


Abstract
Uridylation-dependent RNA decay is a widespread eukaryotic pathway modulating RNA homeostasis. Terminal uridylyltransferases (Tutases) add untemplated uridyl residues to RNA 3′-ends, marking them for degradation by the U-specific exonuclease Dis3L2. In Schizosaccharomyces pombe, Cid1 uridylates a variety of RNAs. In this study, we investigate the prevalence and impact of uridylation-dependent RNA decay in S. pombe by transcriptionally profiling cid1 and dis3L2 deletion strains. We found that the exonuclease Dis3L2 represents a bottleneck in uridylation-dependent mRNA decay, whereas Cid1 plays a redundant role that can be complemented by other Tutases. Deletion of dis3L2 elicits a cellular stress response, upregulating transcription of genes involved in protein folding and degradation. Misfolded proteins accumulate in both deletion strains, yet only trigger a strong stress response in dis3L2 deficient cells. While a deletion of cid1 increases sensitivity to protein misfolding stress, a dis3L2 deletion showed no increased sensitivity or was even protective. We furthermore show that uridylyl- and adenylyltransferases cooperate to generate a 5′-NxAUUAAAA-3′ RNA motif on dak2 mRNA. Our studies elucidate the role of uridylation-dependent RNA decay as part of a global mRNA surveillance, and we found that perturbation of this pathway leads to the accumulation of misfolded proteins and elicits cellular stress responses.
INTRODUCTION
RNA synthesis and degradation are regulated through a variety of mechanisms that amend the transcriptome to match cellular needs throughout the cell cycle and adaptation to environmental changes (1). Messenger RNA (mRNA) degradation can proceed by two general pathways, in either a 5′-3′ or 3′-5′ direction, catalyzed by exonucleases or the exosome complex, respectively. These canonical RNA degradation processes usually commence with an initial deadenylation step, followed by decapping by Dcp-1/Dcp-2 and the Lsm1–7 complex. Decapped mRNA is subsequently accessible to 5′-3′ decay catalyzed by the exonuclease Xrn1, while exosome-catalyzed 3′-5- degradation does not require decapping (2). Recently, a second deadenylation-independent pathway of mRNA decay was discovered and appears to be conserved in many eukaryotes. Here, uridylation of polyadenylated mRNAs recruits the Lsm1–7 complex and subsequently leads to mRNA degradation by designated exonucleases (2). This template-independent addition of nucleotides is catalyzed by terminal RNA nucleotidyltransferases (TENTs), a subfamily of the polymerase beta superfamily of nucleotidyltransferases (3). TENTs add ribonucleoside monophosphates to an RNA substrate through a catalytic process involving two metal ion cofactors (3). Of note, non-templated 3′-end uridylation of a variety of RNA species plays key roles in eukaryotic RNA processing pathways including mRNA and pre-miRNA degradation, pre-miRNA maturation, and miRNA silencing (4–6). RNA uridylation is catalyzed by terminal uridylyltransferases (Tutases), and polyuridylated RNAs are subsequently degraded by the U-specific exonuclease Dis3L2 (6–8). While uridylation and deadenylation-dependent RNA decay show some redundancy, uridylation is conserved in many different species indicating that it is important for RNA turnover (9–11).
Fission yeast Cid1 (caffeine-induced death suppressor protein 1) was first discovered in a genetic screen identifying components of the S-M cell cycle checkpoint in Schizosaccharomyces pombe (12). Although S. pombe Δcid1 strains are viable, they are sensitive to a combination of hydroxyurea, a ribonucleotide reductase inhibitor, and caffeine, which overrides the S-M checkpoint and induces mitosis. Overexpression of Cid1 confers resistance to this combination of stressors (12). Cid1 was originally thought to be a poly(A) polymerase due to its significant in vitro poly(A) polymerase activity (13), but recent evidence characterized it as an efficient Tutase in vitro and in vivo (14–16). Cid1 encodes a catalytic nucleotidyltransferase motif and a poly(A) polymerase-associated motif (17), but lacks an identifiable RNA recognition motif. Interestingly, nucleotide specificity appears to have evolved after RNA specificity, with adenylyltransferases and uridylyltransferases playing opposing roles in promoting RNA stability or degradation in eukaryotes, respectively (18). Nucleotide specificity depends on a critical histidine residue (H336), which is responsible for UTP over ATP preference (19,20) (Figure 1A). A H336N mutation in Cid1 converts the enzyme to an adenylyltransferase (16,20), whereas a histidine insertion in its human adenylyltransferase counterpart Gld2 confers UTP specificity (18).
Figure 1.
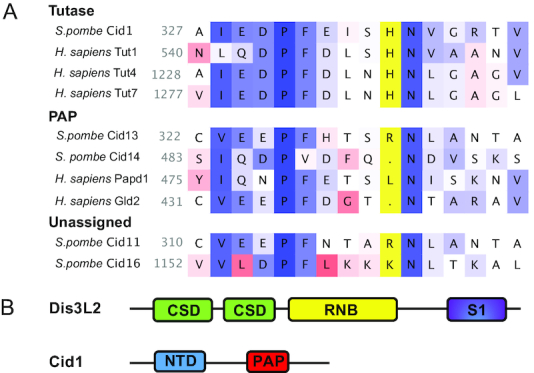
Domain structure and amino acid composition of Cid1 and Dis3L2. (A) Amino acid sequence alignment adapted from (18). Enzymes known to exercise Tutase activity encode a histidine residue (His336 in Cid1, highlighted in yellow), that sterically hinders the larger ATP from entering the active site. Adenylyltransferases (PAPs) do not encode the respective histidine residue. Nucleotide preference for S. pombe Cid11 and Cid16 is undetermined, though Cid16 likely prefers UTP. (B) Dis3L2 displays a typical RNase II domain organisation, encoding two cold shock domains (CSD), an exonucleolytic ribonuclease domain (RNB), and a nonspecific RNA binding domain (S1). Cid1 is composed of a nucleotidyltransferase domain (NTD) and a poly(A) polymerase-associated domain (PAP).
One of the first Cid1 RNA substrates to be identified was actin1 mRNA, which was shown to be uridylated upon S-phase arrest in a Cid1-dependent manner (15). In S. pombe, RNA uridylation mediates mRNA turnover: Cid1 uridylates polyadenylated mRNAs to trigger Lsm1–7-mediated decapping of the RNA 5′-end and subsequent degradation by the U-specific exonuclease Dis3L2 (7,10). Biochemical and structural investigations revealed that despite the absence of a specific RNA recognition motif (Figure 1B), Cid1 is capable of binding and uridylating RNAs in a sequence-independent manner (14). Due to its substrate promiscuity, Cid1 is thought to participate in a widespread mechanism of mRNA decay in S. pombe (11,17,19,21,22), and substrate specificity and selectivity may require accessory proteins, in analogy to the human homologs, Tutases Tut4, Tut7 and the adenylyltransferase Gld2 (18,23–26).
Following uridylation, RNAs are quickly degraded by the U-specific 3′-5′ exonuclease Dis3L2 (6–8,27–29). Recent studies revealed that Dis3L2-catalyzed exonucleolytic RNA degradation constitutes an alternative pathway for RNA decay, independent of exosome and Xrn1-catalyzed decay pathways (7). In S. pombe, Dis3L2 localizes to the cytoplasm and does not associate with the exosome but interacts with components of the cytoplasmic mRNA degradation pathway. While a recent study reported no significant changes in mRNA accumulation in a dis3L2 deletion strain, uridylated mRNAs were found elevated in a dis3L2 and lsm1 double mutant strain, and recombinant Dis3L2 degraded uridylated RNA transcripts in vitro (7). In humans, Dis3L2 is involved in the degradation of uridylated mRNA and miRNA transcripts (6,7,30–32). Mutations in Dis3L2 in humans are associated with the Perlman syndrome of fetal overgrowth, likely due to its role in the degradation of miRNAs and pre-miRNAs of the let-7 family (33). Dis3L2 displays a typical RNase II-like protein domain organization, and encodes two cold shock domains (CSDs), an exonucleolytic ribonuclease domain (RNB), and a nonspecific RNA binding domain (S1) (Figure 1B). Structural analysis of Dis3L2 showed that in the absence of RNA, the enzyme displays an open conformation (28) and RNA binding induces a closed conformation, where three RNA binding domains form a funnel to position the RNA substrate for exonucleolytic degradation (32).
In S. pombe, the Cid1-Dis3L2 RNA degradation pathway constitutes one of three mRNA surveillance pathways. While the individual proteins, Cid1 and Dis3L2, are now recognized and biochemically and structurally characterized, it is unclear whether the three RNA decay pathways (Xrn1, exosome, and Cid1/Dis3L2) are dedicated to specific substrate RNAs or act as three global albeit independent decay mechanisms. In this study, we capture the extent of the Cid1/Dis3L2 mediated RNA degradation and find that depletion of uridylation-dependent RNA decay causes the accumulation of misfolded proteins and an increase in abundance of mRNAs involved in the stress response. Using deep sequencing, we find that while Cid1 depletion has little impact on mRNA homeostasis, Dis3L2 represents a bottleneck in uridylation-dependent RNA decay and its depletion leads to an increase in mRNAs involved in protein folding and degradation pathways. We conclude that perturbation of uridylation-dependent RNA decay elicits a stress response, likely due to the accumulation of misfolded proteins.
MATERIALS AND METHODS
Yeast strains and growth conditions
S. pombe strains were obtained from Bioneer (Alameda, CA, USA): Wildtype (BG_0000H6, ade6-M210 ura4-D18 leu1-32); Δdis3L2 (BG_H0669; orfΔ SPAC2C4.07c: kanMX4/ORF ade6-M210 ura4-D18 leu1-32) and Δcid1 (BG_H0513; orfΔ SPAC19D5.03: kanMX4/ORF ade6-M210 ura4-D18 leu1-32). Liquid cultures were grown at 30°C in YPD supplemented with adenine to an optical density of OD600 = 0.5. For spotting assays, yeast were grown on Edinburgh minimal media (EMM) (3 g/l potassium hydrogen phthalate, 2.2 g/l sodium phosphate dibasic, 5 g/l ammonium chloride, 20 g/l dextrose, 2.1 g/l mineral salts, 0.02 g/l vitamins, 3 mg/l trace elements) supplemented with 12 g/l l-leucine, 2 g/l uracil, 2 g/l adenine.
Spotting assays
S. pombe cells were inoculated in 4 ml EMM-URA-LEU-ADE liquid media overnight in a 30°C incubator shaker. 100 μl of cells were diluted 1:10 in ddH2O to measure the OD600 to determine cell density. Cells were standardized to OD600 = 1 in the first row of wells on a 96-well plate. A 1:5 serial dilution of cells was performed in the subsequent five rows of wells. Cells were spotted on YES, and EMM-uracil-leucine-adenine media agar plates with or without 10 mM azetidine-2-carboxylic acid (AZC), 100 μM H2O2, 5 mM caffeine, 2 mM caffeine, 5 mM hydroxyurea, 2 mM hydroxyurea, and 2.5 mM caffeine + 10 mM hydroxyurea. Plates were incubated in a 30°C incubator. Photographs of plates were taken on different days to document growth. Spotting assays were photographed, and the image was modified to black and white with the background blackened out and the yeast colonies being white. The circular selection tool on ImageJ was used to select an equal area of colonies and the mean gray value (MGV) was measured for density of cell growth. The blackened plate background gave a MGV of 0 and complete colony growth gave a value of 255. Wildtype values were normalized to 1 and the growth of deletion strains were normalized against wildtype to give a fraction of 1. Unpaired t-test was employed to infer statistical significance between wildtype and each deletion strain at 95% confidence interval.
Cid1 cloning, purification, and activity assays
Total RNA was extracted from S. pombe cells using MasterPure Complete DNA and RNA Purification kit (Epicentre) and reverse transcribed with SuperScript II Reverse Transcriptase (Invitrogen) and random hexamer primers. The resulting cDNA was amplified by PCR using gene specific primers (Cid1F 5′-AAGCTTATGAACATTTCTTCTGCACAATTTATTCCTGGTGT-3′ and Cid1R 5′- CTCGAGCTCAGAATTGTCACCATCGGTTTCATTC-3′) and inserted into a pET-20b(+) expression vector with HindIII and XhoI restriction sites. The construct was confirmed by DNA sequencing (London Regional Genomics Centre). Escherichia coli BL21 Codon Plus cells were transformed with pET-20b(+) encoding His-tagged cid1 and grown in LB media with ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml) at 37°C to an OD600 of 0.6. Protein expression was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM and the culture was grown overnight at 18°C. Cells were harvested and resuspended in Buffer A (50 mM HEPES, pH 6.8, 100 mM NaCl, 2 mM 2-mercaptoethanol) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 0.25 mg/ml lysozyme and lysed with a French Pressure Cell. Following 1 hour of centrifugation (41 000 rpm at 4°C), cell free extract was loaded onto a gravity column containing HisPur Ni-NTA resin (Thermo Scientific) pre-equilibrated in Buffer A. The resin was washed with Buffer B (50 mM HEPES, pH 6.8, 100 mM NaCl, 2 mM 2-mercaptoethanol, 50 mM imidazole) and Cid1 was eluted with Buffer C (50 mM HEPES, pH 6.8, 100 mM NaCl, 100 mM imidazole). After concentrating the eluted protein, remaining contaminants were removed by size exclusion chromatography using a Superdex™ 200 Increase 5/150 GL column and Buffer A. Protein concentration was determined using a Bradford assay and purified proteins were stored at −80°C in 10% glycerol. Enzyme activity assays were carried out as described previously (18).
cRACE and Northern blotting
RNA was isolated using the Masterpure RNA purification kit (Epicentre), dephosphorylated with shrimp alkaline phosphatase (NEB), and phenol chloroform extracted. For decapping, RNA was incubated with tobacco acid pyrophosphatase (NEB) and circularized with T4 RNA ligase (NEB). Reverse transcription was carried out using Superscript Reverse Transcriptase (ThermoFisher) and random hexamer primers. Gene specific amplification was carried out using primers listed in Supplementary Table S1 and PCR products cloned into pCR3.1- TopoTA vector (ThermoFisher) and sequenced at the London Regional Genomics Centre. Northern blots were performed as previously described (34) using 5 or 10 μg of total RNA and 5′32P-labeled gene specific probes amplified from genomic DNA using primers listed in Supplementary Table S1.
RT-qPCR
Reverse transcription and quantitative PCR were performed as described (35). Briefly, total RNA was extracted using the Masterpure RNA purification kit (Epicentre) and reverse transcribed using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). PowerUp SYBR Green Master Mix (Applied Biosystems) was used for the qPCR and amplification was performed on the ViiA 7 Real-Time PCR System. Six biological replicates of each S. pombe strain (wildtype, Δdis3L2, and Δcid1) were analyzed in technical triplicates. All primers are listed in Supplementary Table S1.
Yeast sedimentation assay and Western blot
Wildtype, Δdis3L2 and Δcid1 S. pombe strains were streaked onto YPD agar plates supplemented with adenine, uracil, and leucine and incubated at 30°C. Three biological replicates for each strain were grown in 5 ml YPD media supplemented with adenine, uracil, and leucine overnight at 30°C. The OD600 of all cultures was measured the next morning before the cells were centrifuged at 4000 × g for 5 min at 4°C. The sedimentation assay was adapted from (36). Briefly, cell pellets were lysed in 200 μl lysis buffer (100 mM Tris–HCl pH 7.5, 200 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM dithiothreitol (DTT), 5% glycerol, 0.5% Triton-X, 2 mM phenylmethylsulfonyl fluoride (PMSF)) supplemented with protease inhibitors (Roche, 04 693 159 001) using glass beads on a Disruptor Genie. Cells were disrupted with six 30 s bursts followed by 30 s on ice between each burst. The lysates were separated from the glass beads and 50 μl was added to 50 μl SUMEB (8 M urea, 1% sodium dodecyl sulfate (SDS), 10 mM 3-(N-morpholino)propanesulforic acid (MOPS), 10 mM EDTA, 0.01% bromophenol blue). The remaining lysates were centrifuged at 500 × g for 15 mins at 4°C and 100 μl of the supernatant was added to 100 μl of SUMEB. The pellets were resuspended in 100 μl lysis buffer with no PMSF and 100 μl SUMEB. Samples were analyzed on a 10% SDS gel and total protein in each lane was quantified using the Bio-Rad ChemiDoc MP and Image Lab software. Total lysates were blotted for PGK1 (ThermoFisher Scientific, 459250) as loading control.
RNA sequencing and data analysis
Three biological and three technical replicates for each S. pombe strain (WT, Δdis3L2 and Δcid1) were grown in YPD media to an OD600 of 0.6–0.65. RNA was isolated using the Epicentre RNA extraction kit. Ribosomal RNAs were depleted using the Ribo zero RNA kit and the RNA library was generated with the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina. Samples were analyzed on the MiSeq sequencing v2. Sequencing reads were mapped to the S. pombe MT genome (NC_001326.1) using the CLC Genomics Workbench and changes in gene expression were analyzed with the ANOVA-like Differential Expression (ALDEx2) tool (37) and an effect size cut-off of 1.5. The relative expression (abundance) of each gene within a sample was calculated as the median centered log-ratio (clr) from 1000 Monte Carlo Dirichlet instances. Genes were considered to be differentially expressed if the ALDEx2 effect size was greater than 1.5 (i.e. the difference in abundance between two strains was at least 1.5-fold greater than the difference between biological replicates). Genes differentially up or down regulated by at least 1.5-fold were analyzed for enrichments in specific pathways using the Search Tool for the Retrieval of Interacting Genes/proteins (STRING) (38).
RESULTS
Recombinant Cid1 displays ambiguous substrate specificity in vitro
Previous studies have shown that S. pombe Cid1 uridylates a variety of RNA substrates in vivo (15), which may subsequently be degraded by the exonuclease Dis3L2 (7). To assess whether Cid1 uridylation is ambiguous or dedicated to specific substrates, we produced and purified full length Cid1 and assessed its substrate specificity on several RNA substrates in vitro. We found that Cid1 uridylates a poly(A) tail mimic (15A), tRNA, total mRNA, pre-miRNA and miRNAs equally (Figure 2). It appears that Cid1 uridylates RNA substrates regardless of their secondary structure (pre-miRNA hairpin structure, tRNA structure), or sequence (miRNAs and poly(A) RNA). Total yeast tRNA and total human RNA preparations from cell lines HEK 293T and MDA-MB-231 are heterogeneous mixtures by nature and the frequency of poly- versus monouridylation cannot be assessed. In these cases, the observed products are presented in a smear, consistent with either the uridylation of RNA substrates of varying length or also a mixture of poly- and monouridylation events. Substrates 15A RNA, miRNAs and pre-miRNA were either purchased oligoribonucleotides or products of in vitro transcription (pre-let-7a) (18). For these homogenous RNA substrates, the predominant product is consistent in length with a monouridylated RNA product. In the case of the 15A nucleotide poly(A) tail mimic 15A RNA, a second band at ∼35 nucleotides is detectable, indicating the addition of roughly 20 uridines in vitro. For the microRNA substrates miR-122 and let-7a, polyuridylation can be observed in the form of a ladder-like pattern, as a result of multiple nucleotide additions with variable product lengths. Thus, Cid1 displays no substrate preference in vitro and can act as both a distributive or processive Tutase.
Figure 2.
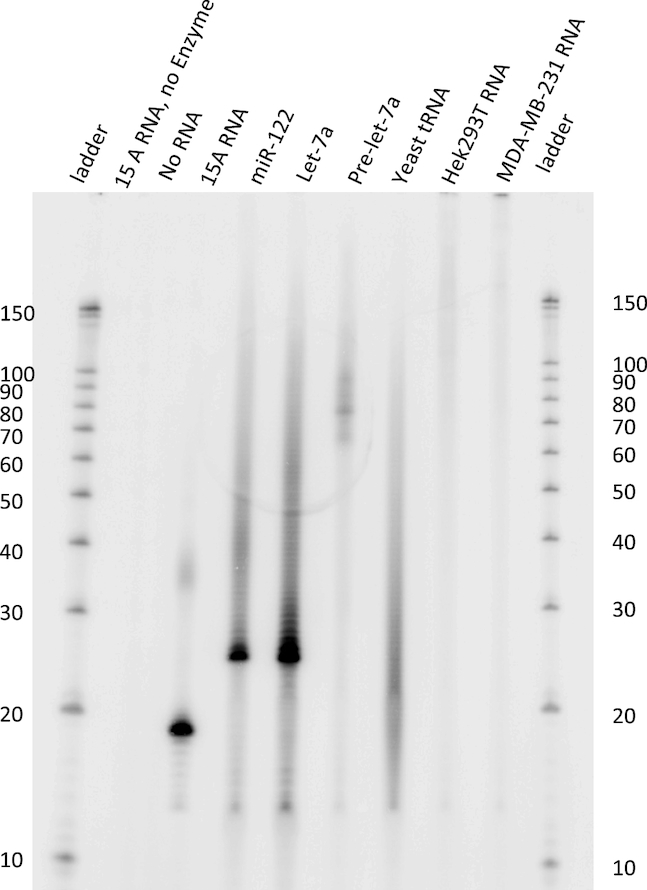
Cid1 displays a promiscuous substrate range in vitro. Cid1 was incubated with different RNA substrates and [α-32P]-UTP as indicated. Formation of [α-32P] labeled RNA products was monitored by electrophoretic separation and subsequent phosphorimaging. Cid1 catalyzed [α-32P]-UTP addition to RNA substrates pre-let-7a (72 nt), mature human miRNAs let-7a-5p (22 nt) and miR122 (22 nt), an oligo(A) tail mimic 15A (15 nt), total yeast tRNA, and total RNA isolated from HEK293T or MDA-MB-321 cells. Radiolabeled RNA Decade marker is used for reference.
RNA uridylation is prominent in wildtype S. pombe and a cid1 deletion strain
To further investigate the function and substrate range of Cid1 in vivo, we purified mRNA from wildtype and Δcid1 S. pombe strains and amplified several mRNA species to sequence their 3′-end by cRACE. Surprisingly, uridylation of RNAs was found in both the wildtype and the deletion strain. The small subunit ribosomal RNA, ScpofMR12 is, as typical for ribosomal RNA, not adenylated, and most samples also did not contain additional uridyl residues at the 3′-end (Figure 3). In S. pombe wildtype cells, one sequence was retrieved with multiple uridines added to the RNA 3′-end (Figure 3A) and in the cid1 deletion strain, monouridylated RNA was found (Figure 3B). Since uridylations are rare and only few sequences were retrieved, no conclusions as to the general uridylation pattern can be drawn from this data. SPBC215.11c, a protein coding RNA, was polyadenylated in both strains, with no 3′-end uridylated RNAs recovered. Interestingly, a uridylation/adenylation pattern was found in several samples of dak2, a protein coding RNA, where a poly(A) tail of differing length was interceded by two uridyl residues, followed by an additional four adenines (5′-NxAUUAAAA-3′). This pattern was found only in dak2 RNA, in eight out of nine sequenced RNA samples, and is not derived from the dak2 5′ or 3′ UTRs (Figure 3C). For another protein coding mRNA, SPAC19G12.09, samples encoding poly(A) tails without uridines were most prevalent. One polyuridylated sample was recovered from the cid1 deletion strain and two monouridylated RNAs from wildtype S. pombe RNA. Again, due to sample size, no conclusions can be drawn whether this represents a general uridylation pattern. Though it appears that uridylation is slightly less prevalent in the cid1 deletion strain, the small sample size and methodology of cRACE does not allow for a quantification of uridylation, but rather the qualitative observation that mono- and polyuridylation occurs in both wildtype and cid1 deletion strains, and that dak2 RNA is prone to an unusual pattern of RNA uridylation and adenylation.
Figure 3.

RNA uridylation of diverse RNA transcripts is found in wildtype and cid1 deletion strains. RNA was extracted from wildtype and cid1 deletion strain, and 3′-ends of selected mRNAs were analyzed by cRACE. In both (A) wildtype and (B) Δcid1 S. pombe cells transcripts containing terminal uridyl residues, and residues incorporated into the poly(A) tail were detected. A slight decrease in uridylated transcripts was detected upon cid1 deletion. (C) Sequence of the dak2 mRNA 3′-UTR (red) and 5′-UTR (blue). The protein coding sequence was omitted and is indicated as dak2 coding sequence.
Deletion of the Dis3L2 exonuclease elicits changes in the transcriptome
To further probe the prevalence of uridylation-dependent RNA decay in S. pombe, we isolated total RNA from wildtype, Δcid1, and Δdis3L2 strains, depleted ribosomal RNA and analysed the RNA content using deep sequencing. Reads were mapped to the S. pombe genome, and differentially expressed genes (effect size > 1.5) with more than a 1.5-fold change in expression between wildtype and Δcid1 or wildtype and Δdis3L2 were considered for the data analysis (Figure 4A and B, Supplementary Tables S2–S6). Gene expression changes in Δcid1 and Δdis3L2 deletion strains followed a similar trend, as outlined in Figure 5A, B and Supplementary Figure S1 and Supplementary Tables S2–S6. Overall, 72 genes were found differentially expressed >1.5-fold between wildtype and Δcid1, and 214 genes were differentially expressed >1.5-fold between wildtype and Δdis3L2 (Supplementary Table S2). 24 of the genes were differentially regulated more than 1.5-fold in both deletion strains (Figure 5B). While changes in the transcriptome of >1.5-fold were more noticeable in Δdis3L2, many of the same genes were similarly up/down regulated in Δcid1 (Figure 5A), albeit to a lesser extent. To verify the results obtained by RNA sequencing, we performed Northern Blotting on several RNAs found to be differentially expressed in the deletion strains. ecl1 was shown to be 1.4-fold upregulated in the cid1 deletion strain (Supplementary file 1), which was confirmed by Northern Blot (Figure 6A), and little to no expression changes were seen in spac19g12.09, spac27e2.11c, thf1 and tdh1, which confirms our sequencing results. We further used RT-qPCR to confirm our sequencing results (Figure 6B and Supplemental file 1), and all data confirmed the data observed in our Next Generation Sequencing data. The qPCR data confirmed no significant change in the expression of pex22 (Wt/dis3L2: qPCR 0.73-fold change, Sequencing 1.0-fold change). Four genes, hsp104 (Wt/dis3L2: qPCR 2.8-fold change, Sequencing 3.9-fold change), hsp78 (Wt/dis3L2: qPCR 1.8-fold change, Sequencing 2.7-fold change), ssa2 (Wt/dis3L2: qPCR 1.8-fold change, Sequencing 2.9-fold change) and tcg1 (Wt/dis3L2: qPCR 1.24-fold change, Sequencing 2.3-fold change) were more abundant in Δdis3L2, confirming our sequencing results. As observed in our next generation sequencing data, the changes in mRNA abundance as measured by RT-qPCR are less pronounced in the cid1 deletion strain than in the dis3L2 deletion strain (Figure 6B).
Figure 4.

Changes in relative abundance of mRNAs in WT, Δcid1, and Δdis3L2 cells. Expression plot comparing relative abundance (log2 centered log ratio, clr) of transcripts from a WT (x-axis) and (A) Δcid1 or (B) Δdis3L2 strain (y-axis). Differentially expressed genes (ALDEx2 effect size > 1.5) are indicated in red, and dotted lines indicated a 2-fold change in expression from the line of best fit for the data (Pearson's r = 0.9805).
Figure 5.
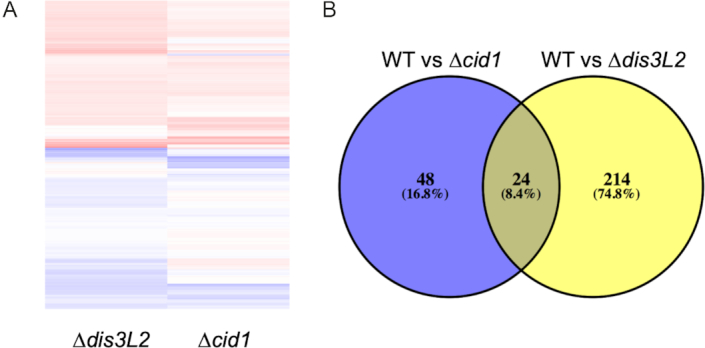
Genes differentially expressed in S. pombe deletion strains compared to wildtype. (A) Heat map showing fold-change for significantly different genes with hits for both WT S. pombe and Δcid1 and WT and Δdis3L2 strains and (B) Venn diagram of genes differentially expressed in Δcid1 and Δdis3L2 deletion strains versus wildtype.
Figure 6.

Northern blot and RT-qPCR showing differential expression of genes in wild-type versus Δcid1 S. pombe. (A) Northern Blots of total RNA extracted from WT and Δcid1 S. pombe was run on a 1% agarose gel in 1X MOPS and capillary blotted onto a Nylon membrane overnight at 4°C. RNA was UV-crosslinked to the membrane and probed with gene-specific oligonucleotideslabeled on the 5′-end with 32P. The blot was exposed to a phosphorimaging screen for two days at −80°C. tdh1 (GAPDH) was used as loading control. Expected sizes are as following: ecl1 with UTRs, 3597 nts; spac19G12.09 with UTRs, 1213 nts; spac27E2.11c with UTRs, 2140 nts; thf1 with UTRs, 3449 nts, with UTRs and introns, 3777 nts; tdh1 with UTRs, 1518 nts. (B) RT-qPCR was performed on WT, Δcid1, or Δdis3L2 S. pombe cells to assess the gene expression changes in the absence of cid1 or dis3L2. Cultures were grown to exponential phase (OD600 = 0.6), harvested and RNA isolated. Expression of each gene was normalized to the WT strain grown to early-exponential phase. Internal control used was rpp0. Error bars show the standard error on the mean (n = 6).
For genes >1.8-fold up or down regulated, we performed STRING analysis for enrichment of specific pathways (38) (Figure 7). In the Δdis3L2 strain, we found significant enrichment in genes upregulated in protein folding and protein degradation pathways (false discovery rate < 0.001, Table 1). Differential expression for genes involved in stress response, especially heat shock proteins, chaperones, and protein degradation were most prominent, but enrichment was also observed in sugar and nucleotide metabolism (false discovery rate < 0.01, Table 1), specifically in galactose metabolism (false discovery rate < 0.02, Table 1). No significant enrichment was found for genes up or down regulated in the cid1 deletion strain (Supplementary Figure S2A and B). Similarly, few genes were down regulated in Δdis3L2, and the gene products did not show enrichment in specific pathways according to our STRING analysis (Supplementary Figure S2C).
Figure 7.

Search tool for the retrieval of interacting genes/proteins (STRING) diagram of RNAs with altered expression levels in a dis3L2 deletion strain compared to wildtype. Respective proteins displayed are upregulated (>1.5-fold) in the Δdis3L2 strain. The diagram was generated using the STRING database. Functional associations between proteins are shown, with confidence of the proposed association denoted by line thickness.
Table 1.
Functional enrichments in genes >1.8-fold upregulated in S. pombe Δdis3L2 compared to wildtype. Gene ontology processes enriched with a false discovery rate <0.01 are listed
| Gene ontology biological process | Observed gene count | False discovery rate | Matching proteins in S. pombe network |
|---|---|---|---|
| Protein folding | 12 | 0.000414 | SPBC1711.08.1, bip1, cdc37, hsp104, hsp78, hsp90, psi1, ssa1, ssa2, ssc1, sti1, trx1 |
| Galactose catabolic process | 4 | 0.0021 | SPBC32F12.10.1, gal1, gal10, gal7 |
| Single-organism catabolic process | 15 | 0.00848 | SPAC26F1.07.1, SPBC32F12.10.1, SPBC3B9.01, SPCC5E4.05c.1, bip1, cdc48, gal1, gal10, gal7, glo1, pgi1, plg7, rpt1, rpt3, trx1 |
| Monosaccharide catabolic process | 6 | 0.00848 | SPAC26F1.07.1, SPBC32F12.10.1, gal1, gal10, gal7, pgi1 |
Deletion of dis3L2 confers resistance to hydroxyurea, whereas deletion of cid1 increases sensitivity to protein misfolding stress
Since our sequencing analysis revealed major changes to the transcriptome of stress related genes in the dis3L2 deletion strain, we assessed phenotypic effects on S. pombe in response to chemical stress (Figure 8). Cid1 was first identified as a protein involved in S-M checkpoint control and cid1 deletion strains were found to be sensitive to caffeine stress (12). For phenotypic analysis, cells were grown on EMM containing 10 mM caffeine, 10 mM hydroxyurea (HU), or a combination of 2.5 mM caffeine and 10 mM HU as described previously (12). Our data shows that the cid1 deletion strain is sensitive to caffeine and HU (Figure 8). Since Cid1 and Dis3L2 function in the same pathway, we next tested the impact of these chemical stressors on the dis3L2 deletion strain. Surprisingly, Δdis3L2 cells were more resistant to HU and caffeine, and a combination of caffeine and HU than wildtype cells (Figure 8). In addition, we tested the sensitivity of strains bearing a deletion of dis3L2 or cid1 to conditions of protein misfolding stress for growth in media containing hydrogen peroxide, which causes oxidative damage. Deletion of dis3L2 did not cause significant changes in sensitivity to hydrogen peroxide compared to wildtype (Figure 8). By contrast, deletion of cid1 resulted in increased sensitivity to oxidative stress.
Figure 8.
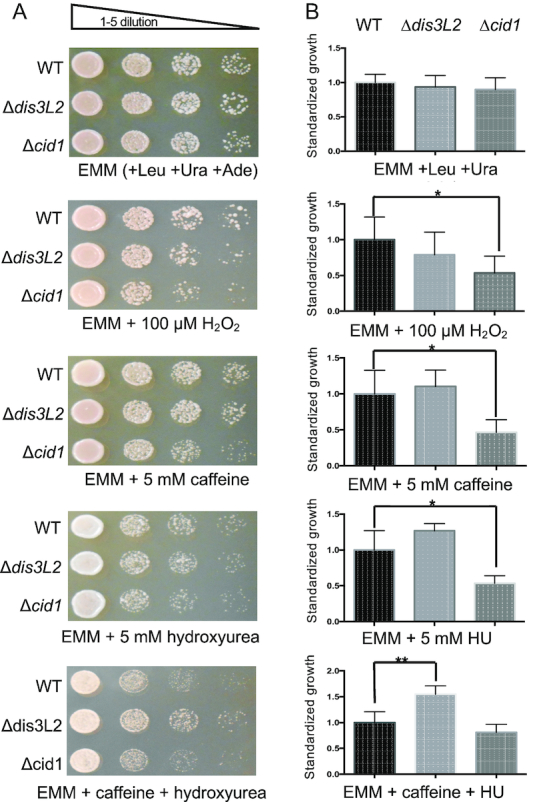
Growth assay of S. pombe wildtype (WT), Δdis3L2 and Δcid1. Cells were grown on EMM with required nutrients and with or without drug treatment at 30°C. (A) Cells were grown overnight in EMM media with Ura, Leu, and Ade and then spotted on media plates. (B) Quantification of standardized growth differences between wildtype and deletion strains. WT growth was normalized to 1 and compared against deletion strains. The P values were calculated using a two-tailed t-test. A P value < 0.05 is indicated by an asterisk (*) and < 0.01 by (**).
Deletion of dis3L2 and cid1 causes the accumulation of misfolded proteins
To test whether the upregulation of stress response genes in the deletion strains was a transcriptional response to cellular stress, we tested WT and deletion strains for the accumulation of misfolded proteins in the cell. Indeed, the overall protein abundance in the insoluble protein fraction was significantly higher in both deletion strains (Figure 9A, C and D). PGK1 (Phosphoglycerate kinase 1) was blotted for in total cell lysates as a loading control (Figure 9B). To investigate whether dis3L2 or cid1 expression is upregulated under stress conditions, we performed a Northern blot on RNAs extracted from WT cells grown under heat, cold, caffeine and hydroxyurea stress (Figure 9E). No changes in abundance were observed.
Figure 9.

Sedimentation assay of aggregated proteins in WT, Δcid1, and Δdis3L2 deletion strains. (A) Representative SDS gel of a sedimentation assay showing total, soluble, and insoluble protein of WT S. pombe cells in triplicate. (B) Western Blot showing similar amount of PGK1 of total cell lysate of WT, Δcid1, and Δdis3L2 deletion strains were used for sedimentation assays. R = replicate. (C) Quantification of total amount of protein in the supernatant and pellet fractions of three biological replicates from the wildtype, Δcid1, and Δdis3L2 strains. D) The ratio between the supernatant and pellet for each strain was calculated and plotted on a bar graph. The P values were calculated using a two-tailed t-test (0.033 between WT and Δcid1, 0.053 between WT and Δdis3L2). A P value <0.05 is indicated by an asterisk (*). (E) Northern blot showing no change in expression of cid1 or dis3L2 RNA in response to different growth conditions.
DISCUSSION
mRNA uridylation does not exclusively depend on Cid1
Uridylation-dependent RNA decay is now well established as an alternative RNA degradation pathway (9). Despite growing knowledge on the biochemical and structural properties of the responsible enzymes Cid1 and Dis3L2 in S. pombe, little is known about the prevalence and substrate-specificity of uridylation-dependent mRNA decay. A truncated Cid1, lacking amino acids 1–31 of the N-terminal domain was previously shown to be highly processive in vitro (15,21), yet only a few U residues are added in vivo (Figure 3 and (15)). By contrast, our full length protein is significantly less processive and its uridylation activity is restricted to few residues (Figure 2). It is therefore possible that the N-terminal domain of Cid1 serves as an auto-inhibitory domain to prevent excessive RNA uridylation.
In the cellular context, our data shows that mRNA uridylation in S. pombe is not exclusive to the founder Tutase Cid1 but is likely also executed by a partially redundant Tutase. We found evidence of mRNA uridylation in a cid1 deletion strain (Figure 3), indicating the activity of an alternate Tutase in this pathway. In addition, less drastic changes were observed in the transcriptome upon cid1 deletion (Figures 4, 5 and Supplemental Figure S2). It remains to be elucidated which TENTs are responsible for RNA uridylation in the absence of Cid1. S. pombe encodes several TENT homologs besides Cid1, namely Cid11, Cid12, Cid13, Cid14 and Cid16. Interestingly, transcriptional levels of the TENT Cid14 are 2-fold down regulated in the cid1 deletion strain. Cid14 is thought to be a poly(A) polymerase and lacks the histidine residue specifying uridine over adenine specificity (Figure 1A). Furthermore, Cid14 has been shown to act as a poly(A) polymerase in ribosomal RNA processing (39), and is thus unlikely to also act as a Tutase. Similarly, Cid11, Cid12 and Cid13 are thought to be adenylyltransferases rather that Tutases (9,40). The most likely candidate for mRNA uridylation appears to be Cid16, an enzyme previously shown to act as a Tutase on small RNAs, targeting Argonaute-bound RNAs to promote their degradation (41). In a recent study, spatially separated activities of the nuclear adenylyltransferase Cid14 and the cytoplasmic Tutase Cid16 were shown to regulate small RNA stability (41). While Cid1 does not compensate for small RNA uridylation in the absence of Cid16 (41), it is possible that Cid16 uridylates mRNAs in the absence of Cid1, or generally functions as a redundant Tutase acting on mRNA.
Mixed mRNA A/U tails
While adenylyltransferases and Tutases have previously been shown to act on the same set of RNAs on separate instances (41), we show here a previously unreported combined 3′ A/U modification in S. pombe. The observed combination of A/U addition suggests that in contrast to the uridylation and adenylation of sRNAs catalyzed by the spatially separate Cid14/Cid16, the dak2 mRNA 5′-NxAUUAAAA-3′ tail most likely occurs in the cytoplasm as a cooperative effort between uridylyl- and adenylyltransferases. The Tutases Cid1 and Cid16, as well as the adenylyltransferases Cid11 and Cid13, localize to the cytoplasm (42) and could interact to form this unusual 3′-end RNA motif. Whether the 5′-NxAUUAAAA-3′motif serves as a specific signal for downstream RNA processing, RNA localization, or degradation remains to be determined. We observed the 5′-NxAUUAAAA-3′ motif on dak2 mRNA, which encodes a dihydroxyacetone kinase. Whether the 5′-NxAUUAAAA-3′motif is unique to this mRNA species is speculative, as we only tested a small sample number of mRNAs. Excitingly, guanylyl- and adenylyltransferases were shown to generate mixed-tails in human cells, and mixed tails were shown to shield mRNA from rapid deadenylation (43). Future research will have to explore the biological function and prevalence of combined A/U tailored transcripts in S. pombe in vivo.
Uridylation-dependent RNA decay is linked to stress response and telomere maintenance
While Cid1 is not essential for the uridylation-dependent RNA decay pathway under non-stress conditions, Dis3L2 appears to represent more of a bottleneck in RNA degradation. Our data shows that the deletion of dis3L2 leads to an accumulation of transcripts predominantly in protein folding and degradation pathways, as well as sugar catabolic processes. While Cid1 does not contain specific RNA recognition motifs, such as Zinc fingers, its crystal structure revealed a positively charged surface area thought to facilitate general, sequence-independent RNA binding (19,20). It has been suggested that RNA binding proteins interact with Cid1 to direct the TENT to substrate RNAs, but these proteins remain to be identified (21).
As previously described for chemical stress, the transcriptome of S. pombe undergoes global changes in response to stress conditions (44) (Supplementary Tables S2-S6). We found most significant changes in the transcriptome in genes related to protein folding, such as heat shock protein genes hsp90, hsp70, co-chaperones cdc37 and wos2, protein remodelling factor hsp104, and chaperone activators aha1 and sti1 with an effect change between 2- and 10-fold in the dis3L2 deletion strain (Supplementary Tables S2, S5, S6). Changes to the Δcid1 transcriptome generally followed the same trend, albeit to a lesser extent, indicating that a partially redundant Tutase reduces the effect of the cid1 deletion (Supplementary Tables S2, S3, S4). Furthermore, protein catabolic processes and protein degradation pathways, including AAA-type ATPase cdc48, and 19S proteasome regulatory subunits rpt1, rpt3 and rpt6, and the ubiquitin C-terminal hydrolase ubp15, were upregulated between 2- and 5.4-fold in Δdis3L2, and up to 2.2-fold in Δcid1. Other accumulated transcripts include RNAs of genes from a variety of metabolic pathways, transcriptional regulators, cell cycle, and the cytoskeleton (Figure 7).
Galactose metabolism genes gal1, gal7 and gal10 are required for using galactose as a carbon or energy source and are usually repressed in wildtype fission yeast in the absence of galactose (45). We found these genes significantly upregulated in the Δdis3L2 strain with effect changes between 3.5 and 8.5 (Supplementary Tables S2, S5 and S6). Expression of gal genes is regulated by telomeric silencing (45). We observed an increased expression of genes involved in telomere organization in the dis3L2 deletion strain, namely DNA replication factor A subunit Ssb1 (effect change = 6.5), and single-stranded telomeric binding protein Tgc1 (effect change = 3.6). Overexpression of proteins involved in telomere organization may be compensatory effects in an effort to counteract a disruption of telomere organization, which is evident from the de-repression of gal genes. Cid1 was initially characterized as a protein involved in S-M checkpoint control, and it may well be that disruption of uridylation-dependent RNA decay interferes with telomere maintenance and repair.
Previous studies in frog oocytes show that uridylation-dependent decay is crucial for clearance of the maternal transcriptome, indicating that this decay pathway plays a role in the degradation of transcripts that are no longer required (46). Similarly, in humans, Dis3L2 depletion is associated with the degradation of damaged RNA transcripts (6). Furthermore, Dis3L2 inhibits global apoptotic mRNA decay and cell death (47). These and other studies indicate that uridylation-dependent RNA decay is part of a global mRNA surveillance, aiding in the clearance of unneeded or damaged RNAs. Our data show that perturbation in uridylation-dependent RNA decay elicits a stress response as evidenced by increased abundance of transcripts enriched in protein folding and degradation pathways. It is possible that some of the other transcriptional changes beyond stress response result in a higher expression rate of those genes. We hypothesise that excess protein production may overwhelm the cellular protein quality control, leading to the accumulation of unfolded proteins and consequently elicit the unfolded protein response. In addition, since the Tut/Dis3L2 pathway in humans functions in damaged transcript decay (6), these transcripts may also be accumulating in S. pombe, but not be detected as a significant change in abundance rates. Stress response genes are likely upregulated as a response to accumulated proteins in the deletion strains (Figure 9).
Dis3L2 depletion increases resistance to hydroxyurea-induced stress
Taking into account that both Cid1 and Dis3L2 are dispensable under normal growth conditions (7,9,12), it is likely that uridylation-dependent RNA decay targets damaged or incomplete RNA transcripts as part of a stress response, similar to the Cid14/16 small RNA surveillance pathway (41). Cid1 was first described as a protein involved in the S-M cell cycle checkpoint (12). A cid1 deletion strain displays a growth retarded phenotype under stress conditions when exposed to caffeine, HU, or a combination of caffeine and HU (Figure 8) and overexpression of Cid1 increases resistance to this stress-inducing combination (12,48). Our data shows that a dis3L2 deletion on the other hand increases viability when exposed to HU and caffeine (Figure 8). Both HU and caffeine are known for their interference with the cell cycle (49). HU inhibits the enzyme ribonucleotide reductase, which is essential for DNA synthesis and its depletion impairs DNA replication and subsequently arrests cells in S phase (50). Interestingly, the RNAs upregulated in the dis3L2 deletion strain included ribonucleotide reductase small subunit suc22 (1.9 fold) and ribonucleoside reductase large subunit cdc22 (3.4 fold), which were previously shown to be upregulated in response to HU (51). The increased expression of ribonucleotide reductase subunits, in combination with an already activated stress response may give Dis3L2 depleted cells the growth advantage over wildtype cells. While overexpression or depletion of enzymes in the uridylation-dependent RNA decay pathway can alter cell viability, we found no evidence that expression of cid1 or dis3L2 is altered at the transcriptional level (Figure 9E). Both cid1 and dis3L2 show similar abundance independent of growth temperature or chemical stress (Figure 8). However, protein production may instead serve as the point of control on the translational level, or enzyme activity could be modulated by posttranslational modification, as described for other nucleotidyltransferases (52,53). It is plausible that enzymes involved in RNA uridylation and decay are constitutively active and degrade damaged RNA transcripts on demand.
We also tested the effect of the deletion of dis3L2 or cid1 on protein quality control. When grown on hydrogen peroxide to induce protein misfolding, the cid1 deletion, but not the dis3L2 deletion strain showed a significant increase in sensitivity (Figure 8). It is possible that the upregulation of the cellular stress responses (e.g. the heat shock response) that we observed in the dis3L2 deletion exerts a protective function compared to the cid1 deletion strain. We further found that both deletion strains accumulated higher proportions of misfolded and insoluble proteins compared to wildtype (Figure 9). These results indicate that the defects in RNA processing in these strains leads to an accumulation of misfolded protein, which in turn appears to induce the expression of many protein quality control genes possibly by activation of the heat shock response.
We conclude that uridylation-dependent RNA decay is part of an RNA surveillance system, and RNA transcripts are not efficiently disposed of in the absence of Dis3L2 or to a lesser extent Cid1. Translation of these potentially damaged or unwanted RNA transcripts leads to the accumulation of misfolded proteins, eliciting the cellular stress response and the increased expression of chaperones and enzymes involved in protein degradation.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Nileeka Balasuriya and Susanna George for their advice, and Patrick O’Donoghue and Gregory Gloor for critical discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
J.P. Bickell Foundation and Natural Sciences and Engineering Research Council of Canada [RGPIN 04776-2014 to I.U.H.]; Ontario Graduate Scholarship and NSERC Canada Graduate Scholarship (to C.Z.C.); NSERC Canada Graduate Scholarship (to M.J.E.). Funding for open access charge: The Natural Sciences and Engineering Research Council of Canada.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cheadle C., Fan J., Cho-Chung Y.S., Werner T., Ray J., Do L., Gorospe M., Becker K.G.. Stability regulation of mRNA and the control of gene expression. Ann. N. Y. Acad. Sci. 2005; 1058:196–204. [DOI] [PubMed] [Google Scholar]
- 2. Norbury C.J. Cytoplasmic RNA: a case of the tail wagging the dog. Nat. Rev. Mol. Cell Biol. 2013; 14:643–653. [DOI] [PubMed] [Google Scholar]
- 3. Martin G., Keller W.. RNA-specific ribonucleotidyl transferases. RNA. 2007; 13:1834–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin C.J., Wen J., Bejarano F., Hu F., Bortolamiol-Becet D., Kan L., Sanfilippo P., Kondo S., Lai E.C.. Characterization of a TUTase/RNase complex required for Drosophila gametogenesis. RNA. 2017; 23:284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Norbury C.J. 3′ Uridylation and the regulation of RNA function in the cytoplasm. Biochem. Soc. Trans. 2010; 38:1150–1153. [DOI] [PubMed] [Google Scholar]
- 6. Ustianenko D., Pasulka J., Feketova Z., Bednarik L., Zigackova D., Fortova A., Zavolan M., Vanacova S.. TUT-DIS3L2 is a mammalian surveillance pathway for aberrant structured non-coding RNAs. EMBO J. 2016; 35:2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Malecki M., Viegas S.C., Carneiro T., Golik P., Dressaire C., Ferreira M.G., Arraiano C.M.. The exoribonuclease Dis3L2 defines a novel eukaryotic RNA degradation pathway. EMBO J. 2013; 32:1842–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pirouz M., Du P., Munafo M., Gregory R.I.. Dis3l2-Mediated decay is a quality control pathway for noncoding RNAs. Cell Rep. 2016; 16:1861–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chung C.Z., Seidl L.E., Mann M.R., Heinemann I.U.. Tipping the balance of RNA stability by 3′ editing of the transcriptome. Biochim. Biophys. Acta. 2017; 1861:2971–2979. [DOI] [PubMed] [Google Scholar]
- 10. Kwak J.E., Wickens M.. A family of poly(U) polymerases. RNA. 2007; 13:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rissland O.S., Norbury C.J.. Decapping is preceded by 3′ uridylation in a novel pathway of bulk mRNA turnover. Nat. Struct. Mol. Biol. 2009; 16:616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang S.W., Toda T., MacCallum R., Harris A.L., Norbury C.. Cid1, a fission yeast protein required for S-M checkpoint control when DNA polymerase delta or epsilon is inactivated. Mol. Cell. Biol. 2000; 20:3234–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Read R.L., Martinho R.G., Wang S.W., Carr A.M., Norbury C.J.. Cytoplasmic poly(A) polymerases mediate cellular responses to S phase arrest. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:12079–12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rissland O.S., Norbury C.J.. The Cid1 poly(U) polymerase. Biochim. Biophys. Acta. 2008; 1779:286–294. [DOI] [PubMed] [Google Scholar]
- 15. Rissland O.S., Mikulasova A., Norbury C.J.. Efficient RNA polyuridylation by noncanonical poly(A) polymerases. Mol. Cell. Biol. 2007; 27:3612–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Munoz-Tello P., Gabus C., Thore S.. A critical switch in the enzymatic properties of the Cid1 protein deciphered from its product-bound crystal structure. Nucleic Acids Res. 2014; 42:3372–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stevenson A.L., Norbury C.J.. The Cid1 family of non-canonical poly(A) polymerases. Yeast. 2006; 23:991–1000. [DOI] [PubMed] [Google Scholar]
- 18. Chung C.Z., Jo D.H., Heinemann I.U.. Nucleotide specificity of the human terminal nucleotidyltransferase Gld2 (TUT2). RNA. 2016; 22:1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Munoz-Tello P., Gabus C., Thore S.. Functional implications from the Cid1 poly(U) polymerase crystal structure. Structure. 2012; 20:977–986. [DOI] [PubMed] [Google Scholar]
- 20. Lunde B.M., Magler I., Meinhart A.. Crystal structures of the Cid1 poly (U) polymerase reveal the mechanism for UTP selectivity. Nucleic Acids Res. 2012; 40:9815–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yates L.A., Durrant B.P., Fleurdepine S., Harlos K., Norbury C.J., Gilbert R.J.. Structural plasticity of Cid1 provides a basis for its distributive RNA terminal uridylyl transferase activity. Nucleic Acids Res. 2015; 43:2968–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yates L.A., Durrant B.P., Barber M., Harlos K., Fleurdepine S., Norbury C.J., Gilbert R.J.. Improved crystallization and diffraction of caffeine-induced death suppressor protein 1 (Cid1). Acta Crystallogr. F Struct. Biol. Commun. 2015; 71:346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aphasizhev R. RNA uridylyltransferases. Cell. Mol. Life Sci.: CMLS. 2005; 62:2194–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burns D.M., D’Ambrogio A., Nottrott S., Richter J.D.. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature. 2011; 473:105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lim J., Ha M., Chang H., Kwon S.C., Simanshu D.K., Patel D.J., Kim V.N.. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell. 2014; 159:1365–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thornton J.E., Chang H.M., Piskounova E., Gregory R.I.. Lin28-mediated control of let-7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7). RNA. 2012; 18:1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gallouzi I.E., Wilusz J.. A DIStinctively novel exoribonuclease that really likes U. EMBO J. 2013; 32:1799–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lv H., Zhu Y., Qiu Y., Niu L., Teng M., Li X.. Structural analysis of Dis3l2, an exosome-independent exonuclease from Schizosaccharomyces pombe. Acta Crystallogr. D Biol. Crystallogr. 2015; 71:1284–1294. [DOI] [PubMed] [Google Scholar]
- 29. Robinson S.R., Oliver A.W., Chevassut T.J., Newbury S.F.. The 3′ to 5′ exoribonuclease DIS3: From structure and mechanisms to biological functions and role in human disease. Biomolecules. 2015; 5:1515–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Towler B.P., Jones C.I., Harper K.L., Waldron J.A., Newbury S.F.. A novel role for the 3′-5′ exoribonuclease Dis3L2 in controlling cell proliferation and tissue growth. RNA Biol. 2016; 13:1286–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lubas M., Damgaard C.K., Tomecki R., Cysewski D., Jensen T.H., Dziembowski A.. Exonuclease hDIS3L2 specifies an exosome-independent 3′-5′ degradation pathway of human cytoplasmic mRNA. EMBO J. 2013; 32:1855–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Faehnle C.R., Walleshauser J., Joshua-Tor L.. Mechanism of Dis3l2 substrate recognition in the Lin28-let-7 pathway. Nature. 2014; 514:252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chang H.M., Triboulet R., Thornton J.E., Gregory R.I.. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature. 2013; 497:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ellis M.J., Trussler R.S., Haniford D.B.. Hfq binds directly to the ribosome-binding site of IS10 transposase mRNA to inhibit translation. Mol. Microbiol. 2015; 96:633–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ellis M.J., Trussler R.S., Charles O., Haniford D.B.. A transposon-derived small RNA regulates gene expression in Salmonella Typhimurium. Nucleic Acids Res. 2017; 45:5470–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shiber A., Breuer W., Brandeis M., Ravid T.. Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when Hsp70 chaperone levels are limiting. Mol. Biol. Cell. 2013; 24:2076–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fernandes A.D., Macklaim J.M., Linn T.G., Reid G., Gloor G.B.. ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLoS One. 2013; 8:e67019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P. et al.. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011; 39:D561–D568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Keller C., Woolcock K., Hess D., Buhler M.. Proteomic and functional analysis of the noncanonical poly(A) polymerase Cid14. RNA. 2010; 16:1124–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saitoh S., Chabes A., McDonald W.H., Thelander L., Yates J.R., Russell P.. Cid13 is a cytoplasmic poly(A) polymerase that regulates ribonucleotide reductase mRNA. Cell. 2002; 109:563–573. [DOI] [PubMed] [Google Scholar]
- 41. Pisacane P., Halic M.. Tailing and degradation of Argonaute-bound small RNAs protect the genome from uncontrolled RNAi. Nat. Commun. 2017; 8:15332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matsuyama A., Arai R., Yashiroda Y., Shirai A., Kamata A., Sekido S., Kobayashi Y., Hashimoto A., Hamamoto M., Hiraoka Y. et al.. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 2006; 24:841–847. [DOI] [PubMed] [Google Scholar]
- 43. Lim J., Kim D., Lee Y.S., Ha M., Lee M., Yeo J., Chang H., Song J., Ahn K., Kim V.N.. Mixed tailing by TENT4A and TENT4B shields mRNA from rapid deadenylation. Science. 2018; 361:701–704. [DOI] [PubMed] [Google Scholar]
- 44. Biswas P., Ghosh S.. Global transcriptomic profiling of Schizosaccharomyces pombe in response to nitrosative stress. Gene. 2015; 558:241–253. [DOI] [PubMed] [Google Scholar]
- 45. Matsuzawa T., Fujita Y., Tanaka N., Tohda H., Itadani A., Takegawa K.. New insights into galactose metabolism by Schizosaccharomyces pombe: isolation and characterization of a galactose-assimilating mutant. J. Biosci. Bioeng. 2011; 111:158–166. [DOI] [PubMed] [Google Scholar]
- 46. Chang H., Yeo J., Kim J.G., Kim H., Lim J., Lee M., Kim H.H., Ohk J., Jeon H.Y., Lee H. et al.. Terminal Uridylyltransferases execute programmed clearance of maternal transcriptome in vertebrate embryos. Mol. Cell. 2018; 70:72–82. [DOI] [PubMed] [Google Scholar]
- 47. Thomas M.P., Liu X., Whangbo J., McCrossan G., Sanborn K.B., Basar E., Walch M., Lieberman J.. Apoptosis triggers specific, rapid, and global mrna decay with 3′ uridylated intermediates degraded by DIS3L2. Cell Rep. 2015; 11:1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang S.W., Norbury C., Harris A.L., Toda T.. Caffeine can override the S-M checkpoint in fission yeast. J. Cell Sci. 1999; 112:927–937. [DOI] [PubMed] [Google Scholar]
- 49. Moser B.A., Brondello J.M., Baber-Furnari B., Russell P.. Mechanism of caffeine-induced checkpoint override in fission yeast. Mol. Cell. Biol. 2000; 20:4288–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koc A., Wheeler L.J., Mathews C.K., Merrill G.F.. Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J. Biol. Chem. 2004; 279:223–230. [DOI] [PubMed] [Google Scholar]
- 51. Fernandez Sarabia M.J., McInerny C., Harris P., Gordon C., Fantes P.. The cell cycle genes cdc22+ and suc22+ of the fission yeast Schizosaccharomyces pombe encode the large and small subunits of ribonucleotide reductase. Mol. Gen. Genet. 1993; 238:241–251. [DOI] [PubMed] [Google Scholar]
- 52. Mohan N., Sudheesh A.P., Francis N., Anderson R., Laishram R.S.. Phosphorylation regulates the Star-PAP-PIPKIalpha interaction and directs specificity toward mRNA targets. Nucleic Acids Res. 2015; 43:7005–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Phatnani H.P., Greenleaf A.L.. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006; 20:2922–2936. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.