

Abstract
4,6-Diamino-5-formamidopyrimidine (Fapy•dG) is an abundant form of oxidative DNA damage that is mutagenic and contributes to the pathogenesis of human disease. When Fapy•dG is in its nucleotide triphosphate form, Fapy•dGTP, it is inefficiently cleansed from the nucleotide pool by the responsible enzyme in Escherichia coli MutT and its mammalian homolog MTH1. Therefore, under oxidative stress conditions, Fapy•dGTP could become a pro-mutagenic substrate for insertion into the genome by DNA polymerases. Here, we evaluated insertion kinetics and high-resolution ternary complex crystal structures of a configurationally stable Fapy•dGTP analog, β-C-Fapy•dGTP, with DNA polymerase β. The crystallographic snapshots and kinetic data indicate that binding of β-C-Fapy•dGTP impedes enzyme closure, thus hindering insertion. The structures reveal that an active site residue, Asp276, positions β-C-Fapy•dGTP so that it distorts the geometry of critical catalytic atoms. Removal of this guardian side chain permits enzyme closure and increases the efficiency of β-C-Fapy•dG insertion opposite dC. These results highlight the stringent requirements necessary to achieve a closed DNA polymerase active site poised for efficient nucleotide incorporation and illustrate how DNA polymerase β has evolved to hinder Fapy•dGTP insertion.
INTRODUCTION
During oxidative stress, oxygen and nitrogen radicals accumulate and damage DNA bases in duplex DNA and the nucleotide pool. These damaged DNA lesions promote cytotoxicity and/or mutagenesis, leading to adverse human health outcomes (1–3). Understanding how damaged nucleotides are inserted into the genome is imperative for improving cancer therapies that utilize DNA damaging mechanisms, such as radiation therapy, nucleotide analog inhibitors, and MTH1 inhibitors (4–9). Under endogenous and exogenous oxidative stress, guanine (dG) in both duplex DNA and the nucleotide pool (dGTP) undergoes increased damage in comparison to other DNA bases because of its oxidation potential (10). Of the possible damaged guanine intermediates, the C8-OH-adduct radical generates two of the most prominent DNA lesions, 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodGuo) and 4,6-diamino-5-formamidopyrimidine (Fapy•dG) (11,12). 8-oxodGuo results from oxidation of the C8-OH intermediate, while Fapy•dG is formed via opening of the imidazole ring and subsequent reduction (Figure 1) (13). The formation of these two species are therefore in competition with each other, with both being prevalent DNA lesions found in cellular DNA (11,14–16). The biological importance of the lesions is highlighted by the elaborate cellular defense mechanisms that have evolved to remove these lesions from DNA and the nucleotide pool (17).
Figure 1.

Formation of formamidopyrimidines and 8-oxopurine lesions comes from a common intermediate, adapted from (26).
Fapy•dG poses a threat to genomic integrity through mismatched base pairing, which can cause transversions and transitions in the genome if not removed. The mutagenic profile of Fapy•dG during replication bypass is that Fapy•dG predominantly promotes G to T transversion mutations, akin to 8-oxodGuo, but also promotes G to A transitions (18–20). Despite the lesion's mutagenicity and the levels at which it is formed, our mechanistic understanding of Fapy•dG mediated mutagenesis lags behind that of 8-oxodGuo due to challenges generating suitable substrates containing the Fapy•dG lesion (21). Although Fapy•dG is believed to exist predominantly as the β-anomer in duplex DNA, monomeric formamidopyrimidines undergo facile epimerization (21–23). Therefore, obtaining a pure sample containing a single Fapy•dG anomer for biochemical assays has been challenging. To bypass these prohibitive synthetic challenges, configurationally stable analogs of Fapy•dG have been used to elucidate biochemical mechanisms that confer the biological consequences of this prevalent DNA lesion (24–27). Accordingly, we utilized a similar approach to generate a nucleotide Fapy•dGTP analog, β-C-Fapy•dGTP, in which the glycosidic nitrogen is replaced with a methylene group to preserve the biologically relevant β-anomer (Supplementary Figure S1). For simplicity, the analog will be referred to as Fapy•dGTP.
Previous studies have elicited that Fapy•dG bypass in prokaryotes is weakly mutagenic (18,25,26,28). In contrast, it is highly mutagenic in eukaryotes, even more so than 8-oxodGuo in some sequence contexts (19,20). 8-OxodGuo is frequently thought of as a significant, highly mutagenic DNA lesion. The contrasting results in different species, and especially the mutagenicity compared to 8-oxodGuo in eukaryotes emphasizes the significance of understanding Fapy•dG-induced effects at the molecular level. Previous structural studies of templating Fapy•dG bypass revealed that Watson–Crick base pairing and base tautomerization confer mutagenicity (27). Here, we expand on the molecular understanding of Fapy•dG mutagenesis by examining the nucleotide form of Fapy•dG. Importantly, Fapy•dGTP is poorly cleansed from the nucleotide pool by the sanitizing enzyme MutT in Escherichia coli and its mammalian counterpart MTH1 (26,29). These enzymes are responsible for removing oxidized nucleotides in the nucleotide pool by hydrolyzing the triphosphate form and generating the monophosphate form (30); this effectively prevents oxidized nucleotides from being inserted in the genome thereby deterring mutagenesis. However, since Fapy•dGTP is not efficiently hydrolyzed by MutT/MTH1, it could accumulate and be utilized by DNA polymerases during DNA replication and repair. To gain mechanistic insight into Fapy•dGTP insertion, we report high resolution pre-catalytic ternary substrate crystallographic structures of Fapy•dGTP opposite templating adenine (dA) or cytosine (dC), as well as kinetic characterization for the insertion reaction of Fapy•dGTP using the model mammalian DNA polymerase (pol) β. These results provide mechanistic molecular insight into how Fapy•dGMP incorporation into DNA is deterred by DNA polymerases.
MATERIALS AND METHODS
DNA sequences
The following DNA sequences were used to generate the 16-mer DNA duplexes used in crystallization studies (the templating base is underlined): template, 5′-CCG ACX GCG CAT CAG C-3′, where X represents the coding templating base (dA or dC); primer, 5′-GCT GAT GCG C-3′; downstream, 5′-pGTC GG-3′. Each oligonucleotide was suspended in 10 mM Tris–HCl, pH 7.4 and 1 mM EDTA, and the concentration was determined from their ultraviolet absorbance at 260 nm. DNA substrates were prepared by annealing the three purified oligonucleotides. The annealing reactions were performed by incubating a solution of primer with downstream and template oligonucleotides (1:1.2:1.2 molar ratio, respectively) at 95°C for 5 min, followed by 65°C for 30 min, and finally cooling 1°C min−1 to 10°C in a PCR thermocycler.
For use in our kinetic experiments, a 34-mer oligonucleotide DNA substrate containing a single nucleotide gap was prepared by annealing 3 gel-purified oligonucleotides to create a single-nucleotide gap at position 16. Each oligonucleotide was resuspended in 10 mM Tris–HCl, pH 7.4, and 1 mM EDTA, and the concentration determined from their UV absorbance at 260 nm. The annealing reactions were carried out by incubating a solution of 10 μM primer with 12 μM each of downstream and template oligonucleotides at 90–100°C for 3 min followed by 30 min at 65°C, and then slow cooling to room temperature. The sequence of the gapped DNA substrate was: primer, 5′-CTG CAG CTG ATG CGC-3′, downstream oligonucleotide, 5′-GTA CGG ATC CCC GGG TAC-3′, and template, 3′-GAC GTC GAC TAC GCG XCA TGC CTA GGG GCC CAT G-5′ where the X represents the coding templating base (dA or dC).
Pol β expression, purification and crystallization with β-C-Fapy•dGTP
Human pol β was overexpressed inE. coli and purified as previously described (31). Pol β was incubated with prepared single-nucleotide gapped DNA (1:1.2) containing either a templating dA or dC. To form binary complex crystals, they were grown in a solution containing 50 mM imidazole, pH 8.0, 13–19% PEG3350 and 350 mM sodium acetate as previously described (32). Binary pol β:DNA complex crystals were then soaked in a cryosolution containing 25% ethylene glycol, 50 mM imidazole, pH 7.5, 19% PEG3350 and 70 mM sodium acetate, 3 mM β-C-Fapy•dGTP (prepared as previously described (26)) and 50 mM CaCl2 for 1 h. This resulted in ‘ground state’ ternary pol β:DNA:β-C-Fapy•dGTP crystals for X-ray crystallography.
Data collection and refinement
Fapy•dGTP insertion opposite dC with either wild-type (WT) or D276G pol β (PDB accession numbers: 6DIA and 6DIC, respectively) were collected at 100 K on an SATURN92 CCD (charge-coupled device) detector system mounted on a MiraMax-007HF rotating anode generator at a wavelength of 1.54 Å. Fapy•dGTP insertion opposite dA with either WT or D276G pol β (6MR7 and 6MR8, respectively) were collected on a Rigaku MicroMax-007 HF rotating anode diffractometer equipped with a Dectris Pilatus3R 200K-A detector system at a wavelength of 1.54 Å. Data were processed and scaled using the HKL3000R software package (33). Initial models were determined using the PHENIX single one-component interface molecular replacement program by searching as a whole with the previously determined open binary (3ISB) or closed ternary (2FMS) structures of pol β bound to DNA with all ligands removed. The Rfree flags were taken from the starting model 2FMS for 6DIC. Rfree flags were generated randomly for the three open-ternary complexes, 6DIA, 6MR7, 6MR8, due to a lack of isomorphism with the open binary model used for molecular replacement (3ISB). Simulated annealing was performed to ensure no model bias. Refinement was performed using PHENIX and model building using Coot (34,35). The metal ligand coordination restraints were generated by ReadySet (PHENIX). The figures were prepared in PyMOL (Schrödinger LLC) and density maps were generated as polder maps within the phenix suite (36,37). Ramachandran analysis determined 100% of non-glycine residues lie in the allowed regions and at least 97% in favored regions.
Single-nucleotide gap filling DNA synthesis
Catalytic efficiencies (kpol/Kd) for single-nucleotide gap filling reactions were determined by single-turnover analysis (i.e. enzyme >> DNA). At least seven time points were gathered for each single-exponential time course determined with at least three sub-saturating concentrations of β-C-Fapy•dGTP. The standard reaction mixture contained 50 mM Tris–HCl, pH 7.4 (37°C), 40 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 100 μg/ml bovine serum albumin, 10% glycerol, 100 nM single-nucleotide gapped DNA, and 500 nM enzyme. Reactions were stopped with 150 mM EDTA and mixed with formamide dye. The substrates and products were separated on 16% denaturing (8 M urea) polyacrylamide gels. Since a 6-carboxyfluorescein 5′-labeled primer was used in these assays, the products were quantified using the GE Typhoon phosphorimager in fluorescence mode. Because sub-saturating concentrations of β-C-Fapy•dGTP were used (5–200 μM, dependent on the templating base), the data were fitted to an alternate form of the Michaelis equation to extract apparent catalytic efficiencies (kpol/Kd): kobs = ((kpol/Kd) * S) / (1 + (S/Kd)), where S refers to the concentration of β-C-Fapy•dGTP (Supplementary Figure S2).
RESULTS
Kinetic characterization of Fapy•dGTP insertion
The catalytic efficiencies for insertion of Fapy•dGTP opposite dC and dA were determined by single-turnover kinetic analysis (Figure 2). These results help contextualize the structural information described below. In comparison to undamaged dGTP opposite dC, the insertion efficiency of Fapy•dGTP is dramatically lower (nearly 4-orders of magnitude), and Fapy•dGTP insertion opposite dA is even less efficient. Consequently, insertion across from dA is only 82-fold less efficient than dC, resulting in a ∼750-fold reduction in discrimination compared to dGTP. Nevertheless, pol β is effective in excluding a Fapy•dGTP analog under circumstances where the oxidatively-generated nucleotide has accumulated in the nucleotide pool. The reduction in catalytic efficiency for this substrate is likely a result of unique and unfavorable binding within the polymerase active site as highlighted by the inability to saturate the polymerase with reasonable (<20 μM) concentrations of oxidized nucleotide (Figure S2). To provide molecular insight into the origin of these observations, we turned to X-ray crystallography.
Figure 2.
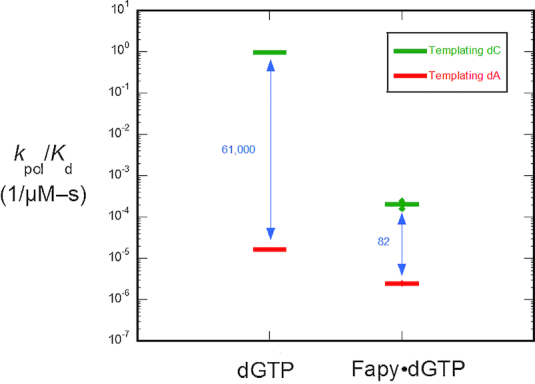
Discrimination plot for insertion of β-C-Fapy•dGTP. The catalytic efficiencies (kpol/Kd) for insertion of β-C-Fapy•dGTP opposite dC (green) or dA (red) for wild-type (WT) pol β are shown. The distance between the respective catalytic efficiencies is a measure of discrimination/fidelity. Each horizontal short bar represents the mean of duplicate independent determinations. Additionally, the value for each determination is plotted (small solid circle) to illustrate the reproducibility of these assays. The insertion efficiencies for dGTP opposite dC and dA were taken from (39).
Crystallographic characterization
The general enzymatic steps of DNA polymerization by pol β are structurally and biochemically characterized, and provide a comparison for evaluating Fapy•dGTP insertion. Upon binding duplex DNA, pol β assumes an ‘open’ conformation. Selection of the correct nucleotide occurs via an induced fit mechanism, requiring closure of the N-subdomain (fingers of replicative polymerases) resulting in proper alignment of active site residues with duplex DNA and incoming nucleotide, as well as proper coordination of active site metals that hasten catalysis. This subdomain repositioning also occurs with replicative polymerases, making pol β an applicable crystallographic model for evaluating nucleotide insertion. The resultant conformational rearrangement is termed the ternary-closed conformation. Catalysis occurs via a two metal mechanism, where nucleophilic attack by the oxyanion of O3′ of the primer terminus on α-phosphate (αP) of the incoming nucleotide results in incorporation of a deoxynucleoside monophosphate and release of pyrophosphate (38). Here, we evaluated the alignment of the catalytic atoms within pre-catalytic Fapy•dGTP insertion complexes containing either a templating dC or dA to provide molecular insight into the insertion of Fapy•dGTP.
Altered position of Fapy•dGTP during insertion opposite dC
To observe the pre-catalytic complex of non-mutagenic insertion of Fapy•dGTP by pol β, we crystallized a binary 1-nucleotide gapped DNA:pol β complex with templating dC. Crystals were placed in a cryoprotectant solution containing CaCl2 and Fapy•dGTP, whereby Ca2+ prevents catalysis, but allows for nucleotide binding (39,40). The resulting complex diffracted to 1.97 Å, Supplementary Table S1. Pol β retains an open conformation, and Fapy•dGTP is located in a stable but distorted position within the nucleotide binding pocket (Figure 3). The open ternary conformation is here-on referred to as ternary-open, as opposed to the canonical ternary-closed conformation. Within the nucleotide binding site, calcium binding is observed in the nucleotide metal site and absent in the catalytic metal site. Despite the absence of a catalytic metal, the O3′ (primer terminus) and αP (Fapy•dGTP) are 3.8 Å apart (Figure 3A), which is similar to the 3.7 Å distance observed in the undamaged pre-catalytic pol β:DNA:dGTP:Ca2+ complex (39). The Watson–Crick edge of Fapy•dGTP faces upstream with its pyrimidine ring in the DNA minor groove perpendicular to the primer terminus base (Figure 3B). The formamide carbonyl oxygen (O8) resulting from the broken imidazole ring interacts weakly with the templating dC (3.3 Å, Figure 3C). The Fapy•dG base is stabilized by several protein contacts and rotated about its glycosidic bond into a syn-conformation. The N-helix residue Asp276 is within hydrogen bonding distance of the O3′ of Fapy•dGTP (2.6 Å), while Arg258 forms stabilizing contacts with the analog through hydrogen bonds with N1 and O6 of the Watson–Crick edge (3.0 and 3.2 Å, respectively; Figure 3C). We also observed a Ca2+ ion coordinating the O2 of the templating dC. The presence of this metal is likely dependent on the identity of the templating base and the open polymerase conformation that accommodates the metal ion within the minor groove of pol β. The altered position of Fapy•dGTP observed in this open complex provides structural justification for previous assertions that damaged or incorrect nucleotides deter incorporation by stabilizing an unfavorable open conformation (41,42).
Figure 3.
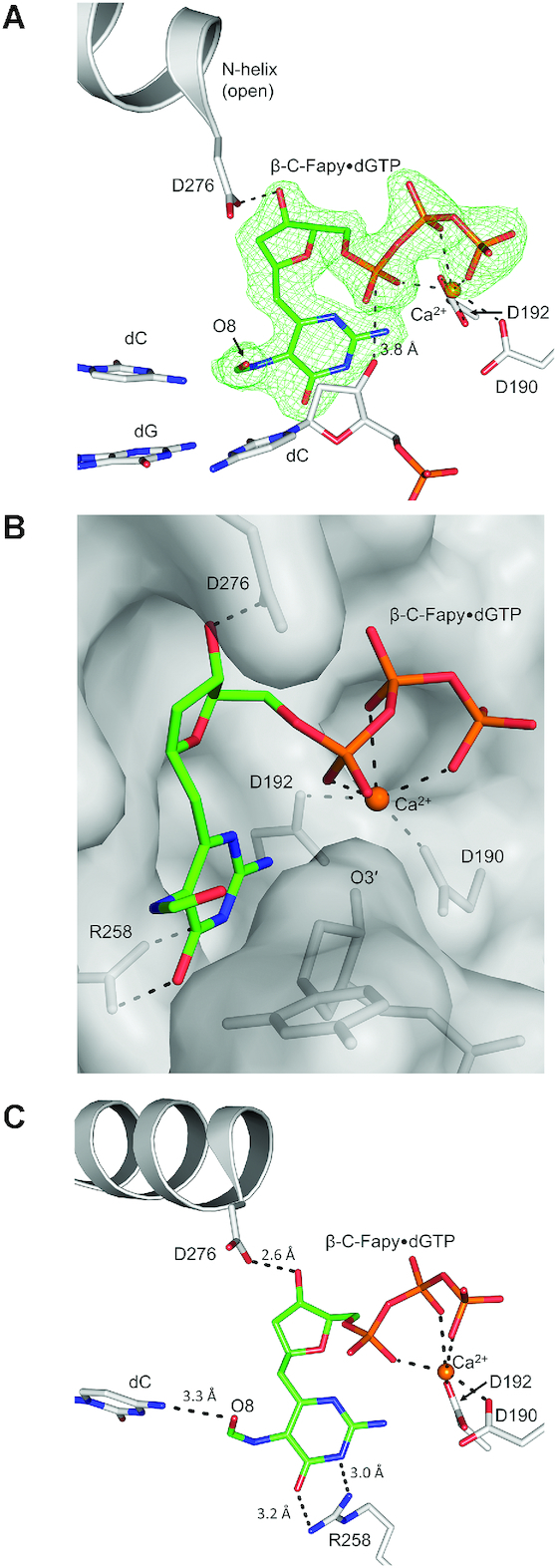
(A) Pre-catalytic ternary-open complex of wild-type pol β (gray) with β-C-Fapy•dGTP (green sticks) bound across from dC; Ca2+ ions are shown in orange. A polder map (green mesh) contoured at 3.0σ is shown for the incoming β-C-Fapy•dGTP. (B) β-C-Fapy•dGTP is shown as green sticks with the nucleotide binding pocket in surface representation looking into the minor groove behind the primer terminus (O3′). The Watson–Crick edge (W–C edge) is indicated. (C) Active site residues (gray sticks) that contact β-C-Fapy•dGTP (green) or Ca2+ (orange) are shown. Potential hydrogen bonds are shown as black dashed lines.
Insertion of Fapy•dGTP opposite dA
Because Fapy•dG has been reported to generate G to T transversion mutations, it is assumed that Fapy•dGTP insertion occurs opposite templating dA. To evaluate the mechanism of this type of insertion of Fapy•dGTP, we crystallized a binary DNA/pol β complex with templating dA in a 1-nucleotide gapped DNA substrate and soaked in CaCl2 and Fapy•dGTP to generate a pre-catalytic ternary complex. The crystal complex diffracted to 2.05 Å, Supplementary Table S1. Similar to what was observed with insertion opposite dC, pol β adopts a ternary-open conformation and Fapy•dGTP is positioned with the Watson–Crick edge in the minor groove facing upstream (Figure 4A). The distance between the O3′ of the primer terminus and αP of Fapy•dGTP increased subtly from 3.8 Å (opposite dC) to 4.1 Å (opposite dA). Additionally, the formamide group of Fapy•dG adopted multiple conformations opposite dA. Among the two conformations, one conformation is positioned within hydrogen bonding distance to N1 of dA via the formamide carbon (3.1 Å) and the formamide oxygen (3.2 Å), Figure 4B. In contrast, the formamide O8 interacts weakly with N4 of the templating dC (3.3 Å) (Figure 3C). When both the template dA and dC structures are overlaid the result is nearly identical ternary-open complexes (Figure 4C). However, slight changes were observed in the contacts between Fapy•dGTP and residues Arg258, Lys234, and the templating base (Figure 4D). In the templating dA complex, Arg258 adopts a different rotamer such that only NH2 comes within hydrogen bonding distance of O6 of the Fapy•dG Watson–Crick edge (2.6 Å). In the template dC complex, these two nitrogens of Arg258, NH1 and NH2, contact N1 (3.0 Å) and O6 (3.2 Å), respectively (Figure 4D). Lastly, a contact between Lys234 and O6 of the Fapy•dG base (3.2 Å) in the templating dC structure is not observed with templating dA.
Figure 4.

(A) Pre-catalytic ternary-open complex of wild-type pol β (gray) with β-C-Fapy•dGTP (green sticks) bound across from dA; Ca2+ ions are shown in orange. A polder map (green mesh) contoured at 3.0σ is shown for the incoming β-C-Fapy•dGTP. (B) An alternate viewpoint demonstrating the two formamide conformations of β-C-Fapy•dGTP (green sticks) opposite adenine (gray sticks) with the polder map contoured to 3.0σ (green mesh). (C) An overlay between the pre-catalytic ternary-open structures of β-C-Fapy•dGTP across from dA (green) or dC (yellow). Key residues are shown as gray sticks and the N-helix in gray cartoon. (D) An overlay between the pre-catalytic ternary-open structures of β-C-Fapy•dGTP across from dA (green) or dC (yellow) highlighting altered amino acids in stick format with the N-helix subdomain shown as gray cartoon and potential hydrogen bonds as dashed lines.
Asp276 guards the incoming nucleotide binding site
As revealed in the structures shown in Figures 3 and 4, the Fapy•dG base and sugar are stabilized by contacts with Asp276 in a fashion that hinders formation of the closed conformation and this, in turn, deters catalytic metal binding and chemistry. We chose to evaluate the significance of these distorting contacts by changing this residue to glycine, i.e. yielding the D276G variant. Consistent with the structural observation, the mutant enzyme partially restored efficiency for Fapy•dGTP insertion opposite dC (Figure 5). In contrast, there was little effect on insertion efficiency opposite template dA (Figure 5). Therefore, Asp276 functionally guards against Fapy•dG insertion opposite template dC.
Figure 5.
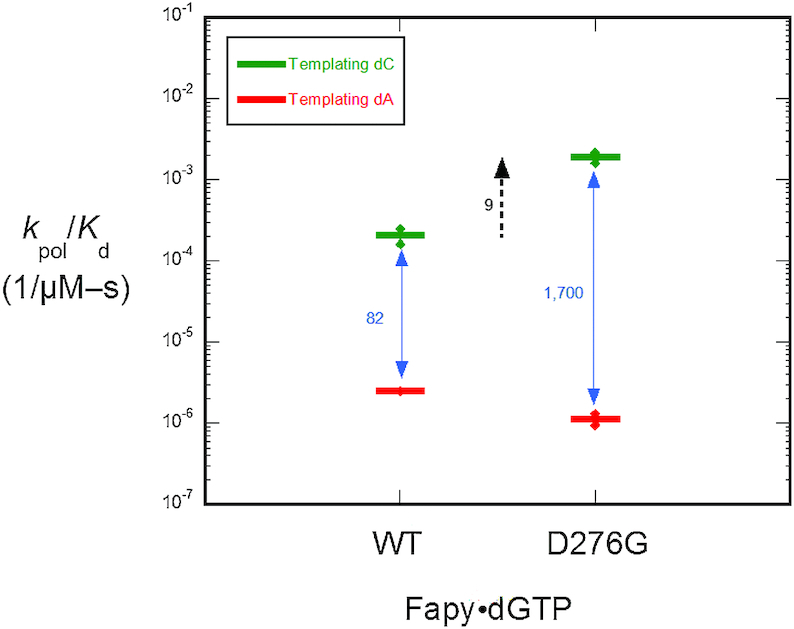
Discrimination plot for insertion of β-C-Fapy•dGTP. The catalytic efficiency of β-C-Fapy•dGTP insertion opposite dC or dA for wild-type pol β and the mutant where Asp276 was substituted with glycine (D276G) are shown. Each horizontal short bar represents the mean of duplicate independent determinations. Additionally, the value for each determination is plotted (small solid circle) to illustrate the reproducibility of these assays.
Active site contact by Asp276 impedes insertion
Both template dA and dC insertions of the Fapy•dGTP analog result in a similarly distorted position of the incoming nucleotide in the active site of wild-type (WT) pol β, and the distortion is stabilized by active site residue Asp276. To further elucidate the role of Asp276 as a ‘guardian’ against the insertion of Fapy•dGTP opposite template dC and dA, we solved structures with the D276G pol β variant. We chose to replace Asp276 with glycine in order to remove the interaction between residue Asp276 in α-helix N of pol β and the sugar moiety of Fapy•dGTP. The loss of the Asp276 sidechain was confirmed via an isomorphous difference map between our D276G pol β complex (PDB accession number: 6DIC) and a WT pre-catalytic complex (PDB accession number: 2FMS), Supplementary Figure S3. Notably, changes in nucleotide and templating base identity between the two structures are also easily observed.
A pre-catalytic complex of Fapy•dGTP insertion by D276G across from dC revealed that removing the aspartate side chain enables stabilization of the ternary-closed conformation (Figure 6A). In this structure, (anti) Fapy•dGTP forms a planar Watson–Crick base pair with the templating dC. Both metal binding sites are occupied and the O3′ (primer terminus) and αP of the incoming damaged nucleotide are in proximity (3.7 Å, Figure 6A) for chemistry. The formamide group of Fapy•dG is located out of plane with the remaining pyrimidine ring, likely interacting via a weak hydrogen bond with Arg40 in one of two conformers (3.2 Å or 3.3 Å; Figure 6A). An overlay of an undamaged ternary-closed complex with a correct nucleotide (PDB accession number: 4UB4) (Figure 6B) reveals that the formamide group (O8, C8, N7) would clash with one of the γ-oxygens, Cγ, and Cβ of the WT location of Asp276 (Figure 6C), suggesting that these Asp276 contacts prevent pol β closure. Furthermore, Asp276 stabilizes the open conformation through a hydrogen bond between Asp276 and the O3′ of the incoming nucleotide.
Figure 6.
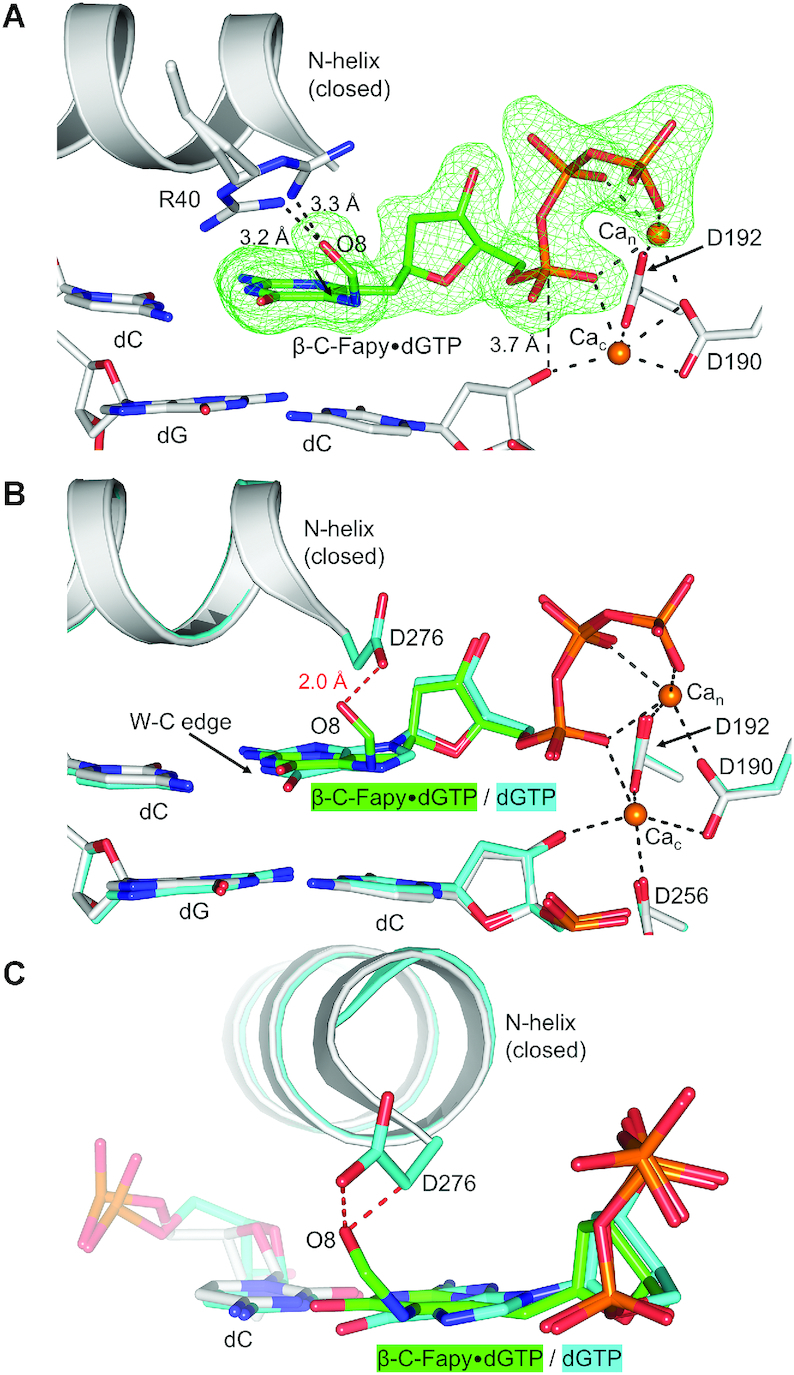
(A) Pol β D276G mutant in a ternary-closed ground state (gray) with incoming β-C-Fapy•dGTP base (green sticks) pairing with dC in a canonical conformation. The polder map is shown as green mesh and contoured to 3.0σ. (B) Overlaid is PDB ID: 4UB4 demonstrating undamaged dGTP pre-catalytic insertion (cyan). The Watson–Crick edge (W–C edge) is indicated. Potential hydrogen bonds are shown as black dashed lines. The D276 and formamide/O8 clash is demonstrated via red dashed lines. (C) A rotated viewpoint of panel B highlighting the extent of the Asp276 and formamide/O8 clashes (red dashed lines).
A pre-catalytic complex of D276G pol β and Fapy•dGTP across from dA revealed that loss of the aspartate side chain alters the nucleotide conformation within the open-ternary complex (Supplementary Figure S4). The coordination of the Fapy•dGTP O3′ by residue Asp276 is lost upon glycine mutation and the incoming nucleotide refines to an occupancy of 85% for the most occupied conformation of the incoming Fapy•dGTP. Per this conformation, the Fapy•dGTP places the Watson–Crick edge of Fapy•dG towards the DNA major groove with the formamide oriented towards the minor groove (Supplementary Figure S4A). The distance between O3′ of the primer terminus and αP of Fapy•dGTP increases from 4.1 Å for the WT structure with Fapy•dGTP opposite dA to 5.8 Å (Supplementary Figure S3A and S4A). We also observed an additional Ca2+ ion whose presence likely arises from the unique triphosphate orientation (Supplementary Figure S4B). These structural changes are consistent with kinetic data (Figure 5).
DISCUSSION
The mutagenic Fapy•dGTP lesion was found to be poorly cleansed from nucleotide pools by E. coli MutT and mammalian MTH1, making it both a menacing and available substrate for DNA polymerases during oxidative stress (26,29). Here, we utilized X-ray crystallography and kinetic analyses to evaluate the insertion of Fapy•dGTP into DNA by the model mammalian DNA polymerase, pol β. The catalytic subdomain of pol β is structurally homologous to the corresponding subdomain (palm) of the bacterial replicative enzymes (43,44). In addition, pol β serves as an excellent model polymerase given that it is structurally well-characterized and exhibits open and closed structural states during transitions between different liganded states, which directly pertain to the mechanisms of eukaryotic replicative B-family polymerases (45). Through our kinetic studies, we observed that pol β inserts Fapy•dGTP with dramatically reduced catalytic efficiency opposite both dC and dA in comparison to undamaged dGTP and 8-oxodGTP (39). This is at least, in part, due to the weak binding affinity of the modified nucleotide (Supplementary Figure S2). Additionally, insertion of Fapy•dGTP opposite dA was decreased relative to the mismatch insertion efficiency of dA:dGTP by nearly an order of magnitude (Figure 2). Our structures revealed that Fapy•dGTP adopts an altered and distorted position in the pol β active site via flexibility of the ring-opened base (Figures 3 and 4). Pol β active-site residues were observed to stabilize an altered position of Fapy•dGTP and prevent closure of the N-subdomain upon nucleotide binding across from either dA or dC.
Mechanistic insight into Fapy•dGTP binding and insertion
Correct and incorrect nucleotides are discriminated from each other on the basis of proper alignment of catalytic atoms within the polymerase active site (39,40). This alignment requires that the polymerase close around the nascent base pair. During Fapy•dGTP insertion, N-subdomain closure of pol β is prevented by interactions made between active site residues and the altered nucleotide position. Previous structures examining pol β active-site assembly have also observed ternary-open complexes which instead contain bound incoming non-damaged nucleotides (42,46). Overlaying the Fapy•dGTP ternary-open complex with a matched base pair ternary-open complex (Figure 7A) and a mismatched base pair ternary-open complex (Figure 7B) provides insight into how pol β accommodates an incoming nucleotide in the open conformation (42). Importantly, this conformation is mediated through binding of the triphosphate moiety. In contrast, the nucleoside base and deoxyribose sugar adopt different orientations depending on their unique interactions with the templating base. Figure 7A shows the nucleoside bases of a matched cytosine analog, dCMP(CF2)PP, and Fapy•dGTP hydrogen bonding differently with their templating base using anti- and syn- conformations, respectively. Figure 7B shows the different conformations adopted by a mismatched adenine analog (dAMPCPP) and Fapy•dGTP nucleoside bases, with the dAMPCPP base failing to hydrogen bond with the templating base. Comparing the matched nucleotide insertion ternary-closed complex (dUMPNPP) with our matched Fapy•dGTP ternary-open complex revealed that the triphosphate moiety, O3′, and catalytic residues are not properly aligned in the Fapy•dGTP complex, likely preventing stable coordination of the catalytic metal (Figure 7C).
Figure 7.
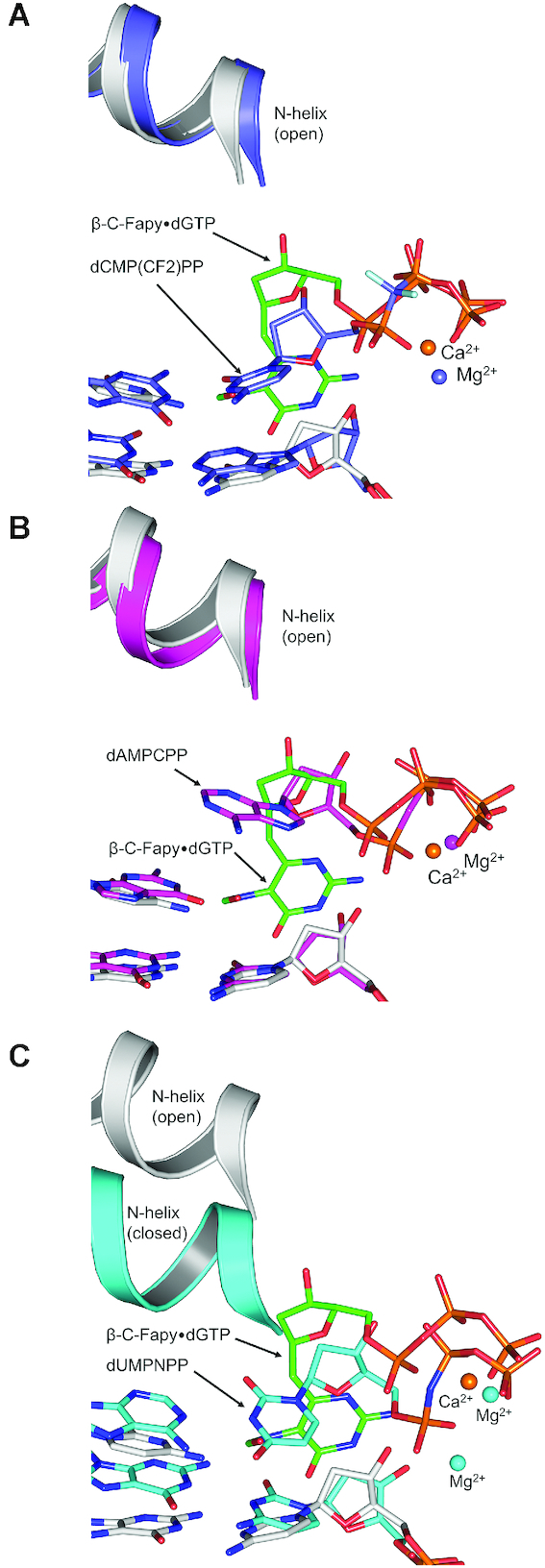
The pre-catalytic ternary-open pol β:β-C-Fapy•dGTP:dC (gray/green) structure is overlaid with the following: (A) A ternary-open complex (indigo) with an incoming nucleotide analog, dCMP(CF2)PP, opposite a templating 8-oxoGuo (PDBID: 4F5O). (B) A ternary-open complex (magenta) with an incoming nucleotide analog, dAMPCPP, opposite a templating dG (PDBID: 4F5P). (C) A ternary-closed complex (cyan) with an incoming analog, dUMPNPP, opposite a templating dA (PDBID: 2FMS).
Both Asp276 and Arg258 stabilize the altered nucleotide position of Fapy•dGTP and have been proposed to influence nucleotide binding and subdomain closure (47,48). Arg258 forms a salt bridge with Glu295 which is key for the stabilization of the closed pol β conformation (48). Substitution of the arginine sidechain with alanine decreases nucleotide binding affinity but increases the insertion rate without altering fidelity, which suggests that Arg258 aids in nucleotide binding but can generate a population of non-productive ternary complexes (49–51). Consistent with this, our ternary-open Fapy•dGTP complex illustrates a possible role for Arg258 in nucleotide binding prior to polymerase closure. The role for Asp276 involves stacking interactions of the sidechain Cβ and the base of the incoming nucleotide to influence ground-state nucleotide binding during template base recognition (47). In the closed conformation, the carboxylate sidechain is neutralized by Arg40. Mutation of the D276 residue to D276V, essentially removing only the charged portion of Asp276, increases the binding affinity for the incoming nucleotide, indicating Asp276 likely plays a role in the sampling and orienting of the incoming nucleotide (47).
We have shown here that during Fapy•dGTP insertion opposite dC, Asp276 stabilizes a non-productive nucleotide position, and in turn hinders subdomain closure and subsequent insertion. We further demonstrated that the D276G mutation allowed for Fapy•dGTP opposite dC to adopt a proper nucleotide orientation, closure of the N-subdomain, and catalytic metal coordination (Figure 6A) providing more evidence for the important role of Asp276 in nucleotide sampling and a guardian role blocking Fapy•dGTP insertion across from dC. However, this is not the case for the insertion of Fapy•dGTP opposite dA. Our structural data show that in the case of a templating adenine, the D276G mutation does not stabilize enzyme closure nor proper nucleotide orientation (Supplementary Figure S4A). In this conformation, the insertion efficiency of Fapy•dGTP opposite dA by the D276G pol β mutant is further reduced (Figure 5).
Although our kinetic data indicates catalysis occurs at a decreased efficiency, Fapy•dGTP can be inserted by pol β. Therefore, either a minor fraction of pol β is eventually able to close or catalysis occurs in the open or intermediate conformation. Let's consider that the ternary-closed structure of D276G pol β with a templating dC shows Fapy•dGTP formamide is placed in the cavity formerly occupied by the Asp276 side chain. In WT pol β, the formamide group clashes with the γ-oxygens and Cβ of Asp276 should the enzyme adopt the closed conformation (Figure 6B). To avoid this clash, the formamide would be forced into the major groove, similar to what was previously observed with other nucleotides containing modifications at the major groove C5 position (52). This transiently closed pol β conformation could be accommodated to a limited extent; however, the stabilizing contact with Asp276 and the templating base in the observed open pol β conformation must be preferred. This suggests Fapy•dGTP might be inserted in a partially-open intermediate state whereby the O3′ proton is abstracted via an interaction with a transient catalytic metal. In this case, the O3′ anion might attack αP from a more distant position (3.8 Å, Figure 3A). In comparison, the O3′ to αP distance is 3.4 Å for the closed non-damaged insertion complex in the presence of MgCl2 (32). Combined, our pre-catalytic structures and kinetic studies provide a rationale for a limited insertion mechanism occurring in a partially open conformation. Notably, a resultant Fapy•dG at the primer terminus has been shown to be extended during subsequent insertion events (26).
Insertion of Fapy•dGTP by pol β, the primary polymerase in base excision repair, is biologically relevant during DNA repair synthesis, since DNA maintenance provides many opportunities for repair polymerases to utilize damaged nucleotide precursors. A robust DNA repair is especially important in non-dividing cells. Although a wide-range of persistent base excision repair intermediates have been detected (53), these steady-state values do not provide an estimate of the cellular rate of DNA repair. Lindahl has estimated that the rate of spontaneous depurination is 10,000/cell/day (54). This translates to >1 trillion abasic sites per adult per second underestimating the true burden since it does not include those generated by damage-specific DNA glycosylases.
Previous reports indicated that pol λ contributes significantly to the Fapy•dG mutagenic signature (18). However, in that study the Fapy•dG lesion was in the DNA template strand. Since pol λ is also an X-family polymerase exhibiting structural similarity to pol β and has been implicated in base excision repair under certain conditions (55,56), it is worth noting that the pol λ residue corresponding to Asp276 of pol β is Ala510. The smaller size and loss of hydrogen bonding capacity with alanine would be expected to relieve some steric and electrostatic interactions that stabilize the altered conformation in the Fapy•dGTP in the ternary-open complex. Previous studies replacing Asp276 with valine (47) or glycine (57) resulted in a mutant enzyme that exhibited an increased affinity for the correct incoming nucleotide. However, a clash might be expected between the Ala510 βC of pol λ and the formamide of Fapy•dG based on the clashes observed in Figure 6B and C, especially since pol λ differs from pol β in that it does not exhibit protein subdomain motions upon binding an incoming nucleotide (58). Additionally, we observed that the pol β D276G mutation still discriminates against insertion opposite dA (Supplementary Figure S4). The similar efficiencies observed with dGTP and Fapy•dGTP insertion opposite dA for the D276G mutant suggests similar discriminating mechanisms.
Pol β has served as a model enzyme for characterizing the nucleotidyl transferase reaction, which is universally employed by DNA polymerases during DNA synthesis (38,59,60). Replicative polymerases in prokaryotic and eukaryotic organisms share structural homology to the corresponding catalytic subdomain (palm) of pol β and similarly adopt multiple subdomain conformations during the catalytic cycle as a result of different liganded states (43–45). With this in mind, the general strategies uncovered here during Fapy•dGTP insertion observed with pol β would be expected to be exploited by replicative polymerases. Specifically, we hypothesize that replicative polymerases alter their open to closed structural transition during Fapy•dGTP insertion to reduce binding and catalysis.
The mechanism of Fapy•dG mutagenesis differs from 8-oxodGuo
8-oxodGuo, a well-studied oxidative lesion that shares a chemical intermediate with Fapy•dG, uses different glycosidic bond conformations (syn/anti) to promote mutagenesis (61,62). The anti-conformation of 8-oxodGuo base pairs with dC through the Watson–Crick edge, while the syn-conformation of 8-oxodGuo base pairs with dA through the Hoogsteen edge. Previous structures of a high-fidelity polymerase with a templating Fapy•dG analog demonstrated that it does not require the syn-conformation to base pair with dA. Instead, the flexibility of the opened ring in Fapy•dG can simply rotate the formamide group orthogonal to the aromatic heterocycle (27).
Structures of 8-oxo-dGTP insertion revealed that pol β adopts a closed conformation and planar base pairing interactions during both mutagenic and non-mutagenic insertion, whereby 8-oxo-dGTP used either an anti- or syn-conformation to base pair with dC or dA, respectively (39). As an incoming base, Fapy•dGTP does not use a similar mechanism as 8-oxodGTP. Instead, we observed pol β in the ‘open’ conformation with Fapy•dGTP in an altered orientation and utilization of the syn-conformation during both template dA and dC insertions. This indicates that the ternary-open complex and the non-planar nucleotide position necessitates the rotation of the glycosidic bond into the syn-conformation during insertion of Fapy•dGTP. Our results provide further evidence that opening of the imidazole ring in Fapy•dG enables a different mechanism compared with that of 8-oxodGuo mutagenesis.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession numbers 6DIA, 6MR7, 6DIC and 6MR8.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Collaborative Crystallography Group at National Institute of Environmental Health Sciences (NIEHS) for help with data collection and analysis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Institute of Health, National Institute of Environmental Health Sciences (NIEHS) [Z01-ES050158, Z01-ES050159 to S.H.W.]; National Institutes of Health [R01-ES027558 to M.G., B.D.F. M.R.S.]. Funding for open access charge: Intramural Research Program of the National Institute of Health, National Institute of Environmental Health Sciences (NIEHS) [Z01-ES050158, Z01-ES050159 to S.H.W.]; National Institutes of Health [R01-ES027558 to M.G., B.D.F. M.R.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ziech D., Franco R., Pappa A., Panayiotidis M.. Reactive oxygen species (ROS)–induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res. 2011; 711:167–173. [DOI] [PubMed] [Google Scholar]
- 2. Perez-Campo R., Lopez-Torres M., Cadenas S., Rojas C., Barja G.. The rate of free radical production as a determinant of the rate of aging: evidence from the comparative approach. J. Comp. Physiol. 1998; 168:149–158. [DOI] [PubMed] [Google Scholar]
- 3. Whitaker A., Schaich M., Smith M., Flynn T., Freudenthal B.. Base excision repair of oxidative DNA damage: from mechanism to disease. Front. Biosci. 2017; 22:1493–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ellermann M., Eheim A., Rahm F., Viklund J., Guenther J., Andersson M., Ericsson U., Forsblom R., Ginman T., Lindstrom J. et al.. Novel class of potent and cellularly active inhibitors devalidates MTH1 as Broad-Spectrum cancer target. ACS Chem. Biol. 2017; 12:1986–1992. [DOI] [PubMed] [Google Scholar]
- 5. Aristizabal Prada E., Orth M., Nolting S., Spottl G., Maurer J., Auernhammer C.. The MTH1 inhibitor TH588 demonstrates anti-tumoral effects alone and in combination with everolimus, 5-FU and gamma-irradiation in neuroendocrine tumor cells. PLoS One. 2017; 12:e0178375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gavande N., VanderVere-Carozza P., Hinshaw H., Jalal S., Sears C., Pawelczak K., Turchi J.. DNA repair targeted therapy: The past or future of cancer treatment. Pharmacol. Therapeut. 2016; 160:65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ewald B., Sampath D., Plunkett W.. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene. 2008; 27:6522. [DOI] [PubMed] [Google Scholar]
- 8. Gad H., Koolmeister T., Jemth A-S., Eshtad S., Jacques S., Ström C., Svensson L., Schultz N., Lundbäck T., Einarsdottir B. et al.. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014; 508:215. [DOI] [PubMed] [Google Scholar]
- 9. Huber K., Salah E., Radic B., Gridling M., Elkins J., Stukalov A., Jemth A., Gokturk C., Sanjiv K., Stromberg K. et al.. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature. 2014; 508:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steenken S., Jovanovic S.V.. How easily oxidizable is DNA?One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem.Soc. 1997; 119:617–618. [Google Scholar]
- 11. Pouget J., Frelon S., Ravanat J., Testard I., Odin F., Cadet J.. Formation of modified DNA bases in cells exposed either to gamma radiation or to High-LET particles. Radiat. Res. 2002; 157:589–595. [DOI] [PubMed] [Google Scholar]
- 12. Dizdaroglu M., Jaruga P.. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012; 46:382–419. [DOI] [PubMed] [Google Scholar]
- 13. Tudek B. Imidazole ring-opened DNA purines and their biological significance. J. Biochem. Mol. Biol. 2003; 36:12–19. [DOI] [PubMed] [Google Scholar]
- 14. Gajewski E., Rao G., Nackerdien Z., Dizdaroglu M.. Modification of DNA bases in mammalian chromatin by radiation-generated free radicals. Biochemistry. 1990; 29:7876–7882. [DOI] [PubMed] [Google Scholar]
- 15. Xue L., Greenberg M.. Facile quantification of lesions derived from 2′-deoxyguanosine in DNA. J. Am. Chem. Soc. 2007; 129:7010–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pouget J., Douki T., Richard M., Cadet J.. DNA damage induced in cells by gamma and UVA radiation as measured by HPLC/GC-MS and HPLC-EC and Comet assay. Chem. Res. Toxicol. 2000; 13:541–549. [DOI] [PubMed] [Google Scholar]
- 17. Beard W., Batra V., Wilson S.. DNA polymerase structure-based insight on the mutagenic properties of 8-oxoguanine. Mutat Res. 2010; 703:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sha Y., Minko I., Malik C., Rizzo C., Lloyd R.. Error-prone replication bypass of the imidazole ring-opened formamidopyrimidine deoxyguanosine adduct. Environ. Mol. Mutagen. 2017; 58:182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pande P., Haraguchi K., Jiang Y., Greenberg M., Basu A.. Unlike catalyzing error-free bypass of 8-oxodGuo, DNA polymerase lambda is responsible for a significant part of Fapy.dG-induced G → T mutations in human cells. Biochemistry. 2015; 54:1859–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalam M., Haraguchi K., Chandani S., Loechler E., Moriya M., Greenberg M., Basu A.. Genetic effects of oxidative DNA damages: comparative mutagenesis of the imidazole ring-opened formamidopyrimidines (Fapy lesions) and 8-oxo-purines in simian kidney cells. Nucleic Acids Res. 2006; 34:2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Greenberg M., Hantosi Z., Wiederholt C., Rithner C.. Studies on N4-(2-Deoxy-d-pentofuranosyl)-4,6-diamino-5-formamidopyrimidine (Fapy•dA) and N6-(2-Deoxy-d-pentofuranosyl)- 6-diamino-5-formamido-4-hydroxypyrimidine (Fapy•dG). Biochemistry. 2001; 40:15856–15861. [DOI] [PubMed] [Google Scholar]
- 22. Patro J., Haraguchi K., Delaney M., Greenberg M.. Probing the configurations of formamidopyrimidine lesions Fapy.dA and Fapy.dG in DNA using endonuclease IV. Biochemistry. 2004; 43:13397–13403. [DOI] [PubMed] [Google Scholar]
- 23. Lukin M., Minetti C., Remeta D., Attaluri S., Johnson F., Breslauer K., de Los Santos C.. Novel post-synthetic generation, isomeric resolution, and characterization of Fapy-dG within oligodeoxynucleotides: differential anomeric impacts on DNA duplex properties. Nucleic Acids Res. 2011; 39:5776–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wiederholt C., Delaney M., Pope M., David S., Greenberg M.. Repair of DNA containing Fapy.dG and its beta-C-nucleoside analogue by formamidopyrimidine DNA glycosylase and MutY. Biochemistry. 2003; 42:9755–9760. [DOI] [PubMed] [Google Scholar]
- 25. Weledji Y., Wiederholt C., Delaney M., Greenberg M.. DNA polymerase bypass in vitro and in E. coli of a C-nucleotide analogue of Fapy•dG. Bioorg. Med. Chem. 2008; 16:4029–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Imoto S., Patro J., Jiang Y., Oka N., Greenberg M.. Synthesis, DNA polymerase incorporation, and enzymatic phosphate hydrolysis of formamidopyrimidine nucleoside triphosphates. J. Am. Chem. Soc. 2006; 128:14606–14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gehrke T., Lischke U., Gasteiger K., Schneider S., Arnold S., Muller H., Stephenson D., Zipse H., Carell T.. Unexpected non-Hoogsteen-based mutagenicity mechanism of FaPy-DNA lesions. Nat. Chem. Biol. 2013; 9:455–461. [DOI] [PubMed] [Google Scholar]
- 28. Patro J., Wiederholt C., Jiang Y., Delaney J., Essigmann J., Greenberg M.. Studies on the replication of the ring opened formamidopyrimidine, Fapy·dG in Escherichia coli. Biochemistry. 2007; 46:10202–10212. [DOI] [PubMed] [Google Scholar]
- 29. Kamiya H., Cadena-Amaro C., Dugue L., Yakushiji H., Minakawa N., Matsuda A., Pochet S., Nakabeppu Y., Harashima H.. Recognition of nucleotide analogs containing the 7,8-dihydro-8-oxo structure by the human MTH1 protein. J. Biochem. 2006; 140:843–849. [DOI] [PubMed] [Google Scholar]
- 30. Fujikawa K., Kamiya H., Yakushiji H., Fujii Y., Nakabeppu Y., Kasai H.. The oxidized forms of dATP are substrates for the human MutT homologue, the hMTH1 protein. J. Biol. Chem. 1999; 274:18201–18205. [DOI] [PubMed] [Google Scholar]
- 31. Beard W., Wilson S.. Purification and domain-mapping of mammalian DNA polymerase beta. Methods in Enzymology. 1995; 262:Academic Press; 98–107. [DOI] [PubMed] [Google Scholar]
- 32. Batra V., Beard W., Shock D., Krahn J., Pedersen L., Wilson S.. Magnesium induced assembly of a complete DNA polymerase catalytic complex. Structure (Camb.). 2006; 14:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Otwinowski Z., Minor W.. [20]Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 34. Adams P., Afonine P., Bunkoczi G., Chen V., Davis I., Echols N., Headd J., Hung L., Kapral G., Grosse-Kunstleve R. et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D, Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D, Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 36. Liebschner D., Afonine P., Moriarty N., Poon B., Sobolev O., Terwilliger T., Adams P.. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D, Struct. Biol. 2017; 73:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. The PyMOL Molecular Graphics System, Version 1.8. Schrödinger, L.L.C. [Google Scholar]
- 38. Beard W., Wilson S.. Structure and mechanism of DNA polymerase β. Biochemistry. 2014; 53:2768–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Freudenthal B., Beard W., Perera L., Shock D., Kim T., Schlick T., Wilson S.. Uncovering the polymerase-induced cytotoxicity of an oxidized nucleotide. Nature. 2015; 517:635–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Freudenthal B., Beard W., Shock D., Wilson S.. Observing a DNA polymerase choose right from wrong. Cell. 2013; 154:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsai Y., Johnson K.. A new paradigm for DNA polymerase specificity. Biochemistry. 2006; 45:9675–9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Freudenthal B., Beard W., Wilson S.. Structures of dNTP intermediate states during DNA polymerase active site assembly. Structure. 2012; 20:1829–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bailey S., Wing R., Steitz T.. The structure of T. aquaticus DNA polymerase III is distinct from eukaryotic replicative DNA polymerases. Cell. 2006; 126:893–904. [DOI] [PubMed] [Google Scholar]
- 44. Lamers M., Georgescu R., Lee S., O’Donnell M., Kuriyan J.. Crystal structure of the catalytic alpha subunit of E. coli replicative DNA polymerase III. Cell. 2006; 126:881–892. [DOI] [PubMed] [Google Scholar]
- 45. Wu S., Beard W., Pedersen L., Wilson S.. Structural comparison of DNA polymerase architecture suggests a nucleotide gateway to the polymerase active site. Chem Rev. 2014; 114:2759–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eckenroth B., Towle-Weicksel J., Sweasy J., Doublie S.. The E295K cancer variant of human polymerase beta favors the mismatch conformational pathway during nucleotide selection. J. Biol. Chem. 2013; 288:34850–34860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vande Berg B., Beard W., Wilson S.. DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase beta. Implication for the identity of the rate-limiting conformational change. J. Biol. Chem. 2001; 276:3408–3416. [DOI] [PubMed] [Google Scholar]
- 48. Pelletier H., Sawaya M., Wolfle W., Wilson S., Kraut J.. Crystal structures of human DNA polymerase β complexed with DNA: Implications for catalytic mechanism, processivity, and fidelity. Biochemistry. 1996; 35:12742–12761. [DOI] [PubMed] [Google Scholar]
- 49. Yang L., Beard W., Wilson S., Broyde S., Schlick T.. Highly organized but pliant active site of DNA polymerase beta: compensatory mechanisms in mutant enzymes revealed by dynamics simulations and energy analyses. Biophys. J. 2004; 86:3392–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Menge K., Hostomsky Z., Nodes B., Hudson G., Rahmati S., Moomaw E., Almassy R., Hostomska Z.. Structure-function analysis of the mammalian DNA polymerase .beta. active site: role of aspartic acid 256, arginine 254, and arginine 258 in nucleotidyl transfer. Biochemistry. 1995; 34:15934–15942. [DOI] [PubMed] [Google Scholar]
- 51. Beard W., Shock D., Batra V., Prasad R., Wilson S.. Substrate-induced DNA polymerase beta activation. J. Biol. Chem. 2014; 289:31411–31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schaich M., Smith M., Cloud A., Holloran S., Freudenthal B.. Structures of a DNA polymerase inserting therapeutic nucleotide analogues. Chem. Res. Toxicol. 2017; 30:1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakamura J., Swenberg J.. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999; 59:2522–2526. [PubMed] [Google Scholar]
- 54. Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993; 362:709–715. [DOI] [PubMed] [Google Scholar]
- 55. Braithwaite E., Kedar P., Lan L., Polosina Y., Asagoshi K., Poltoratsky V., Horton J., Miller H., Teebor G., Yasui A. et al.. DNA polymerase lambda protects mouse fibroblasts against oxidative DNA damage and is recruited to sites of DNA damage/repair. J. Biol. Chem. 2005; 280:31641–31647. [DOI] [PubMed] [Google Scholar]
- 56. Braithwaite E., Kedar P., Stumpo D., Bertocci B., Freedman J., Samson L., Wilson S.. DNA polymerases beta and lambda mediate overlapping and independent roles in base excision repair in mouse embryonic fibroblasts. PLoS One. 2010; 5:e12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lavrik O., Prasad R., Beard W., Safronov I., Dobrikov M., Srivastava D., Shishkin G., Wood T., Wilson S.. dNTP binding to HIV-1 reverse transcriptase and mammalian DNA polymerase beta as revealed by affinity labeling with a photoreactive dNTP analog. J. Biol. Chem. 1996; 271:21891–21897. [DOI] [PubMed] [Google Scholar]
- 58. Garcia-Diaz M., Bebenek K., Krahn J., Kunkel T., Pedersen L.. A closed conformation for the Pol lambda catalytic cycle. Nat. Struct. Mol. Biol. 2005; 12:97–98. [DOI] [PubMed] [Google Scholar]
- 59. Freudenthal B., Beard W., Wilson S.. New structural snapshots provide molecular insights into the mechanism of high fidelity DNA synthesis. DNA Repair (Amst.). 2015; 32:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Whitaker A., Smith M., Schaich M., Freudenthal B.. Capturing a mammalian DNA polymerase extending from an oxidized nucleotide. Nucleic Acids Res. 2017; 45:6934–6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Oda Y., Uesugi S., Ikehara M., Nishimura S., Kawase Y., Ishikawa H., Inoue H., Ohtsuka E.. NMR studies of a DNA containing 8-hydroxydeoxyguanosine. Nucleic Acids Res. 1991; 19:1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Batra V., Shock D., Beard W., McKenna C., Wilson S.. Binary complex crystal structure of DNA polymerase β reveals multiple conformations of the templating 8-oxoguanine lesion. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession numbers 6DIA, 6MR7, 6DIC and 6MR8.