


Abstract
Bacteria use quorum sensing to monitor cell density and coordinate group behaviours. In Vibrio cholerae, the causative agent of the diarrheal disease cholera, quorum sensing is connected to virulence gene expression via the two autoinducer molecules, AI-2 and CAI-1. Both autoinducers share one signal transduction pathway to control the production of AphA, a key transcriptional activator of biofilm formation and virulence genes. In this study, we demonstrate that the recently identified autoinducer, DPO, also controls AphA production in V. cholerae. DPO, functioning through the transcription factor VqmA and the VqmR small RNA, reduces AphA levels at the post-transcriptional level and consequently inhibits virulence gene expression. VqmR-mediated repression of AphA provides an important link between the AI-2/CAI-1 and DPO-dependent quorum sensing pathways in V. cholerae. Transcriptome analyses comparing the effect of single autoinducers versus autoinducer combinations show that quorum sensing controls the expression of ∼400 genes in V. cholerae and that all three autoinducers are required for a full quorum sensing response. Together, our data provide a global view on autoinducer interplay in V. cholerae and highlight the importance of RNA-based gene control for collective functions in this major human pathogen.
INTRODUCTION
To efficiently interact with their environment, bacteria often work in groups to solve complex tasks. Coordination of collective functions requires communication among the members of the group, a process commonly referred to as quorum sensing (QS) (1,2). QS involves the production, release, and subsequent detection of extracellular small molecules called autoinducers.
In Vibrio cholerae, the causative agent of cholera disease, QS is intimately linked to several collective functions, including biofilm formation (3), type VI secretion (4,5), competence (6,7), phage resistance (8) and virulence gene expression (9). The canonical QS pathway of V. cholerae (Figure 1A and B) involves the two autoinducer molecules, CAI-1 ((S)-3-hydroxytridecan-4-one) and AI-2 ((2S,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran borate). CAI-1 and AI-2 are synthesized by the CqsA and LuxS enzymes and accumulate to concentrations of ∼0.3 μM (CAI-1) and ∼1–2 μM (AI-2) in cell-free supernatants of Vibrio species (10,11). Their cognate receptors are the membrane-bound proteins CqsS and LuxPQ, respectively (11–16). Both, CqsS and LuxPQ channel phosphate to the phospho-transfer protein LuxU, which transfers the phosphate to the response regulator LuxO (17). Phosphorylated LuxO together with the alternative sigma factor σN activates the expression of genes encoding four homologous regulatory small RNAs (sRNAs), called Qrr1-4 (18). The Qrr sRNAs act at the heart of the two QS systems by reciprocally controlling the production of the transcriptional regulators HapR and AphA, which regulate biofilm formation and virulence of V. cholerae (19). Importantly, the CqsS and LuxPQ receptors act as kinases in the absence of AI-2 and CAI-1, but convert to phosphatases when the autoinducers are present (20). Thus, expression of the Qrr sRNAs is repressed by AI-2 and CAI-1 (Figure 1B). In addition, two other receptor proteins, CqsR and VpsS, have been reported to channel information through LuxO, indicating the existence of at least four sensory inputs for this pathway (21).
Figure 1.

Quorum sensing in V. cholerae is controlled by three autoinducer molecules. The CAI-1 and AI-2 autoinducers are produced by CqsA and LuxS and detected by the membrane-bound CqsS and LuxPQ receptors, respectively. The DPO autoinducer derives from threonine catabolism, and requires the Tdh (threonine dehydrogenase) enzyme. DPO is released into the environment and binds to and activates the VqmA receptor. (A) At low autoinducer concentrations, CqsS and LuxPQ act as kinases to phosphorylate LuxU. LuxU-P transfers the phosphate to LuxO, and LuxO-P induces the expression of the Qrr1–4 sRNAs. The Qrr sRNAs act post-transcriptionally to repress hapR and activate aphA, promoting virulence gene expression and biofilm formation. AphA also activates the transcription of vpsT. (B) At high autoinducer concentrations, binding of CAI-1 and AI-2 to CqsS and LuxPQ, respectively, converts the receptors to phosphatases, which reduces LuxO-P levels and inhibits qrr1–4 expression. Under these conditions, aphA is repressed and hapR is activated. The VqmA-DPO complex induces the transcription of the VqmR sRNA. VqmR inhibits biofilm formation by repressing VpsT and virulence gene expression by inhibiting AphA. In addition, HapR and AphA antagonize each other at the transcriptional level. Active factors are highlighted in blue, inactive (repressed) factors are shown in gray.
Recently, we discovered another QS system operating in V. cholerae (Figure 1 and (22)). In contrast to CAI-1 and AI-2, this system does not require LuxU, LuxO or the Qrr sRNAs, but rather relies on the catabolic degradation of L-threonine by threonine dehydrogenase (encoded by tdh) and the concomitant synthesis of another autoinducer, called DPO (3,5-dimethylpyrazin-2-ol). DPO is sensed by VqmA, a cytoplasmic LuxR-type transcriptional regulator, which induces the transcription of the VqmR sRNA. VqmR belongs to the ubiquitous class of Hfq-associated regulatory RNAs (23) and we have shown that VqmR inhibits multiple trans-encoded target genes through direct base-pairing with their respective mRNAs (24). The target spectrum of VqmR also includes the transcript encoding VpsT, a key activator of biofilm formation in V. cholerae (24,25). Consequently, DPO, by acting through VqmA and VqmR, inhibits biofilm formation in V. cholerae (22).
Biofilm formation and pathogenicity are closely connected in V. cholerae (26,27). During the initial phases of infection, biofilms allow V. cholerae to survive the acidic environment of the stomach (28) and intravital microscopy revealed the formation of biofilms in the small intestines of infected mice (29). Therefore, perhaps not surprisingly, biofilm formation and intestinal colonization share a large set of co-regulated genes in V. cholerae. The two transcription factors, HapR and AphA, which are also regulated by QS (Figure 1), have overarching roles in both processes as they control the genes for biofilm and virulence regulation in an opposite manner (19). Specifically, AphA, together with another transcriptional regulator, called AphB (30), activates the production of the toxin-co-regulated pilus (TCP) and the cholera toxin (CTX). Both, TCP and CTX are necessary for infections in humans (31). Likewise, AphA activates VpsT production, which enhances biofilm formation (32). HapR antagonizes these functions by inhibiting the production of AphA and VpsT, as well as several other genes related to biofilm formation and virulence gene expression (33). Of note, HapR also controls Type VI secretion in V. cholerae (5), a process which has recently been reported to drive interspecies competition during host colonization (34,35).
In this study, we used RNA-sequencing to identify additional target mRNAs of VqmR in V. cholerae. Our analysis revealed five previously unknown target transcripts, including the aphA mRNA. We show that VqmR inhibits AphA production by interacting with the ribosome binding site (RBS) of the corresponding mRNA and that base-pairing involves the Rho-independent terminator sequence of VqmR. VqmR-mediated repression of AphA is stimulated by DPO and results in reduced virulence gene expression. Reduction of AphA levels by DPO connects the two QS pathways of V. cholerae at a critical regulatory node and suggests a coactive role in gene regulation. Indeed, global RNA-sequencing analysis of autoinducer-treated cells shows that QS controls more than 400 genes in V. cholerae and that AI-2, CAI-1 and DPO work together to control biofilm formation, virulence gene expression, and other collective functions in this major human pathogen.
MATERIALS AND METHODS
Strains, plasmids and growth conditions
Strains are listed in Supplementary Table S2. V. cholerae and E. coli were grown aerobically in LB or M9 minimal medium (0.4% glucose) at 37°C. Antibiotics were used at the following concentrations: 50 U ml−1 polymyxin B, 100 μg ml−1 ampicillin, 50 μg ml−1 kanamycin, 5000 μg ml−1 streptomycin, and 20 μg ml−1 chloramphenicol. Experiments involving AKI growth conditions were performed following previously published protocols (36).
Oligonucleotides and plasmids
Plasmids and DNA oligonucleotides are listed in Supplementary Tables S3 and S4, respectively. Details on plasmid construction are provided in the Supplementary Methods section.
Northern Blot analysis
Total RNA was prepared and transferred as previously described (37). Membranes were hybridized in Roti-Hybri-Quick buffer (Roth) at 42°C with [32P] end-labelled DNA oligonucleotides, or 63°C when using riboprobes. Signals were visualized using a Typhoon phosphorimager (Amersham) and band intensities were quantified using the GelQuant software (biochemlabsolutions). Oligonucleotides for Northern Blot analyses are provided in Supplementary Table S4.
Western Blot analysis and fluorescence assays
Western Blot analyses of GFP and FLAG fusion proteins followed previously published protocols (38). Signals were visualized using a Fusion FX EDGE imager (Vilber) and band intensities were quantified using the BIO-1D software (Vilber). Fluorescence assay of V. cholerae and E. coli strains were performed as previously described (22,37).
Preparation of secreted protein fractions
The cell densities (OD600) of AKI cultures were determined after 16h of continuous shaking. Two milliliter of each culture were centrifuged at 13 000 rpm for 30 min at 4°C and 1.6 ml of the supernatants were transferred to a new reaction tube. To precipitate secreted proteins, 0.4 ml of 25% ice-cold trichloroacetic acid was added (5% final conc.) followed by 15 min incubation on ice. Protein pellets were obtained by centrifugation (13 000 rpm, 30 min, 4°C) and washed two times with ice-cold acetone (13 000 rpm, 15 min, 4°C). The supernatants were carefully removed and the pellets were allowed to air dry. Pellets were resuspended in individual volumes of SDS loading buffer relative to the OD600 measurements of the respective culture.
Sample collection for RNA-seq analyses
RNA-seq experiment to identify VqmR targets: Biological triplicates of ΔvqmR cells carrying either the pBAD-Ctr or the pBAD-vqmR plasmid were grown to OD600 = 0.5 in LB media. Cells were treated with 0.2% (final conc.) l-Arabinose and harvested after 15 min. Addition of Stop Mix (95% [vol/vol] EtOH and 5% [vol/vol] phenol) terminated ongoing transcription and translation. The samples were frozen in liquid nitrogen and stored at –80°C until RNA preparation. Autoinducer RNA-seq: Biological triplicates of a ΔluxS, cqsA, tdh triple mutant strain were grown overnight in M9 minimal media, supplemented with single or combinations of the following autoinducers (5 μM final conc. each): autoinducer 2 (AI-2), cholera-autoinducer-1 (CAI-1), or 3,5-dimethylpyrazin-2-ol (DPO) or water (mock). Bacteria were diluted 1:500 in fresh media containing the same autoinducers and samples were collected at OD600 = 0.2. Stop Mix (95% EtOH, 5% phenol, [vol/vol]) prevented further transcription and translation. Cells were pelleted (4000 rpm, 15 min, 4°C), resuspended in Tri-reagent (Sigma) and stored at –80°C until further processing.
Construction of cDNA libraries and Illumina sequencing
Total RNA was digested with DNaseI and depletion of ribosomal RNA was performed using the Ribo-Zero kit (Epicentre) for Gram-negative bacteria. Integrity of the prepared RNA was confirmed using an Agilent 2100 Bioanalyzer. Directional cDNA libraries were prepared using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB E7760) according to the manufacturer's instructions and cDNA library quality was tested on an Agilent 2100 Bioanalyzer. The libraries were sequenced using a HiSeq 1500 machine (Illumina) in single-read mode with 100 bp read length. The sequencing data has been deposited at Gene Expression Omnibus (GEO) under the GSE115711 accession code.
RESULTS
Identification and validation of additional VqmR target mRNAs
QS regulates hundreds of genes in Vibrios (33) and global transcriptome profiles show that QS-controlled genes can be classified into low- and high-cell density. In our previous work, we used high-cell density cultures and VqmR pulse expression followed by global transcriptome analysis to identify target mRNAs of VqmR in V. cholerae (24). To identify additional targets of VqmR, here we investigated exponentially growing cells (OD600 = 0.5) and scored global transcriptome changes using RNA-sequencing. Differentially expressed genes were determined by comparing cells induced for VqmR expression from a pBAD promoter for 15 min to an empty vector control. These analyses identified 11 mRNAs showing at least 2.5-fold regulation by VqmR (Table 1). VqmR-mediated repression of five transcripts (vpsT, vca0068, vc1865, vc1063 and vca0591-vca0590) was also observed in our previous analyses (24), supporting our approach. Newly identified target candidates included the mRNAs of two conserved hypothetical proteins (vc0789 and vc0865), ulaA (encoding part of an ascorbate transport system), ndk (encoding nucleoside diphosphate kinase), as well as aphA (encoding a major transcriptional regulator of QS in V. cholerae, see Figure 1). All transcripts were repressed by VqmR, which was also confirmed using quantitative real-time PCR (Supplementary Figure S1A).
Table 1.
Genes differentially regulated by VqmR pulse expression
| Gene | Descriptiona | Fold changeb |
|---|---|---|
| vc0789 | Hypothetical protein | −12.0 |
| vc1865 c | Hypothetical protein | −9.6 |
| vca0068 c | Methyl-accepting chemotaxis protein | −5.7 |
| vc0865 | Hypothetical protein | −3.4 |
| ulaA | PTS system ascorbate-specific transporter | −3.3 |
| vca0591 c | Peptide ABC transporter | −3.2 |
| vca0590 c | Peptide ABC transporter permease | −3.0 |
| vc1063 c | Acyl-CoA thioesterase II | −2.9 |
| aphA | PadR family transcriptional regulator | −2.8 |
| ndk | Nucleoside-diphosphate kinase | −2.5 |
| vpsT c | LuxR family transcriptional regulator | −2.5 |
aDescription based on the annotation at KEGG (https://www.genome.jp/kegg).
bFold change obtained by transcriptomic analysis of pBAD-driven VqmR expression using RNA-seq. Genes that were at least 2.5-fold differentially regulated and were statistically significant (Bonferroni ≤ 1E–10) are listed.
cVqmR target genes previously reported in (24).
Previous work focussing on the molecular mechanism of VqmR-mediated gene regulation showed that the VqmR sRNA employs one of two conserved domains (R1 and R2, see Figure 2A) to base-pair with target mRNAs (24). To test if the newly identified targets, i.e. vc0789, vc0865, ulaA, ndk and aphA, were also regulated at the post-transcriptional level by VqmR, we used a well-established GFP-based reporter system tailored to score post-transcriptional gene control in bacteria (39). In this system, the 5′ UTR (untranslated region) and the sequence corresponding to the first 20 amino-acids of the target genes are fused to gfp under the control of the PTetO promoter. These plasmids were introduced into Escherichia coli along with a second plasmid expressing the vqmR gene from a PTac promoter. We discovered significantly reduced GFP production for all five candidate targets when VqmR was present (Figure 2B). We repeated these experiments in an E. coli strain lacking hfq, and no target regulation occurred (Supplementary Figure S1B). To investigate which of the two conserved base-pairing domains of VqmR mediated target repression, we individually deleted the R1 and R2 sequences in vqmR and measured GFP production. We discovered that repression of vc0865 and ulaA was significantly impaired in the absence of domain R2, while down-regulation of ndk and vc0789 was impaired when domain R1 was removed. Unexpectedly, VqmR-mediated repression of aphA did not require either of the two base-pairing domains (Figure 2B).
Figure 2.

VqmR target genes and base-pairing of VqmR with the aphA 5′ UTR. (A) Secondary structure of VqmR (24). The VqmR base-pairing sequences are highlighted in red (R1), blue (R2) and green (R3). Arrows and brackets mark the truncation start sites and the internal deletion regions investigated in C, respectively. (B) E. coli harbouring plasmids carrying the five genes denoted on the x-axis each fused to gfp were co-transformed with a control plasmid (pCtr) or the indicated VqmR expressing plasmids. Transcription of vqmR and gfp was driven by constitutive promoters. Cells were cultivated in LB to OD600 = 0.5 and GFP production was measured. GFP levels of strains carrying the control plasmid were set to 1. Error bars represent the SD of three biological replicates. (C) E. coli cells carrying the aphA::gfp reporter were tested for repression by various VqmR mutants. Cells were grown in LB to OD600 = 0.5 and GFP production was measured. Error bars indicate the SD of three biological replicates. (D) Predicted base-pairing of the VqmR R3 sequence (green) with the 5′ UTR of aphA. The arrows indicate the single nucleotide mutations tested in E and the start codon is underlined. (E) Repression of AphA::GFP and AphA*::GFP (G-3C) by VqmR and VqmR* (C133G). Cells were grown in LB to OD600 = 0.5 and GFP levels were measured using Western Blot. RNAP served as the loading control.
VqmR interacts with aphA via a third base-pairing domain
The data presented in Figure 2B suggested that VqmR inhibits aphA by base-pairing using some unknown sequence element of VqmR. To test this possibility, we generated truncated VqmR variants, i.e. we deleted the first 30, 60 and 90 nucleotides of VqmR (these VqmR variants are called TΔ30, TΔ60, and TΔ90, respectively) and monitored AphA::GFP levels. None of these mutants abrogated VqmR repression (Figure 2C, bars 1–5). In addition, we also constructed internal deletions in vqmR, removing nucleotides 91–101, 91–111 and 91–121. Again, these mutants did not affect repression of AphA::GFP (Figure 2C, bars 6–8).
These results showed that none of the canonical base-pairing sequences of VqmR are involved in aphA repression and led us to conclude that VqmR-mediated repression of aphA possibly depended on a sequence element located in the Rho-independent terminator of VqmR. To probe this hypothesis, we exchanged the terminator of vqmR with the terminator sequence of an unrelated sRNA of V. cholerae, named Vcr089 (24). Although the level of production of this chimeric sRNA was comparable to wild-type VqmR (Supplementary Figure S2A), repression of AphA::GFP was significantly reduced (∼1.7-fold versus ∼13.5-fold; Figure 2C, bar 9 versus 2). In the reciprocal experiment, we exchanged the terminator of vcr089 with the vqmR terminator and discovered that AphA::GFP repression increased to ∼3-fold (Figure 2C, bar 10), while Vcr089 itself had no effect on AphA::GFP levels (Figure 2C, bar 11). Of note, the limited repression of AphA::GFP by the Vcr089 sRNA carrying the VqmR terminator (bar 10) might well be explained by the reduced stability of this chimeric sRNA when compared to the native VqmR sRNA (Supplementary Figure S2A).
These experiments prompted us to search for a possible base-pairing interaction using the RNA hybrid algorithm (40) with the aphA 5′ UTR and the VqmR terminator sequence as inputs. Indeed, these analyses revealed a potential RNA duplex involving the loop of the VqmR terminator element and the sequence directly upstream of the aphA start codon (Figure 2D). To test this prediction, we altered cytosine to guanine at position 133 of VqmR and measured production of AphA::GFP (Figure 2E and Supplementary Figure S2B). Mutation at this position strongly reduced AphA::GFP repression, while sRNA levels were unaffected. Likewise, a compensatory mutation from guanine to cytosine at position –3 of aphA::gfp fully restored repression by the mutated VqmR, whereas repression by wild-type VqmR was inhibited (Figure 2E and Supplementary Figure S2B). Thus, VqmR uses its Rho-independent terminator, and specifically the loop sequence, to repress AphA production. In accordance with the previously defined base-pairing sequences of VqmR, we termed this sequence R3 (Figure 2A).
The data presented in Figure 2D and E suggested that VqmR inhibits aphA by sequestering its RBS, which will block translation initiation and consequently reduce protein levels. To test this hypothesis, we first mutated the Shine-Dalgarno element in the RBS of the aphA::gfp reporter at three consecutive positons and monitored AphA::GFP levels. In all three cases, GFP production was strongly reduced (Supplementary Figure S3A). Next, we performed toeprinting analysis (41) of the aphA mRNA (Supplementary Figure S3B). Addition of purified 30S ribosomes along with initiator tRNAfMet to the aphA mRNA resulted in a termination signal located 16 nucleotides downstream of translation initiation, which is in accordance with the annotated AUG start codon. To mimic base-pairing of VqmR at the predicted position in aphA, we used an LNA (locked nucleic acid) oligonucleotide matching to the VqmR seed sequence of VqmR-aphA RNA duplex (corresponding to nucleotides 131–138 of VqmR; compare Figure 2D and Supplementary Figure S3A). Indeed, titration of the LNA oligonucleotide reduced 30S binding in a concentration dependent manner and led to the detection of a second termination signal corresponding to the VqmR binding site (Supplementary Figure S3B). Together, our in vivo and in vitro data indicate that interaction with the VqmR sRNA inhibits translation initiation of the aphA mRNA and that VqmR competes with 30S ribosomes for binding of the aphA RBS.
VqmR and DPO inhibit AphA protein production
Next, we were interested to test the effect of VqmR on AphA protein production in vivo. To this end, we engineered an aphA::3XFLAG construct and introduced it onto the chromosome of V. cholerae wild-type and ΔvqmR strains at the aphA locus. These strains were transformed with either a vector control (pCtr) or a VqmR over-expression plasmid (pVqmR) and cultivated in M9 minimal medium containing casein acid hydrolysate (casamino acids) as a threonine source for DPO production. At selected time-points, total RNA and protein samples were collected and examined by Northern and Western blotting, respectively. As expected, in wild-type, levels of AphA protein decreased at high cell density along with a reduction in aphA mRNA abundance (Figure 3A, lanes 1–4). In V. cholerae cells lacking vqmR, AphA protein levels remained unaffected at low cell density (OD600 of 0.2), which was also recapitulated at the aphA mRNA level and is in accordance with limited VqmR expression under this condition (Figure 3A, lane 1 vs. 5). At higher cell densities (OD600 of 1.0, 1.5 and 3 h after cells reached an OD600 of 1.5), AphA protein levels were ∼3–4-fold higher in the vqmR mutant when compared to wild-type cells (lanes 2–4 versus 6–8). In contrast, V. cholerae ΔvqmR cells over-expressing VqmR displayed significantly reduced AphA protein and mRNA levels under all conditions (Figure 3A, lane 9–12). Of note, plasmid-borne VqmR expression had a stronger effect on AphA protein levels, when compared to the reduction in aphA mRNA. These results could indicate that, when over-expressed, VqmR-mediated repression of aphA acts predominantly by inhibiting translation initiation.
Figure 3.

DPO inhibits AphA production. (A) V. cholerae wild-type and vqmR mutants carrying the indicated plasmids were cultivated in M9 minimal media supplemented with casamino acids (0.4% final conc.). At the indicated growth phases, total RNA and protein samples were collected. AphA::3XFLAG production was monitored on Western Blots and RNAP served as the loading control. VqmR and aphA::3XFLAG mRNA levels were probed on Northern Blots using 5S rRNA as loading control. (B) Total RNA and protein samples were collected from V. cholerae wild-type, ΔvqmA, ΔvqmR and Δtdh strains at low cell density (OD600 = 0.2). Cells were cultivated in M9 minimal media and one set of cultures was supplemented with DPO (100 μM final conc.). AphA::3XFLAG production was determined using Western Blot. Northern Blot was used to probe the expression of aphA-3XFLAG and VqmR. RNAP and 5S rRNA served as loading controls for the Western and Northern Blot analyses, respectively. (C) V. cholerae Δtdh cells were cultivated in M9 medium supplemented with the indicated DPO concentrations (x-axis) and total RNA and protein samples were harvested at low cell densities (OD600 = 0.2). AphA production was analyzed on Western Blots (left y-axis), sRNA levels (VqmR and Qrr4) were determined on Northern Blots (right y-axis). Error bars represent the SD of five (AphA) and three (sRNAs) biological replicates, respectively.
Elevated levels of AphA in ΔvqmR cells cultivated to high cell density (Figure 3A) indicated that VqmR has a negative effect on AphA levels when DPO accumulates in the environment. To explore this possibility, we cultivated V. cholerae cells in M9 minimal medium lacking amino-acids (eliminating endogenous DPO production) to low cell density (OD600 = 0.2) and compared AphA production in the presence and absence of exogenously supplied synthetic DPO (100 μM final conc.). In wild-type cells, addition of DPO reduced AphA levels by ∼3-fold, which was also corroborated at the aphA mRNA level (Figure 3B, lane 1 versus 2). As expected, AphA protein and its mRNA did not change in response to DPO in V. cholerae cells lacking either vqmA or vqmR (Figure 3B, lanes 3–6). However, DPO-mediated repression of AphA occurred in cells lacking tdh (Figure 3B, lanes 7–8), which is required for DPO synthesis but not for DPO detection or signal transduction (Figure 1). In line with these observations, exogenously added DPO activated VqmR production in wild-type and Δtdh cells, while no VqmR was detected in the vqmA and vqmR mutants. These data show that DPO inhibits AphA production in V. cholerae and that the DPO-receptor, VqmA and the VqmR sRNA mediate this phenotype.
We previously showed that DPO accumulates in cell-free supernatants of V. cholerae at a concentration of ∼1 μM (22). To test if endogenous levels of DPO would also inhibit AphA production, we performed a titration experiment in which we gradually increased the levels of synthetic DPO and tested AphA and VqmR levels on Western and Northern Blots, respectively (Figure 3C). We discovered that DPO concentrations as low as 0.33μM significantly increased VqmR production, which also resulted in a ∼2-fold reduction in AphA production. At a concentration of 1 μM DPO, repression of AphA increased to ∼2.5-fold and reached a maximum of ∼3-fold when higher concentrations of DPO were used. Importantly, this saturation in AphA repression coincided with the maximal VqmR expression, while DPO titration did not affect Qrr4 production (Figure 3C).
DPO down-regulates virulence gene expression in V. cholerae
AphA is a key regulator of virulence gene expression in V. cholerae and required for intestinal colonization in an infant mouse model of infection (30). In concert with AphB, AphA induces the expression of the transmembrane regulators TcpP and TcpH (42). TcpPH and another transmembrane regulator, ToxRS, activate the production of ToxT, which finally induces the expression of ctxAB and tcpA-F (Figure 4A and (43)).
Figure 4.
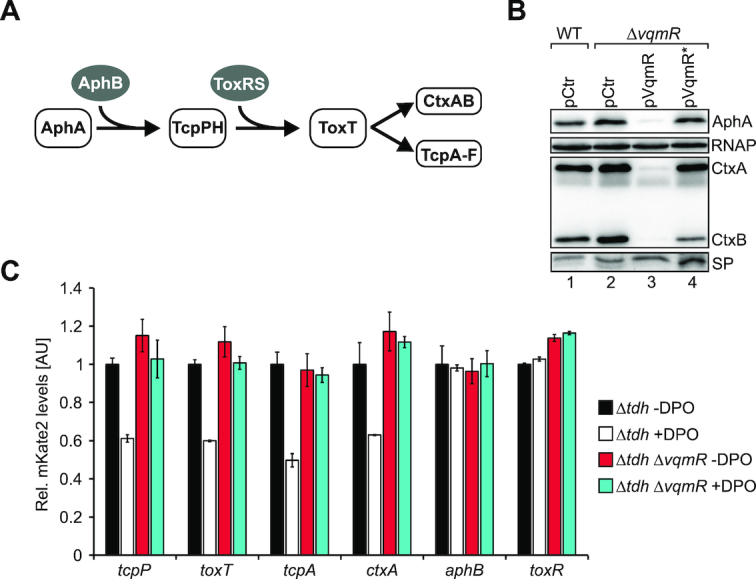
DPO and VqmR inhibit virulence gene expression. (A) The virulence cascade of V. cholerae. (B) V. cholerae wild-type and ΔvqmR strains carrying the indicated plasmids were cultivated under AKI conditions. Cellular and secreted protein (SP) fractions were harvested 2h and 16 h after switching from static to aerating conditions, respectively and tested for AphA-3XFLAG and CtxAB production on Western Blots. RNAP and a coomassie-stained SDS gel (bottom) confirmed equal loading of the two protein fractions. (C) V. cholerae Δtdh or Δtdh, vqmR cells carrying plasmids with the indicated transcriptional reporters were cultivated under AKI conditions in the presence or absence of DPO (100 μM final conc.) and fluorescence was measured 2 h after switching to aerating conditions. mKate2 levels of the Δtdh cells cultivated without DPO were set to 1. Error bars represent the SD of three biological replicates.
To investigate the role of DPO-mediated gene control in virulence gene expression in V. cholerae, we first tested the effect of VqmR on CtxAB and AphA protein production. Specifically, we cultivated V. cholerae wild-type and ΔvqmR cells, both carrying a vector control, in AKI medium to induce virulence factor production (36). We discovered a modest (∼1.5-fold) increase in AphA levels in the absence of vqmR (Figure 4B, lane 1 versus 2). In contrast, a vqmR mutant strain carrying vqmR on a multi-copy plasmid strongly reduced AphA levels (∼13-fold), when compared to wild-type V. cholerae (Figure 4B, lane 1 versus 3). Likewise, VqmR over-production down-regulated CtxA and CtxB levels by ∼18.2-fold and ∼27.6-fold, respectively. To obtain additional evidence that base-pairing of VqmR with the aphA mRNA caused AphA and CtxAB repression, we also tested the effect of the VqmR point-mutant showing strongly reduced AphA::GFP repression (Figure 2D and E) on AphA and CtxAB production. We discovered that the mutated VqmR variant failed to inhibit AphA and CtxAB production (Figure 4B, lane 4). Together, these data suggest that VqmR-mediated repression of aphA prevents virulence gene expression in V. cholerae.
To monitor the activity of V. cholerae virulence genes in the context of DPO, we generated mKate2-based transcriptional reporters to all genes of the cascade (Figure 4A) and transformed these constructs into V. cholerae cells lacking tdh (to eliminate endogenous DPO production) and cells lacking tdh and vqmR. Again, we used AKI medium to induce virulence gene expression, however, for these experiments, one set of cultures was supplemented with synthetic DPO (100 μM final conc.). Our data showed that DPO significantly inhibited the promoter activities of tcpP, toxT, tcpA, and ctxA, however, as expected, did not affect the promoters of aphB and toxR (Figure 4C). These results were specific to DPO-mediated activation of vqmR, since DPO failed to down-regulate the promoters of tcpP, toxT, tcpA, and ctxA in the Δtdh vqmR double mutant (Figure 4C). Therefore, we conclude that the DPO-controlled QS pathway negatively affects the production of virulence factors in V. cholerae.
The three autoinducers of V. cholerae act together to control AphA production
There are currently three autoinducers known in V. cholerae (11,12,22). Whereas AI-2 and CAI-1 act through LuxO and the Qrr sRNAs to activate aphA, DPO functions through VqmA and VqmR to repress aphA (Figure 1B). Importantly, although VqmR and the Qrr sRNAs act antagonistically on aphA, all three autoinducers inhibit aphA production since AI-2 and CAI-1 inhibit production of the Qrr sRNAs which are activators of aphA, and DPO activates VqmR transcription, which directly represses aphA. To test the individual contributions of the autoinducers on AphA production, we cultivated a luxS, cqsA, tdh triple mutant in M9 minimal medium and added AI-2, CAI-1 and DPO at saturating concentrations (5 μM final conc.). We collected total protein and RNA samples at low cell density (OD600 = 0.2) and probed AphA protein levels on Western Blots, as well as Qrr4 and VqmR production on Northern Blots (Figure 5A). We discovered that AI-2, although reducing Qrr4 levels by ∼2.2-fold (Figure 5A, lane 1 versus 2), did not significantly reduce AphA levels (Figure 5A and B). CAI-1 and DPO both inhibited AphA by ∼1.8-fold (Figure 5B). CAI-1 reduced Qrr4 levels by ∼5.6-fold, whereas DPO did not affect Qrr4 (Figure 5A, lanes 1, 3, 4). As expected, VqmR expression was strongly induced by DPO (∼15-fold), but remained unaffected by AI-2 (Figure 5A, lanes 1, 2, 4). Interestingly, CAI-1 also slightly activated (∼2.8-fold) VqmR production (lane 1 versus 3). We currently do not understand the molecular mechanism underlying CAI-1-mediated VqmR induction and whether this regulation is biologically relevant.
Figure 5.
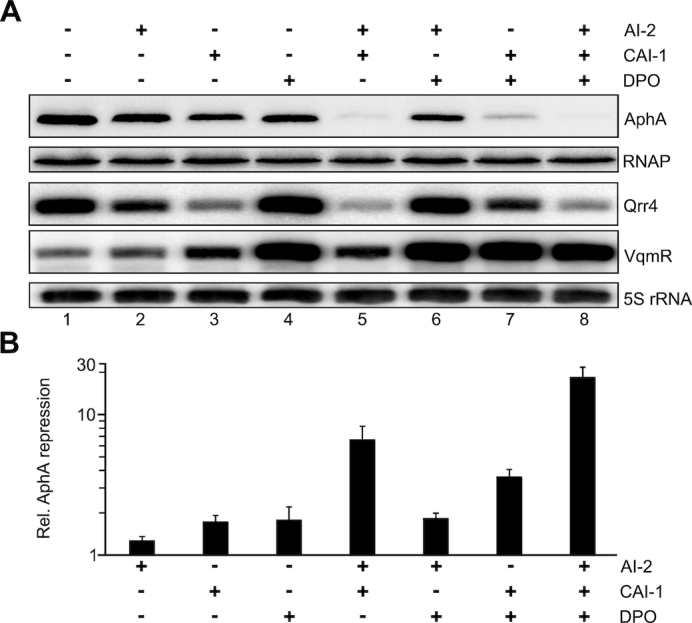
AI-2, CAI-1 and DPO act in concert to repress AphA production. (A) The V. cholerae ΔluxS, cqsA, tdh mutant was cultivated in M9 minimal media containing the indicated autoinducers (5 μM final conc. each) to OD600 = 0.2. AphA::3XFLAG, Qrr4 and VqmR levels were monitored on Western Blots and Northern Blots, respectively. RNAP (Western Blot) and 5S rRNA (Northern Blot) served as loading controls. (B) Quantification of (A). AphA levels in the mock-treated sample was set to 1. Error bars represent the SD of three biological triplicates.
The genetic setup of V. cholerae’s QS circuit (Figure 1) suggested that the three autoinducers act together to control AphA production. To test this hypothesis, we monitored the collective effect of the autoinducers on AphA, Qrr4 and VqmR levels. Combination of AI-2 and CAI-1 inhibited AphA by ∼6.6-fold and reduced Qrr4 levels by ∼8.8-fold when compared to the untreated sample (Figure 5A and B). In contrast, AphA and Qrr4 levels of cells treated with AI-2 and DPO were similar to those treated with DPO only, suggesting that CAI-1 has a stronger effect on Qrr4 production, when compared to AI-2 (16). In line with this observation, the combination of CAI-1 and DPO resulted in a more robust (∼3.6-fold) reduction in AphA, when compared to AI-2 and DPO (Figure 5A, lanes 1, 6, 7). Finally, we also tested the combined effect of all three autoinducers. Indeed, addition of AI-2, CAI-1 and DPO boosted AphA repression to 18.3-fold along with the expected reduction in Qrr4 levels (∼10-fold) and activation of VqmR (∼11-fold).
To corroborate these results, we also performed the reciprocal experiments, i.e. we generated single, double and triple deletion strains of the autoinducer synthase genes (luxS, cqsA and tdh) and monitored AphA production in late stationary phase cells (6 h after cells reached an OD600 of 1.5) using Western Blot analysis (Supplementary Figure S4). When compared to wild-type V. cholerae, all three single mutants displayed a significant increase in AphA levels with ΔcqsA showing the strongest up-regulation (∼7.3-fold, Supplementary Figure S4, lane 1 versus 3). Mutation of luxS or tdh both increased AphA production by ∼3-fold (Supplementary Figure S4, lanes 2 and 4). V. cholerae strains lacking two of the synthases, i.e. ΔluxS, cqsA; ΔluxS, tdh and ΔcqsA, tdh, all showed elevated production of AphA, when compared to the relevant mutants lacking only one of the synthase genes (Supplementary Figure S4, lanes 5–7). Finally, deletion of all three autoinducer synthase genes boosted AphA levels by >15-fold (Supplementary Figure S4, lane 8), which was the strongest effect detected in our panel.
Together our data show that QS-mediated down-regulation of AphA in V. cholerae is enhanced by the combined action of multiple autoinducers acting through LuxO and VqmA to modulate production of the Qrr and VqmR sRNAs.
Global transcriptome analysis of autoinducer function in V. cholerae
The combined regulatory effect of AI-2, CAI-1 and DPO on AphA levels prompted us to probe autoinducer functions in V. cholerae at a larger scale. Specifically, we used the setup of the previous experiments (Figure 5A) and RNA-sequencing to monitor autoinducer-controlled changes at a transcriptome-wide level. Again, we performed these experiments in ΔluxS, cqsA, tdh cells and added single or combinations of the autoinducers at saturating levels (5 μM final conc.) to identify the full set of autoinducer-responsive genes in V. cholerae.
In line with our above results (Figure 5B), autoinducer-mediated repression of aphA was most prominent when all three autoinducers were supplemented (∼11-fold), whereas addition of single autoinducers resulted in modest repression (Supplementary Table S1). In general, transcriptome analysis revealed only very few (4) differentially expressed genes (≥2-fold) in response to AI-2 and moderate changes (40 and 60 genes) when cells were exposed to DPO or CAI-1, respectively ( Figure 6 and Supplementary Table S1). Treatment of V. cholerae with two autoinducers significantly enhanced QS-mediated gene regulation. Combination of AI-2 and CAI-1 had the strongest effect leading to the differential expression of 323 genes, followed by DPO/CAI-1 (151 genes), and AI-2/DPO (59 genes). Together, AI-2, DPO and CAI-1 rendered almost 400 genes (Supplementary Table S1), suggesting that all three autoinducers are required for a full QS response. For example, genes from two genetic islands specific to V. cholerae strains of the 7th pandemic (VSP-1 and VSP-2) were not affected by AI-2, mildly activated by DPO and CAI-1, and strongly induced by combined treatment with the three autoinducers (Figure 6). We discovered similar expression patterns for genes associated with fatty acid metabolism (vca0688-vca0691), as well as genes located in two chemotaxis clusters of V. cholerae (vc1394-vc1403 and vca1088-cheA-3). QS-mediated activation of chemotaxis genes in V. cholerae is in accordance with a previous report (44).
Figure 6.

Genome-wide transcriptome changes in response to the AI-2, DPO and CAI-1 autoinducers. Heatmap displaying 420 genes differentially expressed (≥2-fold) in response to at least one of the autoinducers. V. cholerae ΔluxS, cqsA, tdh cells were cultivated in M9 minimal media containing single or combinations of the autoinducers (5 μM final conc. each). Selected gene clusters showing significant regulation are highlighted on the right. Fold changes of the normalized expression values were calculated relative to the normalized expression values of the mock treated replicates.
We also discovered reciprocal expression patterns, i.e. downregulation in response to the autoinducers. For example, genes relevant for iron transport (vc0199-fhuB), biofilm formation (vpsU, vpsA, rbmC and vpsL) and RTX toxin secretion (rtxA-D) were all repressed in response to the autoinducers. Of note, DPO alone also significantly repressed the rtx operon, which is in accordance with our previous work showing VqmR-mediated repression of rtx (24). Interestingly, DPO by itself seems to function as a repressor of type VI secretion genes, however, this regulation can be overcome by CAI-1 but not by AI-2 (Figure 6).
DISCUSSION
Gene regulation by QS is crucial for virulence factor production and collective functions of various bacterial pathogens, including V. cholerae (26). For many bacterial pathogens, QS relies on multiple signalling molecules, however, how these act together to control gene expression is frequently unknown (2). Given that enteric pathogens such as V. cholerae regularly interact with other species, e.g. during the course of an infection, one can predict that exposure to multiple signalling molecules is the norm rather than the exception. Therefore, studying the influence of autoinducer mixtures on gene expression and behaviour of bacterial pathogens is fundamental to develop a global understanding of QS functions in these organisms.
In this study, we discovered that the DPO autoinducer inhibits the production of AphA, a central regulator of virulence gene expression in V. cholerae. Repression of AphA by DPO requires the VqmA receptor protein, as well as the VqmR sRNA (Figure 3B). VqmR belongs to the large group of Hfq-dependent sRNAs (23). These sRNAs control gene expression by base-pairing with target mRNAs, which can either repress or activate gene expression (45,46). Repression of aphA by VqmR relies on a base-pairing sequence that we discover here to be located in the loop of the Rho-independent terminator stem of the sRNA (Figure 2A). There are only few documented cases of target recognition via the terminator sequence of an Hfq-dependent sRNA. For example, base-pairing of the OxyS sRNA with the fhlA mRNA requires two independent sequence elements, one of which is located in the loop of the terminal stem in the sRNA (47). Rho-independent terminator sequences have been shown to recruit Hfq to sRNAs (48) and are considered important for transcript stability providing protection from 3′-5′ exonucleolytic degradation (49). We speculate that base-pairing with aphA could destabilize the terminator structure of VqmR and thereby facilitate turn-over of the sRNA. Indeed, previous work focussing on the regulatory mechanisms of the Qrr sRNAs in Vibrio harveyi revealed that base-pairing with specific target mRNAs affects Qrr degradation and thereby modulates QS fidelity (50). One of the target mRNAs relevant for Qrr turn-over is aphA, which is also regulated by VqmR. However, the Qrr sRNAs and VqmR have antagonizing effects on aphA, with the Qrr sRNAs acting to increase AphA production (19,51), whereas VqmR reduces AphA levels (Figure 3A). It will be interesting to test how the two sRNAs compete for aphA regulation and if the molecular mechanisms underlying post-transcriptional control of this mRNA will provide priority to regulation by one of the sRNAs. Sequence alignment of the aphA 5′ UTR showed that the base-pairing sites of VqmR and the Qrr sRNAs are conserved among Vibrios (Supplementary Figure S5) and so are the relevant interaction sites in Qrr2-4 (51), as well as VqmR (Figure 2A). Therefore, regulation of aphA by two competing sRNAs species could be relevant for collective functions of many Vibrio strains.
Regulation of aphA by VqmR links the AI-2/CAI-1 and DPO QS pathways at a critical point as AphA controls virulence by activating tcpPH (42), modulates QS by repressing hapR (19), and enhances biofilm formation by inducing vpsT transcription (32). Activation of vpsT transcription by AphA, together with post-transcriptional repression of aphA (Figure 2B) and vpsT (24) by VqmR indicates the presence of a type 2 coherent feed-forward loop (52) controlling VpsT production in V. cholerae (Figure 1B). In this scenario, VqmR acts on top of the cascade repressing vpsT translation by base-pairing to the mRNA, while repressing vpsT transcription by reducing AphA levels. An increasing number of regulatory RNAs are being recognized to participate in mixed network motifs with transcription factors (53). For example, the Spot42 sRNA of E. coli together with the CRP transcriptional regulator forms a multi-output feed-forward loop to decrease leaky expression of target genes (54). In Salmonella, the RprA sRNA activates the expression of RpoS and RicI to prevent plasmid conjugation when the cell membrane is damaged (55). Coherent feed-forward loops typically reduce noise in biological systems (53) and in the case of VqmR-mediated repression of aphA and vpsT might help to facilitate transition between QS states and to coordinate virulence gene expression and biofilm formation in V. cholerae. Of note, VpsT activity is also controlled post-translationally by binding of c-di-GMP (56), which could add an additional layer of regulation.
How and when V. cholerae changes from one QS state to another depends on the accumulation of autoinducers in the environment (57). AI-2 and CAI-1 are recognized by the membrane-bound receptors, LuxPQ and CqsS, respectively and channel information into a shared signalling cascade (Figure 1). Importantly, both receptors function as kinases to phosphorylate LuxU in the absence of AI-2 and CAI-1, but convert to phosphatases when the autoinducers are bound. This logic prevents premature modulation of QS-responsive pathways when only a single autoinducer is present (21). Indeed, global gene expression analysis showed that, although provided at saturating concentrations (16), AI-2 and CAI-1 had only modest effects on the transcriptome of V. cholerae, when compared to a combination of the two autoinducers (Figure 6 and Supplementary Table S1). Our data are also in line with a recent report showing that CqsS exhibits a stronger phosphatase activity than LuxQ and supports the existence of a positive feedback loop upregulating cqsS levels by the autoinducers (16). Activation of cqsS was most prominent (∼2.5-fold) when all three autoinducers were present indicating a combined effect on cqsS expression (Supplementary Figure S6A).
Similarly, we also discovered autoinducer-mediated activation of the mRNAs encoding the VqmA and CqsR receptors (21), while the mRNAs of luxPQ and vpsS remained constant under all tested conditions (Supplementary Figure S6A). Activation of the VqmA production might also explain induction of VqmR expression in CAI-1 treated cells (Figure 5A). However, how CAI-1 influences vqmA expression is currently unclear. Transcripts encoding proteins involved in transduction of QS signals (luxOU and vspV) displayed only minor changes in response to the autoinducers (Supplementary Figure S6B), whereas mRNAs of downstream transcriptional regulators, i.e. aphA, hapR and vpsT, showed the expected expression patterns in response to the autoinducers (Supplementary Figure S6C). Modest upregulation (∼1.5-fold) of luxOU in cells treated with a combination of autoinducers supports previous reports suggesting that Qrr-mediated repression of luxO does not involve significant transcript turn-over (50,58). Strong activation of hapR by AI-2 and CAI-1 is expected due to Qrr-mediated repression of hapR (18), however, hapR levels were also activated by DPO (∼2-fold, Supplementary Figure S6C), which might be explained by negative regulation of the hapR promoter by AphA (19). Therefore, although only the Qrr sRNAs base-pair with hapR to inhibit translation, VqmR can promote similar regulation by repressing aphA. Indeed, adding CAI-1 and DPO to V. cholerae significantly increased hapR abundance when compared to cells treated with CAI-1 only (Supplementary Figure S6C). Together, these data support the hypothesis that the three autoinducer act in concert to modulate QS functions. Of note, naturally occurring frameshifts in hapR have been reported for several toxigenic V. cholerae isolates (59). Mutants lacking HapR are likely to lose most of their QS functions, however, regulation of aphA and vpsT by AI-2/CAI-1 and DPO possibly retains basic QS-mediated gene regulation in these strains.
Besides looking at known QS-mediated responses in V. cholerae, our transcriptomic approach also allowed us to investigate the QS response of additional sets of genes relevant for pathogenicity and collective behavior. Mapping of the transcriptome data to the V. cholerae genome revealed activation of genes located in two chemotaxis clusters (Figure 6 and (44,60)). We also discovered autoinducer-mediated regulation of several methyl-accepting chemotaxis proteins (MCPs), also known as chemoreceptors. The chromosomes of V. cholerae El Tor contain 45 potential MCPs (61), 22 of which were differently regulated by the autoinducers (Supplementary Figure S7A). Whereas 18 MCP genes were upregulated in response to the autoinducers, four genes were repressed. Among these, vca0068 was previously reported to be expressed during the infection process of V. cholerae (62,63) and we have shown that vca0068 is repressed by base-pairing with VqmR (24). The ligand of VCA0068 is currently unknown, but regulation of this gene by both the autoinducers might indicate a role for this MCP in QS transition.
Another key factor for virulence, biofilm formation, and overall physiology of V. cholerae is c-di-GMP (64). Our transcriptomic data revealed differential expression of genes associated with the production, degradation and binding of c-di-GMP, with the majority (19/23) being induced when the autoinducers were supplemented (Supplementary Figure S7B). There was no clear separation between diguanylate cyclases (DGCs) and phosphodiesterases (PDEs) showing upregulation or downregulation by the autoinducers. However, we did find significant overlap with previous work studying QS-mediated regulation of DGCs and PDEs in V. cholerae. For example, expression of vc1086, vc1370, vca0080, vca0848 and vc0965 was induced by the autoinducers, which is in accordance with elevated expression of these genes in luxO deficient V. cholerae, which fail to produce the Qrr sRNAs (3). By the same token, levels of cdgA (vca0074), encoding a DGC involved in biofilm formation of V. cholerae (65,66), were inhibited in cells treated with multiple autoinducers (Supplementary Figure S7B) and were similarly reduced in the luxO mutant (3). How exactly QS signals and cellular c-di-GMP levels are coordinated to control biofilm formation and other collective functions in V. cholerae is currently unknown. However, activation of the aphA promoter by c-di-GMP-bound VpsR (67), together with QS-mediated post-transcriptional control of aphA mRNA (Figure 1), support the idea that the inter- and intracellular signalling pathways are intimately connected in V. cholerae (68).
AphA is a crucial factor for pathogenicity of V. cholerae, and so is biofilm formation (26). Our discovery that DPO inhibits both virulence expression (Figure 4C) and biofilm formation (24) via VqmR-mediated repression of aphA and vpsT, respectively, predicts that DPO could be used to restrict V. cholerae infections. This hypothesis is fuelled by a previous report showing that vqmA mutants outcompete V. cholerae wild-type cells in colonization assays using germ-free mice and the commensal gut bacterium Ruminococcus obeum (69). V. cholerae cells lacking vqmA fail to produce VqmR and display increased VpsT (24) and AphA (Figure 3A) levels, which could reinforce biofilm formation and virulence gene expression. There is significant interest in development of QS manipulation strategies to promote and to terminate beneficial and harmful bacterial behaviours, respectively (2,70). DPO could serve as a scaffold for such therapies since, conceivably, simple DPO-precursors such as l-threonine could be used to enhance DPO production and consequently inhibit biofilm formation and virulence genes expression of V. cholerae in the small intestine.
DATA AVAILABILITY
The sequencing data has been deposited at Gene Expression Omnibus (GEO) under the GSE115711 accession code.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Helmut Blum and Stefan Krebs for help with the RNA sequencing experiments, and Andreas Starick for excellent technical support. We thank Bonnie Bassler for the gift of synthetic AI-2 and CAI-1; Franz Narberhaus, Cynthia Sharma and Gisela Storz for supplying toeprinting reagents. Also, we would like to thank Mona Dotzler for help with plasmid construction. We thank Jörg Vogel, Gisela Storz and Bonnie Bassler for comments on the manuscript and all members of the Papenfort lab for insightful discussions and suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Research Foundation (DFG) [PA2820/1 and Exc114-2]; Human Frontier Science Program [CDA00024/2016-C]; European Research Council [StG-758212]. K.F. and K.P. acknowledge support by the LMU Mentoring program of the LMU Faculty of Biology and Young Scholars’ Programme of the Bavarian Academy of Sciences and Humanities, respectively. Funding for open access charge: DFG.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hawver L.A., Jung S.A., Ng W.L.. Specificity and complexity in bacterial quorum-sensing systems. FEMS Microbiol. Rev. 2016; 40:738–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papenfort K., Bassler B.L.. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016; 14:576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waters C.M., Lu W., Rabinowitz J.D., Bassler B.L.. Quorum sensing controls biofilm formation in Vibrio cholerae through modulation of cyclic di-GMP levels and repression of vpsT. J. Bacteriol. 2008; 190:2527–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shao Y., Bassler B.L.. Quorum regulatory small RNAs repress type VI secretion in Vibrio cholerae. Mol. Microbiol. 2014; 92:921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zheng J., Shin O.S., Cameron D.E., Mekalanos J.J.. Quorum sensing and a global regulator TsrA control expression of type VI secretion and virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21128–21133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lo Scrudato M., Blokesch M.. A transcriptional regulator linking quorum sensing and chitin induction to render Vibrio cholerae naturally transformable. Nucleic Acids Res. 2013; 41:3644–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Antonova E.S., Hammer B.K.. Quorum-sensing autoinducer molecules produced by members of a multispecies biofilm promote horizontal gene transfer to Vibrio cholerae. FEMS Microbiol. Lett. 2011; 322:68–76. [DOI] [PubMed] [Google Scholar]
- 8. Hoque M.M., Naser I.B., Bari S.M., Zhu J., Mekalanos J.J., Faruque S.M.. Quorum regulated resistance of Vibrio cholerae against environmental bacteriophages. Sci. Rep. 2016; 6:37956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu J., Miller M.B., Vance R.E., Dziejman M., Bassler B.L., Mekalanos J.J.. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:3129–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kelly R.C., Bolitho M.E., Higgins D.A., Lu W., Ng W.L., Jeffrey P.D., Rabinowitz J.D., Semmelhack M.F., Hughson F.M., Bassler B.L.. The Vibrio cholerae quorum-sensing autoinducer CAI-1: analysis of the biosynthetic enzyme CqsA. Nat. Chem. Biol. 2009; 5:891–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen X., Schauder S., Potier N., Van Dorsselaer A., Pelczer I., Bassler B.L., Hughson F.M.. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 2002; 415:545–549. [DOI] [PubMed] [Google Scholar]
- 12. Higgins D.A., Pomianek M.E., Kraml C.M., Taylor R.K., Semmelhack M.F., Bassler B.L.. The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature. 2007; 450:883–886. [DOI] [PubMed] [Google Scholar]
- 13. Surette M.G., Miller M.B., Bassler B.L.. Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: a new family of genes responsible for autoinducer production. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:1639–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ng W.L., Perez L.J., Wei Y., Kraml C., Semmelhack M.F., Bassler B.L.. Signal production and detection specificity in vibrio CqsA/CqsS quorum-sensing systems. Mol. Microbiol. 2011; 79:1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neiditch M.B., Federle M.J., Miller S.T., Bassler B.L., Hughson F.M.. Regulation of LuxPQ receptor activity by the quorum-sensing signal autoinducer-2. Mol. Cell. 2005; 18:507–518. [DOI] [PubMed] [Google Scholar]
- 16. Hurley A., Bassler B.L.. Asymmetric regulation of quorum-sensing receptors drives autoinducer-specific gene expression programs in Vibrio cholerae. PLoS Genet. 2017; 13:e1006826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bassler B.L., Wright M., Silverman M.R.. Multiple signalling systems controlling expression of luminescence in Vibrio harveyi: sequence and function of genes encoding a second sensory pathway. Mol. Microbiol. 1994; 13:273–286. [DOI] [PubMed] [Google Scholar]
- 18. Lenz D.H., Mok K.C., Lilley B.N., Kulkarni R.V., Wingreen N.S., Bassler B.L.. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004; 118:69–82. [DOI] [PubMed] [Google Scholar]
- 19. Rutherford S.T., van Kessel J.C., Shao Y., Bassler B.L.. AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev. 2011; 25:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ng W.L., Bassler B.L.. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 2009; 43:197–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jung S.A., Chapman C.A., Ng W.L.. Quadruple quorum-sensing inputs control Vibrio cholerae virulence and maintain system robustness. PLoS Pathog. 2015; 11:e1004837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Papenfort K., Silpe J.E., Schramma K.R., Cong J.P., Seyedsayamdost M.R., Bassler B.L.. A Vibrio cholerae autoinducer-receptor pair that controls biofilm formation. Nat. Chem. Biol. 2017; 13:551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Updegrove T.B., Zhang A., Storz G.. Hfq: the flexible RNA matchmaker. Curr. Opin. Microbiol. 2016; 30:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Papenfort K., Forstner K.U., Cong J.P., Sharma C.M., Bassler B.L.. Differential RNA-seq of Vibrio cholerae identifies the VqmR small RNA as a regulator of biofilm formation. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E766–E775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Casper-Lindley C., Yildiz F.H.. VpsT is a transcriptional regulator required for expression of vps biosynthesis genes and the development of rugose colonial morphology in Vibrio cholerae O1 El Tor. J. Bacteriol. 2004; 186:1574–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Silva A.J., Benitez J.A.. Vibrio cholerae Biofilms and Cholera Pathogenesis. PLoS Negl. Trop. Dis. 2016; 10:e0004330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teschler J.K., Zamorano-Sanchez D., Utada A.S., Warner C.J., Wong G.C., Linington R.G., Yildiz F.H.. Living in the matrix: assembly and control of Vibrio cholerae biofilms. Nat. Rev. Microbiol. 2015; 13:255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tamayo R., Patimalla B., Camilli A.. Growth in a biofilm induces a hyperinfectious phenotype in Vibrio cholerae. Infect. Immun. 2010; 78:3560–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Millet Y.A., Alvarez D., Ringgaard S., von Andrian U.H., Davis B.M., Waldor M.K.. Insights into Vibrio cholerae intestinal colonization from monitoring fluorescently labeled bacteria. PLoS Pathog. 2014; 10:e1004405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kovacikova G., Skorupski K.. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol. Microbiol. 2001; 41:393–407. [DOI] [PubMed] [Google Scholar]
- 31. Almagro-Moreno S., Pruss K., Taylor R.K.. Intestinal colonization dynamics of Vibrio cholerae. PLoS Pathog. 2015; 11:e1004787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang M., Frey E.M., Liu Z., Bishar R., Zhu J.. The virulence transcriptional activator AphA enhances biofilm formation by Vibrio cholerae by activating expression of the biofilm regulator VpsT. Infect. Immun. 2010; 78:697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ball A.S., Chaparian R.R., van Kessel J.C.. Quorum sensing gene regulation by LuxR/HapR master regulators in Vibrios. J. Bacteriol. 2017; 199:e00105-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao W., Caro F., Robins W., Mekalanos J.J.. Antagonism toward the intestinal microbiota and its effect on Vibrio cholerae virulence. Science. 2018; 359:210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Logan S.L., Thomas J., Yan J., Baker R.P., Shields D.S., Xavier J.B., Hammer B.K., Parthasarathy R.. The Vibrio cholerae type VI secretion system can modulate host intestinal mechanics to displace gut bacterial symbionts. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E3779–E3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Iwanaga M., Yamamoto K., Higa N., Ichinose Y., Nakasone N., Tanabe M.. Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 El Tor. Microbiol. Immunol. 1986; 30:1075–1083. [DOI] [PubMed] [Google Scholar]
- 37. Papenfort K., Sun Y., Miyakoshi M., Vanderpool C.K., Vogel J.. Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell. 2013; 153:426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frohlich K.S., Haneke K., Papenfort K., Vogel J.. The target spectrum of SdsR small RNA in Salmonella. Nucleic Acids Res. 2016; 44:10406–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Corcoran C.P., Podkaminski D., Papenfort K., Urban J.H., Hinton J.C., Vogel J.. Superfolder GFP reporters validate diverse new mRNA targets of the classic porin regulator, MicF RNA. Mol. Microbiol. 2012; 84:428–445. [DOI] [PubMed] [Google Scholar]
- 40. Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R.. Fast and effective prediction of microRNA/target duplexes. RNA. 2004; 10:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hartz D., McPheeters D.S., Traut R., Gold L.. Extension inhibition analysis of translation initiation complexes. Methods Enzymol. 1988; 164:419–425. [DOI] [PubMed] [Google Scholar]
- 42. Hase C.C., Mekalanos J.J.. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:730–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. DiRita V.J., Parsot C., Jander G., Mekalanos J.J.. Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:5403–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ringgaard S., Hubbard T., Mandlik A., Davis B.M., Waldor M.K.. RpoS and quorum sensing control expression and polar localization of Vibrio cholerae chemotaxis cluster III proteins in vitro and in vivo. Mol. Microbiol. 2015; 97:660–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Lay N., Schu D.J., Gottesman S.. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J. Biol. Chem. 2013; 288:7996–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Papenfort K., Vanderpool C.K.. Target activation by regulatory RNAs in bacteria. FEMS Microbiol. Rev. 2015; 39:362–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Argaman L., Altuvia S.. fhlA repression by OxyS RNA: kissing complex formation at two sites results in a stable antisense-target RNA complex. J. Mol. Biol. 2000; 300:1101–1112. [DOI] [PubMed] [Google Scholar]
- 48. Otaka H., Ishikawa H., Morita T., Aiba H.. PolyU tail of rho-independent terminator of bacterial small RNAs is essential for Hfq action. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:13059–13064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mohanty B.K., Kushner S.R.. Enzymes involved in posttranscriptional RNA metabolism in Gram-Negative bacteria. Microbiol. Spectr. 2018; 6:19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Feng L., Rutherford S.T., Papenfort K., Bagert J.D., van Kessel J.C., Tirrell D.A., Wingreen N.S., Bassler B.L.. A qrr noncoding RNA deploys four different regulatory mechanisms to optimize quorum-sensing dynamics. Cell. 2015; 160:228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shao Y., Bassler B.L.. Quorum-sensing non-coding small RNAs use unique pairing regions to differentially control mRNA targets. Mol. Microbiol. 2012; 83:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shen-Orr S.S., Milo R., Mangan S., Alon U.. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 2002; 31:64–68. [DOI] [PubMed] [Google Scholar]
- 53. Nitzan M., Rehani R., Margalit H.. Integration of bacterial small RNAs in regulatory networks. Annu. Rev. Biophys. 2017; 46:131–148. [DOI] [PubMed] [Google Scholar]
- 54. Beisel C.L., Storz G.. The base-pairing RNA spot 42 participates in a multioutput feedforward loop to help enact catabolite repression in Escherichia coli. Mol. Cell. 2011; 41:286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Papenfort K., Espinosa E., Casadesus J., Vogel J.. Small RNA-based feedforward loop with AND-gate logic regulates extrachromosomal DNA transfer in Salmonella. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E4772–E4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krasteva P.V., Fong J.C., Shikuma N.J., Beyhan S., Navarro M.V., Yildiz F.H., Sondermann H.. Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science. 2010; 327:866–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Svenningsen S.L., Waters C.M., Bassler B.L.. A negative feedback loop involving small RNAs accelerates Vibrio cholerae's transition out of quorum-sensing mode. Genes Dev. 2008; 22:226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tu K.C., Long T., Svenningsen S.L., Wingreen N.S., Bassler B.L.. Negative feedback loops involving small regulatory RNAs precisely control the Vibrio harveyi quorum-sensing response. Mol. Cell. 2010; 37:567–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Joelsson A., Liu Z., Zhu J.. Genetic and phenotypic diversity of quorum-sensing systems in clinical and environmental isolates of Vibrio cholerae. Infect. Immun. 2006; 74:1141–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Butler S.M., Camilli A.. Going against the grain: chemotaxis and infection in Vibrio cholerae. Nat. Rev. Microbiol. 2005; 3:611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Heidelberg J.F., Eisen J.A., Nelson W.C., Clayton R.A., Gwinn M.L., Dodson R.J., Haft D.H., Hickey E.K., Peterson J.D., Umayam L. et al.. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000; 406:477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lombardo M.J., Michalski J., Martinez-Wilson H., Morin C., Hilton T., Osorio C.G., Nataro J.P., Tacket C.O., Camilli A., Kaper J.B.. An in vivo expression technology screen for Vibrio cholerae genes expressed in human volunteers. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:18229–18234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Camilli A., Mekalanos J.J.. Use of recombinase gene fusions to identify Vibrio cholerae genes induced during infection. Mol. Microbiol. 1995; 18:671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Conner J.G., Zamorano-Sanchez D., Park J.H., Sondermann H., Yildiz F.H.. The ins and outs of cyclic di-GMP signaling in Vibrio cholerae. Curr. Opin. Microbiol. 2017; 36:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Beyhan S., Bilecen K., Salama S.R., Casper-Lindley C., Yildiz F.H.. Regulation of rugosity and biofilm formation in Vibrio cholerae: comparison of VpsT and VpsR regulons and epistasis analysis of vpsT, vpsR, and hapR. J. Bacteriol. 2007; 189:388–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Massie J.P., Reynolds E.L., Koestler B.J., Cong J.P., Agostoni M., Waters C.M.. Quantification of high-specificity cyclic diguanylate signaling. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:12746–12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Srivastava D., Harris R.C., Waters C.M.. Integration of cyclic di-GMP and quorum sensing in the control of vpsT and aphA in Vibrio cholerae. J. Bacteriol. 2011; 193:6331–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Srivastava D., Waters C.M.. A tangled web: regulatory connections between quorum sensing and cyclic Di-GMP. J. Bacteriol. 2012; 194:4485–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hsiao A., Ahmed A.M., Subramanian S., Griffin N.W., Drewry L.L., Petri W.A., Haque R., Ahmed T., Gordon J.I.. Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature. 2014; 515:423–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Whiteley M., Diggle S.P., Greenberg E.P.. Progress in and promise of bacterial quorum sensing research. Nature. 2017; 551:313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data has been deposited at Gene Expression Omnibus (GEO) under the GSE115711 accession code.