

Abstract
Eicosanoids play important roles in modulating inflammation throughout the body. The gastrointestinal (GI) tract, in part because of its intimate relationship with the gut microbiota, is in a constant state of low‐grade inflammation. Eicosanoids like PGs, lipoxins and leukotrienes play essential roles in maintenance of mucosal integrity. On the other hand, in some circumstances, these mediators can become major drivers of inflammatory processes when the lining of the GI tract is breached. Drugs such as nonsteroidal anti‐inflammatories, by altering the production of various eicosanoids, can dramatically impact the ability of the GI tract to respond appropriately to injury. Disorders such as inflammatory bowel disease appear to be driven in part by altered production of eicosanoids. Several classes of drugs have been developed that target eicosanoids.
Linked Articles
This article is part of a themed section on Eicosanoids 35 years from the 1982 Nobel: where are we now? To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.8/issuetoc
Abbreviations
- GI
gastrointestinal
- IBD
inflammatory bowel disease
- LOX
lipoxygenase
- LT
leukotriene
- LX
lipoxin
- NSAID
nonsteroidal anti‐inflammatory drug
The discovery by Sir John Vane that aspirin and other nonsteroidal anti‐inflammatory drugs (NSAIDs) inhibited the formation of PGs led to a burst of research into the role of this group of lipid mediators in gastrointestinal (GI) function (Vane, 1971). Prior to that, Andre Robert and his colleagues at The Upjohn Company, in the USA, had discovered that PGs were potent inhibitors of gastric acid secretion, and these lipid mediators could also prevent experimental ulcer formation (Robert et al., 1968). Robert's group went on to characterize the effects of several longer acting synthetic PG analogues in animals and humans (Konturek et al., 1974; Robert et al., 1979), and in a landmark paper in 1979, described the powerful ‘cytoprotective’ effects of these agents in the stomach and duodenum (Robert et al., 1979). Remarkably, these very low doses of PGs were reported to protect the lining of the rat stomach from damage induced by oral administration of high concentrations of hydrochloric acid or sodium hydroxide, absolute alcohol and even to boiling water (Robert et al., 1979).
The first PG analogue to reach the marketplace was misoprostol, a PGE1 derivative developed by Searle and launched in 1986. Misoprostol was shown to significantly reduce the incidence of NSAID‐induced gastric ulcers, but only at doses that suppressed gastric acid secretion (Graham et al., 1988). At such doses, misoprostol significantly increased the incidence of diarrhoea, consistent with known stimulatory effects of PGs on GI epithelial secretion (Matuchansky and Coutrot, 1978).
COX‐1 and COX‐2
In 1972, another very important discovery was made by Roderick Flower and Sir John Vane that would only be fully appreciated two decades later. They observed that acetaminophen, which was known not inhibit peripheral tissue COX activity at therapeutic doses, was able to suppress PG synthesis in the brain (Flower and Vane, 1972). They speculated that there may be more than one form of COX, differentially affected by anti‐inflammatory drugs. This notion lingered in the background until 1991, when two groups in the USA reported a previously unrecognized, inducible COX enzyme, which would come to be known as ‘COX‐2’ (Kujubu et al., 1991; Xie et al., 1991). These publications resulted in a burst of research into the factors regulating expression of the two COX enzymes and their sensitivity to inhibition by the many NSAIDs already on the market, as well as to many new NSAIDs designed to selectively inhibit COX‐2 (Masferrer et al., 1994; Mitchell et al., 1994; Warner et al., 1999). The latter was based on the notion that PGs derived from COX‐2 accounted for pain and inflammation, while PGs from COX‐1 accounted for the cytoprotective effects that Robert and his colleagues had first observed in the late 1960s (Robert et al., 1968). Several of the world's largest pharmaceutical companies raced to develop selective COX‐2 inhibitors. The first to succeed was Searle in 1999 (celecoxib; Celebrex®), followed less than a year later by Merck (rofecoxib; Vioxx®). Both were hugely successful from a commercial standpoint, but less than 5 years after its launch, Merck removed rofecoxib from worldwide markets because of evidence that it caused serious cardiovascular events (Bombardier et al., 2000; Bresalier et al., 2005). Celecoxib, which was less selective for COX‐2 than rofecoxib (Warner et al., 1999), has remained on the market and is one of the most commercially successful drugs. Concerns that the selective COX‐2 inhibitors increased the risk of serious cardiovascular events led to recommendations from regulators and medical associations for concomitant use of low‐dose aspirin in patients deemed to be at increased risk for serious cardiovascular events. However, there is clear evidence that co‐administration of aspirin with celecoxib greatly diminishes any reduction of significant GI adverse events that may have been achieved through use of celecoxib versus a conventional NSAID (Silverstein et al., 2000; Wallace et al., 2000).
Animal studies clearly demonstrated that selective inhibition of COX‐2 did not result in significant damage in the stomach (Masferrer et al., 1994). However, selective inhibition of COX‐1 also did not result in mucosal damage (Wallace et al., 2000). Selective inhibition of one isoform of COX results in rapid up‐regulation of the other, at least in the GI tract (Davies et al., 1997; Wallace et al., 2000). Thus, suppression of both COX enzymes was necessary for gastric damage to be induced (Wallace et al., 2000) (Figure 1). These observations from animal models were consistent with the clinical observations on co‐administration of celecoxib and low‐dose aspirin (Silverstein et al., 2000). The ‘COX‐2 hypothesis’ was also challenged by evidence that COX‐1‐derived PGs contribute significantly to inflammation (Wallace et al., 1997), and gastric COX‐1 is inducible (Ferraz et al., 1997).
Figure 1.
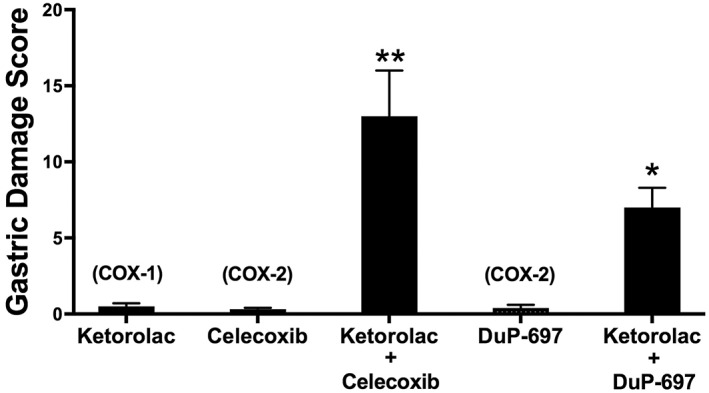
Induction of haemorrhagic, gastric damage by NSAIDs requires inhibition of both COX‐1 and COX‐2. Ketorolac at a dose (3 mg·kg−1) that inhibited COX‐1 but not COX‐2 did not cause significant gastric damage. Likewise, celecoxib or DuP‐697 at doses that selectively inhibit COX‐2 (15 mg·kg−1 and 10 mg·kg−1, respectively) did not cause significant gastric damage. However, the combination of a COX‐1 and COX‐2 inhibitor resulted in extensive haemorrhagic damage in the stomach (*P < 0.05, **P < 0.01 compared with the groups treated only with a single drug). This figure was constructed from previously published data (Wallace et al., 2000).
Most of the research in the early ‘COX‐2 era’ was focused on ulceration and bleeding in the stomach and proximal duodenum. These are the regions most easy to examine with an endoscope and where the damage is clearly dependent upon the presence of acid. However, NSAIDs can cause clinically significant ulceration and bleeding throughout the GI tract (Bjarnason et al., 1993; Graham et al., 2005; Lanas et al., 2009; Watanabe et al., 2013; Washio et al., 2016). Several clinical studies demonstrated that there was no reduction in incidence or severity of more distal small intestinal damage with a selective COX‐2 inhibitor as compared to that with a conventional NSAID (McCarthy, 2009; Graham et al., 2011). These observations were surprising given that selective COX‐2 inhibitors do not block platelet aggregation, which would contribute to GI bleeding. Indeed, endoscopic studies of selective COX‐2 inhibitors in healthy volunteers, who typically exhibit greater resistance to NSAID‐induced GI damage than arthritis patients, have revealed very high rates of intestinal damage and bleeding (Maiden et al., 2005; Maiden et al., 2007). Also, while selective COX‐2 inhibitors were commercially promoted on the basis of vastly improved GI safety, they are commonly co‐prescribed with agents that suppress gastric acid secretion. However, administration of selective COX‐2 inhibitors with proton pump inhibitors or histamine H2receptor antagonists has been shown to markedly increase the severity of damage in the distal intestine (Wallace et al., 2011; Satoh et al., 2012; Blackler et al., 2014, Washio et al., 2016). This is in part due to dramatic shifts in the intestinal microbiome (Wallace et al., 2011).
Leukotrienes and ulcers
When pharmacological tools for studying leukotrienes (LT) first became available, there were numerous studies suggesting a role for these mediators in upper GI injury, but most involved animal models of injury, such as administration of high concentrations of ethanol, with questionable relevance to the injury that is most commonly observed in humans. However, there were some studies identifying increased mucosal levels of LTB4 and LTC4 in biopsies from humans infected with Helicobacter pylori (Fukuda et al., 1990; Ahmed et al., 1992). NSAID‐induced ulceration was shown to be a neutrophil‐dependent process in animal studies (Wallace et al., 1990), leading to the proposal that blockade of COX activity may result in a shunting of arachidonic acid to lipoxygenase (LOX) enzymes and that LTs might be key mediators in activating leukocyte adherence and activation, as well as contributing to mucosal injury. Indeed, NSAID‐induced leukocyte adherence was shown to be markedly reduced in rats by a 5‐LOX inhibitor and by an LTB4(BLT1) receptor antagonist (Asako et al., 1992), and gastric mucosal synthesis of LTB4 was shown to be significantly increased in humans taking NSAIDs (Hudson et al., 1993). The availability of selective antagonists of LT receptors facilitated several studies of the role of LTs in animal models of gastric damage. Suppression of LT synthesis by inhibitors of 5‐LOX was found to significantly reduce the severity of gastric damage induced by an NSAID (Vaananen et al., 1992). However, treatment with LTB4 or LTC4/LTD4 antagonists had no effect in these studies.
There does not appear to have been any clinical testing of LT antagonists or selective 5‐LOX inhibitors with respect to GI safety. Licofelone (ML3000) is a dual COX/5‐LOX inhibitor that was proposed to be a ‘balanced inhibitor’ of 5‐LOX and COX with improved GI safety (Jovanovic et al., 2001), but it ultimately failed in phase III clinical development.
Prostaglandins and intestinal secretion
The ‘cytoprotective’ effects of PGs can be observed throughout the GI tract in various animal models of injury. Some of these effects may be in part attributable to stimulation of secretion in the gut, which can result in dilution of luminal irritants or toxins, and contribute to the process of expelling such agents. Thus, PGE2 can relax colonic circular muscle via actions on EP2 and EP4 receptors (Martinez‐Cutillas et al., 2014), while smooth muscle contraction can be increased by PGs acting through the EP1receptor (Chan and Mashimo, 2013). With respect to the latter, a PGE1 analogue, lubiprostone, has been employed in humans as a treatment for constipation‐predominant irritable bowel syndrome (Owen, 2008) and for opioid‐induced constipation (Jamal et al., 2015). On the other hand, EP2 and EP4 receptor antagonists have been used to suppress diarrhoea, such as that induced by cholera toxin in infected ileal loops (Satitsri et al., 2016).
Duodenal bicarbonate secretion is particularly important for protection of the tissue from high concentrations of gastric acid, and its mechanisms have been well characterized. PGE2 stimulates duodenal bicarbonate secretion in rats via activation of EP3 and EP4 receptors (Takeuchi et al., 2010; Said et al., 2015).
Eicosanoids and inflammatory bowel disease
The ability of PGs to stimulate colonic secretion led some investigators to propose that PGs may play an important role in inflammatory bowel disease (IBD) (ulcerative colitis and Crohn's disease). As outlined above, PGs may indeed contribute significantly to IBD‐associated diarrhoea. However, studies of inhibitors of COX activity in human IBD or in animal models of colitis overwhelmingly suggest important protective (anti‐inflammatory) effects of PGs. Several animal studies convincingly demonstrated that selective inhibition of COX‐2 led to a marked exacerbation of colitis (Reuter et al., 1996; McCartney et al., 1999; Morteau et al., 2000).
Use of NSAIDs has also been shown to be associated with microscopic colitis, which is characterized by watery diarrhoea (Masclee et al., 2015). It is not clear if this was directly related to suppression of prostanoid synthesis. Proton pump inhibitors are similarly associated with an increased risk of microscopic colitis (Verhaegh et al., 2016). Significant alterations in the gut microbiome may contribute to induction of microscopic colitis by both NSAIDs and proton pump inhibitors (Wallace et al., 2011; Wallace, 2013).
Leukotrienes in inflammatory bowel disease
The observation by Musch et al. (1982) that LOX products of arachidonic acid could potently stimulate colonic secretion drew the attention of other researchers to the possibility of LTs contributing significantly to the pathogenesis of IBD. Subsequently, a marked increase in colonic synthesis of LTB4 was documented in patients with IBD (Sharon and Stenson, 1984), as was a marked increase in peptido‐LTs (i.e. LTC4, LTD4, LTE4) (Peskar et al., 1986). The increase in LTB4 levels in the colonic mucosa of IBD patients was shown to be due to a significant decrease in ω‐hydroxylase activity and a marked increase in 5‐LOX activity (Ikehata et al., 1995).
Mesalamine (5‐aminosalicylic acid) is a commonly used, first‐line therapy for IBD, with an unknown mechanism of action. It was shown to markedly reduce peptido‐LT synthesis by colonic tissue from patients with IBD (Peskar et al., 1986). As discussed above, NSAIDs are known to exacerbate colitis in many IBD patients. Hudson et al. (1993) demonstrated that NSAID administration to IBD patients resulted in a significant enhancement of LTB4 synthesis and suggested that this could be the mechanism underlying the exacerbating effects of NSAIDs in colitis.
As more selective inhibitors of LT synthesis became available, they were tested in rodent models of IBD and were shown to be very effective in suppressing LT synthesis and accelerating resolution of colitis (Wallace et al., 1989). On the other hand, intracolonic administration of LTB4 significantly exacerbated experimental colitis (Wallace and Keenan, 1990).
Despite the promising findings in animal models, results from clinical trials of inhibitors of LT synthesis were disappointing. MK‐591 was among the first tested. This inhibitor of LTB4 synthesis was evaluated in a multi‐centre clinical trial in ulcerative colitis patients (Roberts et al., 1997). Patients were treated twice daily for 8 weeks and rectal dialysate LTB4 concentrations and disease activity scores were assessed at the end of that period. The highest dose of MK‐591 tested in the study (100 mg twice daily) reduced rectal dialysate LTB4 levels by ~98%, but lower doses (50 or 12.5 mg twice daily) had no significant effect on LTB4 levels as compared with placebo‐treated patients. None of the doses of MK‐591 produced a significant improvement in clinical activity as compared with the placebo‐treated group.
The role of LTB4 in ulcerative colitis was further examined in a 6‐month clinical trial of zileuton, another selective 5‐LOX inhibitor (Hawkey et al., 1997). The patients were in remission at the start of the study. Treatment with zileuton (600 mg qid) was compared with treatment with placebo or mesalamine (400 mg qid). The primary endpoint was maintenance of remission over the 6‐month period. As in the trial of MK‐571, the results of the zileuton trial did not support a significant role of LTB4 in ulcerative colitis. The remission rate in patients treated with zileuton (54%) was not significantly different from that in patients treated with placebo (43%) or mesalamine (63%). The relapse rate for patients treated with mesalamine was significantly lower than the relapse rate for patients on placebo.
PGD2: a stop signal in colitis?
Experimental and clinical data suggest that PGs, while contributing to diarrhoea, can also play an important role in dampening inflammation during bouts of colitis. On the other hand, NSAIDs can exacerbate colitis, presumably due to suppression of ‘cytoprotective’ PGE2 synthesis. However, while increased PGE2 production is associated with the promotion of oedema formation and pain (Murakami and Kudo, 2006), PGD2 acts in the opposite direction, exerting significant anti‐inflammatory effects (Gilroy et al., 1999; Ajuebor et al., 2000; Rajakariar et al., 2007). For example, we demonstrated that in experimental colitis, markedly elevated PGD2 synthesis was a rapid response to tissue injury, acting as a ‘braking mechanism’ that countered the pro‐inflammatory effects of PGE2 and other chemotaxins (Ajuebor et al., 2000). Remarkably, during the resolution phase of colitis, when tissue structure and function returned to normal, we observed a marked elevation of tissue PGD2 synthesis that persisted for months after the acute colitis had resolved (Ajuebor et al., 2000; Zamuner et al., 2003). The PGD2 produced in this setting was derived primarily via COX‐2, and selective inhibition of that enzyme resulted in marked increase in granulocyte infiltration into the mucosa. This infiltration was significantly reduced by administration of either PGD2 or a DP receptor agonist (Ajuebor et al., 2000). The prolonged up‐regulation of PGD2 synthesis after resolution of a bout of colitis was also accompanied by significant changes in colonic epithelial function. Thus, colonic chloride secretory responses (in vitro) were markedly diminished relative to those in controls, but could be recovered to normal by treatment with a selective COX‐2 inhibitor (celecoxib), but not selective COX‐1 inhibitor (SC‐560) (Zamuner et al., 2003). This hyporesponsiveness could be mimicked in normal colonic tissue by exposure to PGD2, but not to its metabolite, 15‐deoxy‐Δ12‐14PGJ2. There was also further evidence for substantially altered colonic epithelial function in rats that appeared to have fully recovered from colitis: although appearing healthy and normal, these rats exhibited a 10‐fold increase in bacterial colonization of the colon and more than a threefold increase in bacterial translocation. The latter could be reversed by treatment for 1 week with a selective COX‐2 inhibitor (rofecoxib). These studies demonstrate an important role for COX‐2, apparently via generation of PGD2, in mediating the prolonged barrier and epithelial secretory dysfunction that is evident after apparent resolution of colitis in the rat (Zamuner et al., 2003).
There are also human data suggestive of an important role for PGD2 in the resolution of colitis and particularly in maintenance of remission. Vong et al. (2010) analysed colonic biopsies from patients with active ulcerative colitis, looking at pro‐ and anti‐inflammatory PG production, expression of the major enzymes of synthesis and expression of the key receptors. They used two groups as controls: healthy subjects with no prior history of ulcerative colitis and healthy individuals who had previously had ulcerative colitis, but had been in clinical remission for at least 4 years. As expected, the patients with active colitis exhibited significant elevations of colonic synthesis of PGE2 and increased expression (fourfold to sixfold) of COX‐2, IFN‐γ and TNF‐α (Figure 2). However, there were some remarkable findings related to PGD2 when the group of patients who had been in long‐term remission were examined. In that group alone, there were significant increases in PGD2 synthesis and in expression of mRNA for the DP1receptor, but not for the DP2receptor. PGD2 preferentially binds to DP1 receptors, and its activation is largely thought to be responsible for the anti‐inflammatory effects of PGD2. These data suggest that the same long‐term increase in colonic PGD2 synthesis that had been observed in rats after resolution of colitis (Ajuebor et al., 2000; Zamuner et al., 2003) are also evident in humans who are in long‐term remission after resolution of colitis. Sturm et al. (2014) suggested that there may be a role for modulation of colitis through blockade of the other DP receptor, the DP2 receptor also known as chemoattractant receptor‐homologous molecule expressed on T‐helper type 2 cells (CRTH2). They demonstrated that blockade of the DP2 receptor (CRTH2) significantly reduced the severity of colitis in a murine model.
Figure 2.
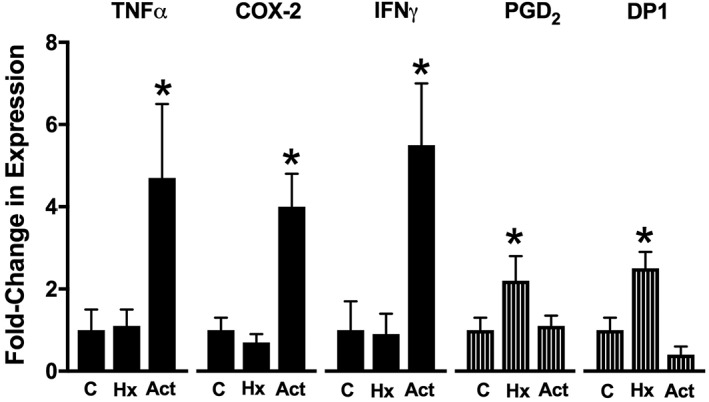
Expression of pro‐ and anti‐inflammatory factors in biopsies from healthy volunteers and ulcerative colitis patients. The ‘healthy’ groups include subjects with no history (Hx) of ulcerative colitis (‘C’) and subjects who had previously had ulcerative colitis, but were in long‐term remission (more than 4 years). Pro‐inflammatory markers such as TNF‐α, COX‐2 and IFN‐γ were significantly elevated in the patients with active (Act) colitis. Anti‐inflammatory markers such as PGD2 and the DP1 receptor (DP1) were significantly elevated in patients in long‐term remission. *P < 0.05 versus controls. This figure was constructed from previously published data (Vong et al., 2010).
Lipoxins in the GI tract: anti‐inflammatory and protective
Lipoxins (LX) are endogenous, anti‐inflammatory molecules that contribute significantly to reducing tissue injury and progression from acute to chronic inflammation (Serhan et al., 2007; 2015). The ‘classical’ LX‐generating pathways involve (1) the sequential actions of 5‐LOX (in leukocytes) and 12‐LOX (in platelets) to produce LXA4 and (2) the sequential actions of 15‐LOX (in epithelial cells) and 5‐LOX (in leukocytes) to produce LXB4 (Serhan et al., 2015). A third pathway for LX synthesis, involving aspirin, is described below.
LXA4 analogues have been synthesized and have shown promising anti‐inflammatory effects in models of colitis. For example, Gewirtz et al. (2002) demonstrated that LXA4 analogues reduced pro‐inflammatory gene expression in intestinal epithelial cells and significantly attenuated the severity of experimental colitis in mice. LXA4 and LX analogues have been shown to down‐regulate leukocyte degranulation (Gewirtz et al., 1999) and chemokine release (Gewirtz et al., 1998). Moreover, LXA4 has been shown to dampen intestinal inflammation by inhibiting NF‐κB activity (Kure et al., 2010). LXA4 has also been reported to be important for driving IL‐10 release, thereby producing additional anti‐inflammatory effects (Souza et al., 2007), as well as mediating the protective effects of an n‐6 fatty acid‐enriched diet in ischaemia–reperfusion injury (Gobbetti et al., 2015).
The oral efficacy of ZK‐192, a β‐oxidation‐resistant analogue of aspirin‐triggered lipoxin, was examined in experimental colitis. Daily oral administration of ZK‐192 at 300 or 1,000 μg·kg−1 markedly reduced the severity of colitis in rodents, whether given in preventive or therapeutic treatment regimens. This included significant reductions of macroscopic and histological colon injury, weight loss and mucosal neutrophil infiltration. The observed beneficial effects of ZK‐192 were comparable with those observed with 3 to 10 mg·kg−1 of prednisolone. Expression of a mRNA for several pro‐inflammatory mediators (e.g. inducible NOS, macrophage inflammatory protein 2, COX‐2) was significantly decreased by treatment with ZK‐192, along with significantly reduced mucosal mRNA and protein levels of TNF‐α, IL‐2 and IFN‐γ.
A significant role for LXA2 in maintenance of remission in patients with ulcerative colitis was suggested by Vong et al. (2012). They observed that there were markedly elevated levels of colonic LXA2 in the patients who were in remission, but not in healthy controls or in patients with active ulcerative colitis (Figure 3). They also observed significant elevations of 5‐LOX expression in the patients with active colitis, but no change in 12‐ or 15‐LOX expression (Vong et al., 2012). These results demonstrated a specific up‐regulation of a pro‐resolution circuit in patients who were in remission from ulcerative colitis, with significant contributions of LXA2 in promoting mucosal homeostasis.
Figure 3.
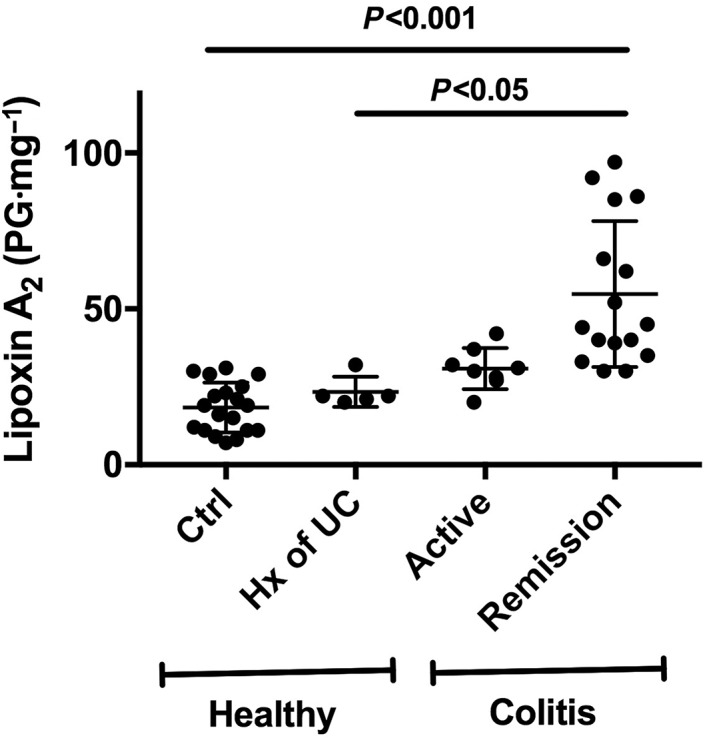
Mucosal lipoxin A2 levels in biopsies from healthy volunteers and ulcerative colitis patients. The ‘healthy’ group included subjects with no history (Hx) of ulcerative colitis (Ctrl) and subjects who previously had ulcerative colitis (UC), but had been in long‐term remission (more than 4 years). The ‘colitis’ patients were subdivided into those with active disease and those in medically induced remission. The patients in remission exhibited markedly elevated lipoxin A2 levels as compared with the two groups of healthy subjects. This figure was constructed from previously published data (Vong et al., 2012).
The ability of aspirin to cause damage in the GI tract, particularly in the stomach, is well known. Acetylation of COX‐1 by aspirin results in an irreversible inhibition of that enzyme, preventing PG synthesis. However, acetylation of COX‐2, while also preventing PG synthesis, does not completely block the function of the enzyme. Arachidonic acid can still be modified by acetylated COX‐2, leading to the formation of 15R–HETE (15R–hydroxyeicosatetraenoic acid), which can be further transformed to ‘aspirin‐triggered lipoxin’ (such as 15‐epi‐LXA4) via the action of 5‐LOX (Serhan et al., 2007). In the GI tract, aspirin‐triggered lipoxins have been shown to contribute significantly to GI mucosal defence, as well as exhibiting significant anti‐inflammatory effects (Fiorucci et al., 2002, 2004).
Closing Comment
As in other tissues, eicosanoids are involved in a wide range of physiological and pathological processes in laboratory animals and humans. There are a wide range of drugs that have been developed to enhance or inhibit the actions of eicosanoids in the GI tract. Despite extensive research for the past five decades, there remains a need for better understanding of the role of eicosanoids in resolution of inflammation in the GI tract. Such research will be an important part of shifting treatment regimens from a target of reducing the magnitude of inflammatory processes (largely symptom control) to an orderly promotion of resolution of inflammation.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2017a,b).
Conflict of interest
The author declares no conflicts of interest.
Acknowledgements
This work was supported by a grant from the Canadian Institutes of Health Research.
Wallace, J. L. (2019) Eicosanoids in the gastrointestinal tract. British Journal of Pharmacology, 176: 1000–1008. 10.1111/bph.14178.
References
- Ahmed A, Holton J, Vaira D, Smith SK, Hoult JR (1992). Eicosanoid synthesis and Helicobacter pylori associated gastritis: increase in leukotriene C4 generation associated with H. pylori colonization. Prostaglandins 44: 75–86. [DOI] [PubMed] [Google Scholar]
- Ajuebor MN, Singh A, Wallace JL (2000). Cyclooxygenase‐2‐derived prostaglandin D2 and resolution of acute inflammation through PGD2 and 15‐deoxy‐Δ12,14 PGJ2 . Am J Physiol Liver Physiol 279: G238–G244. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asako H, Kubes P, Wallace J, Gaginella T, Wolf RE, Granger DN (1992). Indomethacin‐induced leukocyte adhesion in postcapillary venules: role of lipoxygenase products. Am J Physiol 262: G903–G908. [DOI] [PubMed] [Google Scholar]
- Bjarnason I, Hayllar J, MacPherson AJ, Russell AS (1993). Side effects of nonsteroidal anti‐inflammatory drugs on the small and large intestine in humans. Gastroenterology 104: 1832–1847. [DOI] [PubMed] [Google Scholar]
- Blackler RW, Gemici B, Manko A, Wallace JL (2014). NSAID‐gastroenteropathy: new aspects of pathogenesis and prevention. Curr Opin Pharmacol 19: 11–16. [DOI] [PubMed] [Google Scholar]
- Bombardier C, Laine L, Reicin A, Shapiro D, Burgos‐Vargas R, Davis B et al (2000). Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med 343: 1520–1528. [DOI] [PubMed] [Google Scholar]
- Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K et al (2005). Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med 352: 1092–1102. [DOI] [PubMed] [Google Scholar]
- Chan WW, Mashimo H (2013). Lubiprostone increases small intestinal smooth muscle contractions through a prostaglandin E receptor 1 (EP1)‐mediated pathway. J Neurogastroenterol Motil 19: 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies NM, Sharkey KA, Asfaha S, MacNaughton WK, Wallace JL (1997). Aspirin induces a rapid up‐regulation of cyclooxygenase‐2 expression in the rat stomach. Aliment Pharmacol Ther 11: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Ferraz JG, Sharkey KA, Reuter BK, Asfaha S, Tigley AW, Brown ML et al (1997). Induction of cyclooxygenase 1 and 2 in the rat stomach during endotoxemia: role in resistance to damage. Gastroenterology 113: 195–204. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Menezes de Lima O, Mencarelli A, Palazzetti B, Distrutti E, McKnight W et al (2002). COX‐2‐derived lipoxin A4 increases gastric resistance to aspirin‐induced damage. Gastroenterology 123: 1598–1606. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Wallace JL, Mencarelli A, Distrutti E, Rizzo G, Farneti S et al (2004). A beta‐oxidation‐resistant lipoxin A4 analog treats hapten‐induced colitis by attenuating inflammation and immune dysfunction. Proc Natl Acad Sci U S A 104: 20979–20984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower RJ, Vane JR (1972). Inhibition of prostaglandin synthetase in brain explains the anti‐pyretic activity of paracetamol (4‐acetamidophenol). Nature 240: 410–411. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Kimura S, Arakawa T, Kobayashi K (1990). Possible role of leukotrienes in gastritis associated with Campylobacter pylori. J Clin Gastroenterol 12 (Suppl 1): 131–134. [DOI] [PubMed] [Google Scholar]
- Gewirtz AT, Collier‐Hyams LS, Young AN, Kucharzik T, Guilford WJ, Parkinson JF et al (2002). Lipoxin A4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate‐induced colitis. J Immunol 168: 5260–5267. [DOI] [PubMed] [Google Scholar]
- Gewirtz AT, McCormick B, Neish AS, Petasis NA, Gronert K, Serhan CN et al (1998). Pathogen‐induced chemokine secretion from model intestinal epithelium is inhibited by lipoxin A4 analogs. J Clin Invest 101: 1860–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz AT, Fokin VV, Petasis NA, Serhan CN, Madara JL (1999). LXA4, aspirin‐triggered 15‐epi‐LXA4, and their analogs selectively downregulate PMN azurophilic degranulation. Am J Physiol 276: C988–C994. [DOI] [PubMed] [Google Scholar]
- Gilroy DW, Colville‐Nash PR, Willis D, Chivers J, Paul‐Clark MJ, Willoughby DA (1999). Inducible cyclooxygenase may have anti‐inflammatory properties. Nat Med 5: 698–701. [DOI] [PubMed] [Google Scholar]
- Gobbetti T, Ducheix S, le Faouder P, Perez T, Riols F, Boue J et al (2015). Protective effects of n‐6 fatty acids‐enriched diet on intestinal ischaemia/reperfusion injury involve lipoxin A4 and its receptor. Br J Pharmacol 172: 910–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DY, Agrawal NM, Roth SH (1988). Prevention of NSAID‐induced gastric ulcer with misoprostol: multicentre, double‐blind, placebo‐controlled trial. Lancet 2: 1277–1280. [DOI] [PubMed] [Google Scholar]
- Graham DY, Opekun AR, Willingham FF, Qureshi WA (2005). Visible small‐intestinal mucosal injury in chronic NSAID users. Clin Gastroenterol Hepatol 3: 55–59. [DOI] [PubMed] [Google Scholar]
- Graham DY, Jewell NP, Chan FKL (2011). Rofecoxib and clinically significant upper and lower GI events revisited based on documents from recent litigation. Am J Med Sci 342: 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkey CJ, Dube LM, Rountree LV, Linnen PJ, Lancaster JF (1997). A trial of zileuton versus mesalazine or placebo in the maintenance of remission of ulcerative colitis. The European Zileuton Study Group For Ulcerative Colitis. Gastroenterology 112: 718–724. [DOI] [PubMed] [Google Scholar]
- Hudson N, Balsitis M, Everitt S, Hawkey CJ (1993). Enhanced gastric mucosal leukotriene B4 synthesis in patients taking non‐steroidal anti‐inflammatory drugs. Gut 34: 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikehata A, Hiwatashi N, Kinouchi Y, Yamazaki H, Ito K, Toyota T (1995). Altered leukotriene B4 metabolism in colonic mucosa with inflammatory bowel disease. Scand J Gastroenterol 30: 44–49. [DOI] [PubMed] [Google Scholar]
- Jamal MM, Adams AB, Jansen JP, Webster LR (2015). A randomized, placebo‐controlled trial of lubiprostone for opioid‐induced constipation in chronic noncancer pain. Am J Gastroenterol 110: 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic DV, Fernandes JC, Martel‐Pelletier J, Jolicoeur FC, Reboul P, Laufer S et al (2001). The in vivo dual inhibition of cyclooxygenase and lipoxygenase by ML‐3000 reduces the progression of experimental osteoarthritis. Suppression of collagenase‐1 and interleukin‐1β synthesis. Arthritis Rheum 44: 2320–2330. [DOI] [PubMed] [Google Scholar]
- Konturek SJ, Radecki T, Demitrescu T, Kwiecień N, Pucher A, Robert A (1974). Effect of synthetic 15‐methyl analog of prostaglandin E2 on gastric secretion and peptic ulcer formation. J Lab Clin Med 84: 716–725. [PubMed] [Google Scholar]
- Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR (1991). TIS10, a phorbol ester tumor promoter‐inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem 266: 12866–12672. [PubMed] [Google Scholar]
- Kure I, Nishiumi S, Nishitani Y, Tanoue T, Ishida T, Mizuno M et al (2010). Lipoxin A4 reduces lipopolysaccharide‐induced inflammation in macrophages and intestinal epithelial cells through inhibition of nuclear factor‐кB activation. J Pharmacol Exp Ther 332: 541–548. [DOI] [PubMed] [Google Scholar]
- Lanas A, García‐Rodríguez LA, Polo‐Tomás M, Ponce M, Alonso‐Abreu I, Perez‐Aisa MA et al (2009). Time trends and impact of upper and lower gastrointestinal bleeding and perforation in clinical practice. Am J Gastroenterol 104: 1633–1641. [DOI] [PubMed] [Google Scholar]
- Maiden L, Thjodleifsson B, Theodors A, Gonzalez J, Bjarnason I (2005). A quantitative analysis of NSAID‐induced small bowel pathology by capsule endoscopy. Gastroenterology 128: 1172–1178. [DOI] [PubMed] [Google Scholar]
- Maiden L, Thjodleifsson B, Seigal A, Bjarnason II, Scott D, Birgisson S et al (2007). Long‐term effects of nonsteroidal anti‐inflammatory drugs and cyclooxygenase‐2 selective agents on the small bowel: a cross‐sectional capsule enteroscopy study. Clin Gastroenterol Hepatol 5: 1040–1045. [DOI] [PubMed] [Google Scholar]
- Martinez‐Cutillas M, Mañe N, Gallego D, Jimenez M, Martin MT (2014). EP2 and EP4 receptors mediate PGE2 induced relaxation in murine colonic circular muscle: pharmacological characterization. Pharmacol Res 90: 76–86. [DOI] [PubMed] [Google Scholar]
- Masclee GM, Coloma PM, Kuipers EJ, Sturkenboom MC (2015). Increased risk of microscopic colitis with use of proton pump inhibitors and non‐steroidal anti‐inflammatory drugs. Am J Gastroenterol 110: 749–759. [DOI] [PubMed] [Google Scholar]
- Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG et al (1994). Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci U S A 91: 3228–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matuchansky C, Coutrot S (1978). The role of prostaglandins in the study of intestinal water and electrolyte transport in man. Biomedicine 1978: 143–148. [PubMed] [Google Scholar]
- McCarthy DM (2009). GI bleeding: problems that persist. Gastrointest Endosc 70: 225–228. [DOI] [PubMed] [Google Scholar]
- McCartney SA, Mitchell JA, Fairclough PD, Farthing MJ, Warner TD (1999). Selective COX‐2 inhibitors and human inflammatory bowel disease. Aliment Pharmacol Ther 13: 1115–1117. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Belvisi MG, Akarasereenont P, Robbins RA, Kwon OJ, Croxtall J et al (1994). Induction of cyclo‐oxygenase‐2 by cytokines in human pulmonary epithelial cells: regulation by dexamethasone. Br J Pharmacol 113: 1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morteau O, Morham SG, Sellon R, Dieleman LA, Langenbach R, Smithies O et al (2000). Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase‐1 or cyclooxygenase‐2. J Clin Invest 105: 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Kudo I (2006). Prostaglandin E synthase: a novel drug target for inflammation and cancer. Curr Pharm Des 12: 943–954. [DOI] [PubMed] [Google Scholar]
- Musch MW, Miller RJ, Field M, Siegel MI (1982). Stimulation of colonic secretion by lipoxygenase metabolites of arachidonic acid. Science 217: 1255–1256. [DOI] [PubMed] [Google Scholar]
- Owen RT (2008). Lubiprostone–a novel treatment for irritable bowel syndrome with constipation . Drugs Today (Barc) 2008 44: 645–652. [DOI] [PubMed] [Google Scholar]
- Peskar BM, Dreyling KW, Peskar BA, May B, Goebell H (1986). Enhanced formation of sulfidopeptide‐leukotrienes in ulcerative colitis and Crohn's disease: inhibition by sulfasalazine and 5‐aminosalicylic acid. Agents Actions 18: 381–383. [DOI] [PubMed] [Google Scholar]
- Rajakariar R et al (2007). Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15‐deoxyDelta12 14 PGJ2. Proc Natl Acad Sci U S A 101: 15736–15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter BK, Asfaha S, Buret A, Sharkey KA, Wallace JL (1996). Exacerbation of inflammation‐associated colonic injury in rat through inhibition of cyclooxygenase‐2. J Clin Invest 98: 2076–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Nezamis JE, Lancaster C, Hanchar AJ (1979). Cytoprotection by prostaglandins in rats. Prevention of gastric necrosis produced by alcohol, HCl, NaOH, hypertonic NaCl, and thermal injury. Gastroenterology 77: 433–443. [PubMed] [Google Scholar]
- Robert A, Nezamis JE, Phillips JP (1968). Effect of prostaglandin E1 on gastric secretion and ulcer formation in the rat. Gastroenterology 55: 481–487. [PubMed] [Google Scholar]
- Roberts WG, Simon TJ, Berlin RG, Haggitt RC, Snyder ES, Stenson WF et al (1997). Leukotrienes in ulcerative colitis: results of a multicenter trial of a leukotriene biosynthesis inhibitor, MK‐591. Gastroenterology 112: 725–732. [DOI] [PubMed] [Google Scholar]
- Said H, Kaji I, Kaunitz JD (2015). Gastroduodenal mucosal defense mechanisms. Curr Opin Gastroenterol 31: 486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satitsri S, Pongkorpsakol P, Srimanote P, Chatsudthipong V, Muanprasat C (2016). Pathophysiological mechanisms of diarrhea caused by the Vibrio cholerae O1 El Tor variant: an in vivo study in mice. Virulence 7: 789–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Amagase K, Takeuchi K (2012). Exacerbation of nonsteroidal anti‐inflammatory drug‐induced small intestinal lesions by antisecretory drugs in rats: the role of intestinal motility. J Pharmacol Exp Ther 343: 270–277. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Brain SD, Buckley C, Gilroy DW, Haslett C, O'Neill LA et al (2007). Resolution of inflammation: state of the art definitions and terms. FASEB J 21: 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Dalli J (2015). The resolution code of acute inflammation: novel pro‐resolving lipid mediators in resolution. Semin Immunol 27: 200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon P, Stenson WF (1984). Enhanced synthesis of leukotriene B4 by colonic mucosa in inflammatory bowel disease. Gastroenterology 86: 453–460. [PubMed] [Google Scholar]
- Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A et al (2000). Gastrointestinal toxicity with celecoxib vs nonsteroidal anti‐inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: a randomized controlled trial. Celecoxib Long‐term Arthritis Safety Study. JAMA 284: 1247–1255. [DOI] [PubMed] [Google Scholar]
- Souza DG, Fagundes CT, Amaral FA, Cisalpino D, Sousa LP, Vieira AT et al (2007). The required role of endogenously produced lipoxin A4 and annexin‐1 for the production of IL‐10 and inflammatory hyporesponsiveness in mice. J Immunol 179: 8533–8543. [DOI] [PubMed] [Google Scholar]
- Sturm EM, Radnai B, Jandl K, Stancic A, Parzmair GP, Hogenauer C et al (2014). Opposing roles of prostaglandin D2 receptors in ulcerative colitis. J Immunol 193: 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Koyama M, Hayashi S, Aihara E (2010). Prostaglandin EP receptor subtypes involved in regulating HCO3‐secretion from gastroduodenal mucosa. Curr Pharm Des 16: 1241–1251. [DOI] [PubMed] [Google Scholar]
- Vaananen PM, Keenan CM, Grisham MB, Wallace JL (1992). Pharmacological investigation of the role of leukotrienes in the pathogenesis of experimental NSAID gastropathy. Inflammation 16: 227–240. [DOI] [PubMed] [Google Scholar]
- Vane JR (1971). Inhibition of prostaglandin synthesis as a mechanism of action for aspirin‐like drugs. Nature new biology 231: 232–235. [DOI] [PubMed] [Google Scholar]
- Verhaegh BP, de Vries F, Masclee AA, Keshavarzian A, de Boer A, Souverein PC et al (2016). High risk of drug‐induced microscopic colitis with concomitant use of NSAIDs and proton pump inhibitors. Aliment Pharmacol Ther 43: 1004–1013. [DOI] [PubMed] [Google Scholar]
- Vong L, Ferraz JGP, Panaccione R, Beck PL, Wallace JL (2010). A pro‐resolution mediator, prostaglandin D2, is specifically up‐regulated in individuals in long‐term remission from ulcerative colitis. Proc Natl Acad Sci U S A 107: 12023–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong L, Ferraz JG, Dufton N, Panaccione R, Beck PL, Perretti M et al (2012). Spatio‐temporal up‐regulation of annexin‐A1 and lipoxin A4 in individuals with ulcerative colitis may promote mucosal homeostasis. PLoS One 7: e39244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL (2013). Polypharmacy of osteoarthritis: the perfect intestinal storm. Dig Dis Sci 58: 3088–3093. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Keenan CM (1990). An orally active inhibitor of leukotriene synthesis accelerates healing in a rat model of chronic colitis. Am J Physiol 258: G527–G534. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Bak A, McKnight W, Asfaha S, Sharkey KA, MacNaughton WK (1997). Cyclooxygenase‐1 contributes to inflammatory responses in rats and mice: implications for GI toxicity. Gastroenterology 115: 101–109. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Keenan CM, Granger DN (1990). Gastric ulceration induced by non‐steroidal anti‐inflammatory drugs is a neutrophil‐dependent process. Am J Physiol 259: G462–G467. [DOI] [PubMed] [Google Scholar]
- Wallace JL, MacNaughton WK, Morris GP, Beck PL (1989). Inhibition of leukotriene synthesis markedly accelerates healing in a rat model of inflammatory bowel disease. Gastroenterology 96: 29–36. [DOI] [PubMed] [Google Scholar]
- Wallace JL, McKnight W, Reuter BK, Vergnolle N (2000). NSAID‐induced gastric damage in the rat: requirement for inhibition of both cyclooxygenase‐1 and ‐2. Gastroenterology 119: 706–714. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Denou E, Syer S, Vong L, McKnight W, Jury J et al (2011). Proton pump inhibitors exacerbate NSAID‐induced small intestinal injury via induction of dysbiosis. Gastroenterology 141: 1314–1322. [DOI] [PubMed] [Google Scholar]
- Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR (1999). Nonsteroid drug selectivities for cyclo‐oxygenase‐1 rather than cyclo‐oxygenase‐2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci U S A 96: 7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washio E, Esaki M, Maehata Y, Miyazaki M, Kobayashi H, Ishikawa H et al (2016). Proton pump inhibitors increase incidence of nonsteroidal anti‐inflammatory drug‐induced small bowel injury: a randomized, placebo‐controlled trial. Clin Gastroenterol Hepatol 14: 809–815. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Tanigawa T, Nadatani Y, Nagami Y, Sugimori S, Okazaki H et al (2013). Risk factors for severe nonsteroidal anti‐inflammatory drug‐induced small intestinal damage. Dig Liver Dis 45: 390–395. [DOI] [PubMed] [Google Scholar]
- Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL (1991). Expression of a mitogen‐responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci U S A 88: 2692–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamuner SR, Warrier N, Buret AG, MacNaughton WK, Wallace JL (2003). Cyclooxygenase 2 mediates post‐inflammatory colonic secretory and barrier dysfunction. Gut 52: 1714–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]