

Abstract
Platelets are important players in thrombosis and haemostasis with their function being modulated by mediators in the blood and the vascular wall. Among these, eicosanoids can both stimulate and inhibit platelet reactivity. Platelet Cyclooxygenase (COX)‐1‐generated Thromboxane (TX)A2 is the primary prostanoid that stimulates platelet aggregation; its action is counter‐balanced by prostacyclin, a product of vascular COX. Prostaglandin (PG)D2, PGE2 and 12‐hydroxyeicosatraenoic acid (HETE), or 15‐HETE, are other prostanoid modulators of platelet activity, but some also play a role in carcinogenesis. Aspirin permanently inhibits platelet COX‐1, underlying its anti‐thrombotic and anti‐cancer action. While the use of aspirin as an anti‐cancer drug is increasingly encouraged, its continued use in addition to P2Y12 receptor antagonists for the treatment of cardiovascular diseases is currently debated. Aspirin not only suppresses TXA2 but also prevents the synthesis of both known and unknown antiplatelet eicosanoid pathways, potentially lessening the efficacy of dual antiplatelet therapies.
Linked Articles
This article is part of a themed section on Eicosanoids 35 years from the 1982 Nobel: where are we now? To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.8/issuetoc
Abbreviations
- AA
arachidonic acid
- CYP450
cytochrome P450
- EETs
epoxyeicosatrienoic acids
- ECs
endothelial cells
- HETE
hydroxyeicosatraenoic acid
- LOX
lipoxygenase
- NSAIDs
nonsteroidal anti‐inflammatory drugs
- PGI2
prostacyclin
- PUFAs
polyunsaturated fatty acids
- USPSTF
US Preventive Services Task Force
Introduction
Platelets play a fundamental role in maintaining haemostasis. A fine balance exists in which platelets can be rapidly activated to aggregate and form a plug that prevents bleeding. But when platelets get inappropriately activated, thrombi form within the vessel wall which can lead to thrombotic events such as heart attack and stroke. The activation or inhibition of platelets can be modulated by many agents with a central role being played by eicosanoids. TXA2 and prostacyclin (PGI2) are the main eicosanoids affecting the function of platelets. The groups of Vane and Samuelsson were pioneers in their identification and in establishing their action on platelets and on the vasculature (Bunting et al., 1977; Bunting et al., 1983; Moncada et al., 1976; Moncada et al., 1978; Needleman et al., 1976; Svensson et al., 1975; Whittaker et al., 1976).
Since their discovery, and with the continued development of analytical techniques such as mass spectrometry‐based lipidomics, hundreds of structurally and stereochemically distinct eicosanoid families have been identified (Harkewicz and Dennis, 2011).
This review will focus on the production of eicosanoids by platelets and endothelium and their effect on platelet function in the cardiovascular system. We will discuss how aspirin modulates the synthesis of these eicosanoids and the consequences on its anti‐thrombotic efficacy. Laboratory techniques to evaluate response to aspirin will be also presented, and their ability to predict the occurrence of cardiovascular events will be examined. Finally, recent advances in understanding the role of platelet‐related eicosanoids in cancer will be presented.
Eicosanoids and the fine regulation of platelet function and haemostasis
Eicosanoids are mainly derived from arachidonic acid (AA) but can also be generated from other 20 carbon polyunsaturated fatty acids (PUFAs), such as dihomo‐γ‐linolenic acid, an ω‐6‐derived PUFA, or eicosapentaenoic acid (Subhash et al., 2007). These fatty acids are released from the cellular phospholipid membrane via the action of the enzyme phospholipase A2 (PLA2) and subsequently converted via the COXs into TXA2 and PGs, such as PGI2, PGE2 and PGD2, via lipoxygenases (LOXs) into hydroxyeicosatraenoic acids (e.g. 12‐HETE), and via cytochrome P450 (CYP450) enzymes into epoxyeicosatrienoic acids (EETs) (Dennis and Norris, 2015).
Platelets can produce significant amounts of TXA2, PGE2, PGD2, 11‐, 12‐ and 15‐HETE dependent upon the activity of cytosolic group IV A PLA2, a widely expressed PLA2 isoform (Kirkby et al., 2015; Rauzi et al., 2016). Below, we will discuss platelet and non‐platelet‐derived eicosanoids whose actions modulate platelet function and consequentially haemostasis and thrombosis (Figure 1).
Figure 1.
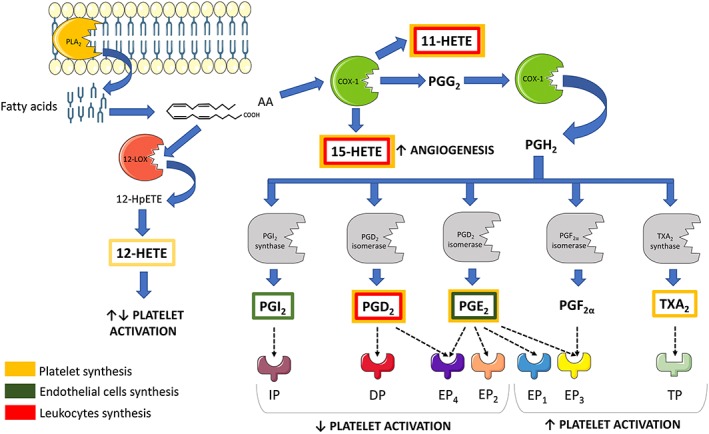
Diagram of the biosynthesis of the main eicosanoids that affect platelet function and where it occurs. The yellow, green and red boxes represent the origin of the eicosanoids as platelets, ECs and leukocytes respectively. The receptors for each eicosanoid are shown as well as the associated effects on platelet activation.
COX‐dependent eicosanoids
COX, more precisely known as PGH synthase, converts AA first into PGG2, via a COX function and then to PGH2 following a peroxidase reaction (Smith and Dewitt, 1996). PGH2 is an unstable molecule and, in platelets, undergoes further transformations catalysed by TX synthase, PGD isomerase or PGE synthase to form TXA2, PGD2 or PGE2 respectively.
Two different isoforms of COX exist in the cardiovascular system, namely, COX‐1 and COX‐2 (Hla and Neilson, 1992; Kujubu et al., 1991; Masferrer et al., 1992; O'Banion et al., 1992; Xie et al., 1991). COX‐1 is usually considered a constitutive form (Kirkby et al., 2012; Langenbach et al., 1997), while COX‐2 is considered to be an inducible enzyme, although a role for constitutive COX‐2 has been shown in the kidneys and the central nervous system (Herschman et al., 1997; Mitchell and Warner, 2006). Platelets mainly express COX‐1, but traces of COX‐2 have been detected, possibly carried over from megakaryocytes, the platelet precursor cells, or as a result of the transcription of residual mRNA into protein (Rocca et al., 2002; Warner et al., 2011).
Thromboxane A2
The most directly important prostanoid for platelet function is COX‐1‐generated TXA2. It was first identified by Vane as a ‘rabbit‐aorta‐contracting substance’ (RCS) produced by the lungs during anaphylaxis (Piper and Vane, 1969). Later, TXA2 was shown to be synthesized by activated platelets and to act in an autocrine and paracrine manner to induce thrombosis (Smith and Willis, 1971). On platelets, TXA2 binds to the thromboxane prostanoid (TP) receptor and initiates an amplification loop leading to further platelet activation, aggregation and TXA2 formation (Reilly and Fitzgerald, 1993). The TP receptor can couple with several G proteins, such as G12/13, leading to platelet shape change via phosphorylation of the myosin light chain, platelet granule release and irreversible aggregation (Smyth, 2010). In the vasculature, TXA2 induces vasoconstriction and the proliferation of vascular smooth muscle cells.
PGI2 (prostacyclin)
When first discovered as an autacoid produced by vascular tissue, PGI2 or prostacyclin was named as PGX and was described as a substance which, in contrast to TXA2, inhibited the clumping of platelets and relaxed vascular strips (Moncada et al., 1976). Now known to be predominantly produced by the endothelium within blood vessels, there has been strong debate as to which isoform of COX catalyses the vascular production of PGI2. Although still controversial, research by ourselves and colleagues strongly suggests that, in the healthy vasculature, PGI2 production is driven by COX‐1 (Bolego et al., 2009; Evangelista et al., 2006; Kirkby et al., 2012; Yu et al., 2012). This is discussed in more detail elsewhere in this issue (Mitchell and Kirkby, 2018).
Endothelium‐produced PGI2 binds to the Gs‐coupled PGI2 receptor (IP) on platelets and generally reduces platelet reactivity, which can be critical to minimizing the risk for atherothrombotic events (Midgett et al., 2011). Binding of PGI2 to the IP receptor results in the activation of adenylate cyclase and a subsequent rise in cAMP levels in platelets (Yang et al., 2002). This stimulates phosphorylation of PKA, which suppresses various signalling pathways involved in platelet function such as adhesion, aggregation and granule secretion. With regard to the subject of this review, PKA activation decreases the release of Ca2+ from internal stores, reducing the activation of cytosolic PLA2 (cPLA2) and the liberation of AA from the phospholipid membrane, and so diminishing the production of platelet‐derived eicosanoids, such as TXA2 (den Dekker et al., 2002).
PGD2
PGD2 is well established as a macrophage product but, in lesser amounts, is also synthesized by platelets. By interaction with platelet DP1 receptors, PGD2 increases adenylyl cyclase activity and so, like PGI2, inhibits platelet activation (Bushfield et al., 1985; Oelz et al., 1977; Whittle et al., 1978).
PGE2
PGE2 is released by endothelial cells (ECs) and, to some extent, by activated platelets. It acts on a range of prostanoid receptors, EP1 ‐ EP4, that differently modulate second messengers, such as cAMP and free Ca2+, within platelets and exert contrasting effects on platelet function (Deeb et al., 2008; Yang et al., 2002). The effects on platelets of PGE2 acting through EP receptors are concentration dependent. At low concentrations (0.1–10 μmol·L−1), PGE2 binds to Gi‐coupled receptors (EP3) to enhance aggregation, whereas at higher concentrations (100 μmol·L−1), it activates Gs‐coupled receptors (EP2, EP4) to inhibit aggregation (Friedman et al., 2015; Glenn et al., 2012; Petrucci et al., 2011). Stimulation of EP3 receptors by PGE2 decreases cAMP levels, thus favouring platelet aggregation, but the full effect is only seen in the presence of another platelet agonist (Fabre et al., 2001; Friedman et al., 2015). On the other hand, the increased cAMP levels which accompany EP4 receptor activation correlate with suppressed platelet aggregation (Glenn et al., 2012).
In addition to PGE2, PGE1, PGF2α and PGD2 can also bind to EP3 and EP4 receptors but with lower affinity and reversible effects (Armstrong et al., 1985; Friedman et al., 2015; Glenn et al., 2012).
As well as the well‐characterized effects of PGE2 mediated through EP3 and EP4 receptors, EP1 receptors are also expressed on platelets (Kauskot and Hoylaerts, 2012; Petrucci et al., 2011). Although the signal transduction pathway is not clear, studies in several cell lines expressing EP1 receptors suggest that its activation increases Ca2+ influx and might thereby stimulate platelet aggregation (Whittle et al., 2012).
While PGE2 seems to both inhibit and potentiate platelet aggregation in vitro, a study by Gross et al. has elegantly shown that, in vivo, PGE2 is produced by the vessel wall or after the rupture of a plaque. Under these conditions, PGE2 activates the EP3 receptors on platelets and clearly enhances, rather than reduces, thrombus formation in the arterial vessel wall (Gross et al., 2007).
LOX‐dependent 12‐HETE
12‐HETE is the major 12‐LOX‐catalysed metabolite and the most abundant eicosanoid produced by platelets upon stimulation (Kirkby et al., 2015; Rauzi et al., 2016), but its effects on platelet function are not completely understood. Initial studies suggested that both 12‐HETE and 14‐hydroxy‐docosahexaenoic acid (14‐OH‐DHA), the 12‐LOX‐derived metabolite of DHA, inhibit platelet aggregation initiated by the TP receptor agonist U46619 (Croset et al., 1988). In agreement with these data, platelet‐specific knockout of 12‐LOX in mice resulted in hypersensitivity to ADP‐induced aggregation, which was reversed by incubation with exogenous 12‐HETE. However, lack of 12‐LOX did not affect collagen‐induced aggregation or platelet adhesion (Johnson et al., 1998). Interestingly, another study reported that inhibition of 12‐LOX led to decreased platelet aggregation that correlated with a significant reduction of 12‐HETE in response to collagen (Maskrey et al., 2014). A recent review concluded that 12‐HETE can exert both pro‐ and anti‐aggregatory effects on platelets that depend crucially on 12‐HETE concentration, stereospecificity and co‐incubation with different agonists (Porro et al., 2014). Platelets also produce hepoxilins from the precursor 12‐hydroperoxyeicosatetraenoic acid. Hepoxilin has shown to exert anti‐thrombotic effects in platelets (Margalit et al., 1995), most likely via inhibition of TXA2 formation and blockade of the TP receptor (Reynaud, 2002).
Platelet‐cellular crosstalk and eicosanoid biosynthesis
Transcellular routes through which platelets exchange eicosanoids with ECs or leukocytes are important to vascular homeostasis as well as to processes such as vascular inflammation. Some of these cellular crosstalk pathways are depicted in Figure 2 and discussed below. For example, ECs can utilize PGH2 released from platelets to produce PGI2. This suggests a counteractive mechanism in which activated platelets that are in direct contact with the vessel wall produce endoperoxide that can in turn be used by ECs to inhibit platelet functions and stimulate the return to homeostasis (Marcus et al., 1980; Porro et al., 2014).
Figure 2.
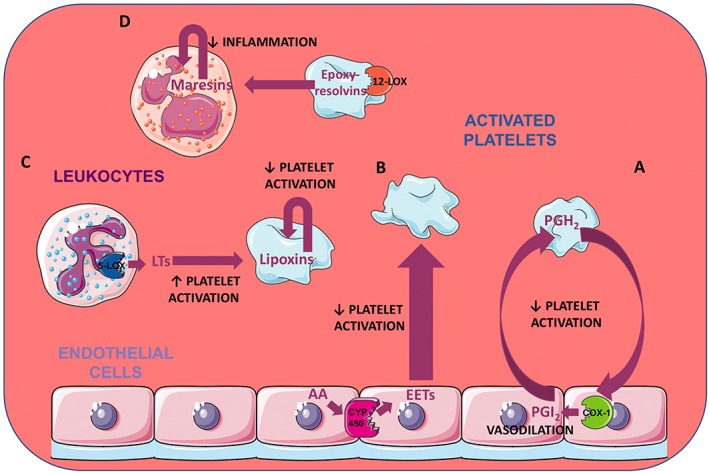
Main pathways of eicosanoid‐mediated crosstalk between platelets and other cells. The eicosanoid exchanges between platelets and ECs and their effects on the vessel homeostasis are illustrated in (A) and (B). Some of the PGH2 released by platelets may be used by COX‐1 in the ECs to produce PGI2 which induces vasodilation and prevents further platelet activation (A). ECs, on the other hand, can synthesize EETs starting from AA, through the action of CYP450. EETs reduce platelet activation (B). (C) and (D) represent some routes of platelet‐leukocyte crosstalk. LTs are synthesized in leukocytes by 5‐LOX and act together with other agonists to potentiate platelet activation. However, platelets can also use LTs to make lipoxins which reduce the activation of platelets (C). 12‐LOX in platelets also produces epoxy‐resolvins that can be used by the leukocytes to make maresins, molecules important for the resolution of inflammation (D).
CYP450 epoxygenases can convert AA into the biologically active EETs. The main producers of EETs are vascular ECs which not only release EETs following stimulation and contribute to vasodilation but also promote anti‐inflammatory effect in the vascular system (Yang, 2015). EETs also have potent anti‐adhesive and anti‐aggregatory activities which they exert by causing hyperpolarization of the platelet membrane (Sudhahar et al., 2010).
In the cardiovascular system, leukocytes represent the main source of 5‐LOX‐derived LTs. These metabolites potentiate adrenaline and thrombin‐induced platelet aggregation, probably by increasing the activity of TXA2 synthetase and thereby TXA2 formation (Mehta et al., 1986). On the other hand, platelets can utilize leukocyte‐derived LTA4 as a precursor for lipoxin production. Following release, lipoxin A4 acts on platelets via the FPR2/ALX receptor (Czapiga et al., 2005) and mediates protective functions by suppressing platelet adhesion, TXA2 formation and platelet–neutrophil interaction (Ortiz‐Muñoz et al., 2014). With regard to inflammation, platelets can transfer eicosanoid precursors to leukocytes which are fundamental for the formation of pro‐resolving mediators. A prominent example is the epoxy‐resolvins, which are produced by platelet 12‐LOX and transferred to neutrophils where they are transformed into maresins, which are molecules with important roles in terminating acute inflammatory responses (Abdulnour et al., 2014).
Modulation of eicosanoid production by platelets and the anti‐thrombotic efficacy of aspirin
John Vane reported for the first time that aspirin inhibits the production of PGs (Vane, 1971). This mechanism was identified as the basis of the therapeutic action of nonsteroidal anti‐inflammatory drugs (NSAIDs) (Vane, 1971) and was confirmed in platelets by Smith and Willis (1971). Many NSAIDs have been developed since then, and we know now that these compounds affect eicosanoid biosynthesis through the inhibition of both COX‐1 and COX‐2. COX‐1 and COX‐2 are expressed to differing levels in different tissues and under different conditions of health and disease. Such differences and their significance has been reviewed extensively (Khan et al., 2002; Mitchell and Warner, 2006; Wallace and Devchand, 2005).
In the context of platelet function, only aspirin produces irreversible inhibition of COX‐1 through its ability to covalently modify the enzyme (Cerletti et al., 1982; Loll et al., 1995). Consequently, aspirin impairs the synthesis of TXA2 for the entire platelet lifespan, and this explains its general antithrombotic action (Ferreira et al., 1971; Smith and Willis, 1971; Vane, 1971), although under some circumstances aspirin‐treated platelets may be able to recover the ability to synthesize TXA2 after de novo synthesis of COX‐1 (Evangelista et al., 2006). Because of its irreversible action, the antiplatelet effects of aspirin are seen with low doses of 50–100 mg·day−1 (Patrignani et al., 1982; Patrono, 2005; Warner et al., 2011). Aspirin is commonly given in combination with antagonists of ADP, acting at P2Y12 receptor, such as clopidogrel, prasugrel or ticagrelor (Bhatt, 2009; Gargiulo et al., 2016; Investigators TCIUaTPRET, 2001; Patrono et al., 2011; Wallentin et al., 2009; Windecker et al., 2014; Wiviott et al., 2007). Despite the proven anti‐thrombotic efficacy of this dual therapy, many studies are currently investigating the benefits of single antiplatelet‐drug therapy, using newer drugs such as ticagrelor (Gargiulo et al., 2016). The hope is to retain the anti‐thrombotic effects of dual antiplatelet therapy while lessening the unwanted side effects. This rationale is not only based on the need to reduce the bleeding risk associated with the dual antiplatelet therapy (Du et al., 2016; Maree and Fitzgerald, 2007) but also because evidence suggests that P2Y12 antagonists alone can decrease platelet TXA2 production and reduce aggregation mediated by TP receptor activation (Armstrong et al., 2010; Armstrong et al., 2011; Bhavaraju et al., 2010; Kirkby et al., 2011). Furthermore, the ability of aspirin to reduce the production of vascular PGI2 directly by inhibiting COX‐1 in ECs or indirectly by inhibiting COX‐1 in other cells supplying precursors of PGI2, such as PGH2, could produce a pro‐thrombotic effect that reduces the overall efficacy of dual antiplatelet therapy (Björkman et al., 2013; FitzGerald et al., 1983; Franchi et al., 2016; Mahaffey et al., 2011; Maree and Fitzgerald, 2007; Warner et al., 2010; Warner et al., 2016). Therefore, it is necessary not only to seek therapeutic strategies apart from aspirin, but also to extensively re‐evaluate the effects of aspirin in vivo. This last goal could be achieved by using more recently developed techniques such as liquid chromatography–tandem mass spectrometry or the genetic manipulation of animals. For example, we have recently found, through the use of mass spectrometry analysis, that aspirin prevents not only the synthesis of TXA2 by platelets but also the production of PGD2, PGE2, 11‐HETE and 15‐HETE. PGD2 and PGE2 are PGs with antiplatelet actions and their inhibition can further contribute to a reduced efficacy of the antithrombotic treatments (Rauzi et al., 2016). In addition, our own recently developed animal models where the expression of COX‐1 is specifically ablated in ECs or in megakaryocytes/platelets will be useful in dissecting the effects of eicosanoids on the cardiovascular system and the outcomes of aspirin treatment.
Eicosanoid measurements and platelet function tests to evaluate the efficacy of aspirin in cardiovascular patients
The way platelets respond to treatment with aspirin can be monitored in the laboratory either by techniques that specifically measure platelet COX‐1 activity or by tests assessing other platelet activation pathways besides COX‐1.
The measurement of platelet‐generated eicosanoids, in particular of TXB2, the stable form of TXA2, either in serum or after in vitro stimulation of platelets, falls in the first category of techniques. With a strong stimulus, the levels of TXB2 can be taken as reflecting the maximal capacity of platelets to synthesize TXA2 via the COX‐1 pathway and this can be regarded as a sensitive measure of the response to aspirin, in the laboratory (Cattaneo, 2007; Maree and Fitzgerald, 2007; Ohmori et al., 2006). On the other hand, the levels of the main TXA2 metabolite found in urine, 11‐ dehydro TXB2, reflect systemic TXA2 generation and may not only reflect the effect of aspirin on platelet COX‐1 (Kirkby et al., 2012; Kirkby et al., 2015; Smith et al., 2012).
Another standard test for studies of platelet inhibition by aspirin is light transmission aggregometry, which measures the ability of platelets to aggregate after being stimulated. Different stimuli can be used in this test to explore different aspects of platelet activation. AA is a substrate for COX‐1, so the aggregation response to this agonist closely reflects platelet COX‐1 activity, while ADP or collagen induces platelet aggregation through pathways that are not exclusively dependent on COX‐1 activation (Thiagarjan and Wu, 2002). Other methodologies, such as flow cytometry evaluation of markers of platelet activation and secretion or of the formation of platelet‐leukocyte aggregates, can also be used to assess platelet inhibition by aspirin. Moreover, semi‐automated point‐of‐care platelet function assays, such as the PFA‐100® system and RPFA‐Verify‐Now Aspirin, have been introduced (Frelinger et al., 2006).
The prevalence of aspirin resistance, that is, lack of effect of aspirin, reported in the literature is largely based on various non‐specific laboratory techniques and, in general, aspirin resistance is much lower when measured with COX‐1 specific methods (Gurbel et al., 2007; Lordkipanidzé et al., 2007).
It is generally held that aspirin should inhibit platelet TXA2 synthesis by at least 95% to reach a functional effect, and this assumption is mainly based on the observation that there is a non‐linear relationship between inhibition of platelet TXA2 synthesis and inhibition of platelet aggregation (Kidson‐Gerber et al., 2010 ; Santilli et al., 2009). However, due to the technical limitations of the tests employed, platelet response to aspirin is usually evaluated using one or two agonists, often at fixed concentration that does not make it possible to properly characterize biological variations in drug response. Recently, we have developed a test using optical multichannel platelet aggregometry in a 96‐well‐plate, that can explore platelet function in response to a broad range of agonists and agonist concentrations (Chan et al., 2011; Lordkipanidzé et al., 2014). This test has indicated that there is a linear relationship between TXA2 synthesis and TXA2‐mediated platelet aggregation, in the presence of different levels of COX‐1 inhibition and could represent a valid alternative method of reliably identifying responders to treatment with aspirin (Armstrong et al., 2008).
The association between a high platelet reactivity while on treatment, and the risk of patients having a thrombotic event is uncertain (Consuegra‐Sánchez et al., 2013; Depta et al., 2012; Li et al., 2014; Tantry et al., 2013). However, four different meta‐analyses have so far indicated that the lack of response to aspirin, as detected in the laboratory, may predict clinical recurrences (Crescente et al., 2008a; Crescente et al., 2008b; Krasopoulos et al., 2008; Reny et al., 2008; Snoep et al., 2007). It also appears, from some of the studies performed in this area, that a combination of tests and of different agonists is better than one single test to establish this type of association (Armstrong et al., 2008; Crescente et al., 2011; Gremmel et al., 2015; Smith et al., 2012) and a summary of these observations is provided in Figure 3 . However, it is essential that additional biomarkers of response to aspirin are identified and larger epidemiological studies performed, before any change of an antiplatelet treatment is made on the basis of laboratory test results. Notably, there have been no clinical trials demonstrating that tailoring antiplatelet therapy to results from ex vivo platelet testing, produces an improvement in patient outcomes (Collet et al., 2012; Depta et al., 2012).
Figure 3.
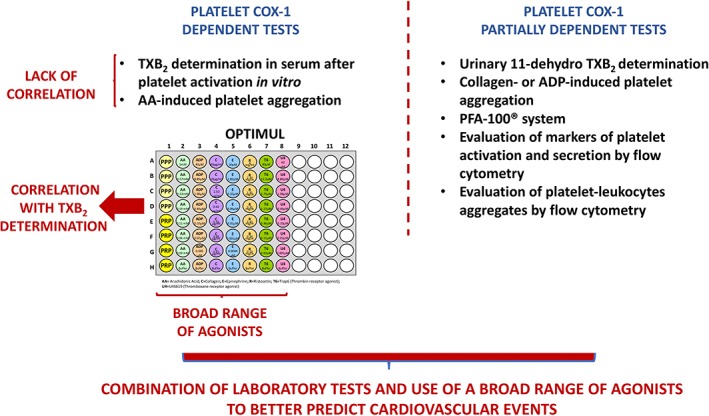
Schematic representation of platelet function tests used to monitor responses to aspirin in cardiovascular patients.
Anti‐cancer effect of aspirin: role for platelet eicosanoids
In 1988, Kune et al. reported for the first time an association between the intake of aspirin and a reduced risk of colorectal cancer, thus extending the therapeutic potential of aspirin beyond its use as an anti‐inflammatory or anti‐thrombotic drug. This observation was confirmed by many subsequent epidemiological studies and by a large meta‐analysis which also showed that aspirin reduced the risk of gastrointestinal cancers in general (Algra and Rothwell, 2012; Burn et al., 2008; Burn et al., 2011; Cole et al., 2009; Cuzick et al., 2015; Rothwell et al., 2012). As well as aspirin, non‐aspirin NSAIDS and, in particular, COX‐2 selective inhibitors, such as celecoxib and rofecoxib, were widely reported to prevent colonic tumourigenesis (Arber et al., 2006; Arber et al., 2011; Baron et al., 2006; Bertagnolli et al., 2006; Cao et al., 2016; Steinbach et al., 2000). However, concerns about the pro‐thrombotic effects of non‐aspirin NSAIDs including COX‐2 inhibitors (Baron et al., 2006; Baron et al., 2008; Collaboration CaTNTC, 2013) have ended cancer prevention trials using COX‐2 inhibitors , and the US Preventive Services Task Force (USPSTF) no longer supports the use of non‐aspirin NSAIDs for the prevention of colorectal cancer.
In contrast, aspirin is the only drug with no cardiovascular risk that is effective in both primary and secondary prevention of colorectal cancer and also reduces the incidence and risk of all‐cause cancer mortality (Cuzick et al., 2015; Rothwell et al., 2011). As aspirin is used in prevention of cardiovascular diseases and the most colorectal cancer cases are diagnosed after the age of 50, the last guidelines from the USPSTF recommend low‐dose aspirin for the primary prevention of colorectal cancer in patients at increased cardiovascular risk (Bibbins‐Domingo, 2016).
The follow‐up studies of many clinical trials indicate that the chemoprotective action of aspirin can be detected at a dose as low as 75 mg·day−1. Furthermore, it is saturable at these low doses and is present when using a controlled‐release aspirin formulation that mainly targets platelet COX‐1 (Patrignani and Patrono, 2016). These findings have been confirmed by studies showing that small doses of aspirin, by blocking the formation of platelet TXA2, PGE2, PG‐containing oxidized phospholipids and sphingosine 1‐phosphate, reduce the exchange of lipid mediators between platelets and cancer cells in the tumour micro‐environment (Aldrovandi et al., 2013; Dovizio et al., 2013; Ulrych et al., 2011).
Strong evidence also suggests that eicosanoids linked to COX‐1 activity act as pro‐angiogenic factors and therefore the anti‐cancer effects of aspirin are also related to a reduction of angiogenesis (Etulain et al., 2013; Rauzi et al., 2016). For example, we have recently found that platelet COX‐1‐derived 15(S)‐HETE induces an angiogenic response in HMEC‐1 cells and rat aortic rings and this effect disappears in presence of aspirin, when the synthesis of 15(S)‐HETE is blocked (Rauzi et al., 2016). In addition to the eicosanoids, platelets can release a variety of pro‐angiogenic factors from their α‐granules and this release can be modulated by treatment with aspirin, as well (Coppinger et al., 2004).
Platelets promote cancer progression also by favouring the metastatic process. In particular, platelets will form aggregates around tumour cells in the bloodstream, that protect tumor cells from being cleared by the immune system (Gay and Felding‐Habermann, 2011). Also, when COX‐1 activity is blocked by aspirin or when a PGE2 antagonist is used, platelets lose the ability to transform human colon carcinoma cells into mesenchymal‐like cancer cells. Moreover, the administration of aspirin to mice prevents the platelet‐induced formation of metastases in the lungs, and this is associated with a reduced systemic synthesis of TXA2 and PGE2 (Guillem‐Llobat et al., 2016).
This evidence suggests that the anti‐cancer efficacy of aspirin resides in its ability to block the biosynthesis of platelet‐derived eicosanoids, which not only serve as substrates for other cells present in the tumour micro‐environment but also promote angiogenesis and the metastatic progression of the tumour (Figure 4). While there is strong evidence for aspirin having beneficial effects in gastrointestinal cancers, the efficacy of aspirin in other cancer types such as gastroesophageal, breast and prostate cancers has still to be evaluated, as well as the most appropriate timings and doses that can be used to maximize its anti‐carcinogenic effects (Patrignani and Patrono, 2016).
Figure 4.
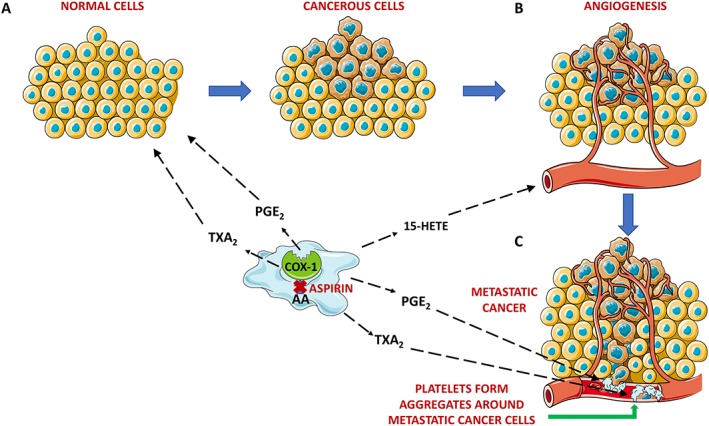
Effects of platelet COX‐1‐derived eicosanoids and of aspirin treatment in the progression of cancer. The preventive role of aspirin in the progression of cancer depends at least in part on its ability to block the formation of eicosanoids by platelet COX‐1. TXA2 and PGE2 are released in the tumour micro‐environment and favour the transformation of cells from a normal to a cancerous phenotype (A). 15‐HETE is another eicosanoid synthesised by COX‐1 in platelets that promotes angiogenesis, a process that further promotes cancer progression (B). TXA2 and PGE2 mediate the formation of platelet aggregates around the metastatic cancer cells, protecting them from the immune system and assisting their spread throughout the body (C).
Conclusions
Eicosanoids produced by platelets, or made from other cells, are important modulators of platelet function and regulate the fine balance between haemostasis and thrombotic disease. The eicosanoid‐mediated crosstalk between platelets and other cells also regulates pathophysiological processes such as cancer. Low doses of aspirin, through their ability to inhibit platelet COX‐1 and the synthesis of pro‐aggregatory TXA2, is still nowadays considered as a first choice treatment to reduce the risk of thrombotic events. Ongoing research may lead to the replacement of aspirin in this role by P2Y12 receptor antagonists, while aspirin continues to be used for protection against the development of a range of cancers.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
All the figures were produced using Servier Medical Art (http://www.servier.com). This work was supported by grants from the British Heart Foundation (PG/15/47/31591; PG/15/79/31777 and PG/17/40/33028) and European Commission (H2020‐MSCA‐ITN‐2015 GA 675111) to T.D.W.
Crescente, M. , Menke, L. , Chan, M. V. , Armstrong, P. C. , and Warner, T. D. (2019) Eicosanoids in platelets and the effect of their modulation by aspirin in the cardiovascular system (and beyond). British Journal of Pharmacology, 176: 988–999. 10.1111/bph.14196.
References
- Abdulnour R‐EE, Dalli J, Colby JK, Krishnamoorthy N, Timmons JY, Tan SH et al (2014). Maresin 1 biosynthesis during platelet–neutrophil interactions is organ‐protective. Proc Natl Acad Sci U S A 111: 16526–16531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrovandi M, Hammond VJ, Podmore H, Hornshaw M, Clark SR, Marnett LJ et al (2013). Human platelets generate phospholipid‐esterified prostaglandins via cyclooxygenase‐1 that are inhibited by low dose aspirin supplementation. J Lipid Res 54: 3085–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174 (Suppl 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174 (Suppl 1): S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algra AM, Rothwell PM (2012). Effects of regular aspirin on long‐term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol 13: 518–527. [DOI] [PubMed] [Google Scholar]
- Arber N, Eagle CJ, Spicak J, Rácz I, Dite P, Hajer J et al (2006). Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med 355: 885–895. [DOI] [PubMed] [Google Scholar]
- Arber N, Spicak J, Racz I, Zavoral M, Breazna A, Gerletti P et al (2011). Five‐year analysis of the prevention of colorectal sporadic adenomatous polyps trial. Am J Gastroenterol 106: 1135–1146. [DOI] [PubMed] [Google Scholar]
- Armstrong PC, Dhanji AR, Tucker AT, Mitchell JA, Warner TD (2010). Reduction of platelet thromboxane A2 production ex vivo and in vivo by clopidogrel therapy. J Thromb Haemost 8: 613–615. [DOI] [PubMed] [Google Scholar]
- Armstrong PC, Leadbeater PD, Chan MV, Kirkby NS, Jakubowski JA, Mitchell JA et al (2011). In the presence of strong P2Y12 receptor blockade, aspirin provides little additional inhibition of platelet aggregation. J Thromb Haemost 9: 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong PCJ, Truss NJ, Ali FY, Dhanji AA, Vojnovic I, Zain ZNM et al (2008). Aspirin and the in vitro linear relationship between thromboxane A2‐mediated platelet aggregation and platelet production of thromboxane A2. J Thromb Haemost 6: 1933–1943. [DOI] [PubMed] [Google Scholar]
- Armstrong RA, Jones RL, Wilson NH (1985). Mechanism of the inhibition of platelet aggregation produced by prostaglandin F2 alpha. Prostaglandins 29: 601–610. [DOI] [PubMed] [Google Scholar]
- Baron JA, Sandler RS, Bresalier RS, Lanas A, Morton DG, Riddell R et al (2008). Cardiovascular events associated with rofecoxib: final analysis of the APPROVe trial. The Lancet 372: 1756–1764. [DOI] [PubMed] [Google Scholar]
- Baron JA, Sandler RS, Bresalier RS, Quan H, Riddell R, Lanas A et al (2006). A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology 131: 1674–1682. [DOI] [PubMed] [Google Scholar]
- Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K et al (2006). Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med 355: 873–884. [DOI] [PubMed] [Google Scholar]
- Bhatt DL (2009). Role of antiplatelet therapy across the spectrum of patients with coronary artery disease. Am J Cardiol 103: 11A–19A. [DOI] [PubMed] [Google Scholar]
- Bhavaraju K, Georgakis A, Jin J, Gartner TK, Tomiyama Y, Nurden A et al (2010). Antagonism of P2Y(1)(2) reduces physiological thromboxane levels. Platelets 21: 604–609. [DOI] [PubMed] [Google Scholar]
- Bibbins‐Domingo K, USPST (2016). Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. preventive services task force recommendation statement. Ann Intern Med 164: 836–845. [DOI] [PubMed] [Google Scholar]
- Björkman J‐A, Zachrisson H, Forsberg G‐B, Von Bahr H, Hansson GI, Warner TD et al (2013). High‐dose aspirin in dogs increases vascular resistance with limited additional anti‐platelet effect when combined with potent P2Y12 inhibition. Thromb Res 131: 313–319. [DOI] [PubMed] [Google Scholar]
- Bolego C, Buccellati C, Prada A, Gaion RM, Folco G, Sala A (2009). Critical role of COX‐1 in prostacyclin production by human endothelial cells under modification of hydroperoxide tone. FASEB J 23: 605–612. [DOI] [PubMed] [Google Scholar]
- Bunting S, Moncada S, Vane JR (1977). Antithrombotic properties of vascular endothelium. Lancet 2: 1075–1076. [DOI] [PubMed] [Google Scholar]
- Bunting S, Moncada S, Vane JR (1983). The prostacyclin–thromboxane A2 balance: pathophysiological and therapeutic implications. Br Med Bull 39: 271–276. [DOI] [PubMed] [Google Scholar]
- Burn J, Bishop DT, Mecklin J‐P, Macrae F, Möslein G, Olschwang S et al (2008). Effect of aspirin or resistant starch on colorectal neoplasia in the lynch syndrome. N Engl J Med 359: 2567–2578. [DOI] [PubMed] [Google Scholar]
- Burn J, Gerdes A‐M, Macrae F, Mecklin J‐P, Moeslein G, Olschwang S et al (2011). Long‐term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. The Lancet 378: 2081–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushfield M, Mcnicol A, Macintyre DE (1985). Inhibition of platelet‐activating‐factor‐induced human platelet activation by prostaglandin D2. Differential sensitivity of platelet transduction processes and functional responses to inhibition by cyclic AMP. Biochem J 232: 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Nishihara R, Wu K, Wang M, Ogino S, Willett WC et al (2016). Population‐wide impact of long‐term use of aspirin and the risk for cancer. JAMA Oncol 2: 762–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo M (2007). Resistance to antiplatelet drugs: molecular mechanisms and laboratory detection. J Thromb Haemost 5: 230–237. [DOI] [PubMed] [Google Scholar]
- Cerletti C, Livio M, De Gaetano G (1982). Non‐steroidal anti‐inflammatory drugs react with two sites on platelet cyclo‐oxygenase. Evidence from “in vivo” drug interaction studies in rats. Biochim Biophys Acta 714: 122–128. [DOI] [PubMed] [Google Scholar]
- Chan MV, Armstrong PCJ, Papalia F, Kirkby NS, Warner TD (2011). Optical multichannel (optimul) platelet aggregometry in 96‐well plates as an additional method of platelet reactivity testing. Platelets 22: 485–494. [DOI] [PubMed] [Google Scholar]
- Cole BF, Logan RF, Halabi S, Benamouzig R, Sandler RS, Grainge MJ et al (2009). Aspirin for the chemoprevention of colorectal adenomas: meta‐analysis of the randomized Trials. J Natl Cancer Inst 101: 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaboration CaTNTC (2013). Vascular and upper gastrointestinal effects of non‐steroidal anti‐inflammatory drugs: meta‐analyses of individual participant data from randomised trials. The Lancet 382: 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collet JP, Cuisset T, Range G, Cayla G, Elhadad S, Pouillot C et al (2012). Bedside monitoring to adjust antiplatelet therapy for coronary stenting. N Engl J Med 367: 2100–2109. [DOI] [PubMed] [Google Scholar]
- Consuegra‐Sánchez L, López‐Palop R, Cano P, Carrillo P, Picó F, Villegas M et al (2013). Assessment of high on‐treatment platelet reactivity in patients with ischemic heart disease: concordance between the Multiplate and VerifyNow assays. J Thromb Haemost 11: 379–381. [DOI] [PubMed] [Google Scholar]
- Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, Mcredmond JP et al (2004). Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 103: 2096–2104. [DOI] [PubMed] [Google Scholar]
- Crescente M, Castelnuovo AD, Iacoviello L, Gaetano GD, Cerletti C (2008a). PFA‐100 closure time to predict cardiovascular events in aspirin‐treated cardiovascular patients: a meta‐analysis of 19 studies comprising 3,003 patients. Thromb Haemost 99: 1129–1131. [DOI] [PubMed] [Google Scholar]
- Crescente M, Castelnuovo AD, Iacoviello L, Vermylen J, Cerletti C, Gaetano GD (2008b). Response variability to aspirin as assessed by the platelet function analyzer (PFA)‐100 – a systematic review. Thromb Haemost 99: 14–26. [DOI] [PubMed] [Google Scholar]
- Crescente M, Mezzasoma AM, Del Pinto M, Palmerini F, Di Castelnuovo A, Cerletti C et al (2011). Incomplete inhibition of platelet function as assessed by the platelet function analyzer (PFA‐100) identifies a subset of cardiovascular patients with high residual platelet response while on aspirin. Platelets 22: 179–187. [DOI] [PubMed] [Google Scholar]
- Croset M, Sala A, Folco G, Lagarde M (1988). Inhibition by lipoxygenase products of TXA2‐like responses of platelets and vascular smooth muscle: 14‐Hydroxy from 22:6N‐3 is more potent than 12‐HETE. Biochem Pharmacol 37: 1275–1280. [DOI] [PubMed] [Google Scholar]
- Cuzick J, Thorat MA, Bosetti C, Brown PH, Burn J, Cook NR et al (2015). Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann Oncol 26: 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czapiga M, Gao J‐L, Kirk A, Lekstrom‐Himes J (2005). Human platelets exhibit chemotaxis using functional N‐formyl peptide receptors. Exp Hematol 33: 73–84. [DOI] [PubMed] [Google Scholar]
- Deeb RS, Upmacis RK, Lamon BD, Gross SS, Hajjar DP (2008). Maintaining equilibrium by selective targeting of cyclooxygenase pathways. Promising Offensives Against Vascular Injury. Hypertension 51: 1–7. [DOI] [PubMed] [Google Scholar]
- den Dekker E, Gorter G, Heemskerk JWM, Akkerman J‐WN (2002). Development of platelet inhibition by cAMP during megakaryocytopoiesis. J Biol Chem 277: 29321–29329. [DOI] [PubMed] [Google Scholar]
- Dennis EA, Norris PC (2015). Eicosanoid storm in infection and inflammation. Nat Rev Immunol 15: 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depta JP, Fowler J, Novak E, Katzan I, Bakdash S, Kottke‐Marchant K et al (2012). Clinical outcomes using a platelet function‐guided approach for secondary prevention in patients with ischemic stroke or transient ischemic attack. Stroke 43: 2376–2381. [DOI] [PubMed] [Google Scholar]
- Dovizio M, Maier TJ, Alberti S, Di Francesco L, Marcantoni E, Münch G et al (2013). Pharmacological inhibition of platelet‐tumor cell cross‐talk prevents platelet‐induced overexpression of cyclooxygenase‐2 in HT29 human colon carcinoma cells. Mol Pharmacol 84: 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G, Lin Q, Wang J (2016). A brief review on the mechanisms of aspirin resistance. Int J Cardiol 220: 21–26. [DOI] [PubMed] [Google Scholar]
- Etulain J, Fondevila C, Negrotto S, Schattner M (2013). Platelet‐mediated angiogenesis is independent of VEGF and fully inhibited by aspirin. Br J Pharmacol 170: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista V, Manarini S, Di Santo A, Capone ML, Ricciotti E, Di Francesco L et al (2006). De novo synthesis of cyclooxygenase‐1 counteracts the suppression of platelet thromboxane biosynthesis by aspirin. Circ Res 98: 593–595. [DOI] [PubMed] [Google Scholar]
- Fabre J‐E, Nguyen M, Athirakul K, Coggins K, Mcneish JD, Austin S et al (2001). Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation. J Clin Invest 107: 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira SH, Moncada S, Vane JR (1971). Indomethacin and aspirin abolish prostaglandin release from the spleen. Nat New Biol 231: 237–239. [DOI] [PubMed] [Google Scholar]
- FitzGerald GA, Oates JA, Hawiger J, Maas RL, Roberts LJ 2nd, Lawson JA et al (1983). Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man. J Clin Invest 71: 676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi F, Rollini F, Aggarwal N, Hu J, Kureti M, Durairaj A et al (2016). Pharmacodynamic comparison of prasugrel versus ticagrelor in patients with type 2 diabetes mellitus and coronary artery disease. Clinical Perspective. The OPTIMUS (Optimizing Antiplatelet Therapy in Diabetes Mellitus)‐4 Study . Circulation 134: 780–792. [DOI] [PubMed] [Google Scholar]
- Frelinger AL, Furman MI, Linden MD, Li Y, Fox ML, Barnard MR et al (2006). Residual arachidonic acid–induced platelet activation via an adenosine diphosphate–dependent but cyclooxygenase‐1– and cyclooxygenase‐2–independent pathway. A 700‐Patient Study of Aspirin Resistance. Circulation 113: 2888–2896. [DOI] [PubMed] [Google Scholar]
- Friedman EA, Ogletree ML, Haddad EV, Boutaud O (2015). Understanding the role of prostaglandin E2 in regulating human platelet activity in health and disease. Thromb Res 136: 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargiulo G, Windecker S, Vranckx P, Gibson CM, Mehran R, Valgimigli M (2016). A critical appraisal of aspirin in secondary prevention: is less more? Circulation 134: 1881–1906. [DOI] [PubMed] [Google Scholar]
- Gay LJ, Felding‐Habermann B (2011). Contribution of platelets to tumour metastasis. Nat Rev Cancer 11: 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn JR, White AE, Iyu D, Heptinstall S (2012). PGE2 reverses Gs‐mediated inhibition of platelet aggregation by interaction with EP3 receptors, but adds to non‐Gs‐mediated inhibition of platelet aggregation by interaction with EP4 receptors. Platelets 23: 344–351. [DOI] [PubMed] [Google Scholar]
- Gremmel T, Koppensteiner R, Panzer S (2015). Comparison of aggregometry with flow cytometry for the assessment of agonists‐induced platelet reactivity in patients on dual antiplatelet therapy. PLoS One 10: e0129666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S, Tilly P, Hentsch D, Vonesch J‐L, Fabre J‐E (2007). Vascular wall‐produced prostaglandin E2 exacerbates arterial thrombosis and atherothrombosis through platelet EP3 receptors. J Exp Med 204: 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillem‐Llobat P, Dovizio M, Bruno A, Ricciotti E, Cufino V, Sacco A et al (2016). Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 7: 32462–32477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurbel PA, Bliden KP, Dichiara J, Newcomer J, Weng W, Neerchal NK et al (2007). Evaluation of dose‐related effects of aspirin on platelet function. Results from the Aspirin‐Induced Platelet Effect (ASPECT) Study. Circulation 115: 3156–3164. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkewicz R, Dennis EA (2011). Applications of mass spectrometry to lipids and membranes. Annu Rev Biochem 80: 301–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschman HR, Reddy ST, Xie W (1997). Function and regulation of prostaglandin synthase‐2. Adv Exp Med Biol 407: 61–66. [DOI] [PubMed] [Google Scholar]
- Hla T, Neilson K (1992). Human cyclooxygenase‐2 cDNA. Proc Natl Acad Sci U S A 89: 7384–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Investigators TCIUaTPRET (2001). Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST‐segment elevation. N Engl J Med 345: 494–502. [DOI] [PubMed] [Google Scholar]
- Johnson EN, Brass LF, Funk CD (1998). Increased platelet sensitivity to ADP in mice lacking platelet‐type 12‐lipoxygenase. Proc Natl Acad Sci U S A 95: 3100–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauskot A, Hoylaerts MF (2012). Platelet receptors In: Gresele P, Born GVR, Patrono C, Page CP. (eds). Antiplatelet Agents. Springer Berlin Heidelberg: Berlin, Heidelberg. [Google Scholar]
- Khan KNM, Paulson SK, Verburg KM, Lefkowith JB, Maziasz TJ (2002). Pharmacology of cyclooxygenase‐2 inhibition in the kidney. Kidney Int 61: 1210–1219. [DOI] [PubMed] [Google Scholar]
- Kidson‐Gerber G, Weaver J, Gemmell R, Prasan AM, Chong BH (2010). Serum thromboxane B2 compared to five other platelet function tests for the evaluation of aspirin effect in stable cardiovascular Disease. Heart Lung Circ 19: 234–242. [DOI] [PubMed] [Google Scholar]
- Kirkby NS, Leadbeater PDM, Chan MV, Nylander S, Mitchell JA, Warner TD (2011). Antiplatelet effects of aspirin vary with level of P2Y12 receptor blockade supplied by either ticagrelor or prasugrel. J Thromb Haemost 9: 2103–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Lundberg MH, Harrington LS, Leadbeater PDM, Milne GL, Potter CMF et al (2012). Cyclooxygenase‐1, not cyclooxygenase‐2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci U S A 109: 17597–17602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby NS, Reed DM, Edin ML, Rauzi F, Mataragka S, Vojnovic I et al (2015). Inherited human group IVA cytosolic phospholipase A2 deficiency abolishes platelet, endothelial, and leucocyte eicosanoid generation. FASEB J 29: 4568–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasopoulos G, Brister SJ, Beattie WS, Buchanan MR (2008). Aspirin “resistance” and risk of cardiovascular morbidity: systematic review and meta‐analysis. BMJ 336: 195–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR (1991). TIS10, a phorbol ester tumor promoter‐inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem 266: 12866–12872. [PubMed] [Google Scholar]
- Kune GA, Kune S, Watson LF (1988). Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res 48: 4399–4404. [PubMed] [Google Scholar]
- Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC et al (1997). Disruption of the mouse cyclooxygenase 1 gene. Characteristics of the mutant and areas of future study. Adv Exp Med Biol 407: 87–92. [PubMed] [Google Scholar]
- Li J, Jian Z, Song M, Guo W, Chen G, Lu W et al (2014). Tailored antiplatelet therapy and clinical adverse outcomes. Heart 100: 41–46. [DOI] [PubMed] [Google Scholar]
- Loll PJ, Picot D, Garavito RM (1995). The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat Struct Biol 2: 637–643. [DOI] [PubMed] [Google Scholar]
- Lordkipanidzé M, Lowe GC, Kirkby NS, Chan MV, Lundberg MH, Morgan NV et al (2014). Characterization of multiple platelet activation pathways in patients with bleeding as a high‐throughput screening option: use of 96‐well Optimul assay. Blood 123: e11–e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lordkipanidzé M, Pharand C, Schampaert E, Turgeon J, Palisaitis DA, Diodati JG (2007). A comparison of six major platelet function tests to determine the prevalence of aspirin resistance in patients with stable coronary artery disease. Eur Heart J 28: 1702–1708. [DOI] [PubMed] [Google Scholar]
- Mahaffey KW, Wojdyla DM, Carroll K, Becker RC, Storey RF, Angiolillo DJ et al (2011). Ticagrelor compared with clopidogrel by geographic region in the platelet inhibition and patient outcomes (PLATO) Trial. Clinical Perspective. Circulation 124: 544–554. [DOI] [PubMed] [Google Scholar]
- Marcus AJ, Weksler BB, Jaffe EA, Broekman MJ (1980). Synthesis of prostacyclin from platelet‐derived endoperoxides by cultured human endothelial cells. J Clin Invest 66: 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maree AO, Fitzgerald DJ (2007). Variable platelet response to aspirin and clopidogrel in atherothrombotic disease. Circulation 115: 2196–2207. [DOI] [PubMed] [Google Scholar]
- Margalit A, Gilutz H, Granot Y (1995). Original article: low regulatory volume decrease rate in platelets from ischemic patients: a possible role for hepoxilin A3 in thrombogenicity. Platelets 6: 371–376. [DOI] [PubMed] [Google Scholar]
- Masferrer JL, Seibert K, Zweifel B, Needleman P (1992). Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc Natl Acad Sci U S A 89: 3917–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskrey BH, Rushworth GF, Law MH, Treweeke AT, Wei J, Leslie SJ et al (2014). 12‐Hydroxyeicosatetraenoic acid is associated with variability in aspirin‐induced platelet inhibition. J Inflamm 11: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P, Mehta J, Lawson D, Krop I, Letts LG (1986). Leukotrienes potentiate the effects of epinephrine and thrombin on human platelet aggregation. Thromb Res 41: 731–738. [DOI] [PubMed] [Google Scholar]
- Midgett C, Stitham J, Martin KA, Hwa J (2011). Prostacyclin receptor regulation – from transcription to trafficking. Curr Mol Med 11: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Warner TD (2006). COX isoforms in the cardiovascular system: understanding the activities of non‐steroidal anti‐inflammatory drugs. Nat Rev Drug Discov 5: 75–86. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Kirkby NS (2018). Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br J Pharmacology. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Gryglewski R, Bunting S, Vane JR (1976). An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature 263: 663–665. [DOI] [PubMed] [Google Scholar]
- Moncada S, Korbut R, Bunting S, Vane JR (1978). Prostacyclin is a circulating hormone. Nature 273: 767–768. [DOI] [PubMed] [Google Scholar]
- Needleman P, Moncada S, Bunting S, Vane JR, Hamberg M, Samuelsson B (1976). Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature 261: 558–560. [DOI] [PubMed] [Google Scholar]
- O'Banion MK, Winn VD, Young DA (1992). cDNA cloning and functional activity of a glucocorticoid‐regulated inflammatory cyclooxygenase. Proc Natl Acad Sci U S A 89: 4888–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelz O, Oelz R, Knapp HR, Sweetman BJ, Oates JA (1977). Biosynthesis of prostaglandin D2. 1. Formation of prostaglandin D2 by human platelets. Prostaglandins 13: 225–234. [DOI] [PubMed] [Google Scholar]
- Ohmori T, Yatomi Y, Nonaka T, Kobayashi Y, Madoiwa S, Mimuro J et al (2006). Aspirin resistance detected with aggregometry cannot be explained by cyclooxygenase activity: involvement of other signaling pathway(s) in cardiovascular events of aspirin‐treated patients. J Thromb Haemost 4: 1271–1278. [DOI] [PubMed] [Google Scholar]
- Ortiz‐Muñoz G, Mallavia B, Bins A, Headley M, Krummel MF, Looney MR (2014). Aspirin‐triggered 15‐epi‐lipoxin A4 regulates neutrophil‐platelet aggregation and attenuates acute lung injury in mice. Blood 124: 2625–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrignani P, Filabozzi P, Patrono C (1982). Selective cumulative inhibition of platelet thromboxane production by low‐dose aspirin in healthy subjects. J Clin Invest 69: 1366–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrignani P, Patrono C (2016). Aspirin and cancer. J Am Coll Cardiol 68: 967–976. [DOI] [PubMed] [Google Scholar]
- Patrono C (2005). Low‐dose aspirin for the prevention of atherothrombosis. N Engl J Med 353: 2373–2383. [DOI] [PubMed] [Google Scholar]
- Patrono C, Andreotti F, Arnesen H, Badimon L, Baigent C, Collet JP et al (2011). Antiplatelet agents for the treatment and prevention of atherothrombosis. Eur Heart J 32: 2922–2932. [DOI] [PubMed] [Google Scholar]
- Petrucci G, De Cristofaro R, Rutella S, Ranelletti FO, Pocaterra D, Lancellotti S et al (2011). Prostaglandin E2 differentially modulates human platelet function through the prostanoid EP2 and EP3 receptors. J Pharmacol Exp Ther 336: 391–402. [DOI] [PubMed] [Google Scholar]
- Piper PJ, Vane JR (1969). Release of additional factors in anaphylaxis and its antagonism by anti‐inflammatory drugs. Nature 223: 29–35. [DOI] [PubMed] [Google Scholar]
- Porro B, Songia P, Squellerio I, Tremoli E, Cavalca V (2014). Analysis, physiological and clinical significance of 12‐HETE: a neglected platelet‐derived 12‐lipoxygenase product. J Chromatogr B 964: 26–40. [DOI] [PubMed] [Google Scholar]
- Rauzi F, Kirkby NS, Edin ML, Whiteford J, Zeldin DC, Mitchell JA et al (2016). Aspirin inhibits the production of proangiogenic 15(S)‐HETE by platelet cyclooxygenase‐1. FASEB J 30: 4256–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly M, Fitzgerald GA (1993). Cellular activation by thromboxane A2 and other eicosanoids. Eur Heart J 14 (Suppl K): 88–93. [PubMed] [Google Scholar]
- Reny JL, De Moerloose P, Dauzat M, Fontana P (2008). Use of the PFA‐100™ closure time to predict cardiovascular events in aspirin‐treated cardiovascular patients: a systematic review and meta‐analysis. J Thromb Haemost 6: 444–450. [DOI] [PubMed] [Google Scholar]
- Reynaud D (2002). The hepoxilin analog PBT‐3 inhibits heparin‐activated platelet aggregation evoked by ADP. FEBS Lett 515: 58–60. [DOI] [PubMed] [Google Scholar]
- Rocca B, Secchiero P, Ciabattoni G, Ranelletti FO, Catani L, Guidotti L et al (2002). Cyclooxygenase‐2 expression is induced during human megakaryopoiesis and characterizes newly formed platelets. Proc Natl Acad Sci U S A 99: 7634–7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PM, Fowkes FGR, Belch JFF, Ogawa H, Warlow CP, Meade TW (2011). Effect of daily aspirin on long‐term risk of death due to cancer: analysis of individual patient data from randomised trials. The Lancet 377: 31–41. [DOI] [PubMed] [Google Scholar]
- Rothwell PM, Price JF, Fowkes FGR, Zanchetti A, Roncaglioni MC, Tognoni G et al (2012). Short‐term effects of daily aspirin on cancer incidence, mortality, and non‐vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. The Lancet 379: 1602–1612. [DOI] [PubMed] [Google Scholar]
- Santilli F, Rocca B, De Cristofaro R, Lattanzio S, Pietrangelo L, Habib A et al (2009). Platelet cyclooxygenase inhibition by low‐dose aspirin is not reflected consistently by platelet function assays: implications for aspirin “resistance”. J Am Coll Cardiol 53: 667–677. [DOI] [PubMed] [Google Scholar]
- Smith JB, Willis AL (1971). Aspirin selectively inhibits prostaglandin production in human platelets. Nat New Biol 231: 235–237. [DOI] [PubMed] [Google Scholar]
- Smith JP, Haddad EV, Taylor MB, Oram D, Blakemore D, Chen Q et al (2012). Suboptimal inhibition of platelet cyclooxygenase‐1 by aspirin in metabolic syndrome. Hypertension 59: 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WL, Dewitt DL (1996). Prostaglandin endoperoxide H synthases‐1 and ‐2. Adv Immunol 62: 167–215. [DOI] [PubMed] [Google Scholar]
- Smyth EM (2010). Thromboxane and the thromboxane receptor in cardiovascular disease. Clin Lipidol 5: 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoep JD, Hovens MC, Eikenboom JJ, Van Der Bom JG, Huisman MV (2007). Association of laboratory‐defined aspirin resistance with a higher risk of recurrent cardiovascular events: A systematic review and meta‐analysis. Arch Intern Med 167: 1593–1599. [DOI] [PubMed] [Google Scholar]
- Steinbach G, Lynch PM, Phillips RKS, Wallace MH, Hawk E, Gordon GB et al (2000). The effect of celecoxib, a cyclooxygenase‐2 inhibitor, in familial adenomatous polyposis. N Engl J Med 342: 1946–1952. [DOI] [PubMed] [Google Scholar]
- Subhash PK, David SG, David RJ, Letts LG (2007). Eicosanoids in Inflammation: biosynthesis, pharmacology, and therapeutic frontiers. Curr Top Med Chem 7: 311–340. [DOI] [PubMed] [Google Scholar]
- Sudhahar V, Shaw S, Imig JD (2010). Epoxyeicosatrienoic acid analogs and vascular function. Curr Med Chem 17: 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson J, Hamberg M, Samuelsson B (1975). Prostaglandin endoperoxides IX. Characterization of rabbit aorta contracting substance (RCS) from guinea pig lung and human platelets. Acta Physiol Scand 94: 222–228. [DOI] [PubMed] [Google Scholar]
- Tantry US, Bonello L, Aradi D, Price MJ, Jeong Y‐H, Angiolillo DJ et al (2013). Consensus and update on the definition of on‐treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J Am Coll Cardiol 62: 2261–2273. [DOI] [PubMed] [Google Scholar]
- Thiagarjan P, Wu KK (2002). In vitro assays for evaluating platelet function In: Gresele P, Page C, Fuster V, Vermylyn J. (eds). Platelets in thrombotic and non thrombotic disorders. Cambridge University Press: Cambridge, UK. [Google Scholar]
- Ulrych T, Böhm A, Polzin A, Daum G, Nüsing RM, Geisslinger G et al (2011). Release of sphingosine‐1‐phosphate from human platelets is dependent on thromboxane formation. J Thromb Haemost 9: 790–798. [DOI] [PubMed] [Google Scholar]
- Vane JR (1971). Inhibition of prostaglandin synthesis as a mechanism of action for aspirin‐like drugs. Nat New Biol 231: 232–235. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Devchand PR (2005). Emerging roles for cyclooxygenase‐2 in gastrointestinal mucosal defense. Br J Pharmacol 145: 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C et al (2009). Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 361: 1045–1057. [DOI] [PubMed] [Google Scholar]
- Warner TD, Armstrong PC, Chan MV, Knowles RB (2016). The importance of endothelium‐derived mediators to the efficacy of dual anti‐platelet therapy. Expert Rev Hematol 9: 223–225. [DOI] [PubMed] [Google Scholar]
- Warner TD, Armstrong PCJ, Curzen NP, Mitchell JA (2010). Dual antiplatelet therapy in cardiovascular disease: does aspirin increase clinical risk in the presence of potent P2Y12 receptor antagonists? Heart 96: 1693–1694. [DOI] [PubMed] [Google Scholar]
- Warner TD, Nylander S, Whatling C (2011). Anti‐platelet therapy: cyclo‐oxygenase inhibition and the use of aspirin with particular regard to dual anti‐platelet therapy. Br J Clin Pharmacol 72: 619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker N, Bunting S, Salmon J, Moncada S, Vane JR, Johnson RA et al (1976). The chemical structure of prostaglandin X (prostacyclin). Prostaglandins 12: 915–928. [DOI] [PubMed] [Google Scholar]
- Whittle BJ, Moncada S, Vane JR (1978). Comparison of the effects of prostacyclin (PGI2), prostaglandin E1 and D2 on platelet aggregation in different species. Prostaglandins 16: 373–388. [DOI] [PubMed] [Google Scholar]
- Whittle BJ, Silverstein AM, Mottola DM, Clapp LH (2012). Binding and activity of the prostacyclin receptor (IP) agonists, treprostinil and iloprost, at human prostanoid receptors: treprostinil is a potent DP1 and EP2 agonist. Biochem Pharmacol 84: 68–75. [DOI] [PubMed] [Google Scholar]
- Windecker S, Kolh P, Alfonso F, Collet J‐P, Cremer J, Falk V et al (2014). 2014 ESC/EACTS Guidelines on myocardial revascularization The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio‐Thoracic Surgery (EACTS)Developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J 35: 2541–2619. [DOI] [PubMed] [Google Scholar]
- Wiviott SD, Braunwald E, Mccabe CH, Montalescot G, Ruzyllo W, Gottlieb S et al (2007). Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 357: 2001–2015. [DOI] [PubMed] [Google Scholar]
- Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL (1991). Expression of a mitogen‐responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci U S A 88: 2692–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wu J, Jiang H, Mortensen R, Austin S, Manning DR et al (2002). Signaling through Gi family members in platelets: redundancy and specificity in the regulation of adenylyl cyclase and other effectors. J Biol Chem 277: 46035–46042. [DOI] [PubMed] [Google Scholar]
- Yang L (2015). The role of epoxyeicosatrienoic acids in the cardiovascular system. Br J Clin Pharmacol 80: 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z et al (2012). Vascular COX‐2 modulates blood pressure and thrombosis in mice. Sci Transl Med 4: 132ra54–132ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]