


Abstract
Adenosine deaminase is involved in adenosine degradation and salvage pathway, and plays important physiological roles in purine metabolism. Recently, a novel type of adenosine deaminase-like protein has been identified, which displays deamination activity toward N6-methyl-adenosine monophosphate but not adenosine or AMP, and was consequently named N6-methyl-AMP deaminase (MAPDA). The underlying structural basis of MAPDA recognition and catalysis is poorly understood. Here, we present the crystal structures of MAPDA from Arabidopsis thaliana in the free and in the ligand-bound forms. The protein contains a conserved (β/α)8 Tim-barrel domain and a typical zinc-binding site, but it also exhibits idiosyncratic local differences for two flexible helices important for substrate binding. The extensive interactions between the N6-methyl-AMP substrate or the inosine monophosphate product and the enzyme were identified, and subsequently evaluated by the deamination activity assays. Importantly, each structure reported here represents a different stage of the catalytic pathway and their structural differences suggested that the enzyme can exist in two distinct conformational states. The open state switches to the closed one upon the binding of ligands, brought about by the two critical helices. Our structural studies provide the first look of this important metabolic enzyme and shed lights on its catalytic pathway, which holds promise for the structure-based drug design for MAPDA-related diseases.
INTRODUCTION
Purine metabolism is implicated in plenty of physiologic and pathologic events in cells. Adenosine is an immunomodulatory molecule that suppresses immune responses in order to protect tissue integrity. The accumulation of dAdo and deoxyguanosine (dGuo) is toxic to lymphocytes (1,2), while elevated dGTP and dATP levels cause problems in DNA repair pathways (3,4). Consequently, ATP concentrations are tightly controlled by purinergic enzymes. Following the combined activities of Ecto-nucleoside triphosphate diphosphohydrolase (E-NTPDase) and Ecto-5′-nucleotidase (e-5NT) that convert ATP to adenosine, adenosine deaminase (ADA) and purine nucleoside phosphorylase (PNP) catalyze the degradation and salvage of purines (5–7). Specifically, ADA is responsible for the irreversible deamination of 2′-deoxyadenosine (dAdo) and adenosine to inosine, which antagonizes the suppressive effects of adenosine in human immune cells (8,9). On the other hand, it was reported that ADA malfunction often results in diabetes mellitus, autoimmune thyroiditis, hepatitis etc. (10–12). In plants, purine metabolism always plays a role in embryo maturation and germination, as well as development of storage organs and sprouting, fruit ripening etc. (13–15).
N 6-methylated adenine (m6A) was discovered in the 1970s (16–19), and is considered the most prevalent posttranscriptional modification in mRNA. The modification has been observed in a wide range of eukaryotic species that range from yeasts, plants, insects to mammals as well as RNA viruses (20–22). m6A homeostasis requires the so-called ‘writers’ (m6A methyltransferases), ‘erasers’ (m6A-demethylating enzymes), and ‘readers’ (m6A-binding proteins) (23–25). The physiological functions of these enzymes are of great interests in recent years due to their possible roles in pathogenesis. Studies have shown that dysregulation of these modifications may be implicated in obesity, brain development abnormalities and other diseases (26–30). For example, the m6A RNA demethylase FTO (fat mass and obesity-associated protein), has been proved to be implicated in tumorigenesis (31–32).
Based on the phylogenetic relationships of various gene products with significant amino acid similarities to ADA using parimony and Bayesian methods, Maier et al. categorized adenyl deaminases into five subfamilies: adenosine deaminase (ADA), adenine deaminase (ADE), AMP deaminase (AMPD), adenosine deaminase-related growth factor (ADGF, or CECR1 in vertebrates), and ADA-like (ADAL) (33). ADE catalyzes the conversion of adenine to hypoxanthine whereas AMPD catalyzes the deamination of AMP to inosine monophosphate (IMP) and implicated in the purine nucleotide cycle. ADGF converts adenosine to inosine or its analogs. Aside from the four families mentioned above, Maier et al. identified a novel family which they named ADA-like (ADAL). In 2006, Schinkmanová et al. isolated and purified the ADAL enzyme from rat liver (34). This enzyme, which they named N6-methyl-adenosine monophosphate aminohydrolase, apparently converted N6-methyl-adenosine monophosphate (N6m-AMP) and N6-methyl-deoxyadenosine monophosphate (N6m-dAMP) to inosine monophosphate (IMP) and deoxyinosine monophosphate (dIMP) respectively. However, previously considered to possess the same function as ADA by Maier et al., due to the conservation of all the key residues required for A-to-I deamination, rat ADAL was proven to be unable to utilize adenosine or AMP as a substrate. Moreover, it was inhibited by deoxycoformycin 5′-phosphate while the adenosine deaminase inhibitor erythro-9-(2-hydroxy-3-nonyl) adenine (EHNA) was ineffective. Five years later, Murakami et al. purified and biochemically characterized the human enzyme, and found that this protein was adenosine deaminase-like protein isoform 1 (ADAL1, i.e. human N6-methyl-AMP deaminase or HsMAPDA). By testing a series of potential substrates, they further demonstrated that ADAL1 specifically acts on N6- or O6-substituted purine or 2-aminopurine nucleoside/nucleotide analogues, but its activity is sensitive to modifications in the phosphate moiety (35).
Recently, the ortholog from Arabidopsis thaliana was identified, and its phylogenetic spread and possible specificity determinants for N6m-AMP were investigated. Chen et al. showed that the plant root growth was barely affected upon the knockout of the gene. However, the subsequent accumulation ofN6m-AMP suggested that it is the main in-vivo substrate of Arabidopsis thaliana MAPDA (AtMAPDA), and might only be metabolized by this enzyme in A. thaliana. Additionally, MAPDA does not belong to the ADA families that we already know (36). Therefore, they renamed the enzyme MAPDA, and also inferred that its function is likely to protect RNA from m6A mis-incorporation.
Crystal structures of ADAs from a variety of organisms have been solved, ranging from lower bacteria to higher eukarya (structures of representative species include PDBs 1ADD, 2ADA, 3IAR, 3MVI, 1VFL, 1WXY, 1KRM, 1NDV and 1NDW) (37–42). The enzymes share the (β/α)8 barrel structure and their core catalytic domains possess a characteristic zinc-binding signature sequence SL(S/N)TDDP (42–44). In addition, plenty of inhibitors were designed and cocrystallized with the enzymes due to their pharmacological values. It was found that ADA might adopt either an ‘open’ or a ‘closed’ conformation, depending upon whether or what inhibitors were bound (41). Compared to the thorough studies conducted on ADAs, the molecular recognition mechanism of MAPDAs is poorly understood due to a lack of structural studies, although the enzymes were identified and characterized biochemically more than a decade ago. In this study, the high-resolution structures of AtMAPDA in the apo form and in complex with various ligands were determined. The interactions between the enzyme and the substrate were investigated by the deaminase assay using the thin layer chromatography (TLC) and UV–Vis spectroscopy. By structural comparison to that of ADAs, two distinct conformational states of AtMAPDA were identified. This study provides insights into the substrate recognition mechanism of MAPDAs and may assist MAPDA-related drug development.
MATERIALS AND METHODS
Cloning, expression and protein purification
The full-length gene encoding At4g04880 (accession number AY099563) was amplified by PCR from the cDNA of the Arabidopsis thaliana accession Col-0 using primers 5′-GCGGCAGCCATATGGAATGGATACAATCACTGCC-3′ and 5′-TTGCACTTCTCGAGCT AAACGTGCTCTGGCGAG-3′. After double digestion by the restriction enzymes NdeI and XhoI, the PCR product was inserted into a modified pET-28a (+) vector. The mutations were created through Quikchange PCRs based on the WT sequence and the primers were listed in Supplementary Table S1. In order to obtain the cocrystals of the enzyme-substrate complex, the D295N mutation was introduced to inactivate the enzyme and to stall the reaction.
The plasmids were transformed into Escherichia coli strain BL21 (DE3) cells for overexpression of the target proteins. The transformed cells were cultured overnight in Luria-Bertani broth containing 50 mg/l kanamycin at 37°C. A 2-l fresh culture medium was inoculated with 20 ml of the overnight culture. When A600 reached 0.8 at 37°C, the expression of AtMAPDA was induced by 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and was kept shaking overnight at 18°C. The E. coli cells were then pelleted by centrifugation at 3500 g for 20 min and resuspended in pre-chilled nickel-nitrilotriacetic acid (Ni-NTA) buffer A containing 40 mM Tris–HCl (pH 8.0), 250 mM NaCl, 10 mM imidazole, 1 mM β-mercaptoethanol (β-ME) and 1 mM phenylmethylsulfonyl fluoride (PMSF). The resuspended cells were disrupted by ultrasonication and the supernatant was obtained by centrifugation at 23 500 g for 1 h at 4°C. The supernatant was then applied onto Ni-NTA affinity resin (Qiagen) pre-equilibrated with Ni-NTA buffer A. The target protein was eluted with Ni-NTA buffer B containing 40 mM Tris–HCl (pH 8.0), 250 mM NaCl, 250 mM imidazole, 1 mM β-ME and 1 mM PMSF. The fractions were further subjected to ion exchange purification by a QHP column (GE Healthcare) using a NaCl gradient, and protein was eluted at ∼150 mM NaCl. The purified protein was pooled and dialyzed against a buffer consisting of 20 mM Tris–HCl (pH 8.0), 150 mM NaCl and 1 mM DTT. AtMAPDA was stored at 3.0 mg/ml at –80°C after being flash frozen by liquid nitrogen.
Crystallization and structure determination
Initial crystallization screens were set up using the sitting-drop vapor diffusion method, and 6.0 mg/ml protein was mixed with equal volume of the reservoir solution at 20°C. Crystals of the apo-protein were obtained in a condition of 26–36% PEG 3350, 0.2 M NaCl, and 0.1 M HEPES (pH 7.5). After optimization, thick crystals were obtained from a condition consisting of 32% PEG 3350, 0.1 M NaCl, and 0.1 M Tris–HCl (pH 9.0) using the hanging-drop vapor diffusion method. The cocrystals with N6m-AMP or IMP were obtained by mixing D295N with 2 mM N6m-AMP, or WT with 2 mM IMP at a molar ratio of 1:15 prior to crystallization and were grown under identical conditions.
The fully-grown crystals were soaked for 1–3 min in a cryoprotective solution containing all the components of the reservoir solution supplemented 20% glycerol (v/v). The soaked crystals were mounted on nylon loops and flash frozen in liquid nitrogen. The native data were collected from frozen crystals at -173°C on ADSC CCD detectors using Beamline 19U1 (BL19U1) at the Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, P.R. China). The data were processed with the program HKL3000 (45). All the space groups of the crystals or the cocrystals were P212121, and there was one molecule in the asymmetric unit, judging from the solvent content. To solve the crystal structures, molecular replacement (MR) was first performed on the dataset of the apo-protein using Phaser (46) with the coordinates of the mouse ADA structure (PDB 2ADA) (37) as the search model. The initial solution was obtained with a high R value (∼0.5), suggesting large structural deviations from the model. Several regions displayed large positive and negative peaks in the difference map, which were manually rebuilt according to the electron density map with COOT (47). The phase was further improved by density modification, and multiple cycles of refinement alternating with model rebuilding was carried out by PHENIX. refine (48). The model was finished with 337 residues built into the density. The final R-factor was 17.4% (Rfree = 22.2%). The final model was validated by Molprobity (49). The Ramachandran plot of the final model has 96.68%, 3.02% and 0.30% of the residues in the most favorable, generously allowed and disallowed region. The structures of the D295N-N6m-AMP and the WT-IMP complexes were solved through a similar fashion, but with the finished model of the apo-protein as the search model for the MR runs. All the data collection and structure refinement statistics were summarized in Table 1. The structural figures were produced with PyMOL (www.pymol.org).
Table 1.
Data collection and refinement statistics
| Apo-WT (6IJM) | D295N-N6m-AMP (6IJN) | WT-IMP (6IJP) | |
|---|---|---|---|
| Data collection | |||
| Resolution (Å) | 50.00-2.00 (2.07–2.00)a | 50.00-1.66 (1.72–1.66) | 50.00–1.85 (1.92–1.85) |
| Space group | P 212121 | P 212121 | P 212121 |
| Cell dimensions(Å) | |||
| a, b, c (Å) | 50.6, 79.9, 86.8 | 49.9, 80.1, 86.8 | 50.9, 81.3, 86.6 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Rmerge | 0.071 (0.55) | 0.10 (0.49) | 0.077 (0.33) |
| CC1/2 | 0.999 (0.876) | 0.998 (0.959) | 0.992 (0.944) |
| Redundancy | 6.4 (6.4) | 12.6 (13.0) | 6.3 (6.5) |
| Completeness (%) | 98.7 (98.7) | 99.7 (99.3) | 99.8 (99.8) |
| I/σ(I) | 20.5 (2.9) | 20.5 (5.1) | 38.2 (4.9) |
| Refinement | |||
| Resolution (Å) | 43.75–2.02 (2.11–2.02) | 43.28–1.66 (1.69–1.66) | 43.89–1.85 (1.90–1.85) |
| No. reflections | 22196 | 42004 | 31411 |
| R work/Rfreeb(%) | 17.4/22.2 | 14.7/17.4 | 15.9/17.8 |
| No. atoms | |||
| Protein | 2613 | 2693 | 2665 |
| Ligand | 1 (Zn2+) | 24 (N6m-AMP) | 1 (Zn2+), 23 (IMP) |
| Water | 193 | 305 | 408 |
| B-factor (Å2) | |||
| Protein | 25.01 | 16.9 | 32.2 |
| Ligand | 24.2 | 10.0 (N6m-AMP) | 26.0 (Zn2+), 26.6 (IMP) |
| Water | 31.60 | 28.7 | 39.4 |
| RMS(bonds) (Å) | 0.011 | 0.009 | 0.015 |
| RMS(angles) (°) | 0.92 | 1.06 | 1.23 |
| Ramachandran favored (%) | 96.68 | 97.33 | 97.62 |
| Outliers (%) | 0.30 | 0.30 | 0.30 |
aValues in parentheses are for the highest-resolution shell.
bRmerge = Σ |(I - < I >)|/σ(I), where I is the observed intensity.
cRwork = Σhkl ||Fo| - |Fc||/ Σhkl |Fo|, calculated from working data set.
d R free is calculated from 5.0% of data randomly chosen and not included in refinement.
Deaminase assay with TLC detection
The assay mixture contained 20 mM Tris–HCl (pH 8.0), 150 mM NaCl and 5 mM N6m-AMP. 50 μM AtMAPDA or mutants were added to initiate the reaction. All the reactions were incubated for 30 min on ice. Aliquots of 0.5 μl were taken and spotted on a cellulose polyethyleneimine plate (Merck Millipore). The spots were 2 cm from the bottom of the plate and 1 cm apart, and the plates were developed with n-BuOH-HOAc-H2O (12:3:5) as the solvents in a glass jar. The TLC plates were then exposed to UV light for visualization.
Deaminase assay with UV–Vis spectroscopy detection
The absorption curves of N6m-AMP and IMP were first measured at the wavelength 230–310 nm range using a UV–Vis spectrophotometer (Hitachi U-2900). Then the individual absorbance values of both N6m-AMP and IMP at different concentrations were measured at 240 nm, and a working curve was generated by plotting the absorbance differences between the two molecules against specific concentrations. The deamination activity of AtMAPDA was assessed by quantifying the conversion of N6m-AMP to IMP at 240 nm and at 25°C. To obtain the kinetic constants, assays were carried out in a buffer containing 20 mM potassium phosphate (pH 7.2) with N6m-AMP in the assay mixture, and the reactions were started by addition of the enzyme. Enzyme with appropriate concentrations (250 nM WT, 500 nM partially impaired mutants, or 1–2 μM completely inactive mutants) and substrates with varying concentrations were employed. Activities of each mutant were repeated in triplicates.
RESULTS
The structure of apo-AtMAPDA
The apo-AtMAPDA protein construct used for crystallization contains 376 residues, with 21 extra amino acids being appended at the N-terminus to assist purification. Each asymmetric unit contains only one monomer, consistent with the solution state as indicated by gel-filtration chromatography analysis (data not shown). The protein is visible from His-1 to His354, and contains two internal disorders for the Lys49-Asp51 and Ala133-Gly147 regions. The region encompassing the former also displays a high temperature factor, indicating local structural flexibility. Therefore, side chains of many residues at this region were not modeled. The refined model contains a total of 337 amino acids, 193 water molecules and a Zn2+ metal ion. The model is of good geometry, with 0.3% residues falling in the outlier region in the Ramachandran plot.
The basic deaminase domain retains the (β/α)8 Tim-barrel fold similar to that of ADAs, with the essential Zn2+ ion tightly bound at the C-terminal end of the barrel (Figure 1A). The high-resolution structure shows that the critical zinc is coordinated by the side chains of four key residues (His13, His15, His217 and Asp295) and adopts an octahedral geometry (Figure 1B). Two water molecules serve as the fifth and sixth ligands respectively, one of which is presumably the catalytic water (Wcat), to be activated for the nucleophilic attack on the C6 position of the substrate.
Figure 1.

The overall structure of apo-AtMAPDA. (A) The structure shown in the ribbon rendition in the front and back views. The N- and C-termini were indicated and the secondary structure elements of AtMAPDA were labeled. The cyan dot indicated the zinc ion. (B) The close-up of the zinc-coordination state. The side chains of the key residues were shown as sticks and labeled. The two water molecules were the water for catalysis (Wcat) and coordination (Wcor) respectively, serving as the fifth and sixth ligands. The distances of the ligands from the metal were shown (units in Å).
The structure of AtMAPDA in complex with the substrate
To find out the structural basis of substrate recognition, we crystallized AtMAPDA in complex with N6m-AMP. To prevent the catalysis on the substrate, we intentionally mutated Asp295, the catalytic residue to an asparagine. In the resulting cocrystal structure, the active site contains anN6m-AMP molecule (Figures 2A and Supplementary Figure S1A). The general shape of the AtMAPDA complex resembles that of the apo-protein, but several local structural changes are evident, namely the α3 and α4 helices (Phe40-His71), which will be further detailed elsewhere. Additionally, the disorder Lys49-Asp51 observed previously on the apo-protein became structured (Figure 2B). At the substrate-binding pocket, the map shows clear density of the methylated AMP, which forms extensive interaction network with surrounding residues (Supplementary Figure S1 and Figure 2C). Specifically, N6 and N1 of the six-membered ring form three hydrogen bonds with Glu220 and Asn295 respectively, whereas N7 from the five-membered ring makes two more with the terminal carboxylate of Asp296 (Figure 2C). Meantime, the base also forms a main chain interaction with the backbone amino group of Gly190 via N3. The recognition on both rings distinguishes purines from pyrimidines. The ribose forms two more hydrogen bonds, including one with a long distance (∼3.4 Å) on the 2′-hydroxyl group from Asp160. Therefore it is not difficult to predict that the enzyme can recognize both AMP and dAMP derivatives. The terminal phosphate group is locked in a pocket by both hydrogen bonds and salt bridges. Particularly, the former involves the main-chain amino groups of Leu16, Asn17 and Thr98, as well as side chains of His65, Asn17 and Thr98. Moreover, the terminal Nϵ atom of Lys100 makes a salt bridge with the phosphate. The tight binding mode accommodates the monophosphate group nicely but also leaves no room for additional phosphates as well, and thus excludes the binding of ADP or ATP. Furthermore, the N6-methyl group is ∼4.0–5.0 Å away from hydrophobic residues Val57 and Phe300, and forms van der Waals contacts with them. These hydrophobic contacts may serve as a ‘sensor’ of the methylation, which partially explains the inactivity of AtMAPDA toward AMP. On the other hand, Val57 is replaced by a cysteine in the human counterpart, which may render the enzyme less sensitive to the methylation and thus confer activity on AMP in both non- and methylated forms (35). Lastly, the aromatic ring of Phe58 is perpendicular to the purine ring of the substrate and forms face-to-edge aromatic interactions, whereas His15 forms π-π stacking interactions. The residues mediating aforementioned interactions are mostly conserved, and we therefore made the mutations on them to test their contribution to the enzymatic activity toward N6m-AMP using TLC. All the mutants were well expressed, purified (Supplementary Figure S2) and behaved normally on a gel-filtration column (data not shown), suggesting that no significant impairment was induced by the mutations to the folding process. When the reaction products were run on cellulose polyethyleneimine plates and developed with designated solvents, the IMP product migrated much slower than the substrate. The TLC results showed that the activity of the WT enzyme towardN6m-AMP was rather strong, which converted all the methylated AMP to IMP in a 30-min reaction (Figure 2D). However, the H15A, E220A, D295A, D295N and D296A mutations almost eliminated deaminase activity, with little amounts of IMP produced. On the other hand, while the N17A, T97A and K100A mutants displayed moderate reduction in activity, the H65A and T98A mutants each had an activity almost comparable to that of WT at an enzyme concentration of 50 μM. Considering that the TLC method was an insensitive method for enzymatic activity characterization, we further conducted the assays and detected the UV absorbance changes accompanying the chemical reaction at 240 nm using UV–Vis spectroscopy, which linearly correlated with the conversion of the substrate to product (Supplementary Figure S3). We discovered that the spectroscopic results agreed with that of TLC (Table 2 and Figure 2D). In particular, the H15A, E220A, D296A, and D295A/N mutations indeed caused the most significant impairment to enzymatic activity, which could not be determined by current spectroscopic method. Of these residues, His15 and Asp295 are not only the ligands for the zinc metal, but also involved in substrate recognition (Figures 1B and 2C). The lesser important residues are residues for base-specific recognition such as Asp296 and Glu220, and the rest of the residues that contact the ribose or the phosphate group were partially active in the deamination assays upon mutations (∼30–70% of WT in terms of catalytic efficiency).
Figure 2.

Substrate interactions with AtMAPDA and enzymatic deaminase activity characterization on mutants. (A) The cocrystal structure of AtMAPDA (rendered in ribbon and surface) in complex with N6m-AMP (in sticks). (B) Structure comparison of the AtMAPDA-N6m-AMP complex (PDB 6IJN, cyan) with apo-AtMAPDA (PDB 6IJM, color scheme as in Figure 1A). The structural changes for the α3 and α4 helices (PheF40-His71) before and after the binding of the substrate were indicated by the red circles. (C) The interaction network of N6m-AMP with AtMAPDA. The residues participating in ligand recognition were depicted as sticks and labeled. The hydrogen bonds were shown by the red dashed lines (distance < 3.5 Å). (D) The deaminase activity assays for the WT and mutants as detected by TLC. (E) The close-up of the zinc-binding site of D295N, bound by the substrate. The closest distances between the N6-methyl group and Val57 or Phe58 were indicated by the red lines and distances were shown (units in Å).
Table 2.
Kinetic constants for AtMAPDA and mutants
| Mutant | K M (μM) | k cat (s−1) | k cat/KM (M−1 s−1) | Relative kcat/KM |
|---|---|---|---|---|
| WT | 4.14 | 0.86 | 0.21 | 1.0 |
| H15A | ND | ND | ND | ND |
| N17A | 7.12 | 1.01 | 0.14 | 0.69 |
| H65A | 5.51 | 0.69 | 0.13 | 0.61 |
| T97A | 8.94 | 0.62 | 0.07 | 0.34 |
| T98A | 4.52 | 0.57 | 0.13 | 0.62 |
| K100A | 6.89 | 0.49 | 0.07 | 0.35 |
| E220A | ND | ND | ND | ND |
| D295A | ND | ND | ND | ND |
| D295N | ND | ND | ND | ND |
| D296A | ND | ND | ND | ND |
ND: not determined.
Interestingly, in contrast to the clear density shown by the N6m-AMP ligand, the zinc density was almost invisible. The planar purine ring of the substrate replaces one of the water molecules (Wcor) and also pushes the catalytic water (Wcat) out of position (Figure 2E). Additionally, the mutated Asn295 residue would not be an appropriate zinc ligand either, because zinc usually prefers histidines, aspartates and cysteines. Therefore, with only three ligands remaining in their positions, the zinc ion became lost due to the insufficient coordination ligands. Although the D295N protein apparently lost its ability to deaminate, it still maintained its intact three-dimensional structure.
The structures of AtMAPDA in complex with the product
Next, we solved the structure of WT AtMAPDA enzyme complexed with IMP in order to investigate the final state of the enzymatic pathway. The product-enzyme structure barely had any changes from that of the initial substrate-enzyme complex, with an RMSD value of 0.20 Å over 336 Cα atoms. The 1.85-Å map indicated that in the active site, O6 does not contact any residues (Figures 3A and Supplementary Figure S1B). The positions and orientation of the pivotal residues are almost identical before and after the reaction, except for residue 295 (aspartate/asparagine) and His240. Side chains of both residues flipped small angles, most likely due to the formation of a hydrogen bond with the catalytic water (data not shown). The water molecule for catalysis (Wcat) is now back in position, probably preparing for the next round of attack since the enzyme is ready for release of the product (Figure 3B). In contrast, Wcor is still missing due to the occupation of its position by the IMP product. Therefore the zinc ion now is coordinated by five ligands and its coordination geometry is trigonal bipyramid. Of note, difference map after the refinement showed weak negative density around the metal, suggesting the partial engagement of the site by zinc, as opposed to the normal density shown by WT in the apo form. In the finished model, the zinc occupancy was refined to 93%. It displayed a temperature factor (26 Å2) comparable to that of the protein chain (32 Å2). Other than that, structure comparison showed that there were minimal changes in the enzyme structure, but we could not rule out the possibility that the enzyme still makes any structural rearrangements as needed for the enzymatic reaction to happen (Figure 3C).
Figure 3.
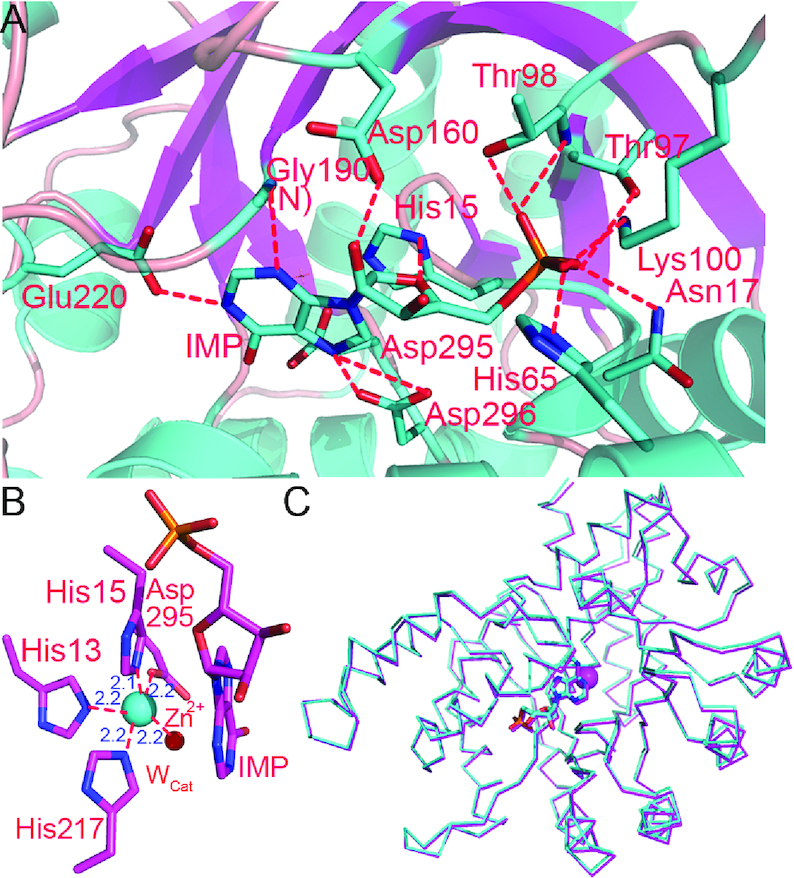
The structure of the enzyme bound by the product. (A) The interaction network of IMP with AtMAPDA. (B) The close-up of the trigonal bipyramidal coordination state of zinc. The distances of the ligands from the metal were shown (units in Å). (C) The structural superimposition of the enzyme-N6m-AMP complex (PDB 6IJN, cyan) and the enzyme-IMP complex (PDB 6IJP, magenta).
Structure comparison to homologs
Dali search for structurally close proteins (50) resulted in similarity to several proteins, all being annotated adenosine deaminases, and the top two are the mouse (MmADA, PDB 2ADA) (37) and murine adenosine deaminases respectively (MuADA, PDB 3MVI) (42). MmADA could be aligned to AtMAPDA with an RMSD of 1.83 Å over 306 Cα atoms, suggesting relatively large variations between the two structures, which may explain the poor solution initially obtained by molecular replacement. By overlaying these structures, we found while the barrel-shaped deaminase domain was preserved among these proteins, large variations were mainly found at four locations flanking the disordered regions at the N-termini: Asp22-Ser53 (I), Val55-Leu67 (II), Lys100-Ile105 (III) and Arg121-Thr132 (IV) (Figure 4A). Especially the two loop regions Lys35-Phe40 and Lys100-Ile105 are substantially shorter, with much simpler connections to the following helices compared to MmADA. Besides the conservation of the Tim-barrel fold, the binding modes of the adenine moieties and the zinc-binding sites are retained as well, suggesting a shared catalytic mechanism between MAPDA and ADA (Figure 4B). However, there are structural variations at Val57-Phe58, which are important for N6-methyl interactions. These two residues are substituted by a phenylalanine (Phe61) and leucine (Leu62) respectively in MmADA. In the overlaid MmADA structure, the methyl group is inappropriately close to the side chain of Phe61 (< 2.1 Å). In addition, our modeling studies as well as that by Chen et al. (36) suggested that in the intermediate stage of MAPDA catalysis where the tetrahedral adduct is formed, the steric hindrance posed by the methyl group might become more severe (Figure 5A). We wonder whether these observations constitute the structural basis that ADA barely catalyzes the deamination of N6m-AMP. Following this idea, we mutated Phe61-Leu62 in MmADA to Val-Phe in order to make more room at the methyl-binding subsite. However, our attempts to switch the substrate specificity was not successful, as our mutant still retained strong activity toward adenosine but displayed no activity toward N6-methyl-2′-deoxyadenosine (data not shown). Therefore, there are more complicated reasons for ADA’s behavior, which require more investigation. Other yet-to-be-identified regions or factors may further contribute to the recognition of the substrate. While inspecting the structures of ADAs, we found that the open and closed conformations reported for ADA were similar to that of AtMAPDA in its ligand-free and ligand-bound forms respectively. Apo bovine ADA adopts the open form while it becomes closed when in complexes with the substrate analogs (41). This process involves the movement of Leu55 and Leu59 towards Glu183 (the fixed point) by as much as 3.0 Å and structural readjustment of the Thr54-Pro67 helix (α3), induced by ligand binding and the removal of a key water coordinated to Asp293 (Figures 5B and C) (PDBs 1VFL and 1KRM) (40,41). The corresponding moving helix in AtMAPDA is α4, along with α3, and it undergoes even larger conformational changes (rotation of a larger angle of ∼10° with Ala70 being fixed) upon binding of the substrate or release of the product: the equivalent residues Leu54, Phe58 move closer to Pro192 (the corresponding residue to Glu183 in bovine ADA), with their closest distances shortened by 6.2 and 4.4 Å respectively (Figures 5D and E). A similar rotation of α3 is also observed. More importantly, these local structural changes also require the dislodgment of the water molecule coordinated to Asp296, a result by the binding of the ligands. In comparison, the rotation in bovine ADA is much smaller and only involves the α3 helix, while no significant structural changes were observed for its preceding helix α2.
Figure 4.
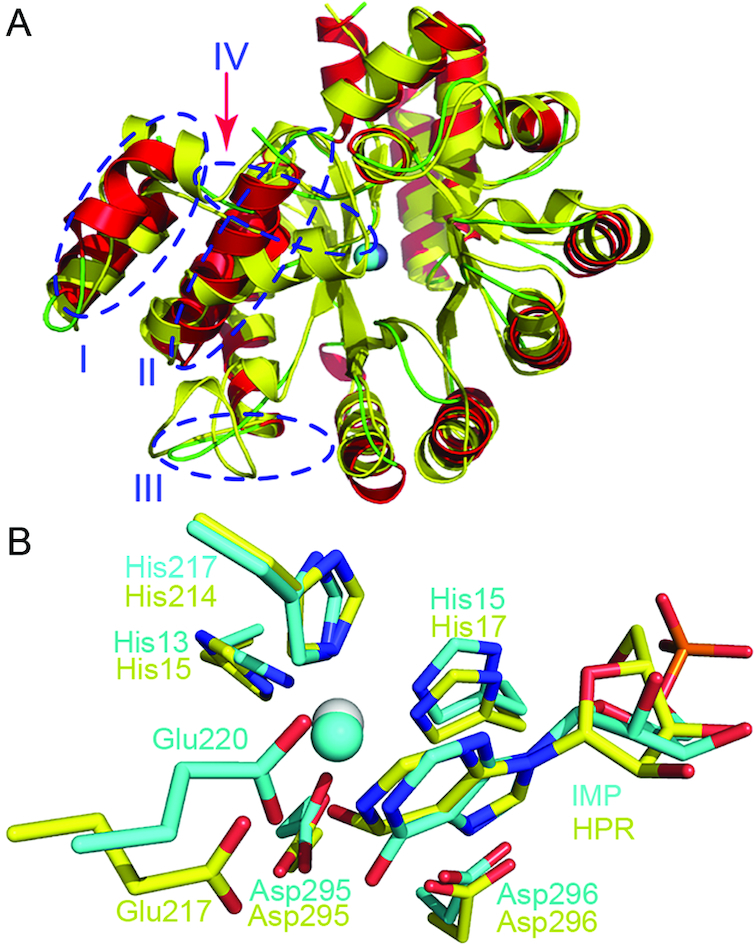
Structure comparison to homologs. (A) The structural superposition of apo-AtMAPDA (PDB 6IJM, color scheme as in Figure 1A) with that of MmADA (PDB 2ADA, yellow). The four regions displaying substantial structural differences were indicated by the blue circles. (B) The shared binding mode between the AtMAPDA-IMP complex (PDB 6IJP, cyan) and the MmADA-HPR complex (yellow, PDB 2ADA). The key residues for the adenine and zinc binding were shown as sticks and the zinc ions were represented as spheres. HPR: 6-hydroxy-7,8-dihydro purine nucleoside.
Figure 5.

The modeled tetrahedral intermediate state and observed alternative conformations for ADA and AtMAPDA. (A) Cartoon showing the possible steric hindrance (shown by arrows) of the hypothetical tetrahedral adduct with Phe61 of ADA (lower panel) and accommodation of the tetrahedral adduct by AtMAPDA (upper panel). (B) The switch of distinct conformational states of bovine ADA (in Cα traces) before and after the binding of the ligand (PDB 1VFL, blue and PDB 1KRM, orange). (C) The close-up of the movements of the local area surrounding the α3 helix (boxed area in (B)) in bovine ADA. (D) AtMAPDA upon the binding of the substrate (PDBs 6IJM, color scheme as in Figures 1A and 6IJN, cyan). The key residues were shown as sticks and their movements were indicated by the arrows. (E) The close-up of the movements of the local area surrounding the α3 and α4 helices (boxed area in (D)) in AtMAPDA.
Model for catalytic pathway
We thus obtained three crystal structures from AtMAPDA, each representing a different stage of the catalytic pathway: the free enzyme, enzyme bound by the substrate, and enzyme bound by the product. The conversion between individual states is accompanied by conformational changes between two distinct states. According to our structural and biochemical analyses, we propose that the MAPDA pathway should proceed as follows, which can be divided into five stages and is well supported by our high-resolution crystal structures. (a) In the apo state, the zinc ion adopts a hexa-coordination state by employing Wcat, Wcor as the fifth and sixth ligands. The zinc geometry is octahedral and the enzyme adopts an open conformation. (b) The substrate binds and induces the conformational transformation to a closed one. This process pushes away Wcor and the zinc metal adopts a trigonal bipyramidal geometry. (c) Asp295 activates Wcat to attack C6 of the substrate and a tetrahedral intermediate forms. (d) The methylamine molecule leaves and the chemical reaction completes. Wcat rebinds and restores the trigonal bipyramidal geometry. (e) IMP is released before Wcor rebinds to reproduce the octahedral geometry. The enzyme goes back to the open state (Figure 6). In stages (c) and (d) of the catalytic pathway, the enzyme barely changes conformations and they are presumably fast processes, whereas the substrate-binding and product-releasing stages (stages (b) and (e)) require large conformational changes, and the processes probably will be slow. Previously proposed mechanisms on ADAs mostly focused on a single aspect of the pathways such as static structural information, conformational transformation, or coordination states etc. occurring in the deamination process. These mechanisms were considered to consist of two major steps involving an addition-elimination type with the direct addition of water on C6 of the purine ring, which subsequently leads to the formation of a tetrahedral intermediate. Our model on the MAPDA enzyme contains more details of the reaction including distinct conformational states, changes, binding and dissociation kinetics as well as the coordination states of the zinc ion, and is consequently more comprehensive with finer steps. In addition, the zinc ion of AtMAPDA mainly adopts a five or six coordination state, in contrast to the four or five coordination states reported on many ADAs, and the purpose of the extra water molecule (Wcor) remains obscure. Lastly, AtMAPDA undergoes even larger conformational changes than that of ADAs during the chemical reaction, which makes it interesting and unusual. Nevertheless, the complete elucidation of the catalytic process awaits more structural models and biochemical characterization. For example, the enzyme bound by an inhibitor mimicking the transition state in combination of solution dynamic characterization will provide a clearer picture of the deamination pathway.
Figure 6.

Proposed catalytic pathway for AtMAPDA. (A) In the apo state, the zinc ion adopts a hexa-coordination state with an open conformation. (B) The substrate binds and induces the conformational transformation to a closed one and a trigonal bipyramidal geometry accordingly. (C) Asp295 activates Wcat for the SN2 reaction and a tetrahedral intermediate forms. (D) The methylamine leaves to finish the reaction and trigonal bipyramidal geometry is reproduced. (E) AMP is released and the enzyme goes back to the open state and the octahedral geometry. The open states were placed to the left of the dashed line whereas the closed states were to the right.
DISCUSSION
In this study, we solved a series of high-resolution structures of AtMAPDA, the first structures of its kind. All three structures reported here display an unstructured region between Ala133 and Gly147, which is not present in ADAs or other MAPDA orthologs (Supplementary Figure S4). The significance of the 15-residue disorder to the enzyme function is unclear, but it appears to play a non-important role, as suggested by its non-conservation among the species.
Our cocrystal structures and biochemical studies not only reveal the substrate recognition mechanism of AtMAPDA, but also provide insight into its catalytic pathway. We first discovered that di- or tri-phosphate adenosines are not suitable substrates due to restrained space at the phosphate-binding subsite, while non-phosphorylated adenosine lacks enough contacts with the enzyme. Secondly, recognition of both rings of purines excludes pyrimidines as substrates. Additionally, HsMAPDA can accommodate modest-sized lipophilic N6-substitutions, which may establish non-polar contacts with residues Val57 and Phe58. In fact, O6-methylguanosine monophosphate is a more effective substrate than N6m-AMP (34). In this aspect, HsMAPDA acts as a demethylase for O6m-GMP. Furthermore, we attempted to switch substrate specificity of ADA to that of AtMAPDA by structure-guided mutations but found simple mutations would not do the ‘trick’. There must be more features yet to be uncovered about this intriguing enzyme. The MAPDA enzymes appear to require the terminal phosphate group for enhanced binding of substrate, whereas ADAs are inactive toward phosphorylated adenosine derivatives. Although higher eukaryotes also have AMP deaminase (AMPD) that carry out the deamination of AMP to IMP, but AMPDs are considerably larger than MAPDAs or ADAs. Additionally, the structures of these enzymes are substantially different, as are their substrate binding modes (PDB 2A3L) (51).
In terms of substrate recognition mechanism, we concluded that the enzyme needs to undergo relative large conformational changes upon the binding of the substrate or at the finish of the catalysis, but it less likely changes conformation during the chemical reaction from the substrate-enzyme complex (ES) to the product-enzyme complex stage (EP). These observations suggested that binding or release of the substrate might be the rate-determining step, while the chemical reaction was a fast step once the ES complex was formed. This deduction was in agreement with the large k2 and k–1 but small k1 values reported on calf ADA (52), which also experiences large conformational changes during substrate binding. Although the two types of enzymes are phylogenetically distinct (33), they share considerable similarities in structure and conformational dynamics as revealed in this study. More importantly, HsMAPDA is a potential pharmacological target for the activation of nucleoside/nucleotide analogs currently approved or in clinical trials. The valuable research experience accumulated from the comprehensive studies on the ADA enzyme during the past provides a frame of reference for future research on MAPDA-based drug design.
DATA AVAILABILITY
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with the accession codes 6IJM, 6IJN and 6IJP.
Supplementary Material
ACKNOWLEDGEMENTS
We thank from BL19U1 beamlines (National Center for Protein Sciences Shanghai (NCPSS)) at Shanghai Synchrotron Radiation Facility, for assistance during data collection.
Author contributions: Wei Xie conceived and designed research; Qian Jia performed research; Wei Xie analyzed data and wrote the paper. All authors reviewed the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31870782, 31170117, 31700657]; Natural Science Foundation of Guangdong province [2018A030313313]. Funding for open access charge: National Natural Science Foundation of China [31870782].
Conflict of interest statement. None declared.
REFERENCES
- 1. Redelman D., Bluestein H.G., Cohen A.H., Depper J.M., Wormsley S.. Deoxyadenosine (AdR) inhibition of newly activated lymphocytes: blockade at the G0-G1 interface. J. Immunol. 1984; 132:2030–2038. [PubMed] [Google Scholar]
- 2. Arpaia E., Benveniste P., Cristofano A.D., Gu Y., Dalal I., Kelly S., Hershfield M., Pandolfi P.P., Roifman C.M., Cohen A.. Mitochondrial basis for immune deficiency: evidence from purine nucleoside phosphorylase-deficient mice. J. Exp. Med. 2000; 191:2197–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohen A., Gudas L.J., Ammann A.J., Staal G.E., Martin D.W. Jr. Deoxyguanosine triphosphate as a possible toxic metabolite in the immunodeficiency associated with purine nucleoside phosphorylase deficiency. J. Clin. Invest. 1978; 61:1405–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shimoyama R.K., Seto S., Mori C.. Protection by various deoxynucleosides against deoxyadenosine-induced DNA damage in adenosine deaminase-inactivated lymphocytes. Mol. Genet. Metab. 1999; 68:455–460. [DOI] [PubMed] [Google Scholar]
- 5. Corriden R., Chen Y., Inoue Y., Beldi G., Robson S.C., Insel P.A., Junger W.G.. Ecto-nucleoside triphosphate diphosphohydrolase 1 (E-NTPDase1/CD39) regulates neutrophil chemotaxis by hydrolyzing released ATP to adenosine. J. Biol. Chem. 2008; 283:28480–28486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zimmermann H. 5′-Nucleotidase: molecular structure and functional aspects. Biochem. J. 1992; 285:345–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taniguchi A. Purine nucleoside phosphorylase (PNP). Nippon Rinsho. 2003; 61(Suppl. 1):85–90. [PubMed] [Google Scholar]
- 8. Alunni S., Orrù M., Ottavi L.. A study on the inhibition of adenosine deaminase. J. Enzym. Inhib. Med. Chem. 2008; 23:182–189. [DOI] [PubMed] [Google Scholar]
- 9. Lambertucci C. Adenosine deaminase: functional implications and different classes of inhibitors. Med. Res. Rev. 2015; 21:105–128. [DOI] [PubMed] [Google Scholar]
- 10. Somech R., Lai Y.H., Grunebaum E., Saux N.L., Cutz E., Roifman C.M.. Polyethylene glycol-modified adenosine deaminase improved lung disease but not liverdisease in partial adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2009; 124:848–850. [DOI] [PubMed] [Google Scholar]
- 11. Aiuti A., Cattaneo F., Galimberti S., Benninghoff U., Cassani B., Callegaro L., Scaramuzza S., Andolfi G., Mirolo M., Brigida I.. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009; 360:447–458. [DOI] [PubMed] [Google Scholar]
- 12. Sauer A.V., Brigida I., Carriglio N., Aiuti A.. Autoimmune dysregulation and purine metabolism in adenosine deaminase deficiency. Front. Immunol. 2012; 3:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. French J. B., Ealick S. E.. Biochemical and structural characterization of a ureidoglycine aminotransferase in the klebsiella pneumoniae uric acid catabolic pathway. Biochemistry. 2010; 49:5975–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramazzina I., Costa R., Cendron L., Berni R., Peracchi A., Zanotti G., Percudani R.. An aminotransferase branch point connects purine catabolism to amino acid recycling. Nat. Chem. Biol. 2010; 6:801–806. [DOI] [PubMed] [Google Scholar]
- 15. Werner A. K., Witte C. P.. The biochemistry of nitrogen mobilization: purine ring catabolism. Trends Plant Sci. 2011; 16:381–387. [DOI] [PubMed] [Google Scholar]
- 16. Desrosiers R., Friderici K., Rottman F.. Identification of methylated nucleosides in messenger RNA from novikoff hepatoma cells. Proc. Natl. Acad. Sci. U.S.A. 1974; 71:3971–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Furuichi Y., Morgan M., Shatkin A.J., Jelinek W., Salditt-Georgieff M., Darnell J.E.. Methylated, blocked 5′ termini in HeLa cell mRNA. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:1904–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adams J.M., Cory S.. Modified nucleosides and bizarre 5′-termini in mouse myeloma mRNA. Nature. 1975; 255:28–33. [DOI] [PubMed] [Google Scholar]
- 19. Wei C.M., Gershowitz A., Moss B.. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell. 1975; 4:379–386. [DOI] [PubMed] [Google Scholar]
- 20. Wei C.M., Moss B.. Methylated nucleotides block 5′-terminus of vaccinia virus messenger RNA. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krug R.M., Morgan M.A., Shatkin A.J.. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J. Virol. 1976; 20:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rottman F.M., Desrosiers R.C., Friderici K.. Nucleotide methylation patterns in eukaryotic mRNA. Prog. Nucleic. Acid. Res. 1977; 19:21–38. [DOI] [PubMed] [Google Scholar]
- 23. Bokar J. A., Shambaugh M. E., Polayes D., Matera A. G., Rottman F. M.. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997; 3:1233–1247. [PMC free article] [PubMed] [Google Scholar]
- 24. Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Tomas L., Pan T., Yang Y., He C.. N 6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011; 7:885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M. et al.. Topology of the human and mouse m6A RNA methylomes revealed by m6a-seq. Nature. 2012; 485:201–206. [DOI] [PubMed] [Google Scholar]
- 26. Zhao X., Yang Y., Sun B.F., Shi Y., Yang X., Xiao W., Hao Y.J., Ping X.L., Chen Y.S., Wang W.J.. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014; 24:1403–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geula S., Moshitch-moshkovitz S., Dan D., Mansour A.F., Kol N., Salmon-divon M., Hershkovitz V., Peer E., Mor N., Manor Y.S.. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015; 347:1002–1006. [DOI] [PubMed] [Google Scholar]
- 28. Liu N., Pan T.. RNA epigenetics. Transl. Res. 2015; 165:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klungland A., Dahl J.A.. Dynamic RNA modifications in disease. Curr. Opin. Genet. Dev. 2014; 26:47–52. [DOI] [PubMed] [Google Scholar]
- 30. Blanco S., Frye M.. Role of RNA methyltransferases in tissue renewal and pathology. Curr. Opin. Cell Biol. 2014; 31:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dina C., Meyre D., Gallina S., Durand E., Körner Antje, Jacobson P. et al.. Variation in FTO contributes to childhood obesity and severe adult obesity. Nature Genet. 2007; 39:724–726. [DOI] [PubMed] [Google Scholar]
- 32. Kaklamani V., Yi N., Sadim M., Siziopikou K., Zhang K., Xu Y., Tofilon S., Agarwal S., Pasche B., Mantzoros C.. The role of the fat mass and obesity associated gene (FTO) in breast cancer risk. BMC Med. Genet. 2011; 12:1471–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maier S.A., Galellis J.R., McDermid H.E.. Phylogenetic analysis reveals a novel protein family closely related to adenosine deaminase. J. Mol. Evol. 2005; 61:776–794. [DOI] [PubMed] [Google Scholar]
- 34. Schinkmanová M., Votruba I., Holý A.. N 6-methyl-AMP aminohydrolase activates N6-substituted purine acyclic nucleoside phosphonates. Biochem. Pharmacol. 2006; 71:1370–1376. [DOI] [PubMed] [Google Scholar]
- 35. Murakami E., Bao H., Mosley R.T., Du J., Sofia M.J., Furman P.A.. Adenosine deaminase-like protein 1 (ADAL1): characterization and substrate specificity in the hydrolysis of N6- or O6-substituted purine or 2-aminopurine nucleoside monophosphates. J. Med. Chem. 2011; 54:5902–5914. [DOI] [PubMed] [Google Scholar]
- 36. Chen M., Urs M.J., Sánchezgonzález I., Olayioye M.A., Herde M., Witte C.P.. m6A RNA degradation products are catabolized by an evolutionarily conserved N6-methyl-AMP deaminase in plant and mammalian cells. Plant Cell. 2018; 30:1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilson D.K., Rudolph F.B., Quiocho F.A.. Erratum: atomic structure of adenosine deaminase complexed with a transition-state analog: understanding catalysis and Iimmunodeficiency mutations. Science. 1991; 253:119–119. [DOI] [PubMed] [Google Scholar]
- 38. Wilson D.K., Quiocho F.A.. A pre-transition-state mimic of an enzyme: X-ray structure of adenosine deaminase with bound 1-deazaadenosine and zinc-activated water. Biochemistry. 1993; 32:1689–1694. [DOI] [PubMed] [Google Scholar]
- 39. Wang Z., Quiocho F.A.. Complexes of adenosine deaminase with two potent inhibitors: X-ray structures in four independent molecules at pH of maximum activity. Biochemistry. 1998; 37:8314–8324. [DOI] [PubMed] [Google Scholar]
- 40. Kinoshita T., Nishio N., Nakanishi I., Sato A., Fujii T.. Structure of bovine adenosine deaminase complexed with 6-hydroxy-1,6-dihydropurine riboside. Acta. Crystallogr. D. Biol. Crystallogr. 2003; 59:299–303. [DOI] [PubMed] [Google Scholar]
- 41. Kinoshita T., Nakanishi I., Terasaka T., Kuno M., Seki N., Warizaya M., Matsumura H., Inoue T., Takano K., Adachi H. et al.. Structural basis of compound recognition by adenosine deaminase. Biochemistry. 2005; 44:10562–10569. [DOI] [PubMed] [Google Scholar]
- 42. Niu W., Shu Q., Chen Z., Mathews S., Di Cera E., Frieden C.. The role of Zn2+ on the structure and stability of murine adenosine deaminase. J. Phys. Chem. B. 2010; 114:16156–16165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang Z., Nygaard P., Chinault A.C., Kellems R.E.. Deduced amino acid sequence of Escherichia coli adenosine deaminase reveals evolutionarily conserved amino acid residues: implications for catalytic function. Biochemistry. 1991; 30:2273–2280. [DOI] [PubMed] [Google Scholar]
- 44. Ribard C., Rochet M., Labedan B., Daignanfornier B., Alzari P., Scazzocchio C., Oestreicher N.. Sub-families of alpha/beta barrel enzymes: a new adenine deaminase family. J. Mol. Biol. 2003; 334:1117–1131. [DOI] [PubMed] [Google Scholar]
- 45. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 46. Mccoy A.J., Grossekunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grossekunstleve R.W.. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta. Crystallogr. D. Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all‐atom structure validation for macromolecular crystallography. Acta. Crystallogr. D. Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Holm L., Sander C.. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 1995; 20:478–480. [DOI] [PubMed] [Google Scholar]
- 51. Han B.W., Bingman C.A., Mahnke D.K., Bannen R.M., Bednarek S.Y., Sabina R.L., Phillips G.N. Jr. Membrane association, mechanism of action, and structure of Arabidopsis embryonic factor 1 (FAC1). J. Biol. Chem. 2006; 281:14939–14947. [DOI] [PubMed] [Google Scholar]
- 52. Porter D.J., Spector T.. Alternative substrates for calf intestinal adenosine deaminase. A pre-steady-state kinetic analysis. J. Biol. Chem. 1993; 268:2480–2485. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with the accession codes 6IJM, 6IJN and 6IJP.