

Abstract
In contrast to the availability of potent and selective antagonists of several prostaglandin receptor types (including DP1, DP2, EP and TP receptors), there has been a paucity of well‐characterized, selective FP receptor antagonists. The earliest ones included dimethyl amide and dimethyl amine derivatives of PGF2α, but these have failed to gain prominence. The fluorinated PGF2α analogues, AL‐8810 and AL‐3138, were subsequently discovered as competitive and non‐competitive FP receptor antagonists respectively. Non‐prostanoid structures, such as the thiazolidinone AS604872, the D‐amino acid‐based oligopeptide PDC31 and its peptidomimic analogue PDC113.824 came next, but the latter two are allosteric inhibitors of FP receptor signalling. AL‐8810 has a sub‐micromolar in vitro potency and ≥2 log unit selectivity against most other PG receptors when tested in several cell‐ and tissue‐based functional assays. Additionally, AL‐8810 has demonstrated therapeutic efficacy as an FP receptor antagonist in animal models of stroke, traumatic brain injury, multiple sclerosis, allodynia and endometriosis. Consequently, it appears that AL‐8810 has become the FP receptor antagonist of choice.
Linked Articles
This article is part of a themed section on Eicosanoids 35 years from the 1982 Nobel: where are we now? To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.8/issuetoc
Abbreviations
- [Ca2+]i
intracellular Ca2+
- BK
bradykinin
- CSD
cortical spreading depression
- ECM
extracellular matrix
- IOP
intraocular pressure
- IPs
inositol phosphates
- KA
kainic acid
- Ki
equilibrium inhibition constant
- OHT
ocular hypertension
- PGA
PG agonist
- POAG
primary open angle glaucoma
Introduction
It is now well established that arachidonic acid (AA) formed by the action of phospholipase A2 on phospholipids is a major substrate for future conversion by lipoxygenase to create leukotrienes and for COX‐1 and COX‐2 to form PGs via PGG2 and PGH2 (Corey and Snider, 1974; Coleman et al., 1994). The five distinct bioactive lipids created by COX‐1 and COX‐2 include PGD2, PGE2, PGF2α, PGI2 and TXA2 (Coleman et al., 1994). The biological actions of these cellular eicosanoids are mediated by separate prostanoid receptors whose names originate from these PGs and are known as DP, EP, FP, IP and TP receptors respectively (Coleman et al., 1994; Alexander et al., 2017a). Some of these major receptors also have sub‐types such as EP1, EP2, EP3 and EP4 receptors. The heptahelical GPCRs associated with these PGs are embedded in the plasma membranes of the majority of the mammalian cells and transduce either an elevation (DP1, EP2, EP4 and IP receptors) or reduction (EP3‐receptors) (Narumiya et al., 1999) of cAMP. Activation of EP1, FP and TXA2 receptors by their cognate ligands results in the production of intracellular inositol phosphates (IPs) that in turn raise intracellular Ca2+ ([Ca2+]i) by releasing it from the endoplasmic reticulum of the cells (Narumiya et al., 1999). These changes in [Ca2+]i and cAMP then evoke downstream signal transduction culminating in the final biological activity of the eicosanoid (e.g. enzyme or hormone release, muscle contraction or relaxation) (Coleman et al., 1994).
Progress in the PG field accelerated when organic chemists began synthesizing not only the endogenous PGs but also analogues and derivatives with arbitrary structural modifications, all in quantities sufficient for biological testing (Corey and Snider, 1974). Additionally, widespread adoption of new physico‐chemical characterization techniques, such as nuclear magnetic resonance spectroscopy and mass spectrometry, enabled researchers to vouchsafe the compositions of what they were testing and to understand the metabolism of the PGs. Additional impetus for new discoveries in the PG arena was provided by the establishment of homogeneous cell cultures with expression of a single or multiple PG receptor sub‐type(s), with analytical technologies measuring well‐defined functional read‐outs such as second messengers (e.g. Griffin et al., 1997; 1998; 1999; Crider et al., 1998; 2000; Crider and Sharif, 2001). These isolated primary cells from normal or diseased animal and human tissues ensured a degree of fidelity and replication of normal physiological and pathological states in vitro respectively. The addition of host cells engineered to express animal or human cloned receptors, which integrated with intracellular signalling machinery, allowed high throughput screening (e.g. Rocha et al., 2016) to be undertaken to facilitate rapid discovery of agents able to activate or inhibit PG receptor‐induced signalling. This technology has been further refined to permit screening and quantification of other phenotypic changes observable in living cells in culture (e.g. cell contraction) using high‐content screening tools (Hu et al., 2017). These and other techniques and assays (e.g. Maddox et al., 1978; Stinger et al., 1982; Crider et al., 1998; Griffin et al., 1999) allowed the discovery of suitable receptor‐selective agonist and antagonists of PG receptors and their subtypes (Coleman et al., 1994; Delaey and Van de Voorde, 1995; Narumiya et al., 1999; Griffin et al., 1999; Sharif et al., 2000a, 2000b; Alexander et al., 2017a).
The first reported FP receptor antagonists
Two general properties are desired for a candidate antagonist useful for probing the role of receptor function in a biological system. The first is that the antagonist should exhibit a relatively high affinity and functional potency (IC50s/K is at least in low μM range; preferably nM). The second characteristic is at least 1‐log‐unit functional potency selectivity against other related PG receptors such that non‐specific effects at other PG receptors or other off‐targets can be avoided. This is very important because most of the endogenous PGs cross‐react with each other's receptors; for example, PGF2α has been reported as having more potent binding at the PG EP3 receptor sub‐type versus its cognate FP receptor (Sharif et al., 2003a,b,c,d).
The first purported FP receptor antagonists were the N,N‐dimethylamine and ‐amide derivatives of PGF2α, in which the C‐1 carboxylic acid has been modified to a non‐acidic function (see Table 1). These compounds were reported as competitive antagonists of the FP receptor that apparently blocked PGF2α‐induced lobar arterial pressure increase in a canine ex vivo lung preparation, with IC50 values in the low micromolar range (Fitzpatrick et al., 1978). The same group then reported on antagonism of the PGF2α‐induced contraction in the gerbil isolated colon ex vivo (Maddox et al., 1978) and rat pulmonary and systemic arterial pressure increase in vivo (Stinger et al., 1982). It should be noted that the investigators included members from Professor E.J. Corey's laboratory, who had been a key player in advancing synthetic methodology and reporting de novo synthesis of endogenous PGs and a number of analogues (Corey and Snider, 1974). This highlights the key role that synthetic discoveries played in advancing molecular level understanding of PG pharmacology. Although these N,N‐dimethylamine and ‐amide derivatives of PGF2α appeared to show promise as potential FP receptor antagonist tools, only a few reports of the use of either compound have appeared in the non‐patent literature (Arnould et al., 2001). A contributing factor may include conflicting reports of no measureable PGF2α – competitive binding (Anderson et al., 1999) or functional antagonism (Sharif et al., 2000a) at the FP receptor.
Table 1.
Some key FP receptor agonists and purported and bona fide FP receptor antagonists
| FP‐R agonist name | Structure |
|---|---|
| Cloprostenol |
|
| Fluprostenol (travoprost acid) |
|
| 16‐Phenoxy‐ω‐tetranor‐PGF2α |
|
| 17‐Phenyl‐ω‐trinor‐PGF2α (bimatoprost acid) |
|
| 13,14‐Dihydro‐17‐phenyl‐ω‐trinor‐PGF2α (latanoprost‐free acid) (PhXA85) |
|
| AFP‐172 (tafluprost acid) |
|
| AL‐12182 acid (AL‐12180) |
|
| FP receptor antagonists name | Structure |
|---|---|
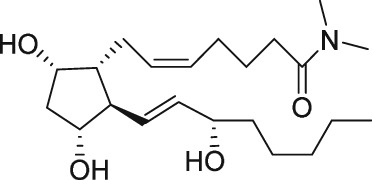
| PGF2α dimethylamide |
|
| PGF2α dimethylamine |
|
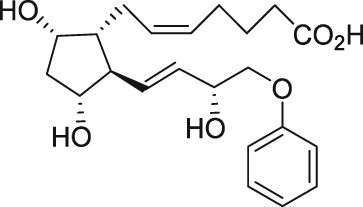
| Phloretin |
|
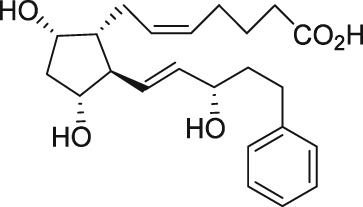
| Glibenclamide |
|
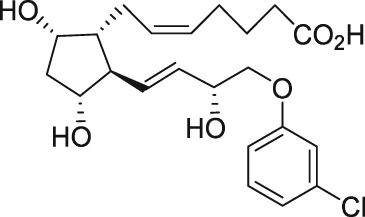
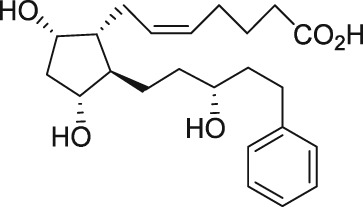
| AL‐8810 |
|
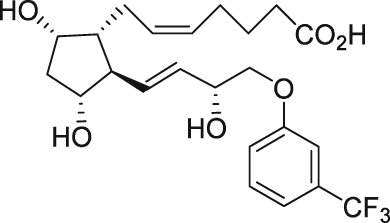
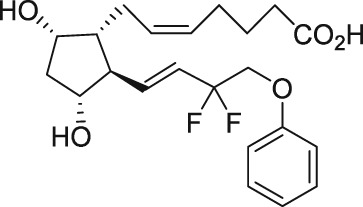
| AL‐3138 |
|
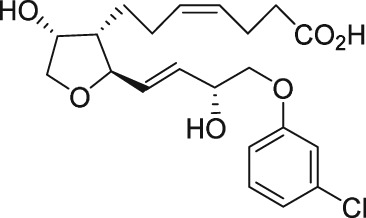
| AS604872 |
|
| THG‐113.31; apparently equiv. to PDC31; a D‐oligopeptide of sequence D‐isoleucyl‐D‐leucylglycyl‐D‐histidyl‐N5‐(aminocarbonyl)‐D‐ornithyl‐D‐aspartyl‐D‐tyrosyl‐D‐lysine |
|
| PDC113.824 |
|
| AGN 211377 |
|
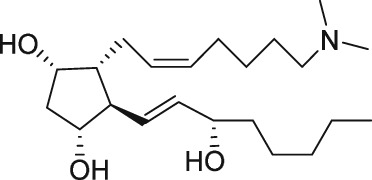
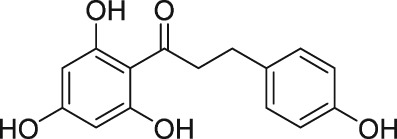
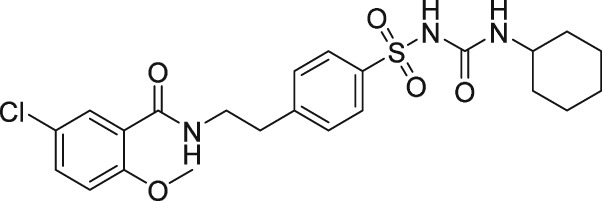
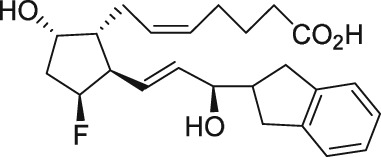
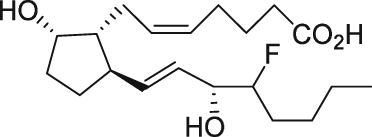
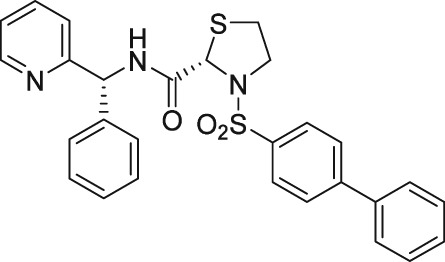
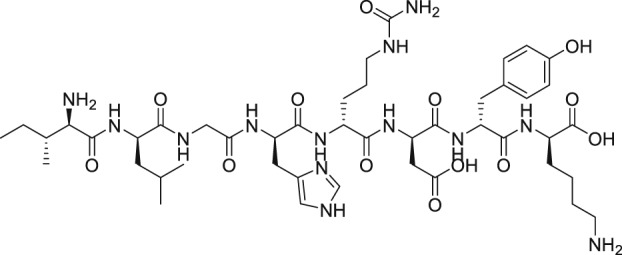
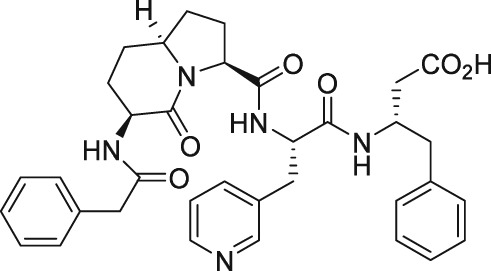
The discovery of AL‐3138 and AL‐8810 as FP receptor antagonists
Serendipity often results in new discoveries, but due vigilance and fast action on novel observations are also vital components. At the time our work in the PG arena began, we were primarily interested in finding new and improved FP‐class PG agonists for the treatment of ocular hypertension (OHT) and primary open angle glaucoma (POAG; Weinreb and Khaw, 2004). One particular relatively selective FP‐class PG analogue (FP‐PGA) agonist of PGF2α, latanoprost isopropyyl ester (Xalatan), had been discovered and reported on (Stjernschantz, 2001). Our medicinal chemists at Alcon (Discovery Research) had been synthesizing numerous analogues of PGF2α for the same purpose, and these had been screened in a battery of PGreceptor radioligand binding assays (Sharif and Davis, 2002; Sharif and Xu, 2004; Sharif et al., 1998, 1999; 2000b, 2002a, 2002b, 2003a), cell‐based functional assays (with second messenger readouts; e.g. Crider et al., 1998, 2000; Crider and Sharif, 2001; Griffin et al., 1997, 1998, 1999; Kelly et al., 2003; Sharif et al., 1998, 1999, 2000a, 2000b, 2001, 2002a, 2002b, 2002c, 2002d, 2003a, 2003b, 2003c, 2004), rat uterine (Sharif, 2008) and in feline iris sphincter muscle (Sharif et al., 2008) contraction assays. During such screening of compounds in mouse Swiss 3T3 fibroblasts (Griffin et al., 1997) and in rat aortic smooth muscle cells (A7r5; Griffin et al., 1998) in particular, it was noticed that unlike many reference FP‐PG agonists (e.g. cloprostenol and fluprostenol) that produced full functional responses akin to those of PGF2α, two newly synthesized analogues of PGF2α, AL‐3138 ((Z)‐7‐((1R,2R,5S)‐2‐((3R,E)‐4‐fluoro‐3‐hydroxyoct‐1‐en‐1‐yl)‐5‐hydroxycyclopentyl)hept‐5‐enoic acid) and AL‐8810 ((Z)‐7‐((1R,2R,3S,5S)‐2‐((R,E)‐3‐(2,3‐dihydro‐1H‐inden‐2‐yl)‐3‐hydroxyprop‐1‐en‐1‐yl)‐3‐fluoro‐5‐hydroxycyclopentyl)hept‐5‐enoic acid; Table 1) exhibited a relatively low in vitro intrinsic activity (efficacy, Emax; only 19% Emax for AL‐8810; Emax of 33–37% for AL‐3138) (Griffin et al., 1999; Sharif et al., 2000a; Figures 1A). Since these compounds did not meet the selection criteria and desired pharmacological characteristics for our OHT/POAG programme, they were removed from the screening funnels. However, having established a multitude of receptor binding assays and functional screening assays for various PG receptors and some of their sub‐types for our drug discovery research programmes (Griffin et al., 1997, 1998; Crider et al., 1998, 2000; Sharif et al., 1998, 1999, 2000a,b, 2001, 2002a,b,c,d, 2003a,b,c; Crider and Sharif, 2001; Kelly et al., 2003), the unavailability of suitable FP receptor antagonists was recalled. It was at that point that the recently ascertained pharmacological properties of AL‐8810 (Griffin et al., 1999; Sharif et al., 2000a) and AL‐3138 (Sharif et al., 2000a) became very important, since pharmacology had taught us that very low‐efficacy partial agonists could behave as ‘antagonists’ under certain experimental conditions (Stephenson, 1956; Kenakin, 1997). These compounds were quickly tested as potential antagonists. [3H]‐myo‐inositol‐loaded A7r5 cells were pretreated with different concentrations of AL‐8810 or AL‐3138 for 15 min and then exposed to a sub‐maximal concentration of a selective FP receptor full agonist, fluprostenol (100 nM), and the total water‐soluble [3H]‐IPs allowed to accumulate in the extracellular medium over an hour as a result of FP receptor activation. The total [3H]‐IPs were isolated by ion‐exchange chromatography and quantified by β‐scintillation spectroscopy (Griffin et al., 1997, 1998; Sharif et al., 1998). It became obvious that both AL‐3138 and AL‐8810 antagonized the effects of fluprostenol in a concentration‐dependent manner (Figure 1B; Griffin et al., 1999; Sharif et al., 2000a). These experiments were repeated several times, and a number of other purported FP receptor blockers were tested simultaneously. Additional experiments included testing AL‐3138 and AL‐8810 against selective agonists of other PG receptors and non‐PG receptors in order to determine their relative selectivities for the FP receptor. Both compounds were found to be relatively selective for the FP receptor (Sharif et al., 2000a, 2003a; Table 2). More detailed pharmacological studies involving the use of Schild‐analysis (Arunlakshana and Schild, 1959) were performed, and it was determined that while AL‐3138 appeared to have non‐competitive antagonist properties (Sharif et al., 2000a), AL‐8810 was a competitive antagonist (Griffin et al., 1999; Sharif et al., 2000a, 2003a) being able to produce a rightward‐shift of the concentration–response curves of FP receptor agonists without diminishing the maximal effect of the agonists (Arunlakshana and Schild, 1959; Figure 1C, D). Since AL‐8810 exhibited a lower efficacy as a partial agonist compared to AL‐3138 and was also a competitive antagonist, it was subsequently out‐licensed to Sigma/Research Biochemicals Inc. and Cayman Chemicals in order that other researchers could verify our findings and start using AL‐8810 as a pharmacological tool. Consequently, there have been over 70 reports published (per Embase/Medline/PubMed searches) in which our original observations of FP receptor antagonist properties of AL‐8810 were confirmed in a variety of in vitro and ex vivo assay systems. Furthermore, the uses of AL‐8810 have been expanded to show antagonist behaviour in animal models of various diseases and to investigate other physiological systems involving endogenous PGs or exogenously administered FP receptor agonists. Some selected key applications of AL‐8810 in biomedical research will now be discussed below.
Figure 1.
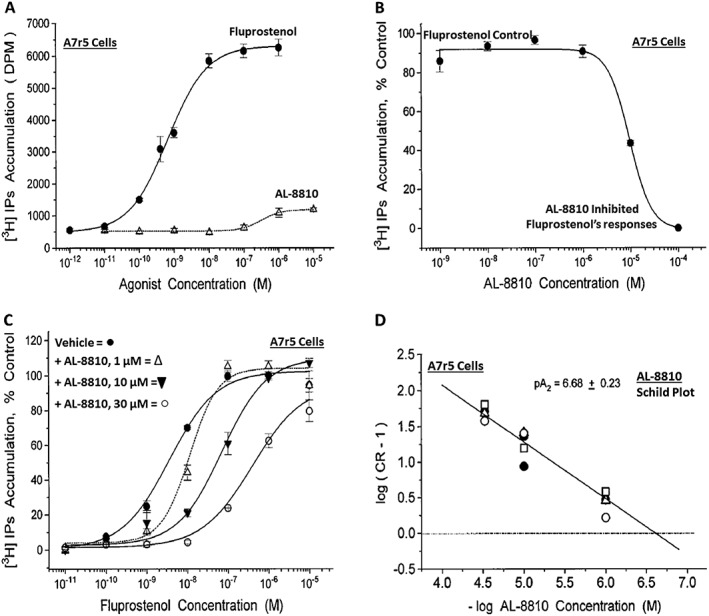
Pharmacological characterization of AL‐8810 in rat aortic smooth muscle cells (A7r5). (A) depicts the ability of AL‐8810 to stimulate the production of [3H]‐IPs as an agonist compared to the full FP receptor PG agonist, fluprostenol. It is clear that AL‐8810 is a very weak low intrinsic activity partial agonist relative to fluprostenol. (B) Shows how increasing concentrations of AL‐8810 are able to antagonize the agonist activity of fluprostenol. (C) Depicts the ability of AL‐8810 (1–30 μM) to rightward‐shift the concentration–response curves of the agonist PG, fluprostenol, in a concentration‐dependent manner, without reducing the overall maximal agonist effects of fluprostenol (Schild analysis). (D) Illustrates a Schild plot of the transformed data from (C). Here, the cumulative pA2 values obtained are shown (all modified from Griffin et al., 1999).
Table 2.
FP receptor antagonist and purported antagonist, affinity and/or functional potencies from various assay systems
| Compound | Antagonist receptor binding affinity or functional potency parameters at prostanoid receptors (IC50 or Ki or Kb, nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| DP | EP1 | EP2 | EP3 | EP4 | FP | IP | TP | |
| AL‐8810 | >30 000 | nd | >30 000 | nd | >30 000 | 285–426 (competitive) | >100 000 | >100 000 |
| AL‐3138 | >100 000 | nd | >100 000 | nd | >100 000 | 86;182–296 (non‐competitive) | >10 000 | nd |
| AS604872 | >10 000 | >10 000 | 650 | >10 000 | >10 000 | 35–323 (undefined) | >10 000 | >10 000 |
| AGN‐211377 | 49 ± 16 | 266 ± 67 | nd | 5008 ± 2571 | 117 ± 29 | 61 ± 16 (apparently non‐competitive) | nd | 11 ± 6 |
| Phloretin | ~4250 | nd | ~4229 | nd | nd | 1400–5248 (non‐competitive) | 2119 | 3383 |
| THG‐113.31 (PDC31) | – | – | – | – | – | ~30 (non‐competitive) | – | – |
AL‐8810 antagonizes in vitro ocular actions of FP receptor agonists
Ocular hypertension (OHT) and primary open angle glaucoma (POAG) adversely affect vision in millions of patients making POAG the most prevalent form of glaucoma and the second leading cause of blindness worldwide (Weinreb and Khaw, 2004; Tham et al., 2014). OHT is caused by the accumulation of excess aqueous humour (AQH) within the anterior chamber of the eye due to poor or incomplete drainage from the trabecular meshwork (TM) into the Schlemm's canal and into the episcleral veinous circulation (Ritch, 2014; Weinreb et al., 2016). A number of FP receptor agonist prodrugs including isopropyl esters of PGF2α, latanoprost‐free acid (Latanoprost; Xalatan®) (Stjernschantz, 2001), (+)‐fluprostenol (travoprost; Travatan®) (Hellberg et al., 2002), tafluprost (Taflutan®; Takagi et al., 2004) and the amide of bimatoprost‐free acid (bimatoprost; Lumigan®) (Woodward et al., 2011) (see Table 1) lower intraocular pressure (IOP) in ocular hypertensive monkeys and humans (Weinreb et al., 2016; Sharif, 2017). The PG‐like unoprostone isopropyl ester has also been introduced into clinical practice as an ocular hypotensive (Toris et al., 2004), but it has a low potency and is a medium‐level partial agonist at the FP receptor (Sharif et al., 2003a,b,c,d). The major effect of these drugs is mediated via the FP receptors located in the ciliary muscle and scleral tissues (Sharif et al., 2003a,b,c,d; Husain et al., 2005) where extracellular matrix (ECM) is digested, thereby enhancing the drainage of AQH via the uveoscleral pathway and thus lowering IOP (Weinreb et al., 2002; Weinreb and Lindsey, 2002). However, since functionally active FP receptors are located on human TM (h‐TM) cells (Sharif et al., 2003c), it has been shown that FP receptor PGAs also promote some TM outflow of the AQH (Toris et al., 2004; Lim et al., 2008). Due to their extraordinary efficacy at lowering and controlling IOP, FP receptor PGAs are now first‐line therapeutics used clinically to treat OHT and POAG (Weinreb et al., 2016; Sharif, 2017, 2018).
Even though it was shown that bimatoprost gets hydrolysed to its free acid in vitro in the presence of ocular tissue enzymes (Maxey et al., 2002; Sharif et al., 2002c; Davies et al., 2003; Hellberg et al., 2003) and also when topical ocularly applied to cause IOP lowering in animals and in humans (Camras et al., 2004, 2008; Faulkner et al., 2010), one research group has invoked an enigmatic ‘prostamide receptor’ to explain the mechanism of action of this amide compound (Woodward et al., 2001; Sharif and Klimko, 2009). Thus, it was considered important to use AL‐8810 and check whether it would interact with the FP receptor at the binding site level and then whether it would block the accumulation of [3H]‐IPs and [Ca2+]i mobilization responses induced by bimatoprost. It was shown that indeed bimatoprost (and many other FP receptor agonists) competed for specific [3H]‐PGF2α and [3H]‐AL‐5848 (travoprost acid) binding (Sharif et al., 1998, 1999) to FP receptors in homogenates of bovine corpus luteum (Sharif et al., 2001, 2003a,b,c,d). Bimatoprost, along with several other FP receptor agonists, was then shown to stimulate [Ca2+]i mobilization and activate MAPK via the FP receptor located on a variety of cell types expressing either endogenous FP receptors [human ciliary muscle (h‐CM; Sharif et al., 2003b), h‐TM cells (Sharif et al., 2003c), A7r5 rat aortic smooth muscle cells (Kelly et al., 2003) and Swiss 3T3 fibroblasts (Sharif et al., 2001; Kelly and Sharif, 2003)] or HEK‐host cells expressing the human cloned ciliary body FP receptor (Kunapuli et al., 1997; Sharif et al., 2003c,d). The inhibition of these various functional responses induced by bimatoprost and the other FP receptor agonists by AL‐8810, in an apparent agonist‐independent manner, confirmed that all these FP‐class PGs were binding to and activating the same classical FP receptor (Sharif et al., 2002a,b,c,d, 2003a,b,c,d). Thus, for instance, AL‐8810 exhibited a similar apparent FP agonist‐independent antagonist potency against the ocular FP receptor cloned from a human ciliary body library (Sharif et al., 2003c,d). The pooled antagonist potencies of AL‐8810 were: mouse 3T3 cell K i 0.2 ± 0.06 μM; rat A7r5 cell K i = 0.4 ± 0.1 μM; human cloned ciliary body‐derived FP receptor K i = 1.9 ± 0.3 M; h‐TM cell K i = 2.6 ± 0.5 μM; h‐CM cell K i = 5.7 μM; using a variety of FP agonists including fluprostenol, travoprost acid, unoprostone (free acid), bimatoprost and bimatoprost‐free acid (Figure 2).
Figure 2.
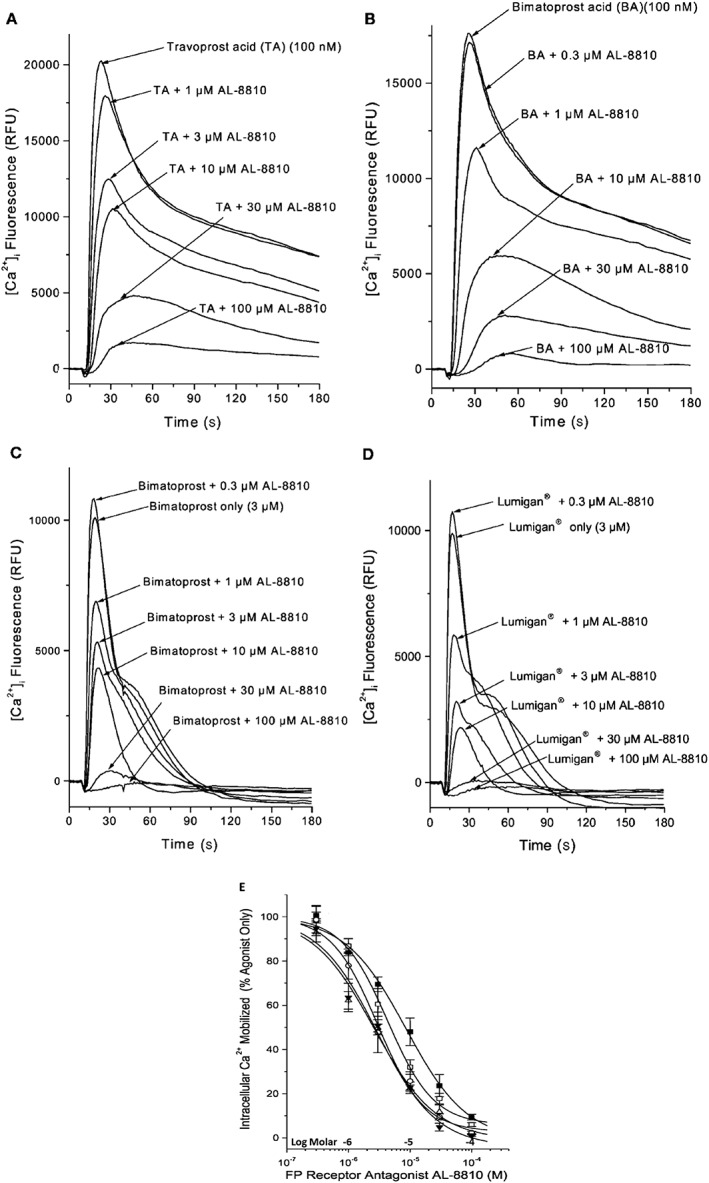
The ability of AL‐8810 to antagonize the signal transduction process in rat aortic smooth muscle A7r5 cells mediated by various FP receptor PGAs is shown. (A) and (B) Illustrate the [Ca2+]i mobilization induced by free acids of travoprost (TA) and bimatoprost (BA) in the absence and presence of increasing concentrations of the FP receptor antagonist, AL‐8810. Note the reduction in [Ca2+]i mobilization evoked by the agonists in the presence of AL‐8810. (C) and (D) Show how AL‐8810 also antagonized [Ca2+]i mobilization induced by the commercially available bimatoprost (amide) and the clinically used version of bimatoprost (Lumigan®). (E) The cumulative concentration–response curves showing the inhibitory effects of AL‐8810 on FP receptor PG agonists‐stimulated generation of [Ca2+]i (all adapted and modified from Sharif et al., 2001, 2002d, 2003a,d).
As mentioned above, it is now well accepted that the FP class of PGs exert their ocular hypotensive activity by releasing a variety of MMPs (mostly MMP‐1 (Hinz et al., 2005) and MMP‐3) that digest ECM within ciliary muscle bundles and create an enlarged uveoscleral pathway for the AQH to drain from the anterior chamber of the eye (Weinreb and Lindsey, 2002). This process is usually a slow and protracted one since the synthesis and secretion of the MMPs takes several hours (Weinreb and Lindsey, 2002). In a study to unravel the acute cellular and molecular mechanism of action of FP‐PGAs in h‐CM cells, Husain et al. (2005) showed that AL‐8810 was able to block the PGF2α‐mediated secretion of MMP‐2. These data strongly suggested that MMP‐2 probably mediates the early phase of IOP lowering via a PKC‐ and ERK‐dependent mechanism, while other MMPs (e.g. MMP‐3) are responsible for the long‐term ocular hypotensive activity of FP receptor agonists. In a similar vein, it was shown that AL‐8810 concentration‐dependently inhibited [Ca2+]i mobilization and also blocked MAPK activity stimulated by a variety of FP receptor PGAs (including bimatoprost and its free acid) in h‐CM cells (Sharif et al., 2003b). Furthermore, Romano and Lograno (2007), showed that in isolated human CM segments, the free acids of travoprost and latanoprost, as well as the intact amide compound (bimatoprost), evoked strong contractile activity. AL‐8810 competitively antagonized the contractions induced by all these PGs (Romano and Lograno, 2007), indicating once again that bimatoprost's actions are indistinguishable from other bona fide FP receptor PGAs that activate the FP receptor either as intact esters or amides or by the more active moieties produced after their hydrolysis to the free acid species. To further elaborate such findings, it was shown that, as expected, a phospholipase C inhibitor prevented contractions induced by all the latter compounds (Romano and Lograno, 2007) as well, indicating that these agents share the same downstream signal transduction mechanism known to be associated with FP receptors (Narumiya et al., 1999; Sharif et al., 2003a,b,c,d). These results further helped explain the molecular mechanisms of action of FP receptor agonists in key cell types (CM and TM) heavily involved in AQH dynamics and ultimately in reducing IOP in vivo.
One possible pathological event leading to OHT/POAG, and perhaps other forms of glaucoma, is the slow but progressive ill‐health and subsequent loss of phagocytic/autophagic activity of TM cells located within the AQH drainage pathway of the anterior chamber of the eye (Weinreb et al., 2014). Oxidative stress caused by local ischaemia/hypoxia leading to a decline in ATP, an energy source, in the TM cells, and in other important cell types within the visual axis [e.g. retinal ganglion cells (RGCs)], could be responsible for causing the eventual demise of TM and RGCs (Thomas et al., 2000; Weinreb et al., 2014; Sharif, 2017, 2018). Thus, it was of some significance that Yu et al. (2009) demonstrated a protective effect of various FP receptor PGAs in isolated human TM cells subjected to oxidative stress via exposure to hydrogen peroxide. Importantly, they showed that AL‐8810 and two other purported FP receptor antagonists (PGF2α dimethyl amine and PGF2α dimethyl amide) reversed such protection afforded by travoprost, latanoprost and bimatoprost (Yu et al., 2009), thereby strongly supporting the involvement of FP receptors in mediating these effects of FP‐PGAs (Sharif et al., 2003c). Once again, these data underscored the probability that bimatoprost behaves just like other FP receptor agonists. These in vitro data were further supported by the observations that whilst all the FP receptor PGA prodrugs tested (e.g. travoprost and bimatoprost) reduced IOP in wild‐type mice, this ocular hypotensive activity was absent in mice where the FP receptor had been genetically deleted (Crowston et al., 2005; Ota et al., 2005).
Functionally active FP receptors were identified and pharmacologically characterized in h‐TM cells using a variety of FP receptor agonists (Sharif et al., 2003c). Significantly, it was demonstrated that AL‐8810 concentration‐dependently antagonized the actions of these PG agonists, including those of bimatoprost and its free acid (Sharif et al., 2003c). Additional studies revealed that both free acids of latanoprost and travoprost increased h‐TM cell membrane potential which could also be blocked by AL‐8810 (Cuppoletti et al., 2007). Likewise, other researchers showed that cultured h‐TM cells respond to PGF2α and fluprostenol by suppressing endothelin‐induced [Ca2+]i mobilization and contraction (Thieme et al., 2006), activities that were sensitive to the FP receptor antagonists, AL‐8810 and PGF2α dimethyl amide. The overall significance of the latter studies extended to the hypothesis that perhaps one way FP receptor PGAs exert their therapeutic effects in OHT/POAG is by suppressing the deleterious actions of locally produced endothelin, a peptide that is elevated in glaucomatous situations in animal models of OHT and in OHT/POAG patients (Choritz et al., 2012).
A subpopulation of myofibroblastic cells in TM impart important contractile property that is important for cell volume changes (Dismuke et al., 2009), the release of local MMPs (Yang et al., 2016) and for affecting closely associated Schlemm's canal cells, which enhance conventional outflow of AQH to reduce IOP. In studying the cellular actions of FP receptor and EP2 receptor agonists in h‐TM cells, Kalouche et al. (2016) showed that latanoprost‐free acid prevented collagen accumulation and contracted TM cells, both processes being antagonized by AL‐8810. In contrast, the EP2 receptor agonist, butaprost, relaxed TM cells and reduced collagen deposition induced by TGFβ2. These actions of latanoprost‐free acid in h‐TM cells appeared to involve the induction of the accumulation of calcipressin, a regulator of calcineurin, by utilization of both intracellular and extracellular Ca2+, an action that was also strongly blocked by AL‐8810 (Fautsch et al., 2011).
The immediate‐early gene Nur77 (also called NGFI‐B, TR3 or NR4A1) encodes an orphan nuclear receptor that is up‐regulated by growth factors and which is involved in cell differentiation and proliferation and perhaps in cellular protection. PGF2α strongly induced Nur77 expression in h‐CM and h‐TM cells, indicating that endogenous PGs acting on the FP receptor impart a beneficial effect to maintain the health of these cell types. The up‐regulation of Nur77 induced by PGF2α in these cells was effectively inhibited by AL‐8810 (Liang et al., 2004).
Pathological neovascularization underlies the action of several types of prostanoids in causing ocular diseases. Indeed, it has been shown that PGF2α is elevated in retinal Muller glial cells and microvascular cells subjected to disease‐relevant stimuli such as ischaemia/hypoxia as in oxygen‐induced retinopathy of prematurity (Barnett et al., 2010; Hu et al., 2017). Accordingly, Savage et al. (2011) showed that latanoprost induced a significant elevation in the secretion of VEGF from isolated Muller cells and increased proliferation of human retinal microvascular endothelial cells that could be blocked by AL‐8810. These data suggested that FP receptor antagonists may have clinical utility as anti‐angiogenic agents, but such findings need additional corroboration and extension.
Numerous PGs modulate contractility of smooth muscle of various ocular tissues (CM, TM and iris sphincter muscle), blood vessels, pulmonary tissues and mammalian uterus (Woodward et al., 2011). AL‐8810 has proven useful in delineating the PG receptors involved in these effects of PGs. Thus, latanoprost, travoprost and AL‐12182‐free acid (AL‐12180; Table 1) potently contracted isolated porcine ciliary arteries, and these effects were effectively antagonized by AL‐8810 (Sharif et al., 2006; Vysniauskiene et al., 2006). Holmgaard and Bek (2010) demonstrated a similar FP receptor inhibition of PGF2α‐induced contraction by AL‐8810 in porcine retinal arterioles mounted in organ baths. In a contrasting study, hypoxia‐induced relaxation of the same porcine tissues was shown to be mediated by nitric oxide and EP4 receptors (but not FP receptors) since GW627368 (an EP4 antagonist) blocked the contraction but AL‐8810 did not (Hansen et al., 2015).
Non‐ocular in vitro utility of the FP receptor antagonist, AL‐8810
The involvement of PGs in cardiac tissue and coronary arteries function/dysfunction is well documented (Zhang et al., 2010). In an additional study, Zhang et al. (2017) recently tried to define the PG receptors involved in the secretion of atrial natriuretic peptide (Bai et al., 2009) and the spontaneous contraction of this cardiac tissue in isolated perfused rat atria. Since both these activities were blocked by perfusion of AH‐6809 (a DP receptor antagonist; Coleman et al., 1994) and AL‐8810 into the atria, they concluded that endogenously released PGD2 and PGF2α were most likely involved. Likewise, since cardiac fibrosis is characterized by collagen I and III deposition in the myocardium, Ding et al. (2012) recently demonstrated that AL‐8810 could prevent this feature of PGF2α and thus suggested a potential clinical utility of AL‐8810 in preventing cardiac fibrosis. This was important since the fibrosis induced by activation of the FP receptor was independent of the fibrotic cytokine, TGFβ1 (Ding et al., 2012). Moreover, Hara et al. (2009) showed that the muscarinic receptor‐mediated positive ionotropic response of mouse isolated left atrium was actually mediated by endogenously released PGs, including PGF2α, since amongst other PG receptor antagonists, AL‐8810 significantly blunted the response to carbachol. In another related study, it was shown that while PGF2α significantly decreased the chronotropic response to cholinergic stimulation of isolated atria from endotoxin‐treated rats (Nikoui et al., 2015), AL‐8810 or indomethacin was able to counteract and reverse this phenomenon.
As mentioned above, a variety of PGs influence venous and arterial blood vessel contractility and/or relaxation in the ocular system. In endotoxin‐treated porcine coronary arteries, the bradykinin (BK) B1 receptor agonist, des‐Arg9‐BK, caused endothelium‐independent contractions of the tissue that were inhibited by COX‐2 inhibitors and a TP receptor antagonist but neither by a COX‐1 inhibitor (aspirin) nor by DP/EP (AH‐6809)‐, FP (AL‐8810)‐ and IP (RO1138452) receptor antagonists (More et al., 2014a,b). However, since AL‐8810 potently and competitively antagonized the contractile effects of free acids of latanoprost, bimatoprost and PGF2α in isolated rings of human umbilical vein, the involvement of FP receptors in causing these responses was confirmed (Errasti et al., 2009). Likewise, AL‐8810 proved useful in ascribing an FP receptor‐mediated vasoconstrictor effect of endogenously released PGF2α in freshly regenerated rat femoral arteries in a rat model of atherosclerosis (Hirao et al., 2008).
The pulmonary system is also a target of endogenous PGs under normal and disease conditions. Airways smooth muscle of various species is contracted by growth factors such as EGF, platelet‐derived growth factor and insulin (Schaafsma et al., 2005, 2007), isoprostanes (Paredes et al., 2007), thromboxanes (Hernandez and Janssen, 2011) and PGE2 (Säfholm et al., 2015). In the bovine trachealis, it appears that E‐ring isoprostanes augment the cholinergic neurotransmission via an FP receptor‐mediated mechanism since AL‐8810 significantly inhibited the response evoked by 15‐E2t–isoprostane (Paredes et al., 2007). Similarly, since guinea pig airway smooth muscle contractions induced by insulin were abolished by AL‐8810, the authors concluded that endogenously released PGF2α plays a major role in airways constriction (Schaafsma et al., 2007). However, in the other aforementioned studies (Hernandez and Janssen, 2011; Säfholm et al., 2015), AL‐8810 proved helpful in ruling out the involvement of FP receptors.
Due to the ubiquity of endogenous PGs in the mammalian body, these agents have numerous other functions that can be classified as physiological or pathological depending on the nature of the tissue and the prevailing circumstances. In many instances, it is either PGE2 and/or PGF2α that are the benefactors or culprits. Isolated uterus strips from mice responded strongly to exogenously added PGF2α and 17‐phenyl‐PGF2α (bimatoprost‐free acid) (Hutchinson et al., 2003). The contractile activity of both FP receptor agonists was competitively antagonized by AL‐8810 since it produced rightward shifts of the concentration–response curves to both agonists (Hutchinson et al., 2003). Basal tone recordings of rat uterus and full contractions of this tissue can also be achieved using a variety of FP receptor agonists (e.g. PGF2α, latanoprost‐free acid and cloprostenol) (Sharif, 2008). The rat uterus appeared to be uniquely sensitive to these PGs, since fine gradations of both potency and efficacy of FP receptor agonists was observed and their agonist actions fully antagonized by AL‐8810 (Sharif, 2008).
In concordance with the heterogeneous distribution of various PG receptors in the gastrointestinal tract, PGE2, PGF2α and TXA2 enhance chloride secretion in human and mouse colon in vitro (see Collins et al., 2009). However, while PGD2 stimulates chloride release in the guinea pig, it inhibits this function in the rat colon. PGF2α also stimulates chloride secretion in porcine small intestine and increases rat, guinea pig and human colonic smooth muscle contractility (see Collins et al., 2009). The effects of PGF2α on chloride secretion were mediated in a cAMP‐dependent fashion in isolated human colon, and AL‐8810 effectively antagonized these effects (IC50 = 190 nM) (Collins et al., 2009). Since PGF2α concentration is raised in Crohn's disease, and FP receptor levels are strikingly elevated in colorectal carcinoma tumors (see Collins et al., 2009), the pharmacological and clinical utility of FP receptor antagonists to combat or down‐regulate these intestinal disorders appears warranted.
The discovery of the phenomenon of receptor dimerization and crosstalk between heterologous receptors has opened potential new avenues for novel drug discovery and treatment of diseases (Rozenfeld and Devi, 2010). Such allosteric functional interactions amongst FP receptors and angiotensin‐II (AT) receptors (AT1 sub‐type) by formation of receptor dimers have been recently described (Goupil et al., 2013, 2015). Here, AL‐8810 (FP receptor antagonist) and AS604872 (allosteric inhibitor; see above) produced trans‐inhibition of IPs production in A7r5 cells and antagonized mouse aortic contraction induced by either angiontensin II or PGF2α (Goupil et al., 2015). Thus, AL‐8810 was able to apparently enter the FP receptor binding pocket to block the activity of the FP receptor and the co‐joined AT1 receptor within the heterodimer (Sleno et al., 2017).
Utility and therapeutic effects of AL‐8810 in animal models of disease
As is often the case, in vitro observations of compound activity is not always replicated in animal models in vivo. This is not necessarily unexpected since compounds can be easily pronounced inactive or non‐efficacious due to limitations of adequately delivering the agents to the target tissues/cells at an effective concentration. Such problems are further exacerbated by potential rapid enzymatic degradation of the compound administered or by sequestration by non‐target cells by active uptake mechanisms and/or potential extrusion of the compound from the target or adjacent cells by P‐glycoprotein multidrug transporter proteins. Nevertheless, it is imperative that a potential drug candidate exhibits the necessary pharmacological and potentially therapeutic effect in animal models of disease for which the compound has been synthesized. Accordingly, it would appear that AL‐8810 has met such expectations in a number of in vivo studies that will be discussed below.
An area where AL‐8810, as an FP receptor antagonist, has made a significant impact is in the CNS‐related studies. Mechanical allodynia induced by intrathecal administration of PGF2α and ATP is mediated via a capsaicin‐insensitive primary afferent pathway within the spinal cord (Kunori et al., 2009; Suzuki‐yamamoto et al., 2009). In FP receptor knockout mice, neither αβ‐methylene ATP nor PGF2α elicited allodynia. However, since AL‐8810 blocked the pain response induced by intrathecal αβ‐methylene ATP in naive wild‐type mice, the authors concluded that activation of the P2X2/3 receptors to cause allodynia is ultimately transduced via the FP receptors that are co‐located with P2X2/3 receptors in the spinal cord (Kunori et al., 2009). Extending these observations, Gatta et al. (2012) showed that direct spinal administration of PGF2α into healthy mice excited nociceptive neurons and this activity was also abolished by AL‐8810.
Brain damage caused by ischaemic insults, such as during stroke, and by traumatic/mechanical injury are debilitating disorders of the brain. In a mouse model of stroke, induced by permanent middle cerebral artery occlusion, pretreatment with i.v. AL‐8810 significantly reduced the cerebral cortical infarct volume (Figure 3A–C), thereby providing protection against this ischaemic insult (Kim et al., 2012). Commensurate with this structural protection, mice also displayed a much greater recovery of behavioural responses as a functional outcome. Both the cortical protection and functional results were confirmed in mice whose FP receptors had been knocked‐out (Figure 3B). Interestingly, these authors also demonstrated these neuroprotective effects of AL‐8810 in vitro using mouse hippocampal slices and cultured hippocampal neurons that were subjected to an oxygen–glucose‐deprivation paradigm (Kim et al., 2012). Mechanistically, AL‐8810 reduced the production of reactive oxygen species and abolished the pathological elevation of excess [Ca2+]i in cortical neurons exposed to the latter ischaemic challenge (Kim et al., 2012) (Figure 3B).
Figure 3.
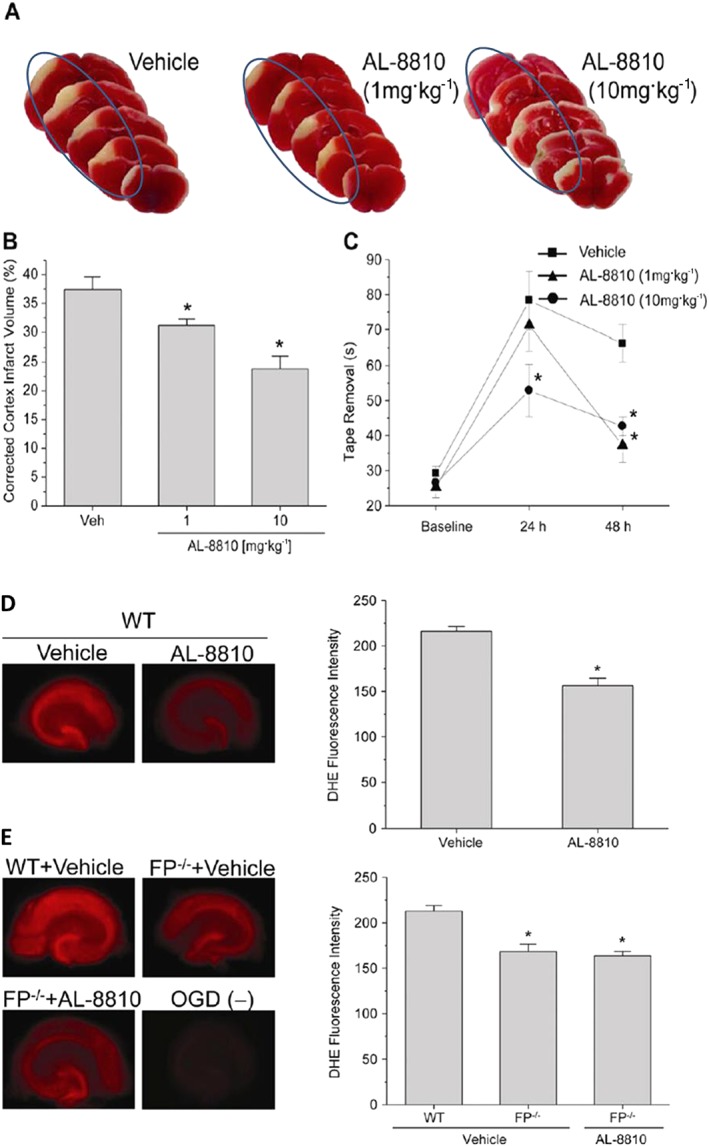
Protective effects of AL‐8810 in experimental brain traumatic injury‐induced changes in mouse brain cortex and in isolated hippocampal slices subjected to oxygen‐glucose‐deprivation (OGD). (A–C) Depict how systemically dosed AL‐8810 (1 and 10 mg·kg−1) was able to reduce the cortical infarct size relative to that of vehicle‐treated mice in an animal model of stroke (A and B). A corresponding faster recovery from neurological dysfunctions was observed in mice treated with AL‐8810 (reduced time for mice to remove a sticky tape from their paws; AL‐8810‐treated versus vehicle‐treated mice; *P < 0.05; n = 10–13 mice). (D) Illustrates how AL‐8810 (10 μM) was able to reduce the formation of ROS and thus protect mouse hippocampal slices [obtained from wild‐type (WT) mice] from the detrimental effects of OGD in vitro. As can be seen in (E), OGD‐induced ROS formation was significantly lower in hippocampal slices from FP−/− mice than in those obtained from WT mice. AL‐8810 had no additional effect on slices from FP−/− mice. *P < 0.05 (n = 20–22 slices) (all figures were adapted and modified from Kim et al., 2012).
It is well known that one major cause of brain damage and ensuing death, especially in elderly people who tend to fall more easily and injure themselves, is traumatic brain injury (TBI). This is characterized by an initial contusion, haematoma, subarachnoid haemorrhage and diffuse injury to the axons of brain neurons resulting in subsequent neuronal demise. The secondary phase of TBI is neuronal inflammation/oedema coupled with oxidative stress and Ca2+ overloading due to excitotoxicity and death of neurons. Endogenous production and release of pro‐inflammatory PGs during and after TBI has been supported by observations of elevated AA release and enhanced COX‐2 activity (Glushakov et al., 2013a,b). Furthermore, elevated COX‐2 has been reported in ischaemic neonatal and adult human brain, and in an experimental mouse models of TBI, Glushakov et al. (2013a,b) showed that a post‐TBI i.p. injection of AL‐8810 significantly reduced hippocampal swelling and improved neurological deficit scores 1 and 2 days post‐insult (Figure 4A, B). They concluded that pharmacological blockade of the FP receptor may represent a novel means to blunt and lower the risk of brain damage after acute physical TBI.
Figure 4.
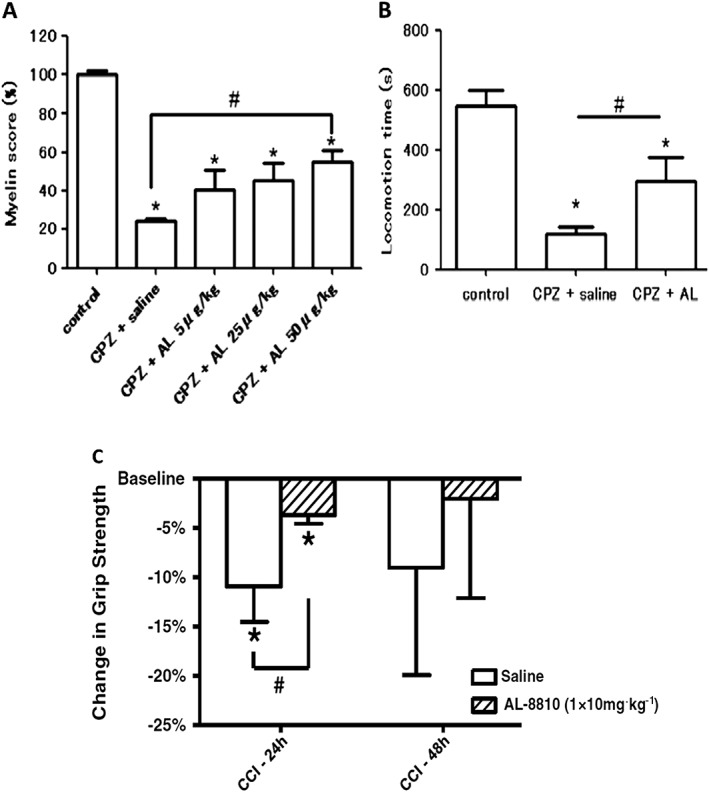
Protective effects of AL‐8810 in an animal model of MS and in a mouse model of TBI. (A) and (B) demonstrate how AL‐8810 (AL) was able to reduce the loss of myelin (A) and thus help retain motor function (B) in a cuprizone (CPZ)‐induced mouse model of MS; (*P < 0.05 vs. control; #P < 0.05 vs. CPZ + saline; modified from Iwasa et al., 2014). (C) Shows the protective effects of AL‐8810 in a controlled cortical impact (CCI) traumatic injury model in mice. Note the improvement of grip strength in mice who received AL‐8810 versus the control saline group at 24 h post CCI (*P < 0.05 vs. control group; modified from Glushakov et al., 2013a,b).
PGF2α can be formed by constitutively expressed PGF synthase in the myelin sheath and by isolated cultured oligodendrocytes, and it is believed that inflammation of the myelin sheath of nerves involves an up‐regulation of PGF2α production. This information indicates PGs are involved in the demyelination process and thus in the aetiology of multiple sclerosis (MS) (Iwasa et al., 2014). In support of this hypothesis, in a rodent model of MS induced by dietary intake of cuprizone, i.c.v. administration of AL‐8810 attenuated cytokine expression in the corpus callosum, prevented brain glial activation, attenuated the demyelination process and helped improve the motor function of the mice (Iwasa et al., 2014) (Figure 4). Therefore, pharmacological antagonism of the FP receptor appears beneficial for reducing the pathological signs and symptoms associated with experimentally‐induced MS. Whether these observations can be translated to the human MS patients remains to be determined, but FP receptor antagonists such as AL‐8810 hold some promise in this regard.
Reduced cerebral blood (oligaemia) can be caused by ischaemic events due to a variety of vascular abnormalities including atherosclerosis, vasospasm and aberrant vasoconstriction. Such persistent cortical oligaemia has been implicated in the development of cortical spreading depression (CSD) in humans. In an experimental model of CSD using urethane‐anaesthetized rats, Gariepy et al. (2017) demonstrated that long‐lasting oligaemia consists of two phases; the initial phase involves activation of COX‐1 and induction of TXA2, while the later phase is mediated by PGF2α derived from stimulation of COX‐2 activity. In correspondence with these results, AL‐8810 was able to prevent only the second phase of oligaemia and thus enhance cerebral blood flow. These findings provide an insight into possible treatment of CSD induced by pathologically reduced brain blood flow and suggest that in order to overcome the majority of the blood‐flow deficit in CSD patients, a combination therapy approach using TXA2 and FP receptor antagonists may be necessary.
Early studies focused on epileptogenic aspects of endogenous and exogenously administered PGs and COX‐1 and COX‐2 inhibitors in relation to kainic acid (KA)‐induced seizures. Inhibitors of PG synthesis or AL‐8810 administered intracisternally (i.c.) prior to induction of seizures exacerbated the condition of these animals by potentiating the seizure activity (Kim et al., 2008). However, i.c. injection of PGF2α, but not PGD2 or PGE2, alleviated KA‐induced seizures, thereby suggesting the involvement of FP receptors in the beneficial effects of endogenously released FP‐PGAs in combating epilepsy. However, such findings need to be confirmed by other researchers.
There is a high degree of complexity surrounding the aetiology and progression of endometriosis, an oestrogen‐driven disease. One important recent finding concerns the up‐regulation of PGF2α synthesizing enzymes in eutopic and ectopic endometria of women suffering from endometriosis (Ahmad et al., 2015). Following up on these observations, the same authors developed a heterologous animal model of endometriosis by implanting human endometrial tissue into the peritoneal cavity of nude mice. Ahmad et al. (2015) subsequently showed that administration of AL‐8810 resulted in a marked reduction in the number and size of the endometriotic lesions by inhibiting biomarkers of inflammation, cell proliferation, angiogenesis and tissue remodelling and by readjusting the levels of pro‐ and anti‐apoptotic factors. Interestingly, in a more detailed study using the same animal model as above, Ahmad et al. (2015) demonstrated that an exogenously administered FP receptor agonist, fluprostenol, elevated all the aforementioned biomarkers. In contrast, AL‐8810 caused a highly statistically significant reduction of secreted MMP‐9 and VEGF and reduced the levels of cell proliferation and capillary formation nuclear antigen and vonWillebrand factor immunoreactivity in the endometriotic tissue (Ahmad et al., 2015). These various beneficial effects of AL‐8810 are summarized in Table 3.
Table 3.
Utility and therapeutic actions of AL‐8810 in various animal models of disease
| Disease/disorder | Naive animal studies or experimentally‐induced disorder/disease | Therapeutic effect of AL‐8810 | Reference |
|---|---|---|---|
| Allodynia (Pain) | Spinal injection of PGF2α caused nociceptive neuronal excitation in mice. | AL‐8810 blocked the pain‐producing signalling of PGF2α. | Gatta et al., 2012 |
| Allodynia was induced in mice by ATP or PGF2α. | AL‐8810 abolished the pain induced by both agents. | Kunori et al., 2009 | |
| Stroke | Middle cerebral artery occlusion in mouse model of stroke (cerebral ischaemia). | AL‐8810 significantly reduced the cortical infarct volume and accelerated the recovery of behavioural function. | Kim et al., 2012 |
| Mouse hippocampal neurons and slices subjected to oxygen–glucose‐deprivation (hypoxia/aglycaemia). | AL‐8810 inhibited the generation of free radicals (reactive oxygen species) and prevented excess [Ca2+]i. | Kim et al., 2012 | |
| TBI | Controlled mechanical brain injury was induced in mice that caused contusion, subarachnoid haemorrhage and diffuse brain axonal injuries, followed by neuronal inflammation/oedema, oxidative stress and neuronal excitotoxicity. Elevated COX‐2 activity and production of PGs was observed. | I.p. administration of AL‐8810 (post‐brain injury) decreased hippocampal swelling and accelerated neurological and behavioural recovery in mice. | Glushakov et al., 2013a,b |
| Ischaemic neonatal and adult human brains display the same features as above. | Mechanistically, AL‐8810 blunted pro‐inflammatory effects of released PGs and prevented Ca2+‐overloading and death of neurons/dendrites and axons. | Glushakov et al., 2013a,b | |
| Oligaemia (reduced cerebral blood) | Aberrant cerebro‐vasoconstriction, vasospasm and atherosclerosis can reduce the amount of blood reaching the cerebral cortex. This causes ischaemia and results in CSD in humans. In a rat model of CSD, oligaemia was induced that exhibited two‐phases. Phase‐1 involved COX‐1 activation with TXA2 being released, while phase‐2 was mediated by released PGF2α. | AL‐8810 potently and effectively prevented the development of 2nd phase of oligaemia and thus increased cerebral blood‐flow thereby reducing the structural and functional cerebral damage. | Gariepy et al., 2017 |
| MS | Dietary intake of cuprizone, a copper chelator, in mice produced signs and symptoms akin to human MS pathology. | I.c.v. injection of AL‐8810 abolished brain glial activation and cytokine production, reduced demyelination, and accelerated recovery of motor function in the mice. | Iwasa et al., 2014 |
| Endometriosis | PGF2α is involved in eutopic and ectopic endometriosis in women that causes cramping and pain. Samples of human diseased endometrial tissue implanted in peritoneal cavity of nude mice capitulated the human disease (blood and cells accumulating in pelvic cavity and causing inflammation, swelling, pain and tissue scaring). | AL‐8810 administration to the mice (with the implanted human diseased endometrial tissue) significantly decreased the number and size of the lesions by inhibiting inflammation, cell proliferation / angiogenesis, and by also reducing pro‐apoptotic factors. | Ahmad et al., 2015 |
| Exogenous administration of an FP agonist, fluprostenol, exacerbated the situation in the above mouse model. | AL‐8810 reduced levels of secreted MMP‐9 and VEGF (pro‐angiogenic factors), with resultant decreased cell proliferation and capillary formation. | Ahmad et al., 2015 |
Utility of other FP receptor antagonists in biological studies
An antioxidant and anti‐inflammatory flavonoid compound, phloretin (de Oliveira, 2016), was historically dubbed as an FP receptor antagonist (Kitanaka et al., 1993). Likewise, the sulfonylurea anti‐diabetic agent, glibenclamide, was also claimed to block PG‐induced tissue contractions via the FP receptor (Delaey and Van de Voorde, 1995). However, even though glibenclamide and tolbutamide competed for [3H]‐PGF2α binding to FP receptors and they exhibited very low affinity, phloretin inexplicably potentiated the binding of this radioligand (Sharif et al., 2000a). All these latter compounds very weakly inhibited the generation of second messengers induced by PGF2α in rat aortic smooth muscle cells but were also weak antagonists at DP, EP and vasopressin receptors (Sharif et al., 2000a). Thus, there appears to be little evidence supporting the true FP receptor antagonist properties of phloretin and glibenclamide.
There have been a few other attempts to discover FP receptor antagonists beyond AL‐8810. Such discovery efforts led to the identification of a non‐PG small molecule (AS604872) that had selective activity against the FP receptor (Chollet et al., 2007; Cirillo et al., 2007; Fukuda et al., 2014). These authors showed that AS604872 binds well to the FP receptor (K i = 35–323 nM), a little less weakly to EP2 receptors (K i = 650 nM) and much less weakly to the other PG receptors (K i > 10 μM) (Cirillo et al., 2007; Table 2) rendering it selective for the FP receptor. In functional studies, AS604872 inhibited PGF2α‐induced [3H]‐IPs production and rat uterine contraction in vivo after i.v. infusion into pregnant female rats near term (Chollet et al., 2007; Cirillo et al., 2007). Despite its apparent high potency and receptor selectivity, it is curious that AS604872 has not been utilized as much as AL‐8810. One possible reason could be that it is not commercially available. However, a more pertinent issue could be the side effects associated with AS604872. One such example concerns the exacerbation of intracranial aneurysm and aortic lesions observed in hypertensive rats that received AS604872 (Fukuda et al., 2014). In rats rendered hypertensive by salt‐loading, an infusion of AS604872 significantly accelerated the degeneration of the aorta and cerebral artery due to an up‐regulation of genes for pro‐inflammatory mediators and due to increased infiltration of macrophages (Fukuda et al., 2014). It would have been much more informative if the authors had also tested AL‐8810 in this rat hypertension model in order to determine if the deleterious effects of AS604872 were a compound‐specific effect or a class effect of FP receptor antagonists.
As mentioned above, endogenous PGF2α contracts myometrium and is heavily involved in labour and birth in mammals (Deaver et al., 2015). During a search for agents that would prevent pre‐term labour, Peri et al. (2002) found an octapeptide ligand (THG113; Ile‐Leu‐Gly‐His‐Arg‐Asp‐Tyr‐Lys) that apparently interacted with FP receptors (Table 2) and which inhibited the generation of intracellular IPs in cells expressing FP receptors (IC50 = 27 nM). However, this blockade of the FP receptor seems to have been not directly at the FP receptor binding site since THG113 failed to displace [3H]‐PGF2α binding from human‐cloned FP receptors expressed in HEK‐293 cells (Peri et al., 2002). As far as it can be determined, a full receptor binding or functional profile of this compound against other PG receptors was not performed. When tested against PGF2α‐induced contractions of retinal microvascular tissue, THG113 behaved as a non‐competitive antagonist, but it did not affect EP1 or TP receptor‐mediated responses. Contractions of murine and porcine myometrial strips isolated from non‐pregnant animals were largely blocked by THG113, and it delayed preterm delivery of fetuses of pregnant mice when an endotoxin was used to induce preterm labour (Peri et al., 2002; Hirst et al., 2005). However, the exact nature of the inhibition of exogenous and endogenous PGF2α‐induced functional activities by THG113 remains undefined but appears to be beyond the extracellular components of the FP receptor protein. Nevertheless, perhaps in an attempt to enhance the FP receptor antagonist potency and/or selectivity of the parent peptide, various derivatives of THG113 were synthesized (Chemtob and Peri, 2006; Peri et al., 2006). However, another THG113‐derived peptidic agent (THG‐113.31) exhibited minimal affinity for the FP receptor PGF2α binding pocket, like the parent molecule (Chemtob and Peri, 2006; Peri et al., 2006). THG113.31 insurmountably antagonized the contraction of pig retinal blood vessels evoked by PGF2α but lacked the ability to block PGF2α‐induced contractions of isolated pregnant human myometrium; and it only weakly inhibited sheep myometrial contractile activity caused by PGF2α (Friel et al., 2005). Curiously, THG113.31 (also apparently known as PDC31; Böttcher et al., 2014) exerted a relaxant effect on both oxytocin‐evoked and spontaneous contractions of human myometrial tissue in vitro, indicating the action of this compound is non‐selective (Friel et al., 2005). However, Doheny et al. (2007) demonstrated that THG113.31 apparently increased the opening of a Ca2+‐dependent large‐conductance K+‐channel in human myometrial cells and caused the subsequent relaxation of this tissue. Despite the uncertainty of the exact mechanism of action of PDC31 (THG113.31), its incomplete pharmacological profile and the potential for significant side effects it could cause, the effects of i.v. administration of the latter compound were tested in a small clinical trial. Böttcher et al. (2014) showed that PDC31was capable of decreasing the strength and duration of uterine contractions in vivo and that it reduced the pain associated with excessive uterine contractility in women suffering from this disorder. As far as we know, no follow‐up studies with this compound have been reported to‐date. However, a small peptide based on the N‐terminal region of the second extracellular loop of the FP receptor (PDC113.824) appeared to antagonize smooth muscle contractions elicited by PGF2α by an undefined mechanism (Peri et al., 2006). Regardless, Goupil et al. (2010) proceeded to show that PDC113.824 behaves as an allosteric inhibitor that blocks the PGF2α‐induced Gα12‐dependent Rho kinase/ROCK signalling pathway and, in this manner, inhibited both endotoxin‐ and PGF2α‐stimulated preterm labour and delayed normal parturition in mice. Further studies with these peptide and allied peptidomimetics are required to ascertain their future utility as tools for studying the actions of FP receptor‐mediated functions/dysfunctions.
A small molecule compound (AGN211377) capable of binding to the FP receptor (K i = 61 nM) and suppressing the [Ca2+]i‐mobilization elicited by 17‐phenyl‐PGF2α via the human cloned FP receptor was recently described (Wang et al., 2016; Tables 1 and 2). Unfortunately, this agent was not selective for the FP receptor since it potently and efficaciously inhibited the signal transduction and functional responses mediated by TP, DP1, EP4, EP1 and DP2 receptor agonists in a variety of cells in culture and in animal studies (Wang et al., 2016). Perhaps analogues of this compound may be more FP receptor‐selective, and such synthetic and pharmacological studies are much warranted.
Conclusions
The authors hope that the present discourse on the discovery, characterization and utility of purported and bona fide FP receptor antagonists, albeit of low affinity and potency, spurs on and inspires others to seek and find better next‐generation antagonists. Such novel compounds are eagerly awaited.
Nomenclature of targets and ligands
Key proteins targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c).
Conflict of interest
The authors declare no conflicts of interest. They only wished to gather and summarize pertinent public domain information on the subject matter and to disseminate this to the scientific community, thereby promoting scientific exchange. The ultimate aim is to help foster and enhance further biomedical research on the role of PGs in health and disease and to find suitable remedies for the diseases discussed in this review.
Sharif, N. A. , and Klimko, P. G. (2019) Prostaglandin FP receptor antagonists: discovery, pharmacological characterization and therapeutic utility. British Journal of Pharmacology, 176: 1059–1078. 10.1111/bph.14335.
References
- Ahmad SF, Akoum A, Horne AW (2015). Selective modulation of the prostaglandin F2α pathway markedly impacts on endometriosis progression in a xenograft mouse model. Mol Hum Reprod 21: 905–916. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al, (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kellyr E, Marrionr NV, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LE, Schultz MK, Wiltbank MC (1999). PG moieties that determine receptor binding specificity in the bovine corpus luteum. J Reprod Fert 116: 133–144. [DOI] [PubMed] [Google Scholar]
- Arnould T, Thibaut‐Vercruyssen R, Bouaziz N, Dieu M, Remacle J, Michiels C (2001). PGF(2alpha), a prostanoid released by endothelial cells activated by hypoxia, is a chemoattractant candidate for neutrophil recruitment. Am J Pathol 159: 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO (1959). Some quantitative use of drug antagonists. Br J Pharmacol Chemother 257: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Gao S, Shah A, Yuan K, Park WH, Kim SH (2009). Regulation of ANP secretion from isolated atria by prostaglandins and cyclooxygenase‐2. Peptides 30: 1720–1728. [DOI] [PubMed] [Google Scholar]
- Barnett JM, McCollum GW, Penn JS (2010). Role of cytosolic phospholipase A(2) in retinal neovascularization. Invest Ophthalmol Vis Sci 51: 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher B, Laterza RM, Wildt L, Seufert RJ, Buhling KJ, Singer CF et al (2014). A first‐in‐human study of PDC31 (prostaglandin F2α receptor inhibitor) in primary dysmenorrhea. Hum Reprod 29: 2465–2473. [DOI] [PubMed] [Google Scholar]
- Camras CB, Sharif NA, Wax MB, Stjernshantz J (2008). Bimatoprost, the prodrug of a prostaglandin analogue. Br J Ophthalmol 92: 862–863. [PubMed] [Google Scholar]
- Camras CB, Toris CB, Sjoquist B, Milleson M, Thorngren JO, Hejkal TW et al (2004). Detection of the free acid of bimatoprost in aqueous humor samples from human eyes treated with bimatoprost before cataract surgery. Ophthalmology 111: 2193–2198. [DOI] [PubMed] [Google Scholar]
- Chemtob S, Peri AG (2006). Peptide antagonists of prostaglandin F2α receptor. US Patent # 6,984,719.
- Chollet A, Tos EG, Cirillo R (2007). Tocolytic effect of a selective FP receptor antagonist in rodent models reveals an innovative approach to the treatment of preterm labor. BMC Preg Childbirth 7 (Suppl 1): S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo R, Tos EG, Page P, Missotten M, Quattropani A, Scheer A et al (2007). Arrest of preterm labor in rat and mouse by an oral and selective nonprostanoid antagonist of the prostaglandin F2α receptor (FP). Am J Obstet Gynecol 197: e1–e9. [DOI] [PubMed] [Google Scholar]
- Choritz L, Machert M, Thieme H (2012). Correlation of endothelin‐1 concentration in aqueous humor with intraocular pressure in primary open angle and pseudoexfoliation glaucoma. Invest Ophthalmol Vis Sci 53: 7336–7342. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S (1994). International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev 46: 205–229. [PubMed] [Google Scholar]
- Collins D, Hogan AM, Skelly MM, Baird AW, Winter DC (2009). Cyclic AMP‐mediated chloride secretion is induced by prostaglandin F2α in human isolated colon. Br J Pharmacol 158: 1771–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey EJ, Snider BB (1974). Preparation of an optically active intermediate for the synthesis of PGs. Tetrahed Lett 39: 256–258. [DOI] [PubMed] [Google Scholar]
- Crider JY, Xu SX, Griffin BW, Sharif NA (1998). Use of a semi‐automated, robotic radioimmunoassay to measure cAMP generated by activation of DP‐, EP2‐ and IP‐prostaglandin receptors in human ocular and other cell‐types. Prostagland Leukotri Essent Fatty Acids 59: 77–82. [DOI] [PubMed] [Google Scholar]
- Crider JY, Griffin BW, Sharif NA (2000). Endogenous EP4 prostaglandin receptors coupled positively to adenylyl cyclase in Chinese hamster ovary cells: pharmacological characterization. Prostaglandins Leukot Essent Fatty Acids 62: 21–26. [DOI] [PubMed] [Google Scholar]
- Crider JY, Sharif NA (2001). Functional pharmacological evidence for EP2 and EP4 prostanoid receptors in immortalized human trabecular meshwork and N‐PCE cells. J Ocular Pharmacol Ther 17: 35–46. [DOI] [PubMed] [Google Scholar]
- Crowston JG, Lindsey JD, Morris CA, Wheeler L, Medeiros FA, Weinreb RN (2005). Effect of bimatoprost on intraocular pressure in prostaglandin FP receptor knockout mice. Invest Ophthalmol Vis Sci 46: 4571–4577. [DOI] [PubMed] [Google Scholar]
- Cuppoletti J, Malinowska DH, Tewari KP, Chakrabarti J, Ueno R (2007). Cellular and molecular effects of unoprostone as a BK channel activator. Biochim Biophys Acta 1768: 1083–1092. [DOI] [PubMed] [Google Scholar]
- Davies SS, Ju WK, Neufeld AH, Abram D, Chemtob S, Robert LJ II (2003). Hydrolysis of bimatoprost (Lumigan) to its free acid by ocular tissue in vitro. J Ocul Pharmacol Ther 19: 45–54. [DOI] [PubMed] [Google Scholar]
- de Oliveira MR (2016). Phloretin‐induced cytoprotective effects on mammalian cells: a mechanistic view and future directions. Biofactors 42: 13–40. [DOI] [PubMed] [Google Scholar]
- Deaver SE, Felix AM, Rhoads ML (2015). Reproductive performance of lactating dairy cattle after intrauterine administration of a prostaglandin F2α receptor antagonist 4 days after insemination. Theriogenology 83: 560–566. [DOI] [PubMed] [Google Scholar]
- Delaey C, Van de Voorde J (1995). Prostanoid‐induced contractions are blocked by sulfonylureas. Eur J Pharmacol 280: 179–184. [DOI] [PubMed] [Google Scholar]
- Ding WY, Ti Y, Wang J, Wang ZH, Xie GL, Shang YY et al (2012). Prostaglandin F2α facilitates collagen synthesis in cardiac fibroblasts via an F‐prostanoid receptor/protein kinase C/Rho kinase pathway independent of transforming growth factor β1. Int J Biochem Cell Biol 44: 1031–1039. [DOI] [PubMed] [Google Scholar]
- Dismuke WM, Sharif NA, Ellis DZ (2009). Human trabecular meshwork cell volume decrease by NO‐independent soluble guanylate cyclase activators YC‐1 and BAY‐58‐2667 involves the BKCa ion channel. Invest Ophthalmol Vis Sci 50: 3353–3359. [DOI] [PubMed] [Google Scholar]
- Doheny HC, O'Reilly MJ, Sexton DJ, Morrison JJ (2007). THG113.31, a specific PGF2alpha receptor antagonist, induces human myometrial relaxation and BKCa channel activation. Reprod Biol Endocrinol 5: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errasti AE, del‐Rey G, Cesio CE, Souza G, Nowak W, Pelorosso FG et al (2009). Expression and functional evidence of the prostaglandin F2α receptor mediating contraction in human umbilical vein. Eur J Pharmacol 610: 68–74. [DOI] [PubMed] [Google Scholar]
- Faulkner R, Sharif NA, Orr S, Sall K, Dubiner H, Whitson JT et al (2010). Aqueous humor concentrations of bimatoprost free acid, bimatoprost and travoprost free acid in cataract surgical patients administered multiple topical ocular doses of LUMIGAN® or TRAVATAN®. J Ocular Pharmacol Ther 26: 147–156. [DOI] [PubMed] [Google Scholar]
- Fautsch MP, Bahler CK, Resch ZT (2011). Latanoprost‐mediated calcipressin accumulation in human trabecular meshwork cells occurs through calcium signaling pathways. Invest Ophthalmol Vis Sci 52: 4638. [Google Scholar]
- Fitzpatrick TM, Alte I, Corey EJ, Ramwell PW, Rose JC, Kot PA (1978). Antagonism of the pulmonary vasoconstrictor response to PGF2α by N‐dimethylamino substitution of PGF2α . J Pharmacol Expt Ther 206: 139–142. [PubMed] [Google Scholar]
- Friel AM, O'Reilly MW, Sexton DJ, Morrison JJ (2005). Specific PGF2α receptor (FP) antagonism and human uterine contractility in vitro. Br J Obstr Gynocol 112: 1034–1042. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Aoki T, Manabe T, Maekawa A, Shirakawa T, Kataoka H et al (2014). Exacerbation of intracranial aneurysm and aortic dissection in hypertensive rat treated with the prostaglandin F–receptor antagonist AS604872. J Pharmacol Sci 126: 230–242. [DOI] [PubMed] [Google Scholar]
- Gariepy H, Zhao J, Levy D (2017). Differential contribution of COX‐1 and COX‐2 derived prostanoids to cortical spreading depression‐Evoked cerebral oligemia. J Cereb Blood Flow Metab 37: 1060–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta L, Piscitelli F, Giordano C, Boccella S, Lichtman A, Maione S et al (2012). Discovery of prostamide f2a and its role in inflammatory pain and dorsal horn nociceptive neuron hyperexcitability. PLoS One 7: e31111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glushakov AV, Robbins SW, Bracy CL, Narumiya S, Doré S (2013a). Prostaglandin F2α FP receptor antagonist improves outcomes after experimental traumatic brain injury. J Neuroinflammation 10: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glushakov AV, Robbins SW, Bracy CL, Narumiya S, Doré S (2013b). Prostaglandin F2α FP receptor antagonist improves outcomes after experimental traumatic brain injury. J Neurotrauma 30: 15 (A163). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goupil E, Tassy D, Bourguet C, Quiniou C, Wisehart V, Pétrin D et al (2010). A novel biased allosteric compound inhibitor of parturition selectively impedes the prostaglandin F2α‐mediated Rho/ROCK signaling pathway. J Biol Chem 285: 25624–25636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goupil E, Laporte SA, Hébert TE (2013). GPCR heterodimers: asymmetries in ligand binding and signaling output offer new targets for drug discovery. Br J Pharmacol 168: 1101–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goupil E, Fillion D, Clément S, Luo X, Devost D, Sleno R et al (2015). Angiotensin II type 1 and prostaglandin F2α receptors cooperatively modulate signaling in vascular smooth cells. J Biol Chem 290: 3137–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin BW, Williams GW, Crider JY, Sharif NA (1997). FP prostaglandin receptors mediating inositol phosphates generation and calcium mobilization in Swiss 3T3 Cells: a pharmacological study. J Pharmacol Exp Ther 281: 845–854. [PubMed] [Google Scholar]
- Griffin BW, Magnino P, Pang I‐H, Sharif NA (1998). Pharmacological characterization of an FP prostaglandin receptor on rat vascular smooth muscle cells (A7r5) coupled to phosphoinositide turnover and intracellular calcium mobilization. J Pharmacol Exp Ther 286: 411–418. [PubMed] [Google Scholar]
- Griffin BW, Klimko P, Crider JY, Sharif NA (1999). AL‐8810: a novel PGF2α analog with selective antagonist effects at the FP prostaglandin receptor. J Pharmacol Exp Ther 290: 1278–1284. [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen PO, Kringelholt S, Simonden U, Bek T (2015). Hypoxia‐induced relaxation of porcine retinal arterioles in vitro depends on inducible NO synthase and EP4 receptor stimulation in the perivascular retina. Acta Ophthalmol 93: 457–463. [DOI] [PubMed] [Google Scholar]
- Hara Y, Ike A, Tanida R, Okada M, Yamawaki H (2009). Involvement of cyclooxygenase‐2 in carbachol‐induced positive inotropic response in mouse isolated left atrium. J Pharmacol Exp Ther 331: 808–815. [DOI] [PubMed] [Google Scholar]
- Hellberg MR, McLaughlin MA, Sharif NA, DeSantis L, Dean TR, Kyba EP et al (2002). Identification and characterization of the ocular hypotensive efficacy of Travoprost, a potent and selective FP prostaglandin receptor agonist, and AL‐6598, a DP prostaglandin receptor agonist. Surv Ophthalmol 47 (Suppl #1): S13–S33. [DOI] [PubMed] [Google Scholar]
- Hellberg MR, Ke T‐L, Haggard K, Klimko PG, Dean TR, Graff G (2003). The hydrolysis of the prostaglandin analog prodrug bimatoprost to 17‐phenyl‐trinor PGF2α by human and rabbit ocular tissue. J Ocul Pharmacol Ther 19: 97–103. [DOI] [PubMed] [Google Scholar]
- Hernandez JM, Janssen LJ (2011). Thromboxane prostanoid receptor activation amplifies airway stretch‐activated contractions assessed in perfused intact bovine bronchial segments. J Pharmacol Exp Ther 339: 248–256. [DOI] [PubMed] [Google Scholar]
- Hinz B, Rösch S, Ramer R, Tamm ER, Brune K (2005). Latanoprost induces matrix metalloproteinase‐1 expression in human nonpigmented ciliary epithelial cells through a cyclooxygenase‐2‐dependent mechanism. FASEB J 19: 1929–1931. [DOI] [PubMed] [Google Scholar]
- Hirao A, Kondo K, Takeuchi K, Inui N, Umemura K, Ohashi K et al (2008). Cyclooxygenase‐dependent vasoconstricting factor(s) in remodelled rat femoral arteries. Cardiovasc Res 79: 161–168. [DOI] [PubMed] [Google Scholar]
- Hirst JJ, Parkington HC, Young IR, Palliser HK, Peri KG, Olson DM (2005). Delay of preterm birth in sheep by THG113.31, a prostaglandin F2α receptor antagonist. Am J Obstet Gynecol 193: 256–266. [DOI] [PubMed] [Google Scholar]
- Holmgaard K, Bek T (2010). ATP‐induced relaxation of porcine retinal arterioles in vitro depends on prostaglandin E synthesized in the perivascular retinal tissue. Invest Ophthalmol Vis Sci 51: 5168–5175. [DOI] [PubMed] [Google Scholar]
- Hu J, Geyer A, Dziumbla S, Awwad K, Zeldin DC, Schunck WH et al (2017). Role of Müller cell cytochrome P450 2c44 in murine retinal angiogenesis. Prostagland Other Lipid Mediat 133: 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain S, Jafri F, Crosson CE (2005). Acute effects of PGF2α on MMP‐2 secretion from human ciliary muscle cells: a PKC‐ and ERK‐dependent process. Invest Ophthalmol Vis Sci 46: 1706–1713. [DOI] [PubMed] [Google Scholar]
- Hutchinson J, Marshall KM, Senior J (2003). Preliminary studies using a putative FP‐receptor antagonist, Al‐8810, on isolated mouse uterus. Proceed Br Pharmacol Soc pA21: Abst 38P.
- Iwasa K, Yamamoto S, Takahashi M, Suzuki S, Yagishita S, Awaji T et al (2014). Prostaglandin F2α FP receptor inhibitor reduces demyelination and motor dysfunction in a cuprizone‐induced multiple sclerosis mouse model. Prostaglandins Leukot Essent Fatty Acids 91: 175–182. [DOI] [PubMed] [Google Scholar]
- Kalouche G, Beguier F, Bakria M, Melik‐Parsadaniantz S, Leriche C, Debeir T et al (2016). Activation of prostaglandin FP and EP2 receptors differently modulates myofibroblast transition in a model of adult primary human trabecular meshwork cells. Invest Ophthalmol Vis Sci 57: 1816–1825. [DOI] [PubMed] [Google Scholar]
- Kelly CR, Williams GW, Sharif NA (2003). Real‐time intracellular Ca2+‐mobilization by travoprost acid, bimatoprost, unoprostone and other analogs via endogenous mouse, rat and cloned human FP prostaglandin receptors. J Pharmacol Exp Ther 304: 238–245. [DOI] [PubMed] [Google Scholar]
- Kenakin TP (1997). The Pharmacologic Analysis of Drug Receptor Interaction, 3rd edn. Lippincott‐Raven: New York, pp. 1–491. [Google Scholar]
- Kim HJ, Chung JI, Lee SH, Jung YS, Moon CH, Baik EJ (2008). Involvement of endogenous prostaglandin F2alpha on kainic acid‐induced seizure activity through FP receptor: the mechanism of proconvulsant effects of COX‐2 inhibitors. Brain Res 1193: 153–161. [DOI] [PubMed] [Google Scholar]
- Kim YT, Moon SK, Maruyama T, Narumiya S, Doré S (2012). Prostaglandin FP receptor inhibitor reduces ischemic brain damage and neurotoxicity. Neurobiol Dis 48: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitanaka J, Ishibashi T, Baba A (1993). Phloretin as an antagonist of prostaglandin F2α receptor in cultured rat astrocytes. J Neurochem 60: 704–708. [DOI] [PubMed] [Google Scholar]
- Kunapuli P, Lawson JA, Rokach J, FitzGerald GA (1997). Functional characterization of the ocular prostaglandin F2alpha (PGF2α) receptor. Activation by the isoprostane, 12‐iso‐PGF2α . J Biol Chem 272: 27147–27154. [DOI] [PubMed] [Google Scholar]
- Kunori S, Matsumura S, Mabuchi T, Tatsumi S, Sugimoto Y, Minami T et al (2009). Involvement of prostaglandin F2α receptor in ATP‐induced mechanical allodynia. Neuroscience 163: 362–371. [DOI] [PubMed] [Google Scholar]
- Liang Y, Li C, Guzman VM, Chang WW, Evinger AJ, Pablo JV et al (2004). Upregulation of orphan nuclear receptor Nur77 following PGF(2alpha), Bimatoprost, and Butaprost treatments. Essential role of a protein kinase C pathway involved in EP(2) receptor activated Nur77 gene transcription. Br J Pharmacol 142: 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KS, Nau CB, O'Byrne MM, Hodge DO, Toris CB, McLaren JW et al (2008). Mechanism of action of bimatoprost, latanoprost, and travoprost in healthy subjects. A crossover study. Ophthalmology 115: 790–795.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox YT, Ramwell PW, Shiner CS, Corey EJ (1978). Amide and i‐amino derivatives of F PGs as PG antagonists. Nature 273: 549–552. [DOI] [PubMed] [Google Scholar]
- Maxey KM, Johnson JL, LaBreque J (2002). The hydrolysis of bimatoprost in corneal tissue generates a potent prostanoid FP receptor agonist. Surv Ophthalmol 47 (suppl 1): 534–540. [DOI] [PubMed] [Google Scholar]
- More AS, Kim HM, Khang G, Hildebrandt T, Bernlöhr C, Doods H et al (2014a). Des‐Arg9‐bradykinin causes kinin B1 receptor mediated endothelium‐independent contractions in endotoxin‐treated porcine coronary arteries. Pharmacol Res 90: 18–24. [DOI] [PubMed] [Google Scholar]
- More AS, Kim HM, Zhao R, Khang G, Hildebrandt T, Bernlöhr C et al (2014b). COX‐2 mediated induction of endothelium‐independent contraction to bradykinin in endotoxin‐treated porcine coronary artery. J Cardiovasc Pharmacol 64: 209–217. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F (1999). Prostanoid receptors: structures, properties, and functions. Physiol Rev 79: 1193–1226. [DOI] [PubMed] [Google Scholar]
- Nikoui V, Ejtemaei Mehr S, Jazaeri F, Ostadhadi S, Eftekhari G, Dehpour AR et al (2015). Prostaglandin F2α modulates atrial chronotropic hyporesponsiveness to cholinergic stimulation in endotoxemic rats. Eur J Pharmacol 748: 149–156. [DOI] [PubMed] [Google Scholar]
- Ota T, Aihara M, Narumiya S, Araie M (2005). The effects of prostaglandin analogues on IOP in prostanoid FP‐receptor‐deficient mice. Invest Ophthalmol Vis Sci 46: 4159–4163. [DOI] [PubMed] [Google Scholar]
- Paredes C, Tazzeo T, Janssen LJ (2007). E‐ring isoprostane augments cholinergic neurotransmission in bovine trachealis via FP prostanoid receptors. Am J Respir Cell Mol Biol 37: 739–747. [DOI] [PubMed] [Google Scholar]
- Peri KG, Quiniou IC, Hou X, Abran D, Varma DR, Lubell WD et al (2002). THG 113: a novel selective FP antagonist that delays preterm labor. Semin Perinatol 26: 389–397. [DOI] [PubMed] [Google Scholar]
- Peri K, Polyak F, Lubell W, Thouin E, Chemtob S (2006). Peptides and peptidomimetics useful for inhibiting the activity of prostaglandin F2α receptor. US Patent Application 20060211626 A1.
- Ritch R (2014). Ocular and systemic manifestations of exfoliation syndrome. J Glaucoma 23 (8 Suppl 1): S1–S8. [DOI] [PubMed] [Google Scholar]
- Rocha DN, Carvalho ED, Pêgo AP (2016). High‐throughput platforms for the screening of new therapeutic targets for neurodegenerative diseases. Drug Discov Today 21: 1355–1366. [DOI] [PubMed] [Google Scholar]
- Romano MR, Lograno MD (2007). Evidence for the involvement of cannabinoid CB1 receptors in the bimatoprost‐induced contractions on the human isolated ciliary muscle. Invest Ophthalmol Vis Sci 48: 3677–3682. [DOI] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA (2010). Receptor heteromerization and drug discovery. Trends Pharmacol Sci 31: 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Säfholm J, Manson ML, Bood J, Delin I, Orre AC, Bergman P et al (2015). Prostaglandin E2 inhibits mast cell‐dependent bronchoconstriction in human small airways through the E prostanoid subtype 2 receptor. J Allergy Clin Immunol 136: 1232–1239. [DOI] [PubMed] [Google Scholar]
- Savage SR, Yang R, Yanni SE, Penn JS (2011). The PGF2α FP receptor mediates retinal angiogenic cell behaviors in vitro. Invest Ophthalmol Vis Sci 52: 4860. [Google Scholar]
- Schaafsma D, Gosens R, Bos IS, Meurs H, Zaagsma J, Nelemans SA (2005). Role of contractile prostaglandins and Rho‐kinase in growth factor‐induced airway smooth muscle contraction. Respir Res 6: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaafsma D, Gosens R, Ris JM, Zaagsma J, Meurs H, Nelemans SA (2007). Insulin induces airway smooth muscle contraction. Br J Pharmacol 150: 136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif NA, Davis TL (2002). Cloned human EP1 prostanoid receptor pharmacology characterized using radioligand binding techniques. J Pharm Pharmacol 54: 539–547. [DOI] [PubMed] [Google Scholar]
- Sharif NA (2017). Ocular hypertension and glaucoma: a review and current perspectives. Int J Ophthalmol Vis Sci 2: 22–36. [Google Scholar]
- Sharif NA (2008). Synthetic FP‐class prostaglandin‐induced contraction of rat uterus smooth muscle in vitro . Prostagland Leukotri Essent Fatty Acids 78: 199–207. [DOI] [PubMed] [Google Scholar]
- Sharif NA (2018). idrug and idevices discovery and development – preclinical assays, techniques and animal models studies for ocular hypotensives and neuroprotectants. J Ocular Pharmacol Ther 34: 7–39. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Xu SX (2004). Pharmacological characterization and identification of EP3 prostanoid receptor binding sites in hamster uterus homogenates. J Pharm Pharmacol 56: 197–203. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Klimko P (2009). Update and commentary on the prodrug bimatoprost and a putative prostamide receptor. Expert Rev Ophthalmol 4: 477–489. [Google Scholar]
- Sharif NA, Williams GW, Xu SX, Crider JY, Griffin BW, Davis TL (1998). Pharmacological analysis of [3H]PGE1 / [3H]PGE2 and [3H]PGF2α binding in bovine corpus luteum: identification of EP3 and FP prostaglandin receptors and correlation with functional data. J Pharmacol Exp Ther 286: 1094–1102. [PubMed] [Google Scholar]
- Sharif NA, Davis TL, Williams GW (1999). [3H]AL‐5848 (9‐β‐[+]fluprostenol): carboxylic acid of Travoprost (AL‐6221), a novel FP‐prostaglandin to study the pharmacology and autoradiographic localization of the FP receptor. J Pharm Pharmacol 51: 685–594. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Crider JY, Davis TL (2000a). AL‐3138 antagonizes FP prostanoid receptor‐mediated inositol phosphates generation: comparison with some purported FP antagonists. J Pharm Pharmacol 52: 1529–1539. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Crider JY, Xu SX, Williams GW (2000b). Affinities, selectivities, potencies and intrinsic activities of natural and synthetic prostanoids using endogenous receptors: focus on DP class prostanoids. J Pharmacol Exp Ther 293: 321–328. [PubMed] [Google Scholar]
- Sharif NA, Williams GW, Kelly CR (2001). Bimatoprost and its free acid are prostaglandin FP receptor agonists. Eur J Pharmacol 432: 211–213. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Williams GW, Davis TL (2002a). Pharmacology and autoradiography of human DP prostanoid receptors using [3H]‐BWA868C, a DP receptor‐selective antagonist radioligand. Br J Pharmacol 131: 1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif NA, Ke T, Haggard K, Kelly C, Williams G, Hellberg M et al (2002b). Bimatoprost hydrolysis to 17‐phenyl‐PGF2α by human and rabbit ocular tissues and agonist activity of bimatoprost and 17‐phenyl‐PGF2α . Invest Ophthalmol Vis Sci 43 Abst # 4080. [Google Scholar]
- Sharif NA, Senchyna M, Xu SX (2002c). Pharmacological and molecular biological (RT‐PCR) characterization of functional TP prostanoid receptors in immortalized human non‐pigmented ciliary epithelial cells. J Ocular Pharmacol Ther 18: 141–162. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Kelly CR, Crider JY (2002d). Agonist activity of bimatoprost, travoprost, latanoprost, unoprostone isopropyl ester and other prostaglandin analogs at the cloned human ciliary body FP prostaglandin receptor. J Ocular Pharmacol Ther 18: 313–324. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Kelly CR, Crider JY, Williams GW, Xu SX (2003a). Ocular hypotensive FP prostaglandin (PG) analogs: PG receptor subtype binding affinities and selectivities, and agonist potencies at FP and other PG receptors in cultured cells. J Ocular Pharmacol Ther 19: 501–515. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Crider JY, Husain S, Kaddour‐Djebbar I, Ansari HR, Abdel‐Latif AA (2003b). Human ciliary muscle responses to FP‐class prostaglandin analogs: phosphoinositide hydrolysis, intracellular Ca2+ mobilization and MAP kinase activation. J Ocular Pharmacol Ther 19: 437–455. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Kelly CR, Crider JY (2003c). Human trabecular meshwork cell responses induced by bimatoprost, travoprost, unoprostone, and other FP prostaglandin receptor agonist analogues. Invest Ophthalmol Vis Sci 44: 715–721. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Kelly CR, Williams GW (2003d). Bimatoprost (Lumigan®) is an agonist at the cloned human ocular FP receptor: real‐time FLIPR‐based intracellular Ca2+ mobilization studies. Prostagland Leukotri Essent Fatty Acids 68: 27–33. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Williams GW, Crider JY, Xu SX, Davis TL (2004). Molecular pharmacology of the ocular hypotensive DP/EP2 class prostaglandin AL‐6598 and localization of DP and EP2 receptor sites in human eyes. J Ocular Pharmacol Ther 20: 489–508. [DOI] [PubMed] [Google Scholar]
- Sharif NA, McLaughlin MA, Kelly CR, Xu SX, Crider JY, Parker J (2006). Preclinical pharmacology of AL‐12182, a new ocular hypotensive 11‐oxa‐prostaglandin analog. J Ocular Pharmacol Ther 22: 291–309. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Kaddour‐Djebbar I, Abdel‐Latif AA (2008). Cat iris sphincter smooth muscle contraction: comparison of FP‐class prostaglandin analog agonist activities. J Ocular Pharmacol Ther 24: 152–163. [DOI] [PubMed] [Google Scholar]
- Sleno R, Devost D, Pétrin D, Zhang A, Bourque K, Shinjo Y et al (2017). Conformational biosensors reveal allosteric interactions between heterodimeric AT1 angiotensin and prostaglandin F2α receptors. J Biol Chem 292: 12139–12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson RP (1956). A modification of receptor theory. Br J Pharmacol 11: 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinger RB, Fitzpatrick TM, Corey EJ, Ramwell PW, Rose JC, Kot PA (1982). Selective antagonism of PGF2α‐mediated vascular responses by N‐dimethylamino substitution of PGF2α . J Pharmacol Expt Ther 220: 521–525. [PubMed] [Google Scholar]
- Stjernschantz JW (2001). From PGF2α‐isopropyl ester to latanoprost: a review of the development of Xalatan®: the Proctor Lecture. Invest Ophthalmol Vis Sci 42: 1134–1145. [PubMed] [Google Scholar]
- Suzuki‐Yamamoto T, Toida K, Sugimoto Y, Ishimura K (2009). Colocalization of prostaglandin F2α receptor FP and prostaglandin F synthase‐I in the spinal cord. J Lipid Res 50: 1996–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi Y, Nakajima T, Shimazaki A, Kageyama M, Matsugi T (2004). Pharmacological characteristics of AF‐168 (tafluprost), a new prostanoid FP receptor agonist, as an ocular hypotensive drug. Exp Eye Res 78: 767–776. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Yamasaki A, Segi E, Tsuboi K, Aze Y, Nishimura T et al (1997). Failure of parturition in mice lacking the prostaglandin F receptor. Science 277: 681–683. [DOI] [PubMed] [Google Scholar]
- Tham Y‐C, Li X, Wong TY, Quigley HA, Aung T, Cheng C‐Y (2014). Global prevalence of glaucoma and projections of glaucoma burden through 2040. Ophthalmology 121: 2081–2090. [DOI] [PubMed] [Google Scholar]
- Thieme H, Schimmat C, Münzer G, Boxberger M, Fromm M, Pfeiffer N et al (2006). Endothelin antagonism: effects of FP receptor agonists prostaglandin F2alpha and fluprostenol on trabecular meshwork contractility. Invest Ophthalmol Vis Sci 47: 938–945. [DOI] [PubMed] [Google Scholar]
- Thomas D, Papadopoulo O, Doshi R, Kapin MA, Sharif NA (2000). Retinal ATP and phosphorus metabolites: reduction by hypoxia and recovery with MK‐801 and diltiazem. Med Sci Res 28: 87–91. [Google Scholar]
- Toris CB, Zhan G, Camras CB (2004). Increase in outflow facility with unoprostone treatment in ocular hypertensive patients. Arch Ophthalmol 122: 1782–1787. [DOI] [PubMed] [Google Scholar]
- Vysniauskiene I, Allemann R, Flammer J, Haefliger IO (2006). Vasoactive responses of U46619, PGF2alpha, latanoprost, and travoprost in isolated porcine ciliary arteries. Invest Ophthalmol Vis Sci 47: 295–298. [DOI] [PubMed] [Google Scholar]
- Wang JW, Woodward DF, Martos JL, Cornell CL, Carling RW, Kingsley PJ et al (2016). Multi‐targeting of selected prostanoid receptors provides agents with enhanced anti‐inflammatory activity in macrophages. FASEB J 30: 394–404. [DOI] [PubMed] [Google Scholar]
- Weinreb RN, Aung T, Medeiros FA (2014). The pathophysiology and treatment of glaucoma: a review. JAMA 311: 1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb RN, Khaw PT (2004). Primary open‐angle glaucoma. Lancet 363: 1711–1720. [DOI] [PubMed] [Google Scholar]
- Weinreb RN, Lindsey JD (2002). Metalloproteinase gene transcription in human ciliary muscle cells with latanoprost. Invest Ophthalmol Vis Sci 43: 716–722. [PubMed] [Google Scholar]
- Weinreb RN, Toris CB, Gabelt BT, Lindsey JD, Kaufman PL (2002). Effects of prostaglandins on the aqueous humor outflow pathways. Surv Ophthalmol 47 (Suppl. 1): S53–S64. [DOI] [PubMed] [Google Scholar]
- Weinreb RN, Leung CK, Crowston JG, Medeiros FA, Friedman DS, Wiggs JL et al (2016). Primary open‐angle glaucoma. Nat Rev Dis Primers 2: 16067. [DOI] [PubMed] [Google Scholar]
- Woodward DF, Jones RL, Narumiya S (2011). International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev 63: 471–538. [DOI] [PubMed] [Google Scholar]
- Woodward DF, Krauss AH, Chen J, Lai RK, Spada CS, Burk RM et al (2001). The pharmacology of bimatoprost (Lumigan®). Surv Ophthalmol 45: S337–S345. [DOI] [PubMed] [Google Scholar]
- Yang YF, Sun YY, Acott TS, Keller KE (2016). Effects of induction and inhibition of matrix cross‐linking on remodeling of the aqueous outflow resistance by ocular trabecular meshwork cells. Sci Rep 6: 30505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Lucitt MB, Stubbe J, Cheng Y, Friis UG, Hansen PB et al (2009). Prostaglandin F2α elevates blood pressure and promotes atherosclerosis. Proc Natl Acad Sci U S A 106: 7985–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Gong Y, Yu Y (2010). PGF2α receptor: a promising therapeutic target for cardiovascular disease. Front Pharmacol 1: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li X, Liu LP, Hong L, Liu X, Zhang B et al (2017). Peroxisome proliferator‐activated receptor γ is essential for secretion of ANP induced by prostaglandin D2 in the beating rat atrium. Korean J Physiol Pharmacol 21: 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]