives (Figure 5).
Role of genetics in cardiovascular protection by COX‐2 and potential of genetic biomarkers
In addition to classical hypothesis testing studies such as those described above, this field needs hypothesis‐generating approaches. With this in mind, we have performed a first of its kind genome‐wide association study using data from a set of ~1000 samples from participants in the Adenoma Prevention with Celecoxib trial (Solomon et al., 2005) and identified four loci exceeding a nominal discovery P‐value threshold of 10−6 but not predicting general cardiovascular risk (CARDIOGRAMplusC4D Consortium), thus suggesting they may have a specific, causative role in NSAID cardiovascular toxicity (Kirkby et al., 2017b). Two of these loci map to viable candidate genes, AHNAK and NCKX2, which we showed to be functionally relevant in renal vessels from knockout mice (Kirkby et al., 2017b). These data, while preliminary, are the first to deliver potential genetic biomarkers of NSAID‐induced cardiovascular adverse events, thereby paving the way for precision medicine in this area.
Summary and future directions
Prostacyclin is a fundamental cardio‐protective hormone whose therapeutic potential will, as we decipher its signalling pathways, inevitably be realized. NSAIDs are here to stay despite the cardiovascular side‐effects that have blighted their recent history. We need to understand the mechanisms by which these occur in order to protect those at risk and so that COX‐2 inhibitors can be used to prevent cancer. This should include testing the translational potential of targeting the COX‐2/prostacyclin/ADMA/eNOS axis to identify those at risk (by measuring ADMA levels) and to protect those taking NSAIDs (with L‐arginine supplementation). Hypothesis‐testing approaches should be complemented by the integration of genetic data and other ‘‐omic’ approaches to find biomarkers and generate new hypotheses to extend our understanding of COX, prostacyclin and eicosanoids in the cardiovascular system.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/2018 (Alexander et al., 2017a,b,c).
Conflict of interest
Within the last 5 years, J.A.M. has acted a consultant and expert witness in cases relating to prostacyclin drugs and non‐steroidal anti‐inflammatory drugs. J.A.M. has received unconditional educational awards from pharmaceutical companies including United Therapeutics and Actelion and is on the scientific advisory board of Antibe Therapeutics. The authors report no other conflicts of interest.
Acknowledgements
N.S.K. is supported by a British Heart Foundation Intermediate Fellowship (FS/16/1/31699).
Mitchell, J. A. , and Kirkby, N. S. (2019) Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. British Journal of Pharmacology, 176: 1038–1050. 10.1111/bph.14167.
Contributor Information
Jane A Mitchell, Email: j.a.mitchell@ic.ac.uk.
Nicholas S Kirkby, Email: n.kirkby@ic.ac.uk.
References
- Adler DH, Cogan JD, Phillips JA 3rd, Schnetz‐Boutaud N, Milne GL, Iverson T et al (2008). Inherited human cPLA(2alpha) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J Clin Invest 118: 2121–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmetaj‐Shala B, Kirkby NS, Knowles R, Al'Yamani M, Mazi S, Wang Z et al (2015). Evidence that links loss of cyclooxygenase‐2 with increased asymmetric dimethylarginine: novel explanation of cardiovascular side effects associated with anti‐inflammatory drugs. Circulation 131: 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali FY, Armstrong PC, Dhanji AR, Tucker AT, Paul‐Clark MJ, Mitchell JA et al (2009a). Antiplatelet actions of statins and fibrates are mediated by PPARs. Arterioscler Thromb Vasc Biol 29: 706–711. [DOI] [PubMed] [Google Scholar]
- Ali FY, Hall MG, Desvergne B, Warner TD, Mitchell JA (2009b). PPARbeta/delta agonists modulate platelet function via a mechanism involving PPAR receptors and specific association/repression of PKCalpha – brief report. Arterioscler Thromb Vasc Biol 29: 1871–1873. [DOI] [PubMed] [Google Scholar]
- Bally M, Dendukuri N, Rich B, Nadeau L, Helin‐Salmivaara A, Garbe E et al (2017). Risk of acute myocardial infarction with NSAIDs in real world use: bayesian meta‐analysis of individual patient data. BMJ 357: j1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron JA, Sandler RS, Bresalier RS, Quan H, Riddell R, Lanas A et al (2006). A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology 131: 1674–1682. [DOI] [PubMed] [Google Scholar]
- Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K et al (2006). Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med 355: 873–884. [DOI] [PubMed] [Google Scholar]
- Bishop‐Bailey D, Larkin SW, Warner TD, Chen G, Mitchell JA (1997). Characterization of the induction of nitric oxide synthase and cyclo‐oxygenase in rat aorta in organ culture. Br J Pharmacol 121: 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombardier C, Laine L, Reicin A, Shapiro D, Burgos‐Vargas R, Davis B et al (2000). Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med 343: 1520–1528, 1522 p following 1528. [DOI] [PubMed] [Google Scholar]
- Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K et al (2005). Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med 352: 1092–1102. [DOI] [PubMed] [Google Scholar]
- Burke JE, Dennis EA (2009). Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res 50 (Suppl): S237–S242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon PJ (1984). Eicosanoids and the blood vessel wall. Circulation 70: 523–528. [DOI] [PubMed] [Google Scholar]
- Catella‐Lawson F, McAdam B, Morrison BW, Kapoor S, Kujubu D, Antes L et al (1999). Effects of specific inhibition of cyclooxygenase‐2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J Pharmacol Exp Ther 289: 735–741. [PubMed] [Google Scholar]
- Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA (2006). Cyclooxygenases, microsomal prostaglandin E synthase‐1, and cardiovascular function. J Clin Invest 116: 1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colazzo F, Gelosa P, Tremoli E, Sironi L, Castiglioni L (2017). Role of the cysteinyl leukotrienes in the pathogenesis and progression of cardiovascular diseases. Mediators Inflamm 2017 2432958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coxib , traditional NTC , Bhala N, Emberson J, Merhi A, Abramson S et al (2013). Vascular and upper gastrointestinal effects of non‐steroidal anti‐inflammatory drugs: meta‐analyses of individual participant data from randomised trials. Lancet 382: 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescente M, Menke L, Chan MV, Armstrong PC, Warner TD (2019). Eicosanoids in platelets and the effect of their modulation by aspirin in the cardiovascular system (and beyond). Br J Pharmacol 176: 988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuinas A, Garcia‐Morales V, Vina D, Gil‐Longo J, Campos‐Toimil M (2016). Activation of PKA and Epac proteins by cyclic AMP depletes intracellular calcium stores and reduces calcium availability for vasoconstriction. Life Sci 155: 102–109. [DOI] [PubMed] [Google Scholar]
- DeWitt DL, Smith WL (1983). Purification of prostacyclin synthase from bovine aorta by immunoaffinity chromatography. Evidence that the enzyme is a hemoprotein. J Biol Chem 258: 3285–3293. [PubMed] [Google Scholar]
- DeWitt DL, Smith WL (1988). Primary structure of prostaglandin G/H synthase from sheep vesicular gland determined from the complementary DNA sequence. Proc Natl Acad Sci U S A 85: 1412–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan NA (2007). Balancing prostanoid activity in the human vascular system. Trends Pharmacol Sci 28: 106–110. [DOI] [PubMed] [Google Scholar]
- Funk CD, FitzGerald GA (2007). COX‐2 inhibitors and cardiovascular risk. J Cardiovasc Pharmacol 50: 470–479. [DOI] [PubMed] [Google Scholar]
- Grosser T, Theken KN, FitzGerald GA (2017). Cyclooxygenase inhibition: pain, inflammation, and the cardiovascular system. Clin Pharmacol Ther 102: 611–622. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LS, Moreno L, Reed A, Wort SJ, Desvergne B, Garland C et al (2010). The PPARbeta/delta agonist GW0742 relaxes pulmonary vessels and limits right heart hypertrophy in rats with hypoxia‐induced pulmonary hypertension. PLoS One 5: e9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RC (2006). COX‐2 and the kidney. J Cardiovasc Pharmacol 47 (Suppl 1): S37–S42. [DOI] [PubMed] [Google Scholar]
- He T, Lu T, d'Uscio LV, Lam CF, Lee HC, Katusic ZS (2008). Angiogenic function of prostacyclin biosynthesis in human endothelial progenitor cells. Circ Res 103: 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemler ME, Lands WE (1977). Biosynthesis of prostaglandins. Lipids 12: 591–595. [DOI] [PubMed] [Google Scholar]
- Hla T, Neilson K (1992). Human cyclooxygenase‐2 cDNA. Proc Natl Acad Sci U S A 89: 7384–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan KB, Evans TW, Curzen NP, Mitchell JA (1997). Evidence for a dilator function of 8‐iso prostaglandin F2 alpha in rat pulmonary artery. Br J Pharmacol 120: 1280–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent RS, Kitchell BB, Shand DG, Whorton AR (1981). The ability of vascular tissue to produce prostacyclin decreases with age. Prostaglandins 21: 483–490. [DOI] [PubMed] [Google Scholar]
- Kirkby NS, Chan MV, Lundberg MH, Massey KA, Edmands WM, MacKenzie LS et al (2013a). Aspirin‐triggered 15‐epi‐lipoxin A4 predicts cyclooxygenase‐2 in the lungs of LPS‐treated mice but not in the circulation: implications for a clinical test. FASEB J 27: 3938–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Chan MV, Zaiss AK, Garcia‐Vaz E, Jiao J, Berglund LM et al (2016). Systematic study of constitutive cyclooxygenase‐2 expression: role of NF‐kappaB and NFAT transcriptional pathways. Proc Natl Acad Sci U S A 113: 434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Jiao J, Herschman HR, Mitchell JA (2017a). Production of high levels of PGI‐M in the kidney and bladder explains the renal origin of urinary markers of prostacyclin. Circulation 136: A18556–A18556. [Google Scholar]
- Kirkby NS, Lundberg MH, Chan MV, Vojnovic I, Solomon AB, Emerson M et al (2013b). Blockade of the purinergic P2Y12 receptor greatly increases the platelet inhibitory actions of nitric oxide. Proc Natl Acad Sci U S A 110: 15782–15787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Lundberg MH, Harrington LS, Leadbeater PD, Milne GL, Potter CM et al (2012). Cyclooxygenase‐1, not cyclooxygenase‐2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci U S A 109: 17597–17602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Lundberg MH, Wright WR, Warner TD, Paul‐Clark MJ, Mitchell JA (2014). COX‐2 protects against atherosclerosis independently of local vascular prostacyclin: identification of COX‐2 associated pathways implicate Rgl1 and lymphocyte networks. PLoS One 9: e98165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Morris AP, Lytton J, Seong JK, Bae YS, Chu JA et al (2017b). Genome‐wide association study links variants with occurrence of cardiovascular events in people taking the COX‐2 inhibitor celecoxib: identification of NCKX2 as a novel protective pathway in renal vessels. Circulation Circulation 136: A15111. [Google Scholar]
- Kirkby NS, Reed DM, Edin ML, Rauzi F, Mataragka S, Vojnovic I et al (2015). Inherited human group IVA cytosolic phospholipase A2 deficiency abolishes platelet, endothelial, and leucocyte eicosanoid generation. FASEB J 29: 4568–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Sampaio W, Etelvino G, Alves DT, Anders KL, Temponi R et al (2018). Cyclooxygenase‐2 selectively controls renal blood flow through a novel PPARbeta/delta‐dependent vasodilator pathway. Hypertension 71: 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Zaiss AK, Urquhart P, Jiao J, Austin PJ, Al‐Yamani M et al (2013c). LC‐MS/MS confirms that COX‐1 drives vascular prostacyclin whilst gene expression pattern reveals non‐vascular sites of COX‐2 expression. PLoS One 8: e69524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojonazarov B, Luitel H, Sydykov A, Dahal BK, Paul‐Clark MJ, Bonvini S et al (2013). The peroxisome proliferator‐activated receptor beta/delta agonist GW0742 has direct protective effects on right heart hypertrophy. Pulm Circ 3: 926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR (1991). TIS10, a phorbol ester tumor promoter‐inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem 266: 12866–12872. [PubMed] [Google Scholar]
- Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM et al (2003). Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science 302: 453–457. [DOI] [PubMed] [Google Scholar]
- Levin RI, Weksler BB, Jaffe EA (1982). The interaction of sodium nitroprusside with human endothelial cells and platelets: nitroprusside and prostacyclin synergistically inhibit platelet function. Circulation 66: 1299–1307. [DOI] [PubMed] [Google Scholar]
- Lezoualc'h F, Fazal L, Laudette M, Conte C (2016). Cyclic AMP sensor EPAC proteins and their role in cardiovascular function and disease. Circ Res 118: 881–897. [DOI] [PubMed] [Google Scholar]
- Li Y, Connolly M, Nagaraj C, Tang B, Balint Z, Popper H et al (2012). Peroxisome proliferator‐activated receptor‐beta/delta, the acute signaling factor in prostacyclin‐induced pulmonary vasodilation. Am J Respir Cell Mol Biol 46: 372–379. [DOI] [PubMed] [Google Scholar]
- Lidbury PS, Antunes E, de Nucci G, Vane JR (1989). Interactions of iloprost and sodium nitroprusside on vascular smooth muscle and platelet aggregation. Br J Pharmacol 98: 1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Luo W, Zhang Y, Li H, Zhu N, Huang D et al (2012). Involvement of cyclo‐oxygenase‐1‐mediated prostacyclin synthesis in the vasoconstrictor activity evoked by ACh in mouse arteries. Exp Physiol 97: 277–289. [DOI] [PubMed] [Google Scholar]
- Luo W, Liu B, Zhou Y (2016). The endothelial cyclooxygenase pathway: insights from mouse arteries. Eur J Pharmacol 780: 148–158. [DOI] [PubMed] [Google Scholar]
- Macdonald PS, Read MA, Dusting GJ (1988). Synergistic inhibition of platelet aggregation by endothelium‐derived relaxing factor and prostacyclin. Thromb Res 49: 437–449. [DOI] [PubMed] [Google Scholar]
- MacDonald TM, Hawkey CJ, Ford I, McMurray JJ, Scheiman JM, Hallas J et al (2017). Randomized trial of switching from prescribed non‐selective non‐steroidal anti‐inflammatory drugs to prescribed celecoxib: the Standard care vs. Celecoxib Outcome Trial (SCOT). Eur Heart J 38: 1843–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majed BH, Khalil RA (2012). Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol Rev 64: 540–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos P, Jordan P (2015). Beyond COX‐inhibition: 'side‐effects' of ibuprofen on neoplastic development and progression. Curr Pharm Des 21: 2978–2982. [DOI] [PubMed] [Google Scholar]
- McAdam BF, Catella‐Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA (1999). Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)‐2: the human pharmacology of a selective inhibitor of COX‐2. Proc Natl Acad Sci U S A 96: 272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGettigan P, Henry D (2011). Cardiovascular risk with non‐steroidal anti‐inflammatory drugs: systematic review of population‐based controlled observational studies. PLoS Med 8: e1001098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade EA, Smith WL, DeWitt DL (1993). Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non‐steroidal anti‐inflammatory drugs. J Biol Chem 268: 6610–6614. [PubMed] [Google Scholar]
- Mitchell J, Knowles RB, Kirkby NS, Reed DM, Edin ML, White WE et al (2017). Kidney transplantation in a patient lacking cytosolic phospholipase A2 proves renal origins of urinary PGI‐M and TX‐M. Circ Res 122: 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Ahmetaj‐Shala B, Kirkby NS, Wright WR, Mackenzie LS, Reed DM et al (2014). Role of prostacyclin in pulmonary hypertension. Glob Cardiol Sci Pract 2014: 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR (1993). Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci U S A 90: 11693–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Evans TW (1998). Cyclooxygenase‐2 as a therapeutic target. Inflamm Res 47 (Suppl 2): S88–S92. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Shala F, Ahmetaj‐Shala B, Jiao J, Armstrong PC, Chan MV et al (2017). Novel tissue‐specific cyclooxygenase‐1 knockout mice demonstrate a dominant role for endothelial cyclooxygenase‐1 in prostacyclin production. Circulation Abstract 18520: http://circ.ahajournals.org/content/136/Suppl_131/A18520. [Google Scholar]
- Mitchell JA, Warner TD (2006). COX isoforms in the cardiovascular system: understanding the activities of non‐steroidal anti‐inflammatory drugs. Nat Rev Drug Discov 5: 75–86. [DOI] [PubMed] [Google Scholar]
- Mitchell J (2016). Kidney transplantation in a patient lacking cytosolic phospholipase A2 leads to urinary prostacyclin and thromboxane A2 metabolites within normal ranges. Circulation Circulation 134: A20347. [Google Scholar]
- Moncada S, Gryglewski R, Bunting S, Vane JR (1976). An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature 263: 663–665. [DOI] [PubMed] [Google Scholar]
- Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K (2011). Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog Lipid Res 50: 152–192. [DOI] [PubMed] [Google Scholar]
- Murakami M, Taketomi Y, Miki Y, Sato H, Yamamoto K, Lambeau G (2014). Emerging roles of secreted phospholipase A2 enzymes: the 3rd edition. Biochimie 107 (Pt A): 105–113. [DOI] [PubMed] [Google Scholar]
- Nakayama T (2010). Genetic polymorphisms of prostacyclin synthase gene and cardiovascular disease. Int Angiol 29: 33–42. [PubMed] [Google Scholar]
- Nissen SE, Yeomans ND, Solomon DH, Luscher TF, Libby P, Husni ME et al (2016). Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. N Engl J Med 375: 2519–2529. [DOI] [PubMed] [Google Scholar]
- Pratico D, Dogne JM (2009). Vascular biology of eicosanoids and atherogenesis. Expert Rev Cardiovasc Ther 7: 1079–1089. [DOI] [PubMed] [Google Scholar]
- Raouf J, Kirkby NS, Ahmetaj‐Shala B, Liu B, Mazi SI, Korotkova M et al (2016). MPGES‐1 deletion increases prostacyclin and evades the elevated systemic ADMA associated with COX‐2 inhibitors: relevance to cardiovascular safety of mpges‐1 inhibitors. Ann Rheum Dis 75: A11. [Google Scholar]
- Rosen GD, Birkenmeier TM, Raz A, Holtzman MJ (1989). Identification of a cyclooxygenase‐related gene and its potential role in prostaglandin formation. Biochem Biophys Res Commun 164: 1358–1365. [DOI] [PubMed] [Google Scholar]
- Scarpignato C, Lanas A, Blandizzi C, Lems WF, Hermann M, Hunt RH (2015). Safe prescribing of non‐steroidal anti‐inflammatory drugs in patients with osteoarthritis – an expert consensus addressing benefits as well as gastrointestinal and cardiovascular risks. BMC Med 13: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabb JB (2001). Physiological substrates of cAMP‐dependent protein kinase. Chem Rev 101: 2381–2411. [DOI] [PubMed] [Google Scholar]
- Sharma J, Turk J, McHowat J (2010). Endothelial cell prostaglandin I(2) and platelet‐activating factor production are markedly attenuated in the calcium‐independent phospholipase A(2)beta knockout mouse. Biochemistry 49: 5473–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P et al (2005). Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 352: 1071–1080. [DOI] [PubMed] [Google Scholar]
- Stefanini L, Bergmeier W (2016). RAP1‐GTPase signaling and platelet function. J Mol Med (Berl) 94: 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toral M, Romero M, Perez‐Vizcaino F, Duarte J, Jimenez R (2017). Antihypertensive effects of peroxisome proliferator‐activated receptor‐beta/delta activation. Am J Physiol Heart Circ Physiol 312: H189–H200. [DOI] [PubMed] [Google Scholar]
- Torres‐Quesada O, Mayrhofer JE, Stefan E (2017). The many faces of compartmentalized PKA signalosomes. Cell Signal 37: 1–11. [DOI] [PubMed] [Google Scholar]
- Trelle S, Reichenbach S, Wandel S, Hildebrand P, Tschannen B, Villiger PM et al (2011). Cardiovascular safety of non‐steroidal anti‐inflammatory drugs: network meta‐analysis. BMJ 342: c7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR, Mitchell JA, Appleton I, Tomlinson A, Bishop‐Bailey D, Croxtall J et al (1994). Inducible isoforms of cyclooxygenase and nitric‐oxide synthase in inflammation. Proc Natl Acad Sci U S A 91: 2046–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, McKnight W, Reuter BK, Vergnolle N (2000). NSAID‐induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology 119: 706–714. [DOI] [PubMed] [Google Scholar]
- Wang P, Liu Z, Chen H, Ye N, Cheng X, Zhou J (2017). Exchange proteins directly activated by cAMP (EPACs): emerging therapeutic targets. Bioorg Med Chem Lett 27: 1633–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner TD, Mitchell JA (2008). COX‐2 selectivity alone does not define the cardiovascular risks associated with non‐steroidal anti‐inflammatory drugs. Lancet 371: 270–273. [DOI] [PubMed] [Google Scholar]
- Whelton A, Hamilton CW (1991). Nonsteroidal anti‐inflammatory drugs: effects on kidney function. J Clin Pharmacol 31: 588–598. [DOI] [PubMed] [Google Scholar]
- Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL (1991). Expression of a mitogen‐responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci U S A 88: 2692–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan K, Gao LN, Cui YL, Zhang Y, Zhou X (2016). The cyclic AMP signaling pathway: exploring targets for successful drug discovery (review). Mol Med Rep 13: 3715–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama C, Yabuki T, Shimonishi M, Wada M, Hatae T, Ohkawara S et al (2002). Prostacyclin‐deficient mice develop ischemic renal disorders, including nephrosclerosis and renal infarction. Circulation 106: 2397–2403. [DOI] [PubMed] [Google Scholar]
- Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z et al (2012). Vascular COX‐2 modulates blood pressure and thrombosis in mice. Sci Transl Med 4 132ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J et al (2005). Genome‐wide analysis of cAMP‐response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A 102: 4459–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]